Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 10 » Issue 1
Effect of a single nucleotide polymorphism in miR-146a on COX-2 protein expression and lung function in smokers with chronic obstructive pulmonary disease
Authors Wang R, Li M, Zhou S, Zeng D, Xu X, Xu R, Sun G
Received 15 September 2014
Accepted for publication 22 October 2014
Published 4 March 2015 Volume 2015:10(1) Pages 463—473
DOI https://doi.org/10.2147/COPD.S74345
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Prof. Dr. Richard Russell
Ran Wang,1,* Min Li,2,* Sijing Zhou,1,3 Daxiong Zeng,4 Xuan Xu,5 Rui Xu,1 Gengyun Sun1
1Department of Respiratory Medicine, The First Affiliated Hospital of Anhui Medical University, Hefei, People’s Republic of China; 2Department of Oncology, The First Affiliated Hospital of Anhui Medical University, Hefei, People’s Republic of China; 3Hefei Prevention and Treatment Center for Occupational Diseases, Hefei, People’s Republic of China; 4Department of Respiratory Medicine, The First Affiliated Hospital of Soochow University, Suzhou, People’s Republic of China; 5Division of Pulmonary/Critical Care Medicine, Cedars Sinai Medical Center, Los Angeles, CA, USA
*These authors contributed equally to this work
Objective: To evaluate the effect of a single nucleotide polymorphism (rs2910164) in the miR-146a precursor on the expression level of miR-146a, cyclooxygenase-2 (COX2), and production of prostaglandin E2 (PGE2) in lung tissue harvested from smokers with chronic obstructive pulmonary disease, as well as the lung function and disease stages from the same patient population.
Methods and results: One-hundred and sixty-eight smokers with diagnosed chronic obstructive pulmonary disease were recruited. The patients were genotyped for rs2910164 polymorphism using Sanger sequencing, and their lung function/disease stages were evaluated following Global Initiative for Chronic Obstructive Lung Disease (GOLD) criteria. Meanwhile, messenger ribonucleic acid and protein expression levels of miR-146a and COX2 as well as PGE2 production were determined in 66 lung tissue samples collected in the patients who received surgical treatment. We confirmed that COX2 is a validated target of miR-146a in human fibroblast cells, and identified the differential expression patterns of miR-146a and COX2 in each rs2910164 genotype group. We observed a significant association between rs2910164 in miR-146a and the levels of either COX2 or PGE2 using real-time polymerase chain reaction and Western blot. Consistently, we were able to demonstrate that the rs2910164 single nucleotide polymorphism has a functional effect on the baseline lung function in the study population.
Conclusion: In the present study, the rs2910164 CC and GC genotype was found to be associated with an improved lung function and milder disease stages, at least partially, mediated by its ability to increase in COX2 expression and PGE2 production.
Keywords: SNP, miR-146a, COX-2, COPD, lung function
Introduction
Chronic obstructive pulmonary disease (COPD) is the fourth leading cause of death worldwide.1 Cigarette smoking is the top risk factor, and smoking cessation in the early stage of the disease may reverse or slow the course and loss of lung function.2 COPD is characterized by a persistent reactive inflammatory response of the lung, even after cessation of smoking,3–6 and this inflammation has been reported to play an important role in the development and progression of COPD.1,7 The histological damages observed in COPD result not only from environmental stimuli, but also from the persistent uncontrolled inflammatory reaction to the stimuli, and abnormally repaired tissue structure.1,7,8
Cyclooxygenase-2 (COX2) is an enzyme that acts to speed up the production of prostaglandins, especially prostaglandin E2 (PGE2). Lovgren et al9 reported that COX2−/− mice presented exacerbated fibrosis in conjunction with severe loss of pulmonary function in response to environmental stimuli. Another study demonstrated that administration of the PGE2 analog, 16,16-dimethyl PGE2, protected against bleomycin-induced lung inflammation and fibrosis in mice.10 The beneficial effect of PGE2 in the lung is also supported by the results obtained from other experimental models of pulmonary fibrosis and inflammation. For example, PGE2 has been reported to suppress fibroblast cells’ growth and collagen synthesis in airway epithelium, and inhibit TGF−/− induced fibroblast to myofibroblast transition.11,12 PGE2 also inhibits LPS-induced NF-κB activation in monocytes, thereby suppressing TNF-α production while increasing the anti-inflammatory cytokine, interleukin (IL)-10.13–15 Consistently, multiple studies have shown that PGE2 is involved in regulating lymphocyte trafficking and differentiation in a variety of models.16,17 In this context, Sato et al demonstrated that micro ribonucleic acid (MiRNA)-146a expression was inhibited, resulting in an enhanced expression of COX2 and increased production of PGE2 in the fibroblast cells harvested from smokers with COPD,18 and it has been further confirmed by another report, showing the expression of miR-146a was down-regulated by activated inflammatory signaling pathway in smokers.19
MiRNAs are an abundant class of ~22-nucleotide-long non-protein coding RNAs, which regulate up to a third of all protein-coding genes by binding to the 3′ untranslated region (UTR) of target gene messenger RNA (mRNA), leading to translational repression and/or mRNA degradation.20 MiRNAs have been reported to be involved in the regulation of various biological processes such as cell cycle, differentiation, inflammation, and damage repair.20 A growing body of evidence indicated that variants in miRNA sequence may influence disease susceptibility by changing miRNA expression, maturation or miRNA–miRNA interaction.21,22 A polymorphism (rs2910164) in the miR-146a precursor has been reported to be associated with a variety of malignancies including breast or ovarian cancer, papillary thyroid cancer, hepatocellular cancer, esophageal squamous cell cancer, gastric cancer, and prostate cancer.23–28 Jazdewski et al23 showed that the C allele of rs2910164 may cause a reduction of mature miR-146a and less inhibition of its target genes such as tumor necrosis receptor-associated factor 6 (TRAF6) and IL-1 receptor-associated kinase 1 (IRAK1) by interfering with the processing and maturing of pre-MiRNA.
Based on the above evidence, we hypothesized that rs2910164 polymorphism may affect the production of PGE2, lung function, and disease severity by altering the expression levels of miR-146a as well as its target, COX2, in COPD patients. To test it, we evaluated the differentially inhibitory effect of miR-146a on expression of COX2 and PGE2 respectively in lung tissues harvested from COPD smokers. Simultaneously, we also performed genotyping analyses for miR-146a rs2910164 polymorphism and evaluated their associations with COX2 expression, PGE2 production, and lung function.
Materials and methods
Patients
One-hundred and sixty-eight smokers with diagnosed COPD were recruited in our hospitals from May 2013 to December 2013. Among them, resected specimens were available in 66 patients, who had received surgery for lung tumor resection except for eight of the subjects with COPD undergoing volume reduction surgery. COPD is a lung disease characterized by chronic obstruction of lung airflow that interferes with normal breathing and is not fully reversible (Defined by World Health Organization). The disease was staged with the following criteria: classification of severity of airflow limitation in COPD. Global Initiative for Chronic Obstructive Lung Disease (GOLD)1 (mild): forced expiratory volume in 1 second (FEV1) >≧80% predicted; GOLD2 (moderate): 50%≤ FEV1<80% predicted; GOLD3 (severe): 30%≤ FEV1<50% predicted; GOLD4 (very severe): FEV1 <30% predicted. In case of lung cancer, tissue samples were taken at a site more than 2 cm away from carcinoma. Each tissue specimen was fixed in 4% paraformaldehyde. After dehydration, it was embedded in paraffin and selected for 5 μm thick serial sectioning. Sections were deparaffnized, immunostained, and observed under a light microscope. Four milliliters of peripheral blood was obtained from all participants. Human lung fibroblasts were cultured as described29,30 from normal-appearing areas of the pulmonary parenchyma in a region as far as possible from the tumor (at least 2 cm) that was free of pleura or large airways. All participants were ethnic Han Chinese people. The study was approved by investigational review committees at Anhui Medical University. Written informed consent for research was obtained from each participant. Demographic data and information on known and potential risk factors were collected through interviewer administered questionnaires.
Measurement of PGE2
PGE2 production from lung tissues was determined by enzyme immunoassay (Jimian, Shanghai, People’s Republic of China) following the manufacturer’s instructions.
Real-time polymerase chain reaction (PCR)
Total RNA was isolated by Trizol one-step RNA isolation kit. The miR-146a, COX2 and U6 cDNA were synthesized from total RNA according to the TaqMan real-time assays protocol (Applied Biosystems, Foster City, CA, USA). Relative quantification of the target was determined by the delta delta cycle threshold (△△Ct) method. Each sample was examined in triplicate and the raw data were presented as the relative quantity of the target, normalized by U6.
Luciferase assay
The COX2 wild-type 3′UTR were PCR amplified and cloned into a modified version of pcDNA3.1(+) that contained a firefly luciferase reporter gene, at a position downstream of the luciferase reporter. The vectors were named wild-type 3′UTRs of COX2. The following primer set was used to subclone the 3′UTR of COX2. 5′-CGGGATCCAAGTCTAATGATCATATTTATTTA-3′ and 5′-ATAAGAATGCGGCCGCTCTTCAGAAAAGATCTGTCAA-3′.
Site-directed mutagenesis of the miRNAs’ binding sites in the 3′UTRs was performed using Site-Directed Mutagenesis Kit (SBS Genetech, Beijing, People’s Republic of China) and named as mutant 3′UTRs. The cells grown in a 48-well plate were co-transfected with 400 ng of either miR-146a, 40 ng of the firefly luciferase reporter plasmid including the 3′UTR of COX2, and 4 ng of pRL-TK, a plasmid expressing rellina luciferase (Promega Corporation, Fitchburg, WI, USA). After 24 h, the cells were collected, and the luciferase reporter assay was performed in TD-20/20 luminometer (Turner Biosystems, Sunnyvale, CA, USA).
miR-146 mimics and inhibitors transfection
The miR-146a mimics, inhibitors and negative control were synthesized by RIBOBIO (Ribobio Co. Ltd, Guangzhou, People’s Republic of China). The sequences for the miR-146a mimics, inhibitor and negative control were described as follows: miR-146a mimics (5′-UGAGAACUGAAUUCCAUGGGUU-3′), miR-146a inhibitor (5′-AACCCAUGGAAUUCAGUUCUCA-3′), negative control (5′-UUGUACUACACAAAAGUACUG-3′). For transfection, cells were seeded into plates, incubated overnight, and then transfected using Lipofectamine 2000 transfection reagent according to the manufacturer’s instructions at 60%–70% cell confluence.
Genotyping of miR-146a rs2910164 single nucleotide polymorphism (SNP)
Four milliliters of peripheral venous blood collected from all participants, and genomic DNA samples for genotyping were isolated using DNA extraction kit (Shunhua Bioengineer [Shanghai] Co Ltd, People’s Republic of China) before being stored at −20°C for future use. DNA specimens were PCR amplified using miR-146a specific primer: 5′-ATTTTACAGGGCTGGGACAG-3′ and 5′-TCTTCCAAGCTCTTCAGCAG-3′. The PCR products were purified with ExoSAP-IT purification kit (USB, London, UK) and subsequently sent to the core facility for sequencing.
Lung function evaluation
The spirometry test is performed to measure the volume of air forcibly exhaled from the point of maximal inspiration (forced vital capacity, FVC), and the volume of air exhaled during the first second of this maneuver (FEV1) using a device called a spirometer. Generally, the patient is asked to take the deepest breath as they possibly can, and then exhale into the spirometer as hard as possible, for as long as possible, at least 6 seconds.
Gas diffusion capacity test is performed by having the subjects blow out all of the air (as much as possible). The subjects then inhale a test gas mixture rapidly and completely. The test gas is held in the lungs for about 10 seconds, and then the subjects exhale. All of the tests were performed prior to the treatment with bronchodilators.
Cell culture and oligonucleotides transfection
Human fibroblast cells were harvested from the resected lung tissues (genotyped as homogenous wild-type for rs2910164), and cultured as previously described.29,30 Human pulmonary smooth muscle cells were purchased from the Shanghai Cell Bank of the Chinese Academy of Sciences (Shanghai, People’s Republic of China). MiR-146a mimics and inhibitors were purchased from Ribobio, and the Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) was used to transfect the oligonucleotides into the fibroblast cells and pulmonary smooth muscle cells.
Western blot analysis
The protein concentration of the cell lysates was quantitatively determined before the lysates were loaded onto 10% SDS-polyacrylamide gel, and the proteins were next transferred to a PVDF membrane. The membrane was then incubated with primary antibodies and horseradish peroxidase-conjugated secondary antibody according to manufacturer’s instruction, and all the antibodies were purchased from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). The signals were detected using ECL chemofluorescent detection kit (Pierce, Rockford, IL, USA). The band density of the target protein was densitometrically analyzed after normalization with the density of β-actin.
Statistical analysis
Data are expressed as mean ± standard deviation. Analyses were performed using the software SPSS (version 19; IBM Corporation, Armonk, NY, USA), and a P-value <0.05 was considered significant. Analysis of variance (ANOVA) was performed with the use of one-way ANOVA. When applicable, the Mann–Whitney U-test was used for comparisons between groups. For the primary outcomes including PGE2 production, COX2 expression, and miR-146a expression, all subjects whose tissue samples were available were evaluated. The chi-square test was used to compare the frequency distribution of sex, and GOLD stages. Analysis of co-variance or logistic regression analysis was used to control for the potential confounding factors in estimating the effect of miR-146a polymorphisms on the lung function and disease stages in the presence of other known prognostic factors, including age, sex, and smoking status.
Results
A total of 168 smokers with COPD (111 males and 57 females) were recruited in this study. Information on the known risk factors of COPD such as age, height, weight, and smoking status, and lung function such as FVC, FEV1, and diffusion capacity for carbon monoxide (DLCO), stratified by miR-146a rs2910164 genotype, are described in Table 1. The COPD patients were classified as stage I to IV (mild to very severe COPD) according to GOLD classification. The genotype frequency of the SNP in the study population was not compatible with Hardy-Weinberg equilibrium. As this was a case-only cross-sectional study, no normal control was enrolled, but the recruited COPD patients showed expected physiologic alterations including significantly lower FEV1 and lower DLCO.
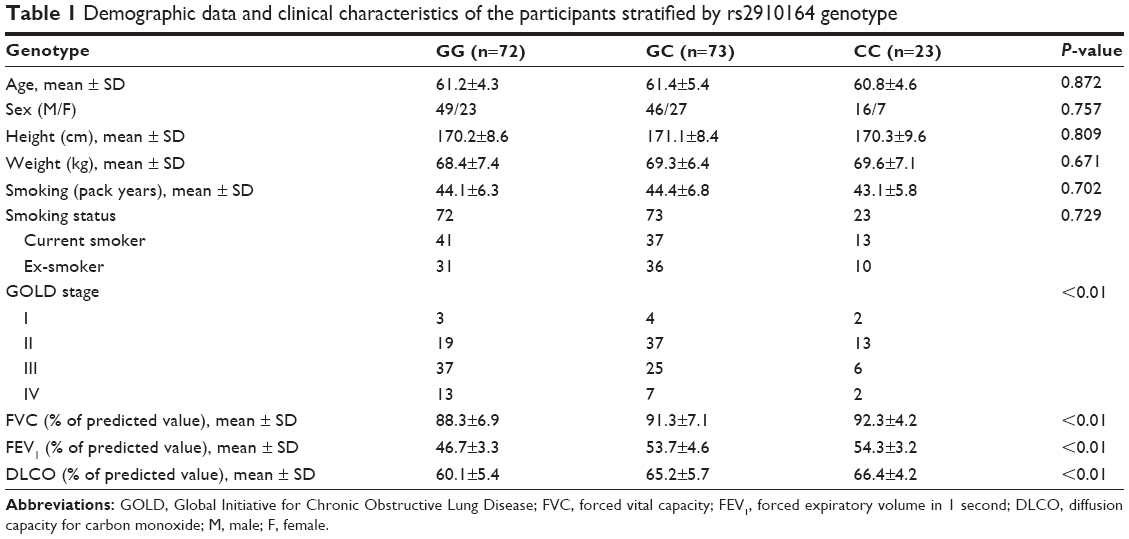 | Table 1 Demographic data and clinical characteristics of the participants stratified by rs2910164 genotype |
Lung tissues were harvested from 66 participants (rs2910164 genotype, GG:28, GC:26, CC:12) out of a total of 168 patients, and the expression of miR-146a and rs2910164 genotype were determined in those samples. Using real-time PCR, we found that the expression levels of miR-146a were comparable between the participants carrying rs2910164 CC and GC genotype groups, both of which had a significantly decreased expression of miR-146a compared with GG genotype group. The rs2910164 C allele showed a dominant influence on the expression of miR-146a in our lung tissue samples, as shown in Figure 1A.
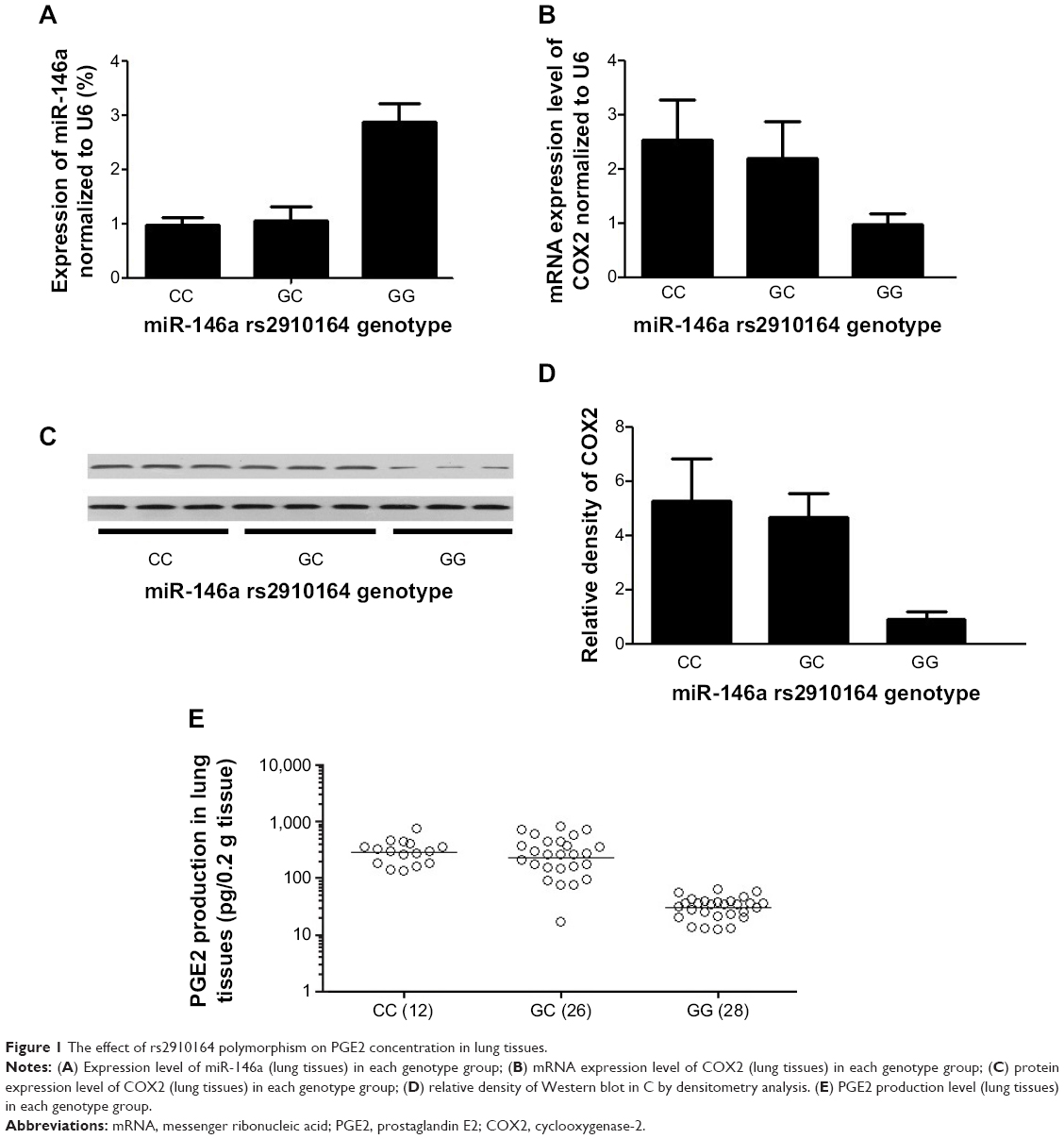 | Figure 1 The effect of rs2910164 polymorphism on PGE2 concentration in lung tissues. |
Furthermore, based on computational screening (www.mirdb.org) and previous reports, COX2 was identified as a virtual target gene of miR-146a (Figure 2A), which was next confirmed using luciferase assay. Luciferase activity in the cells co-transfected with miR-146a mimics and vector containing wild-type 3′UTR of COX2 was substantially lower than the control, while the luciferase activity in the cells transfected with miR-146a mimics and vector containing mutant 3′UTR of COX2 showed no difference compared with the control, as shown in Figure 2B. We examined the expression patterns of COX2, and PGE2 in the lung tissues collected from the patients, and found that, consistent with the pattern of miR-146a, significantly higher levels of COX2 mRNA/protein expression and PGE2 were noted in individuals with CC or GC genotype than GG, as shown in Figure 1B–D. Meanwhile, using immunohistochemical staining, we found that COX2 was universally distributed in lung tissues and the protein expression level of COX2 was significantly higher in CC/GC than GG genotype group, as shown in Figure 3.
 | Figure 2 Luciferase assay with wild-type and mutant COX2 3’UTR. |
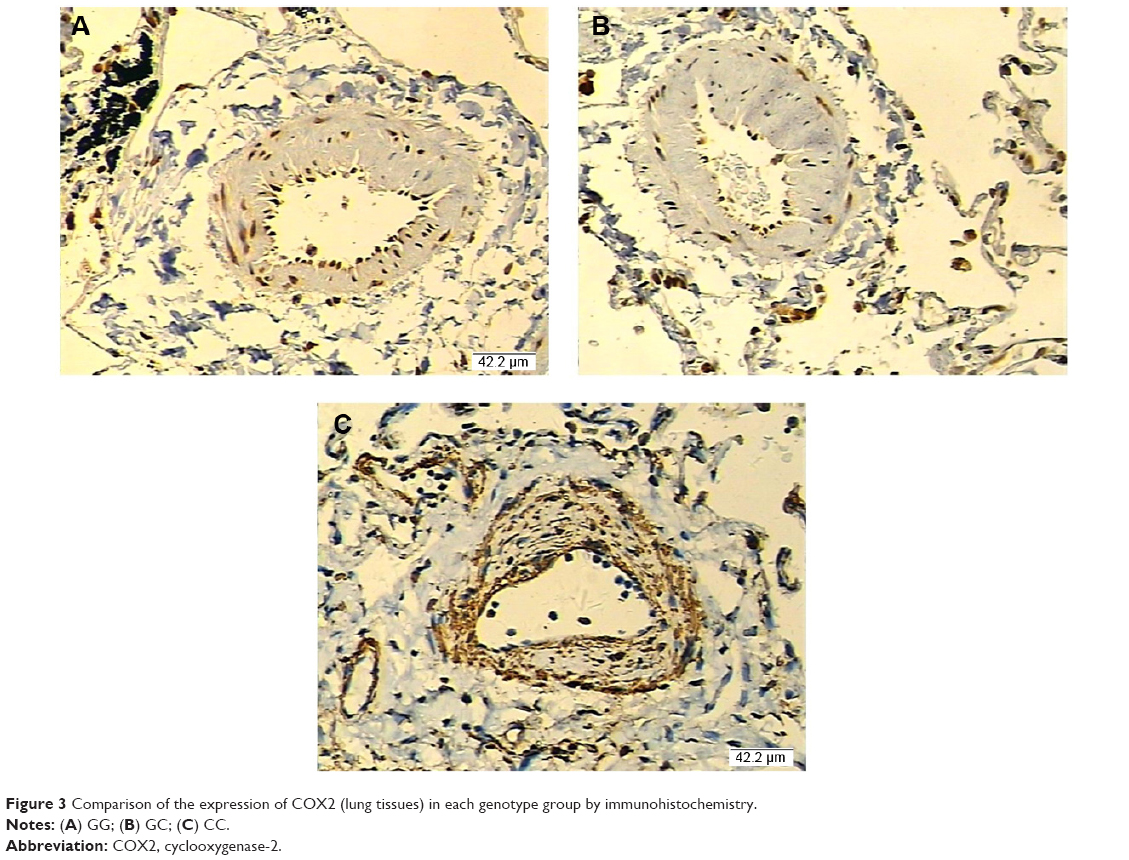 | Figure 3 Comparison of the expression of COX2 (lung tissues) in each genotype group by immunohistochemistry. |
To further confirm the regulatory role of miR-146a in the control of COX2 expression, we performed gain-of-the-function analysis in the human lung fibroblast cells by transfecting the cells with miR-146a mimics. As shown in Figure 4A, the expression level of miR-146a was significantly up-regulated in the cells transfected with the mimics. Meanwhile, we examined the expression level of COX2 in lung fibroblast cells and smooth muscle cells by using real-time PCR and Western blot, and found that up-regulation of miR-146a, caused by transfection of the mimics, substantially suppressed both mRNA and protein expression levels of COX2 in both lung fibroblast cells and pulmonary smooth muscle cells (Figure 4B–D, and Figure 6A–C). In addition to the gain-of-the-function, we conducted loss-of-the-function analysis as well. We showed that transfection of miR-146a inhibitors markedly suppressed the expression level of the miR (Figure 5A), and dramatically promoted the mRNA and protein expression of COX2 in the human lung fibroblast cells and pulmonary smooth muscle cells, as shown in Figure 5B–D, and Figure 6D–F.
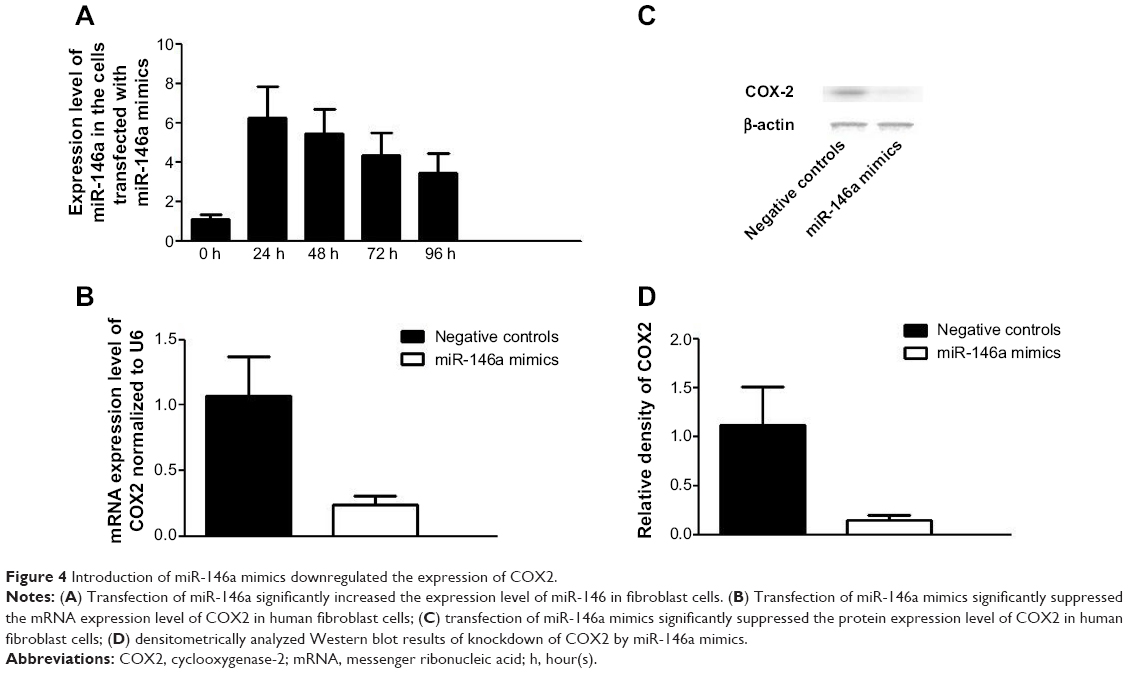 | Figure 4 Introduction of miR-146a mimics downregulated the expression of COX2. |
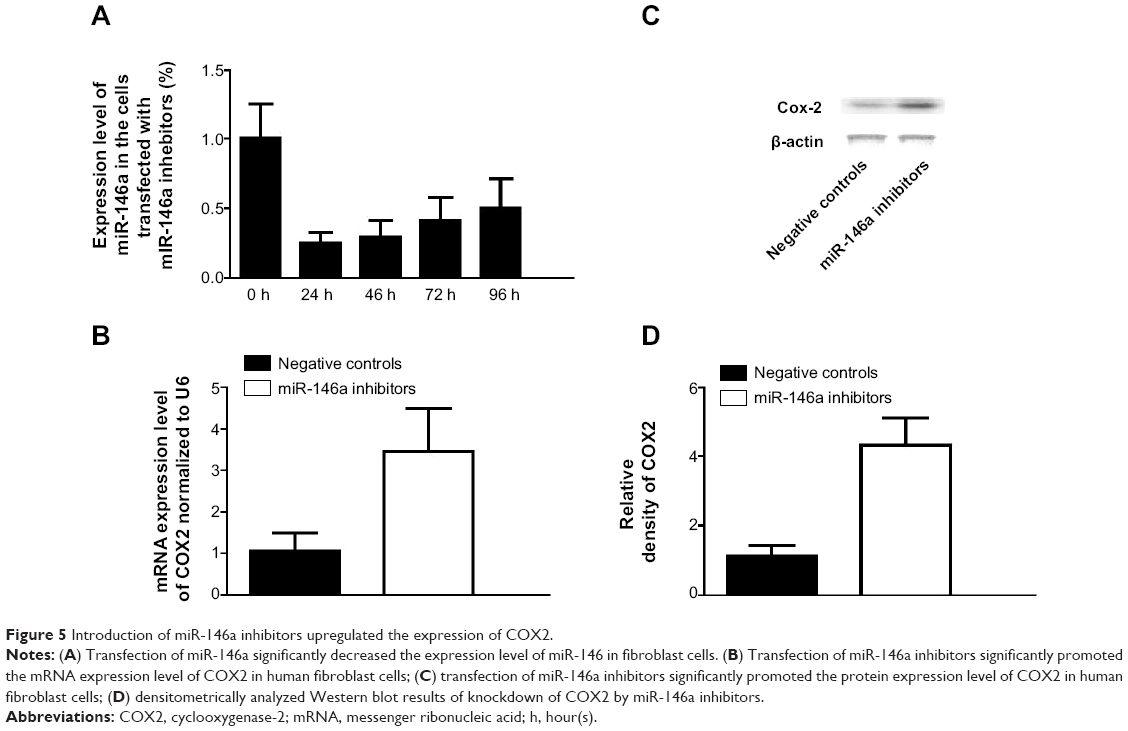 | Figure 5 Introduction of miR-146a inhibitors upregulated the expression of COX2. |
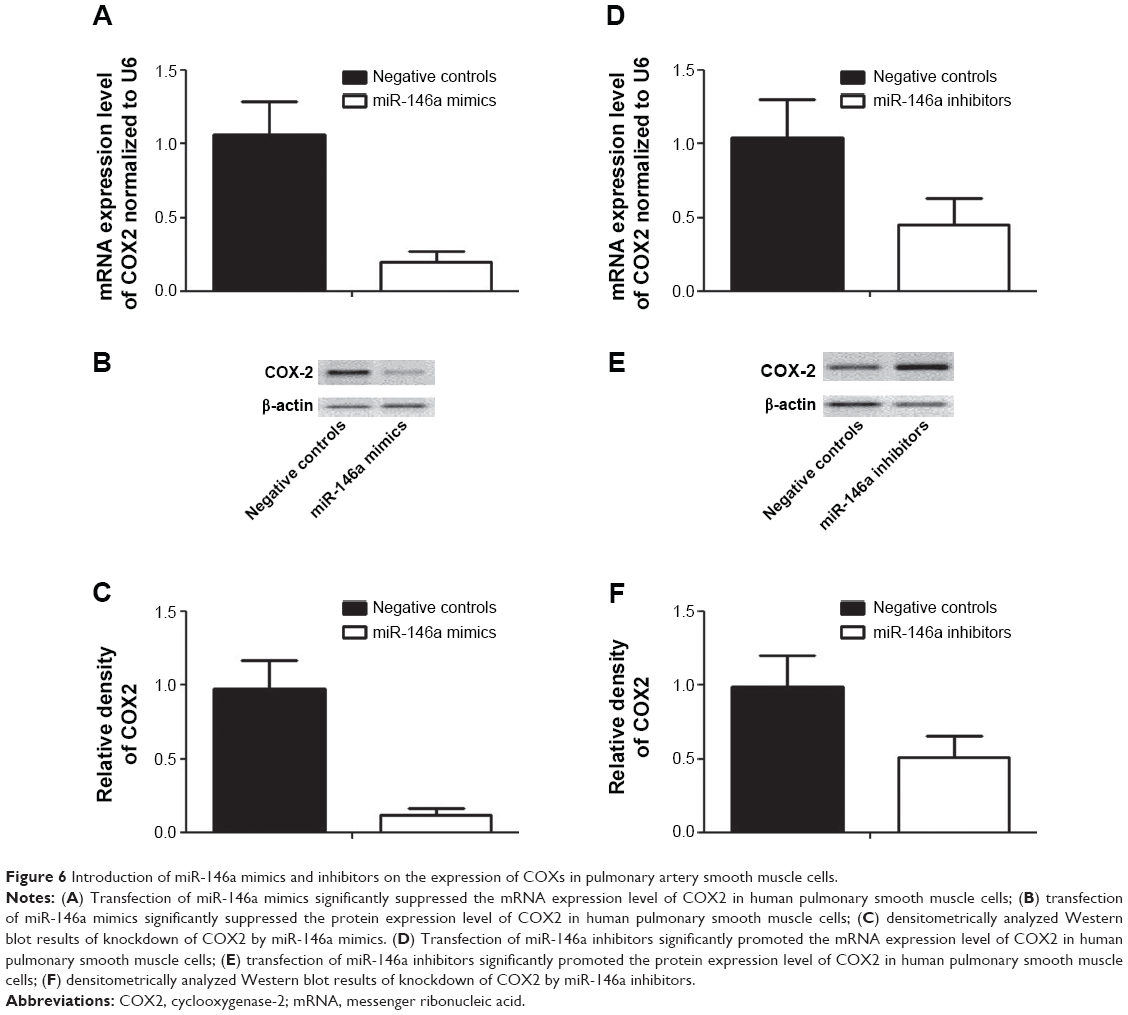 | Figure 6 Introduction of miR-146a mimics and inhibitors on the expression of COXs in pulmonary artery smooth muscle cells. |
Additionally, we measured the lung function of all recruited 168 COPD patients including FEV1, FVC, and DLCO, and showed that, in line with the distribution of COX2 and PGE2, no difference was identified between CC and GC genotype groups regarding FVC, FEV1, and DLCO, whereas FVC, FEV1, and DLCO in CC and GC groups were significantly higher than in GG group (Table 1). Furthermore, the COPD patients were classified as stage I to IV (mild to very severe COPD) according to GOLD classification, and we observed a significant association between the disease stage and rs2910164 genotype, ie, more mild cases (stage I and II) were noted in the COPD patients with CC/GC genotype compared with GG subjects (Table 1).
Discussion
In this study we investigated an SNP in miR-146a genes in relation to cross-sectional lung function in patients with COPD. We further confirmed that COX2 is a validated target of miR-146a in human fibroblast cells, and identified the differential expression patterns of miR-146a and COX2 in each rs2910164 genotype group. In addition, we observed a significant association with rs2910164 in miR-146a with the levels of either COX2 or PGE2 by using real-time PCR and Western blot. Consistently, we were able to demonstrate that the rs2910164 SNP had a functional effect on the baseline lung function in the study population.
Long-term cigarette smoking is still a major global health issue, causing approximately 5–6 million deaths every year worldwide.31 Cigarette smoke is composed of a complex mixture of chemicals including carcinogens and reactive oxygen species, that may cause inflammation by sparkling the relevant signaling pathway, mediated by reactive increase in numerous pulmonary and systemic inflammatory factors, such as IL-6, IL-8, TNF-α, and COX-2. COX2 is an immediate/early protective gene in response to various stimuli, and has been reported to be elevated in patients with COPD,32,33 the leading cause of death amongst smokers. COX2 is responsible for the production of PGE2 in vivo, and it has been reported that fibroblasts from patients with COPD produced more PGE2 than fibroblasts from the normal controls.29 Subsequently the report from the same research group demonstrated that enhanced expression of COX2/PGE2 resulted from the stimulation by pro-inflammatory mediators such as IL-1β and TNF-α, which was believed to be mediated by corresponding alternation of miR-146a in the fibroblast cells harvested from the smokers with COPD,18 and it has been further confirmed by another report, showing the expression of miR-146a was subject to the regulation of inflammatory signaling pathway.19 Several miRs, including miR-16,34 miR-101,35,36 miR-146a,18,19 and miR-199a*,36 have been indicated to be involved in the control of production of PGE2, but the results were quite conflicting. Meanwhile, miRs were recently reported to be involved in the pathogenesis of COPD.37,38 In the present study, we showed a direct effect of miR-146a on COX2 expression level, by verifying the inhibitory influence using luciferase assay and differential expression patterns of COX2 and miR-146a in human lung tissues.
A growing body of evidence showed that miRNAs, serving as key regulators, participated in various important biological processes such differentiation, proliferation, tissue repair, and fibrosis.39–41 Variants in miRNAs have been consistently shown to be associated with a wide spectrum of human diseases.42–44 Jazdzewski et al demonstrated that the C allele of rs2910164 SNP in the pre-miR-146a significantly compromised the processing and maturing of the MiRNA, leading to an approximately two-fold reduction of mature miR-146a compared with the G allele in thyroid cell line.23 The C allele of the SNP, located in a position about 60 bases away from the first nucleotide on the passenger strand of pre-miR-146a, was computationally predicted to cause mispairing within the mature hairpin. Whereas, we examined the mRNA expression level of miR-146a in the 66 patients, whose lung tissue samples were available, and found pre-miR-146a expression level in those who carried rs2910164 CC genotype was comparable with GC, and both CC and GC genotype groups were approximately three times lower than GG carriers (P<0.001), such discrepancy could be attributed, at least in part, to the distinct genetic background of the study population as well as different tissue types.
The anti-inflammatory and anti-fibrotic effects of PGE2 in lungs have been repeatedly reported, and it is logical that inhibited inflammation and fibrosis would contribute to the improved FEV1 and DLCO. A recent animal study showed that mice lacking COX2 had significantly more severe lung dysfunction in response to harmful stimuli compared with the controls.45 In line with this, some other studies presented exacerbated lung fibrosis in COX-2−/− mice in response to various stimuli.9,46 In this study, we measured the lung function of the COPD patients including FEV1, FVC, and DLCO, and showed that, in line with the distribution of COX2 and PGE2, FVC, FEV1, and DLCO were significantly higher in CC/GC than in GG group (Table 1). The COPD patients were classified as stage I to IV (mild to very severe COPD) according to GOLD classification, and we observed a significant association between the disease stage and rs2910164 genotype, ie, more mild cases (stage I and II) were noted in the COPD patients with CC/GC genotype compared with GG subjects (Table 1).
Our findings verified for the first time that rs2910164 polymorphism altered the expression level of COX2 as well as its catalytic product PGE2 via affecting the processing of miR-146a, and the decrease in miR-146a caused an increase in PGE2 production, which improved the lung function in COPD patients and was associated with milder severity of the disease. Such investigation on SNP-miR-COX2-PGE2 axis may improve our understanding of the molecular mechanism underlying the pathogenesis of the disease, and shed light on the development of a novel therapeutic tool in the treatment of COPD. Nevertheless, even though pre-miR-146a rs2910164 polymorphism genotypes were apparently associated with the production of PGE2 and lung function status, the complex nature of COPD also indicates that the pathogenesis of the disease, just like other polygenic medical disorders, might heterogeneously involve multiple variants at different gene loci. Moreover, the relatively small sample size and the fact that only Han Chinese subjects were recruited in this study limited the interpretation of the results. Meanwhile, lack of a comprehensive fine mapping or haplotype analysis involving the chromosome segment where miR-146a is located prevented us ruling out the possibility that rs2910164 polymorphism might be in linkage disequilibrium with some other functional variants mediating its apparent inhibitory effect on the expression level of the MiRNA. Further larger, multi-ethnicity, preferably prospective comprehensive studies are warranted to validate the role of this polymorphism in the development of COPD.
Conclusion
Taken together, we identified minor allele of rs2910164 polymorphism is associated with a decrease in miR-146a level, an increase of COX2 and its catalytic product, PGE2, an improved lung function and a milder severity of COPD. rs2910164 polymorphism could be a new marker to predict the clinical outcome of the disease, and miR-146a could a novel therapeutic target in the treatment of COPD.
Acknowledgments
This research was supported by the fund from the Natural Science Foundation of China (No 81300041, No 81100038), the fund from the Natural Science Foundation of the Anhui Higher Education Institutions of China (No KJ2012Z184), the fund for the academic backbone of the excellent young and middle-age people of Anhui Medical University (2013), the fund from the First Affiliated Hospital of Anhui Medical University for reserve talents (2014).
Disclosure
The authors report no conflict of interest.
References
goldcopd.com [homepage on the Internet]. Global Strategy for Diagnosis, Management, and Prevention of COPD; 2012 [updated January 2014]. Evidence-based strategy document for COPD diagnosis, management, and prevention, with citations from the scientific literature. Available from: www.goldcopd.com. Accessed December 2, 2014. | ||
Anthonisen NR, Connett JE and Murray RP. Smoking and lung function of Lung Health Study participants after 11 years. Am J Respir Crit Care Med. 2002;166(5):675–679. | ||
Turato G, Di Stefano A, Maestrelli P, et al. Effect of smoking cessation on airway inflammation in chronic bronchitis. Am J Respir Crit Care Med. 1995;152(4 Pt 1):1262–1267. | ||
Rutgers SR, Postma DS, ten Hacken NH, et al. Ongoing airway inflammation in patients with COPD who do not currently smoke. Chest. 2000;117(5 Suppl 1):262S. | ||
Hogg JC, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350(26):2645–2653. | ||
Willemse BW, ten Hacken NH, Rutgers B, Lesman-Leegte IG, Postma DS, Timens W. Effect of 1-year smoking cessation on airway inflammation in COPD and asymptomatic smokers. Eur Respir J. 2005;26(5):835–845. | ||
Kim V, Sternberg AL, Washko G, et al. Severe chronic bronchitis in advanced emphysema increases mortality and hospitalizations. COPD. 2013;10(6):667–678. | ||
Coping with Chronic Obstructive Pulmonary Disease. Library Journal. 2003;128:108–108. | ||
Bozinovski S, Anthony D, Vlahos R. Targeting pro-resolution pathways to combat chronic inflammation in COPD. J Thorac Dis. 2014;6(11):1548–1556. | ||
Failla M, Genovese T, Mazzon E, et al. 16,16-Dimethyl prostaglandin E2 efficacy on prevention and protection from bleomycin-induced lung injury and fibrosis. Am J Respir Cell Mol Biol. 2009;41(1):50–58. | ||
Kolodsick JE, Peters-Golden M, Larios J, Toews GB, Thannickal VJ, Moore BB. Prostaglandin E2 inhibits fibroblast to myofibroblast transition via E. prostanoid receptor 2 signaling and cyclic adenosine monophosphate elevation. Am J Respir Cell Mol Biol. 2003;29(5):537–544. | ||
Saltzman LE, Moss J, Berg RA, Hom B, Crystal RG. Modulation of collagen production by fibroblasts. Effects of chronic exposure to agonists that increase intracellular cyclic AMP. Biochem J. 1982;204(1):25–30. | ||
Conte E, Bonaiuto C, Nesci C, Crimi N, Vancheri C, Messina A. Nuclear factor-kappaB activation in human monocytes stimulated with lipopolysaccharide is inhibited by fibroblast conditioned medium and exogenous PGE2. FEBS Lett. 1997;400(3):315–318. | ||
Vancheri C, Mastruzzo C, Tomaselli V, et al. Normal human lung fibroblasts differently modulate interleukin-10 and interleukin-12 production by monocytes: implications for an altered immune response in pulmonary chronic inflammation. Am J Respir Cell Mol Biol. 2001;25(5):592–599. | ||
Vancheri C, Sortino MA, Tomaselli V, et al. Different expression of TNF-alpha receptors and prostaglandin E(2) Production in normal and fibrotic lung fibroblasts: potential implications for the evolution of the inflammatory process. Am J Respir Cell Mol Biol. 2000;22(5):628–634. | ||
Okamoto T, Hino O. Expression of cyclooxygenase-1 and -2 mRNA in rat tissues: tissue-specific difference in the expression of the basal level of mRNA. Int J Mol Med. 2000;6(4):455–457. | ||
Takahashi HK, Liu K, Wake H, et al. Prostaglandin E2 inhibits advanced glycation end product-induced adhesion molecule expression, cytokine production, and lymphocyte proliferation in human peripheral blood mononuclear cells. J Pharmacol Exp Ther. 2009;331(2):656–670. | ||
Sato T, Liu X, Nelson A, et al. Reduced miR-146a increases prostaglandin E(2) in chronic obstructive pulmonary disease fibroblasts. Am J Respir Crit Care Med. 2010;182(8):1020–1029. | ||
Zago M, Rico de Souza A, Hecht E, et al. The NF-kappaB family member RelB regulates microRNA miR-146a to suppress cigarette smoke-induced COX-2 protein expression in lung fibroblasts. Toxicol Lett. 2014;226(2):107–116. | ||
Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6(11):857–866. | ||
Ryan BM, Robles AI, Harris CC. Genetic variation in microRNA networks: the implications for cancer research. Nat Rev Cancer. 2010;10(6):389–402. | ||
Yang Q, Jie Z, Ye S, et al. Genetic variations in miR-27a gene decrease mature miR-27a level and reduce gastric cancer susceptibility. Oncogene. 2014;33(2):193–202. | ||
Jazdzewski K, Murray EL, Franssila K, Jarzab B, Schoenberg DR, de la Chapelle A. Common SNP in pre-miR-146a decreases mature miR expression and predisposes to papillary thyroid carcinoma. Proc Natl Acad Sci U S A. 2008;105(20):7269–7274. | ||
Chen L, Zhang R, Li P, et al. P53-induced microRNA-107 inhibits proliferation of glioma cells and down-regulates the expression of CDK6 and Notch-2. Neurosci Lett. 2013;534:327–332. | ||
Zhang X, Chen T, Zhang J, et al. Notch1 promotes glioma cell migration and invasion by stimulating beta-catenin and NF-kappaB signaling via AKT activation. Cancer Sci. 2012;103(2):181–190. | ||
Mei J, Bachoo R, Zhang CL. MicroRNA-146a inhibits glioma development by targeting Notch1. Mol Cell Biol. 2011;31(17):3584–3592. | ||
Treanor LM, Volanakis EJ, Zhou S, Lu T, Sherr CJ, Sorrentino BP. Functional interactions between Lmo2, the Arf tumor suppressor, and Notch1 in murine T-cell malignancies. Blood. 2011;117(20):5453–5462. | ||
Permuth-Wey J, Thompson RC, Burton Nabors L, et al. A functional polymorphism in the pre-miR-146a gene is associated with risk and prognosis in adult glioma. J Neurooncol. 2011;105(3):639–646. | ||
Togo S, Holz O, Liu X, et al. Lung fibroblast repair functions in patients with chronic obstructive pulmonary disease are altered by multiple mechanisms. Am J Respir Crit Care Med. 2008;178(3):248–260. | ||
Holz O, Zuhlke I, Jaksztat E, et al. Lung fibroblasts from patients with emphysema show a reduced proliferation rate in culture. Eur Respir J. 2004;24(4):575–579. | ||
Huang RY, Chen GG. Cigarette smoking, cyclooxygenase-2 pathway and cancer. Biochim Biophys Acta. 2011;1815(2):158–169. | ||
Chen Y, Chen P, Hanaoka M, Droma Y, Kubo K. Enhanced levels of prostaglandin E2 and matrix metalloproteinase-2 correlate with the severity of airflow limitation in stable COPD. Respirology. 2008;13(7):1014–1021. | ||
Montuschi P, Kharitonov SA, Ciabattoni G, Barnes PJ. Exhaled leukotrienes and prostaglandins in COPD. Thorax. 2003;58(7):585–588. | ||
Shanmugam N, Reddy MA, Natarajan R. Distinct roles of heterogeneous nuclear ribonuclear protein K and microRNA-16 in cyclooxygenase-2 RNA stability induced by S100b, a ligand of the receptor for advanced glycation end products. J Biol Chem. 2008;283(52):36221–36233. | ||
Strillacci A, Griffoni C, Sansone P, et al. MiR-101 downregulation is involved in cyclooxygenase-2 overexpression in human colon cancer cells. Exp Cell Res. 2009;315(8):1439–1447. | ||
Chakrabarty A, Tranguch S, Daikoku T, Jensen K, Furneaux H, Dey SK. MicroRNA regulation of cyclooxygenase-2 during embryo implantation. Proc Natl Acad Sci U S A. 2007;104(38):15144–15149. | ||
Ezzie ME, Crawford M, Cho JH, et al. Gene expression networks in COPD: microRNA and mRNA regulation. Thorax. 2012;67(2):122–131. | ||
Savarimuthu Francis SM, et al. MicroRNA-34c is associated with emphysema severity and modulates SERPINE1 expression. BMC Genomics. 2014;15:88. | ||
Bartels CL, Tsongalis GJ. MicroRNAs: novel biomarkers for human cancer. Clin Chem. 2009;55(4):623–631. | ||
Harfe BD. MicroRNAs in vertebrate development. Curr Opin Genet Dev. 2005;15(4):410–415. | ||
Liang H, Gu Y, Li T, et al. Integrated analyses identify the involvement of microRNA-26a in epithelial-mesenchymal transition during idiopathic pulmonary fibrosis. Cell Death Dis. 2014;5:e1238. | ||
Gao LB, Bai P, Pan XM, et al. The association between two polymorphisms in pre-miRNAs and breast cancer risk: a meta-analysis. Breast Cancer Res Treat. 2011;125(2):571–574. | ||
Hu Z, Chen J, Tian T, et al. Genetic variants of miRNA sequences and non-small cell lung cancer survival. J Clin Invest. 2008;118(7):2600–2608. | ||
Pardini B, Rosa F, Barone E, et al. Variation within 3′-UTRs of base excision repair genes and response to therapy in colorectal cancer patients: A potential modulation of microRNAs binding. Clin Cancer Res. 2013;19(21):6044–6056. | ||
Card JW, Voltz JW, Carey MA, et al. Cyclooxygenase-2 deficiency exacerbates bleomycin-induced lung dysfunction but not fibrosis. Am J Respir Cell Mol Biol. 2007;37(3):300–308. | ||
Bonner JC, Rice AB, Ingram JL, et al. Susceptibility of cyclooxygenase-2-deficient mice to pulmonary fibrogenesis. Am J Pathol. 2002;161(2):459–470. | ||
Keerthisingam CB, Jenkins RG, Harrison NK, et al. Cyclooxygenase-2 deficiency results in a loss of the anti-proliferative response to transforming growth factor-beta in human fibrotic lung fibroblasts and promotes bleomycin-induced pulmonary fibrosis in mice. Am J Pathol. 2001;158(4):1411–1422. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.