Back to Journals » Infection and Drug Resistance » Volume 10
Effect of a small molecule Lipid II binder on bacterial cell wall stress
Authors Malin J ![]() , Shetty AC, Daugherty SC, de Leeuw EPH
, Shetty AC, Daugherty SC, de Leeuw EPH
Received 1 November 2016
Accepted for publication 20 December 2016
Published 28 February 2017 Volume 2017:10 Pages 69—73
DOI https://doi.org/10.2147/IDR.S126254
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Suresh Antony
Jakob Malin,1,2 Amol C Shetty,3 Sean Daugherty,3 Erik PH de Leeuw,1,2
1Institute of Human Virology, 2Department of Biochemistry and Molecular Biology, 3Institute for Genome Sciences, University of Maryland Baltimore School of Medicine, Baltimore, MD, USA
Abstract: We have recently identified small molecule compounds that act as binders of Lipid II, an essential precursor of bacterial cell wall biosynthesis. Lipid II comprised a hydrophilic head group that includes a peptidoglycan subunit composed of N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc) coupled to a short pentapeptide moiety. This headgroup is coupled to a long bactoprenol chain via a pyrophosphate group. Here, we report on the cell wall activity relationship of dimethyl-3-methyl(phenyl)amino-ethenylcyclohexylidene-propenyl-3-ethyl-1,3-benzothiazolium iodide (compound 5107930) obtained by functional and genetic analyses. Our results indicate that compounds bind to Lipid II and cause specific upregulation of the vancomycin-resistance associated gene vraX. vraX is implicated in the cell wall stress stimulon that confers glycopeptide resistance. Our small molecule Lipid II inhibitor retained activity against strains of Staphylococcus aureus mutated in genes encoding the cell wall stress stimulon. This suggests the feasibility of developing this new scaffold as a therapeutic agent in view of increasing glycopeptide resistance.
Keywords: defensin, Lipid II, antibiotics, bacterial membrane, vancomycin
Introduction
Bacteria, and in particular Gram-positive species, surround themselves with cell walls, structures that are essential for survival and growth.1 Interference with the biosynthesis of the cell wall has been a successful antibacterial strategy, and numerous classes of antibiotics have been developed that affect assembly. Penicillin and vancomycin are arguably the two most well-known examples of such therapeutics. Penicillin represents the class of β-lactam agents that interfere with cross-linking of peptide stems of peptidoglycan units outside of the bacterial cell.2 Vancomycin specifically binds to the l-Ala–l-Ala motif of the pentapeptide moiety of Lipid II, an essential precursor of peptidoglycan transport and assembly and acts at the membrane interface. Resistance against agents such as next-generation β-lactams and last-resort antibiotics including vancomycin is evolving at an alarming rate.3 Thus, there is a great need for alternative strategies to combat multidrug-resistant organisms.
We recently identified an interaction between a class of antimicrobial peptides in humans, termed defensins, and bacterial Lipid II.4 Based on these initial findings, we further characterized this interaction in molecular detail and identified small molecule Lipid II inhibitors for the first time.5 In this report, we further characterize one small molecule Lipid II inhibitor, termed compound 5107930. Compound 5107930 was examined for its effects on bacterial cell wall stress, binding to Lipid II and antibacterial killing in the context of glycopeptide resistance.
Materials and methods
Materials
Unless stated otherwise, chemicals were obtained from Sigma–Aldrich (St. Louis, MO, USA). Staphylococcus aureus ATCC 29213 was obtained from Microbiologics (St. Cloud, MN, USA). Dimethyl-3-methyl(phenyl)amino-ethenylcyclohexylidene-propenyl-3-ethyl-1,3-benzothiazolium iodide was purchased from Chembridge Ltd. (San Diego, CA, USA). S. aureus strains carrying deletions in the cell wall stress stimulon (CWSS) were generously provided by M. Senn and C. Qublier (Institute for Medicinal Microbiology, University of Zurich) and were described earlier.6
Antibacterial activity assay
Determination of the minimal inhibitory concentrations (MICs) by dilution was carried out by broth dilution according to CLSI standards.7
Lipid II purification
Short-chain water-soluble Lipid II containing a lipid tail of three isoprene units was generated and purified essentially as described.8
Surface plasmon resonance
Binding experiments were carried out on a BIAcore T100 system (BIAcore, Inc., Piscataway, NY, USA) at 25°C. The assay buffer was 10 mM 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid, 150 mM NaCl, 0.05% surfactant P20, and pH 7.4 (±3 mM EDTA). 3-Lipid II (50 response units) was immobilized on CM5 sensor chips using the amine-coupling chemistry recommended by the manufacturer.
RNA isolation
Staphylococcus aureus strain 29213 was exposed to compound 5107930 for 30 min at 10 µM or left untreated. Total mRNA was subsequently isolated using the RNEasy mini kit (Qiagen; Germantown, MD, USA) according to manufacturer’s instructions.
Sequencing
The mRNA samples were sequenced using the HiSeq platform (Illumina; San Diego, CA, USA) for the four S. aureus samples (one control replicate and three treated replicates) in order to generate 101 bp reads. These paired end reads were populated into two separate FASTQ format files, and the quality of the reads was tested using the FastQC toolkit to ensure quality of the sequencing reads. The reads were trimmed based on the base quality score if necessary.9
Read alignment using bowtie
The sequenced reads were used as input to Bowtie version 0.12.7 to align the reads to the S. aureus 29213 reference genome for each sample.10 The output from Bowtie was obtained as binary alignment map (BAM) format files that consist of information on the alignment of each individual read within the reference genome. In the alignment phase, we allowed two mismatches over the entire read alignment. The BAM alignment files obtained from the Bowtie alignment tool were analyzed to generate the alignment statistics for each sample, namely, the total number of reads, the number of mapped reads, the percent of total mapped reads, and the percentage of reads that mapped to genic and intergenic regions. The coverage analysis aided in determining the success of the sequencing process, the coverage across the reference genome, and the presence of any outliers in the samples.
Differential expression analysis
The alignment BAM files from Bowtie are further utilized to compute gene expression levels and test each gene for differential expression.11 The reference annotation for S. aureus 29213 in GFF3 format was used. The number of reads that mapped to each gene described in the annotation was calculated using the python package HTSeq. The read count represents the expression of the gene. Differential gene expression analysis was conducted using the DESeq R package. The DESeq analysis resulted in the determination of potential differentially expressed genes (DEGs) when compared between the control samples and the treated samples. The read counts for each sample were normalized for sequencing depth and distortion caused by highly DEGs. Then, the negative binomial (NB) model was used to test the significance of differential expression between the control and the treated conditions. The DEGs were deemed significant if the normalized gene expression value was greater than the 10th percentile in either of the conditions and showed >1.5× fold-change difference (upregulated or downregulated) between the conditions.
Results and discussion
In our initial study, we found that compound 5107930 interacted with Lipid II and selectively killed Gram-positive organisms, most notably S. aureus.5 We confirmed the binding of our compound to Lipid II by surface plasmon resonance and found that it binds to Lipid II with a 2 µM binding affinity (Figure 1). Binding to Lipid II and a preferential killing of Gram-positive organisms by this compound could indicate targeting of the bacterial cell wall. To further investigate this possibility, we examined the mRNA expression profile of S. aureus exposed to compound 5107930. Analysis of DEGs with a fold-change cut-off at 1.5× (i.e., absolute (Log2 fold change)≥0.58) revealed a list of 490 genes that were upregulated or downregulated compared to control expression (Figures 2 and 3; Table S1). Downregulated expression was predominantly located to genes involved in protein translation, including ribosomal proteins L6, L18, L24 and S18, and translation elongation factor P. Upregulated gene expression was primarily located to bacteriophage S13 loci as well as the stress response protein GrpE. Most notably, compared to control, exposure to compound 5107930 for 30 min resulted in highly significant (a p value of 2.6×10−14) and robust (Log2 fold change of 3.55) overexpression of the vraX gene (Table S1).
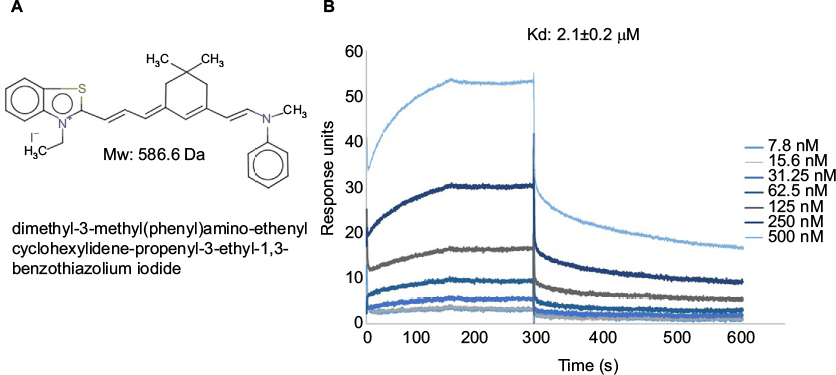 | Figure 1 (A) Chemical structure, molecular weight and nomenclature of the 5107930 compound. (B) Binding kinetics of 5107930 to Lipid II as determined by surface plasmon resonance. Abbreviations: Kd, binding affinity; Mw, molecular weight. |
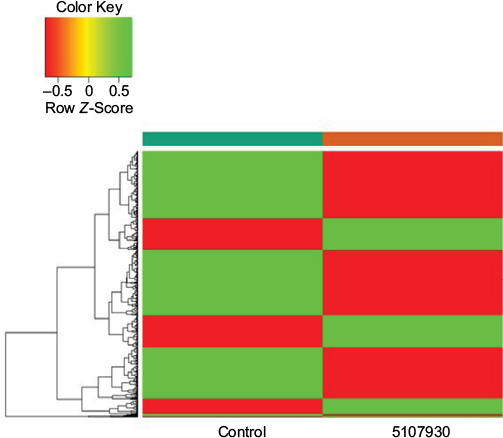 | Figure 2 Heatmap of the 146 upregulated (green) genes and 344 downregulated (red) genes identified to be differentially expressed between the 5107930-treated sample and the control sample. |
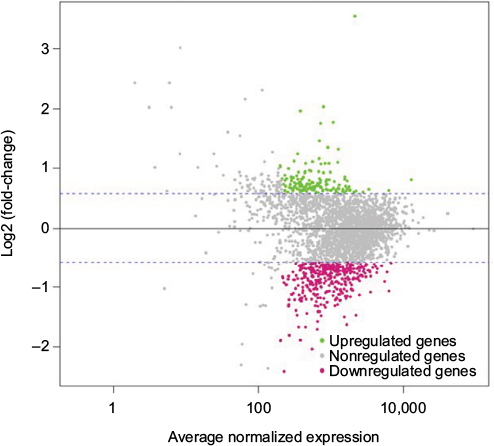 | Figure 3 M–A plot illustrating the average normalized expression values (x-axis) for each gene compared to its Log2 (fold-change) between the 5107930-treated sample and the control sample. Note: Upregulated genes are highlighted in green while downregulated genes are highlighted in red. Abbreviations: A, mean average; M, log ratio. |
The vraX gene encodes a 55-aa protein with a putative signal sequence, suggesting that the protein is exported.12 Overexpression of Vrax has been reported upon exposure to agents that act against the bacterial cell wall, including antimicrobial peptides, bacitracin, fosfomycin, vancomycin, daptomycin and teicoplanin.13–15 Expression of Vrax is regulated by vancomycin-resistance associated regulator (VraR), which together with vancomycin-resistance associated sensor (VraS), orf1 and yvqF, forms a four-component CWSS.6 Both VraR and VraS are overexpressed upon exposure to vancomycin, and overexpression results in decreased vancomycin susceptibility.16,17 Deletion of vraR and vraS was shown to result in increased susceptibility to vancomycin.18 In addition, VraRS contributes to decreased susceptibility to daptomycin in clinical isolates.19 To test if the VraRS regulon is involved in susceptibility to compound 5107930, we determined the MICs against S. aureus strains deleted for each individual gene of the stimulon (Table 1). We find that compound MIC against strains deleted for vraR, vraS, orf1, or yvqF remained unaltered, whereas the MIC of vancomycin varied slightly, but not significantly. Our findings are in agreement with previous findings that showed little effect on vancomycin sensitivity upon CWSS deletion.6 This study and our findings differ from results showing increased susceptibility to vancomycin upon deletion of vraR and vraS.18 A possible explanation for these observed differences could be that the strains we examined have the methicillin-resistant background.6 In conclusion, we show that compound 5107930 is a small Lipid II binder with cell wall targeting specificity. Importantly, we did not observe overexpression of the vraR or vraS genes upon exposure to this compound as reported for many other antibacterial agents. Compound 5107930 thus represents a new chemical scaffold for potential optimization as an antibacterial agent that acts against strains with decreased vancomycin or daptomycin susceptibility due to changes in the cell wall stress regulon.
 | Table 1 Minimal inhibitory concentration (µg/mL) of compound 5107930 and vancomycin against Staphylococcus aureus strains with cell wall stress stimulon deletions |
Acknowledgments
The authors gratefully acknowledge Drs Maria Senn and Chantal Qublier of the Institute for Medicinal Chemistry, University of Zurich, Switzerland, for providing the bacterial strains carrying mutations in the cell wall stress stimulon. This work was supported by National Institutes of Health grant AI092033 and a University of Maryland Ventures grant to EPHdL.
Disclosure
The authors report the following competing interests: US patent number 8,796,323 and US nonprovisional patent application number 13/911,234. The authors report no other conflicts of interest in this work.
References
Vollmer W, Blanot D, de Pedro M.A. Peptidoglycan structure and architecture. FEMS Microbiol Rev. 2008;32(2):149–167. | ||
Bush K, Bradford PA. Beta-lactams and beta-lactamase inhibitors: an overview. Cold Spring Harb Perspect Med. 2016;6(8):a025247. | ||
Munita JM, Bayer AS, Arias CA. Evolving resistance among gram-positive pathogens. Clin Infect Dis. 2015;61(Suppl 2):S48–S57. | ||
de Leeuw E, Li C, Zeng P, et al. Functional interaction of human neutrophil peptide-1 with the cell wall precursor lipid II. FEBS Lett. 2010;584(8):1543–1548. | ||
Varney KM, Bonvin AM, Pazgier M, et al. Turning defense into offense: defensin mimetics as novel antibiotics targeting lipid II. PLoS Pathog. 2013;9(11):e1003732. | ||
McCallum N, Meier PS, Heusser R, Berger-Bächi B. Mutational analyses of open reading frames within the vraSR operon and their roles in the cell wall stress response of Staphylococcus aureus. Antimicrob Agents Chemother. 2011;55(4):1391–1402. | ||
CLSI. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically. Approved standard. 8th ed. Wayne, PA: Clinical and Laboratory Standards Institute; 2009. | ||
Breukink E, van Heusden HE, Vollmerhaus PJ, et al. Lipid II is an intrinsic component of the pore induced by nisin in bacterial membranes. J Biol Chem. 2003;278(22):19898–19903. | ||
Anders S, Pyl PT, Huber W. HTSeq – a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2):166–169. | ||
Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. | ||
Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11(10):R106. | ||
Scherl A, François P, Charbonnier Y, et al. Exploring glycopeptide-resistance in Staphylococcus aureus: a combined proteomics and transcriptomics approach for the identification of resistance-related markers. BMC Genomics. 2006;7:296. | ||
Dengler V, Meier PS, Heusser R, Berger-Bächi B, McCallum N. Induction kinetics of the Staphylococcus aureus cell wall stress stimulon in response to different cell wall active antibiotics. BMC Microbiol. 2011;11:16. | ||
Pietiainen M, François P, Hyyryläinen HL, et al. Transcriptome analysis of the responses of Staphylococcus aureus to antimicrobial peptides and characterization of the roles of vraDE and vraSR in antimicrobial resistance. BMC Genomics. 2009;10:429. | ||
Overton IM, Graham S, Gould KA, et al. Global network analysis of drug tolerance, mode of action and virulence in methicillin-resistant S. aureus. BMC Syst Biol. 2011;5:68. | ||
Kuroda MK, Kuwahara-Arai K, Hiramatsu K. Identification of the up- and down-regulated genes in vancomycin-resistant Staphylococcus aureus strains Mu3 and Mu50 by cDNA differential hybridization method. Biochem Biophys Res Commun. 2000;269(2):485–490. | ||
Kuroda M, Kuroda H, Oshima T, Takeuchi F, Mori H, Hiramatsu K. Two-component system VraSR positively modulates the regulation of cell-wall biosynthesis pathway in Staphylococcus aureus. Mol Microbiol. 2003;49(3):807–821. | ||
Chen H, Xiong Z, Liu K, et al. Transcriptional profiling of the two-component regulatory system VraSR in Staphylococcus aureus with low-level vancomycin resistance. Int J Antimicrob Agents. 2016;47(5):362–367. | ||
Mehta S, Cuirolo AX, Plata KB, et al. VraSR two-component regulatory system contributes to mprF-mediated decreased susceptibility to daptomycin in in vivo-selected clinical strains of methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2012;56(1):92–102. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.