Back to Journals » Drug Design, Development and Therapy » Volume 15
Effect of a 2-HP-β-Cyclodextrin Formulation on the Biological Transport and Delivery of Chemotherapeutic PLGA Nanoparticles
Authors Zheng K, Huang Z, Huang J, Liu X ![]() , Ban J
, Ban J ![]() , Huang X, Luo H
, Huang X, Luo H ![]() , Chen Z, Xie Q, Chen Y, Lu Z
, Chen Z, Xie Q, Chen Y, Lu Z
Received 5 April 2021
Accepted for publication 4 June 2021
Published 17 June 2021 Volume 2021:15 Pages 2605—2618
DOI https://doi.org/10.2147/DDDT.S314361
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Kangyu Zheng,1,2,* Zeju Huang,1,2 Jiaying Huang,1,2 Xiangmei Liu,3,* Junfeng Ban,1,2,* Xin Huang,1,2 Haosen Luo,1,2 Zhicong Chen,1,2 Qingchun Xie,1,2 Yanzhong Chen,1,2 Zhufen Lu1,2
1Guangdong Provincial Key Laboratory of Advanced Drug Delivery Systems, Guangdong Pharmaceutical University, Guangzhou, People‘s Republic of China; 2Guangdong Provincial Engineering Center of Topical Precision Drug Delivery System, Center for Drug Research and Development, Guangdong Pharmaceutical University, Guangzhou, People‘s Republic of China; 3Guangzhou Quality Supervision and Testing Institute, Guangzhou, People‘s Republic of China
*These authors contributed equally to this work
Correspondence: Yanzhong Chen; Zhufen Lu
Guangdong Provincial Key Laboratory of Advanced Drug Delivery Systems, Guangdong Pharmaceutical University, Guangzhou, 510006, People’s Republic of China
Tel +86 20 39352506
Email [email protected]; [email protected]
Background: The aim of this work was to develop a novel and feasible modification strategy by utilizing the supramolecular effect of 2-hydroxypropyl-beta-cyclodextrin (2-HP-β-CD) for enhancing the biological transport efficiency of paclitaxel (PTX)-loaded poly(lactide-co-glycolide) (PLGA) nanoparticles.
Methods: PTX-loaded 2-HP-β-CD-modified PLGA nanoparticles (2-HP-β-CD/PLGA NPs) were prepared using the modified emulsion method. Nano-characteristics, drug release behavior, in vitro cytotoxicity, cellular uptake profiles and in vivo bio-behavior of the nanoparticles were then characterized.
Results: Compared with the plain PLGA NPs, 2-HP-β-CD/PLGA NPs exhibited smaller particle sizes (151.03± 1.36 nm), increased entrapment efficiency (∼ 49.12% increase) and sustained drug release. When added to A549 human lung cancer cells, compared with PLGA NPs, 2-HP-β-CD/PLGA NPs exhibited higher cytotoxicity in MTT assays and improved cellular uptake efficiency. Pharmacokinetic analysis showed that the AUC value of 2-HP-β-CD/PLGA NPs was 2.4-fold higher than commercial Taxol® and 1.7-fold higher than plain PLGA NPs. In biodistribution assays, 2-HP-β-CD/PLGA NPs exhibited excellent stability in the circulation.
Conclusion: The results of this study suggest that the formulation that contains 2-HP-β-CD can prolong PTX release, enhance drug transport efficiency and serve as a potential tumor targeting system for PTX.
Keywords: PLGA nanoparticle, 2-hydroxypropyl-β-cyclodextrin, sustained release, pH-responsive, long-circulating, cancer treatment
Background
When an antitumor nanomedicine is administered in the body, it undergoes 5 main steps, including blood circulation, tumor accumulation, deep penetration, cell internalization, and drug release to effectively kill tumor cells. Any hindrance in this process will affect the overall therapeutic effect of the drug.1,2 Abnormal tumor blood vessels, high interstitial pressure, large tumor cell density, and dense extracellular matrix often hinder the penetration of nanomedicine into the tumor sites.3–5 In tumor tissues, as new blood vessels are generated to supply excess nutrients and oxygen to cells, the permeability of tumor capillaries increases; therefore, the plasma proteins and other macromolecules leach out, causing lymphatic circulation, and thus, interstitial hydraulic pressure is elevated,6,7 causing chaotic arrangement of capillaries and distortion of structure.8 Also, increased levels of proteoglycan, growth factors, and hyaluronic acid in the extracellular matrix of tumors, increased collagen levels,9 increased lysine oxidase content, and enhanced covalent cross-linking ability with collagen,10,11 etc., cause accumulation of drugs in the marginal tissues of the tumor; the drug spreads slowly and faces difficulties to reach the central part,12–15 which hinders drug delivery.16,17 Recently, the anti-tumor nanomedicine research is focusing on understanding the mechanism of nanomedicine’s penetration into the capillary wall to reach the surface layer of tumor tissue by penetrating the complex microenvironment matrix of the tumor to reach the deep layer of the tumor under high interstitial pressure.17 However, it remains necessary to develop new strategies that can simultaneously enhance the tumor targeting and biological transport abilities of therapeutic agents. In this study, we aimed to adjust the size and properties of PLGA nanoparticles delivery system to enhance their penetration at the tumor site and accumulation near the tumor capillaries for their effective entry to the tumor cells. The antitumor efficacy of the formulations was assessed in vitro with cancer cell and in vivo with animals. To investigate the multi-factor influence on bioavailability, the relevance between the physicochemical (size, morphology, dissolution behavior et al) and biochemical properties (cytotoxicity, cellular uptake et al) of NPs was analyzed in detail.
Materials and Methods
Materials
Paclitaxel (PTX) was purchased from InnoChem Science & Technology Co., Ltd. (Beijing, People’s Republic of China); Free paclitaxel (commercial Taxol®) was purchased from Mei Lai Pharmaceutical Co., Ltd. (Chongqing, People’s Republic of China); PLGA (LA:GA =50:50, MW=10 k, 20 k and 30 k, ester terminated) was purchased from Dai Gang Bioengineering Co., Ltd. (Jinan, People’s Republic of China); Polyvinyl alcohol (PVA) was supplied by Cola International Trade Co., Ltd. (Shanghai, People’s Republic of China); 2-hydroxypropyl-β-cyclodextrin (2-HP-β-CD) was a gift from Qian Hui Biotechnology Co., Ltd. (Zibo, People’s Republic of China); Rhodamine-B was purchased from Da Mao Chemical Reagent Co., Ltd. (Tianjin, People’s Republic of China); 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was purchased from Sigma-Aldrich Co. (St Louis, MO, USA). All other reagents were of the highest commercial grade available. Human lung cancer cell lines A549 was purchased by the American Type Culture Collection (ATCC, Manassas, VA, USA).
Water-Oil –Water Emulsion Solvent Evaporation Method
PTX-PLGA NPs were prepared using a water-oil-water (W/OW) emulsion solvent evaporation method.18,19 Briefly, 2 mg of PTX and 60 mg of PLGA were dissolved in 3 mL of acetone: ethyl alcohol (8: 2, v/v) solution. The organic phase of the solvent was then slowly injected into 20 mL 2% PVA solution with ultrasonication (150 kHz of energy output, JY88-IIN, Scientz Biotechnology Co., Ltd, People’s Republic of China) for 10 min. The emulsion solution was stirred overnight to evaporate the organic solvent and the NPs were centrifuged at 15,000 rpm (4°C) for 30 min (5424R, Eppendorf, Germany) and washed three times in 20 mL of distilled water to remove unencapsulated drug and PVA emulsifier. To investigate the effects of different PLGA molecular weights on particle size, zeta potentials and the encapsulation efficiency of the nanoparticles, drug-loaded NPs of different molecular weights (10 k, 20 k and 30 k) were prepared and surveyed.
For preparation of 2-HP-β-CD/PLGA NPs,a mixed solution consisting of 2-HP-β-CD (2%,w/v,in 2% PVA in distilled water) was used as the dispersing phase for the emulsion solvent diffusion process. Fluorescence-labeled NPs were prepared by the replacement of PTX with 2 mg of rhodamine-B (RDM). All the formulations as shown in Table 1.
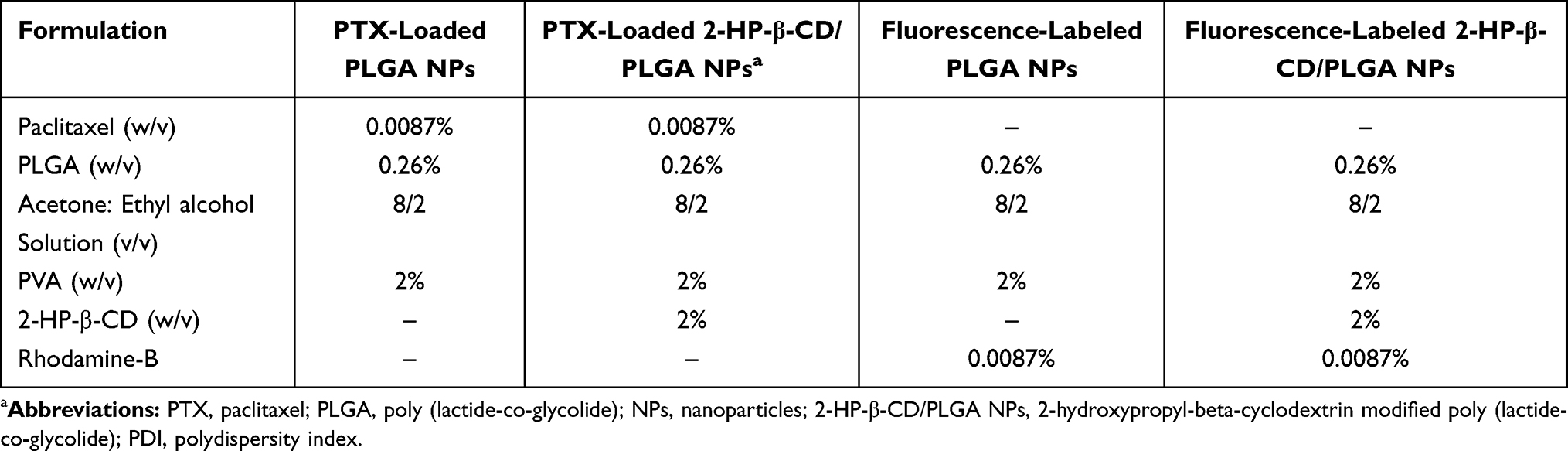 |
Table 1 Formulation Composition |
Nanoparticle Characterization
Particle Size and Zeta Potential
The hydrodynamic size and zeta potential of the PLGA NPs and 2-HP-β-CD/PLGA NPs were measured by dynamic light scattering (DLS; Delsa nano C, Beckman Coulter Inc., California, USA). The NPs were suspended in distilled water prior to experiments and multiple runs (>3) were performed to avoid erroneous results.
Encapsulation Efficiency
The encapsulation efficiency of the NPs was determined by Sephadex Chromatography. Briefly, 0.5 mL of the sample was loaded onto a Sephadex G-50 column (1.5 cm×15 cm) to which 30 mL of sterile water and 30 mL of 5% sodium dodecyl sulfate (SDS) were used for elution. The drug EE (%) of PTX-loaded NPs was determined by HPLC and calculated using the following equation:
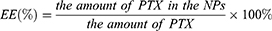
The HPLC analysis was performed using Waters (e2695 HPLC system, Waters, USA) HPLC equipment, fitted with a Kromasil 100-5-C18 column (4.6×250 mm, dp=5 µm). The mobile phase consisted of a mixture of methyl alcohol and purified water (65:35). All HPLC experiments were carried out in the isocratic mode. The injection volume was 20 μL with a flow rate of 1 mL/min. The column temperature was maintained at 25°C, and elution was monitored at 227 nm.
Surface Morphology
The nanoparticles were characterized through the assessment of their morphology via transmission electron microscopy (TEM; JEM-1400, JEOL Co., Tokyo, Japan). Samples for TEM observations were prepared through the drop-wise addition of diluted sample solutions onto a copper grid coated with a carbon membrane. The grid was then dried, dyed with 2% phosphotungstic acid and air-dried. The surface morphology of PLGA and 2-HP-β-CD/PLGA NPs was then examined by TEM.
X-Ray Diffraction Analysis
X-ray diffraction analysis (D8-Advance, Bruker Co., Germany) was used to determine the crystalline state of PTX in different PLGA and 2-HP-β-CD/PLGA NPs. These analyses were carried out using copper targets that were adjusted to a tube pressure of 40 Kv. The tube flow was set to 40 mA and scanning diffraction peaks between 5°and 40°at 2θ were recorded.
Fourier Transform Infrared Spectroscopy Analysis
Fourier transform infrared (FT-IR) spectroscopy analysis of the PLGA NPs and 2-HP-β-CD/PLGA NPs were recorded on a FT-IR spectrophotometer (Spectrum 100, PerkinElmer Co., Boston, USA) using KBr.
In vitro Release Profiles
The pH of the blood stream is ~7.4 compared to ~6.8 in the tumor extracellular microenvironment and ~5.0 in late endosomes/lysosomes.20,21 To verify the pH-dependent kinetics of drug release, PLGA and 2-HP-β-CD/PLGA NPs were incubated in 2% SDS solutions at pH 7.4, pH 6.8 or pH 5.0 at 37°C for 72 h and the release of PTX was monitored using HPLC.
In vitro Cytotoxicity
The cytotoxicity of nanoparticles was evaluated by MTT assays in A549 cells. Cells were seeded in a 96-well plate (4000 cells/well) for 24 h prior to analysis and free PTX, PLGA and 2-HP-β-CD/PLGA NPs with PTX concentrations equivalent to 0.125, 0.25, 0.5, 1, 2, 4, 8, 16 ug/mL were added. Cells were incubated in a 5% CO2 incubator at 37°C for 24, 48 and 72 h. MTT reagent (100 µL, 5 mg/mL in medium) was added for 4 h, following the addition of MTT stop solution and the measurement of optical densities using a microplate reader (800TS Absorbance Reader, BioTek Inc).
Cellular Uptake
The human lung cancer cell line A549 was selected for assessments. The extent of internalization of the modified NPs was visualized under a fluorescence inversion microscope and quantified by flow cytometry (Cytoflex, Beckman Coulter Inc). The cellular uptake and intracellular distribution of free-Rhodamine B (free-RDM), PLGA NPs and 2-HP-β-CD/PLGA NPs in A549 cells were evaluated through assessment of the fluorescence intensity of rhodamine-B (RDM). The concentration of rhodamine-B for each formulation was fixed and unified.
For fluorescence visualization, A549 cells were seeded onto six-well culture plates at a density of 1.7×105 cells. The medium was then replaced with fresh medium containing plain or modified PLGA NPs for 2 h. Cells were then washed three times with cold PBS to terminate cell uptake and cells were fixed with 4% para-formaldehyde for 15 min. Cell nuclei were stained with 4ʹ,6ʹ-diamidino-2-phenylindole (DAPI) for 5 min followed by washing with PBS (3 × 5 min washes) to remove free DAPI. Cells were visualized on a fluorescence inversion microscope.
Flow cytometry was performed to quantify the cellular uptake of NPs. A549 cells were plated in 6-well plates at a density of ~50,000 cells per well overnight and treated with free-RDM or NPs suspended in serum-free medium. Negative control groups were treated with fresh medium containing no NPs. After 1, 2 and 8 h of incubation, cells were collected by trypsinization and analyzed on a flow cytometer. A total of 10,000 gated events were acquired for each analysis.
Pharmacokinetic Studies in Rats
The study was approved by the Institutional Animal Care and Use Committee of Guangdong Pharmaceutical University, which ensured that the care and use of animals conformed to the National Institutes of the Health guide for the care and use of laboratory animals. Half male and half female Sprague–Dawley (SD) rats weighting 180–220 g were used for in vivo pharmacokinetic studies. Male and female rats were randomly divided into 3 groups (3 female and 3 male rats per group) and intravenously administered free PTX, PLGA NPs and 2-HP-β-CD/PLGA NPs at doses of 20 mg/kg via the caudal vein. At 0.0833, 0.25, 0.5, 1, 2, 4, 8 and 12 h post-administration, 1 mL of the blood samples were collected from the ophthalmic venous plexus into heparinized centrifuge tubes. Blood samples were centrifuged at 6000 rpm for 10 min to separate the plasma. The obtained plasma samples were processed as follows: 100μL of which was extracted in 200 μL of ethyl acetate through vortexing and ultrasonification for 1 min. After centrifugation at 6000 rpm for 30 min, supernatants were collected and evaporated under nitrogen. Residues were dissolved in 100 μL of methanol and used for HPLC analysis. Pharmacokinetic parameters were calculated using DAS 2.0 software (Chinese Pharmacological Society, Shanghai, China).
In vivo Biodistribution
To investigate the biodistribution of plain and 2-HP-β-CD modified NPs, Rhodamine-B (RDM)-loaded NPs were prepared by replacing PTX with RDM. The NPs were then injected into Kunming mice at doses of 20 mg/kg via the tail vein. Ex vivo imaging signals of RDM in the major organs were monitored through in vivo imaging at 5 min, 30 min, 2 h, 4 h and 6 h post-injection. After 6 h of administration, mice were sacrificed and the major organs (heart, liver, spleen, lung, kidney and brain) were excised. Tissues were washed three times with normal saline and placed in a surface dish for imaging.
Statistical Analysis
Data are expressed as the mean ± standard deviation (SD) of three independent experiments. Differences amongst the groups were assessed by an analysis of variance. Intragroup group differences were assessed using an independent sample t-test, using SPSS software (SAS Institute, Cary, NC, USA). P<0.05 was considered statistically significant.
Results and Discussion
Fabrication of PTX-Loaded 2-HP-β-CD/PLGA Nanoparticles
The preparation of PTX-loaded 2-HP-β-CD/PLGA NPs is shown in Figure 1. Through ultrasonication, some of the hydrophobic segments of PLGA in the organic phase were captured by 2-HP-β-CD via its supramolecular effects (Process I) and owing to its hydrophilic surface coating (2-HP-β-CD). The formerly hydrophobic segments of PLGA were reversed to hydrophilic segments and partly or entirely embedded into the outer hydrophilic nanostructure during the self-assembly of the NPs (Process II). The remaining uncoated hydrophobic PLGA segments formed an internal lipophilic nanostructure that encapsulated the PTX.
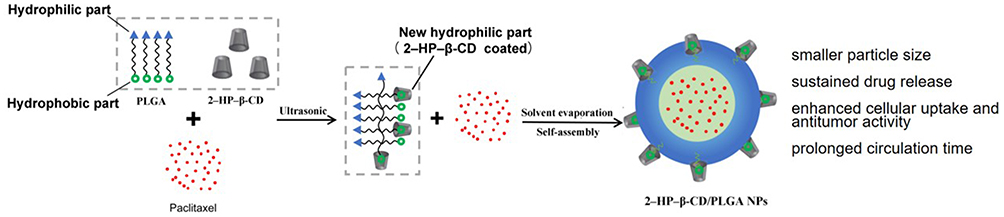 |
Figure 1 Schematic representation of 2-HP-β-CD/PLGA NPs. |
Characterization of the PLGA Nanoparticles
The size and surface properties of the NPs are the important profiles in cellular uptake and drug release processes, and in vivo pharmacokinetic and biodistribution analysis.22 As shown in Table 2 and Figure 2, both PLGA NPs and 2-HP-β-CD/PLGA NPs had particle sizes ranging from 130 nm to 200 nm, which were appropriate size ranges for their accumulation in the tumor vasculature under the influence of the EPR effect.4 Moreover, to determine the effects of the PLGA molecular weight (MW) on the characterization of NPs, PLGA and 2-HP-β-CD/PLGA NPs with an MW of 10 k, 20 k and 30 k were prepared and studied. As shown in Figure 2A, with an increasing MW of the PLGA, the particle sizes of the plain PLGA NPs decreased, whilst 2-HP-β-CD/PLGA NPs increased. Compared with the plain PLGA NPs with an MW of 10 k and 20 k, 2-HP-β-CD/PLGA NPs had smaller average particle sizes. This decreased size (~30–40 nm) may have contributed to 2-HP-β-CD coating that improved the hydrophilic nature of the organic solvent during NP fabrication, evidenced as a successful surface modification. Regarding NPs with an MW of 30 k, the average particle size of the 2-HP-β-CD/PLGA NPs were larger than those of plain PLGA NPs. This reversed result may be due to the steric effects between PLGA and 2-HP-β-CD and the hydrophobic areas of large MW PLGA (30 k) that fails to bond the hydrophobic core of 2-HP-β-CD. As such, 2-HP-β-CD may only partially embed in the outer structure of the NPs or combine with the NPs through peripheral hydrogen bonding, resulting in an increased particle size. During the experiment, PLGA nanoparticles with different molecular weight and the same copolymerization ratio were prepared. The results showed that the hydrophilicity of PLGA nanoparticles decreased and the D90 value of PLGA nanoparticles increased with the increase of copolymerization ratio. When the copolymerization ratio is 50/50 and 75/25, the molecular weight of the uncoated nanoparticles increases, but the particle size becomes smaller. This may be due to the strong hydrophilicity of glycolide, and the molecular chain can be extended in the aqueous phase. With the increase of molecular weight, the molecular chain entangles with each other, but the particle size decreases. When the copolymerization ratio of PLGA is 90/10, the change of particle size is not obvious with the increase of molecular weight. The copolymerization ratio of PLGA is large, the hydrophilicity is weakened, and the molecular chains cannot extend freely in the aqueous phase, so the change of particle size is not obvious with the increase of molecular weight.
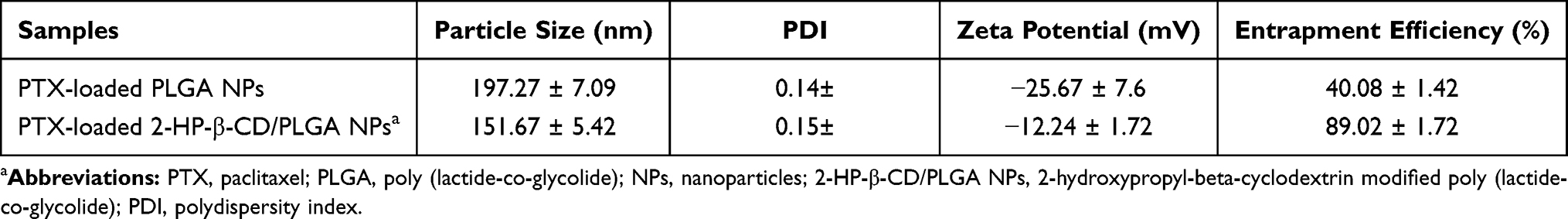 |
Table 2 Characteristics of the PTX-Loaded PLGA NPs (MW=10 k) |
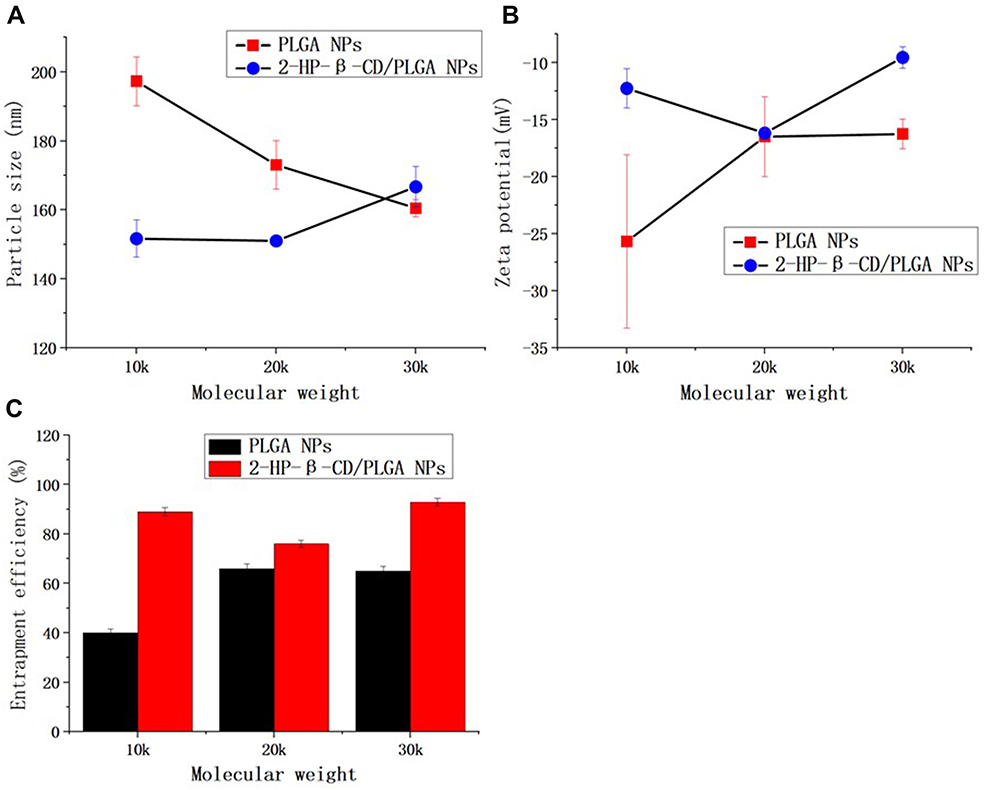 |
Figure 2 The effect of PLGA molecular weight on particle characteristics. (A) particle size, (B) zeta potential and (C) entrapment efficiency. Data are expressed as the means ± standard deviations (n=3). Abbreviations: PLGA NPs, poly(lactide-co-glycolide) nanoparticles; 2-HP-β- CD/PLGA NPs, 2-hydroxypropyl-beta-cyclodextrin modified poly(lactide-co-glycolide) nanoparticles. |
As shown in Table 2 and Figure 2B, plain and modified NPs with an MW of 20 k showed similar zeta potentials (ZP). The ZP of 2-HP-β-CD/PLGA NPs with an MW of 10 k and 30 k were significantly higher than those of PLGA NPs (p<0.05). As a result of the negative charge of NPs, unwanted clearance through the reticuloendothelial system (RES) could be reduced.1–3
As shown in Table 2 and Figures 2C, 2-HP-β-CD significantly enhanced the encapsulation efficiency (EE %) of PLGA NPs at all three surveyed MWs (10 k, 20 k and 30 k). The mean EE% of the 2-HP-β-CD/PLGA NPs (MW=10 k) was 48.94% higher than the plain PLGA NPs (p<0.05). This result may be due to the additional amounts of PTX captured by the lipophilic cavity of 2-HP-β-CD.
As shown in Figure 3A and D, compared with the plain PLGA NPs, the appearance of 2-HP-β-CD/PLGA NPs were more transparent. TEM was used to assess the NP surface morphology, revealing the NPs as spherical under x 3000 magnification (Figure 3B and E). Moreover, under x 50,000 magnification, a rough particle surface could be observed on the PLGA NPs (Figure 3C), which may have formed through the absorbance of PTX onto PLGA. As shown in Figure 3F, compared with plain PLGA NPs, 2-HP-β-CD/PLGA NPs were round in shape, uniform in size, and possessed a layer of translucent material around the NP core. This highlighted that surface modification using 2-HP-β- CD can improve the smoothness of the NP surface and reduce particle size.
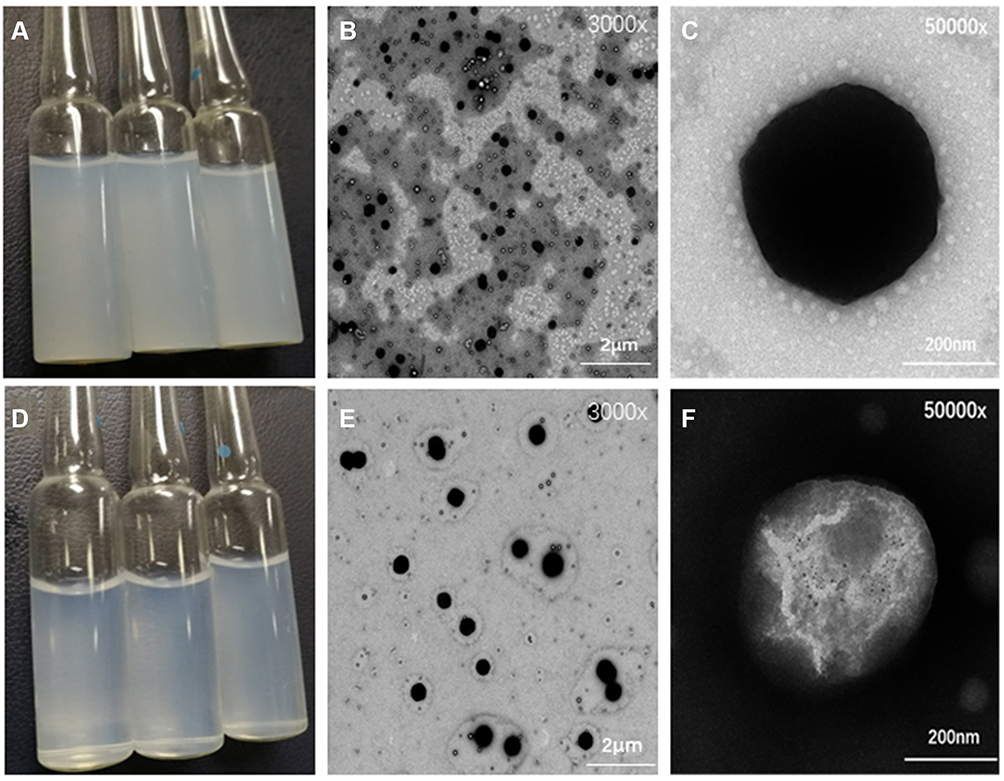 |
Figure 3 Morphology of NPs observed under transmission electron microscopy. (A) Appearance of PLGA NPs; (B) PLGA NPs TEM×12,000; (C) PLGA NPs TEM×50,000; (D) Appearance of 2-HP-β- CD/PLGA NPs; (E) 2-HP-β- CD/PLGA NPs TEM×12,000; (F) 2-HP-β- CD/PLGA NPs TEM×50,000. Abbreviations: TEM, transmission electron microscopy; PLGA NPs, poly(lactide-co-glycolide) nanoparticles; 2-HP-β- CD/PLGA NPs, 2-hydroxypropyl-beta-cyclodextrin modified poly(lactide-co-glycolide) nanoparticles. |
The phase purity of the incorporated components and nanoparticle of the prepared samples were examined using XRD analytical technique (Figure 4). The X-ray powder diffractograms of pure PTX exhibited a series of intense sharp peaks, indicating its crystalline character. As shown in Figure 4, no PTX crystal peaks were present in either PLGA NPs or 2-HP-β-CD/PLGA NPs. These data demonstrated that PTX exists in an amorphous state in both the plain and modified NPs.
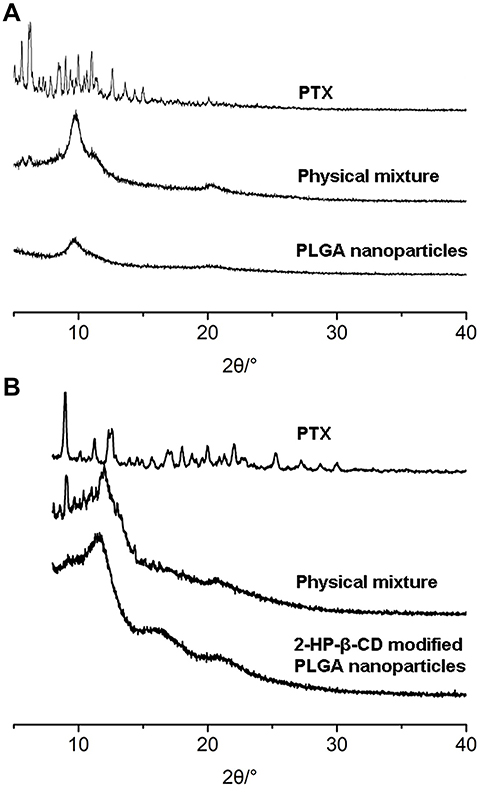 |
Figure 4 X-ray diffraction pattern of PLGA nanoparticles and 2-HP-β-CD/PLGA nanoparticles. (A) PLGA NPs; (B) 2-HP-β- CD/PLGA NPs. Abbreviations: PLGA NPs, poly(lactide-co-glycolide) nanoparticles; 2-HP-β- CD/PLGA NPs, 2-hydroxypropyl-beta-cyclodextrin modified poly(lactide-co-glycolide) nanoparticles. |
The successful encapsulation of PTX and 2-HP-β-CD modifications was further confirmed through FT-IR absorption spectra. According to Figure 5, PTX was characterized by bands of ~1450 cm−1, 1714.4–1733.8 cm−1 and 1646.4 cm−1, corresponding to the benzene ring, ketone carbonyl and amide carbonyl, respectively. The spectra of 2-HP-β-CD were characterized by bands of 3600–3200 cm−1 due to hydroxyl stretching vibrations. In physical mixtures of PLGA NPs and 2-HP-β-CD/PLGA NPs, the spectra obtained were akin to the superimposition of the individual spectra of PTX, PLGA and 2-HP-β-CD. However, in the 2-HP-β-CD/PLGA and PLGA NP spectra, the characteristic absorption peaks of PTX disappeared, indicating that the PTX had been successfully encapsulated by PLGA, and that the unencapsulated PTX was mostly removed through centrifugation. Moreover, compared with the plain PLGA NPs, the spectra of 2-HP-β-CD/PLGA NP showed a broad absorbance between 3600 and 3250 cm−1, which corresponded to the stretching vibrations of the hydroxyl (-OH) of 2-HP-β-CD.
 |
Figure 5 Infrared spectrogram of PLGA nanoparticles and 2-HP-β-CD/PLGA nanoparticles (A) PLGA NPs; (B) 2-HP-β- CD/PLGA NPs. Abbreviations: PTX, paclitaxel; PLGA NPs, poly(lactide-co-glycolide) nanoparticles; 2-HP-β-CD/PLGA NPs, 2-hydroxypropyl-beta-cyclodextrin modified nanoparticles. |
In summary, the decrease in particle size and change of zeta potential, morphology, XRD spectra and FT-IR absorption spectra, suggested that the NPs modified with 2-HP-β-CD were successfully prepared. Since both plain and modified NPs with an MW of 10 k showed the most significant differences in particle size, ZP and EE%, they were selected for analysis in subsequent experiments.
In vitro Drug Release Profiles
The controllable drug release of the NPs and their pH dependency were investigated at 37 ± 1°C in 2% SDS (pH 7.4, 6.4, and 5.0). As shown in Figure 6A, compared with the plain PLGA NPs, 2-HP-β-CD modified NPs exhibited smoother release curves and showed more sustained release characteristics at pH 7.4. The release rates of the plain NPs were ~2-fold faster than those of 2-HP-β-CD/PLGA NPs during the initial 12 h (p<0.05). It is likely that at pH 7.4, PTX was released from the PLGA matrix and bound to the outer 2-HP-β-CD shells.9 The binding was likely mediated by interactions between PTX and the hydrophilic hydroxyls and hydrophobic segments of 2-HP-β-CD in the outer layer of the NPs. In addition, owing to the surface modifications of 2-HP-β-CD, the initial burst release of the NPs were reduced at all of the three surveyed pH conditions, with the total drug release of plain NPs at 72 h typically higher than that of 2-HP-β-CD modified NPs. This may also prove the delay effects and drug control release capability of 2-HP-β-CD modified NPs. Moreover, as shown in Figure 6B and C at pH 6.8 and 5.0, the drug release profiles of 2-HP-β-CD/PLGA NPs were notably faster than those of pH 7.4 (p<0.05) indicating excellent pH-responsive properties for tumor delivery.
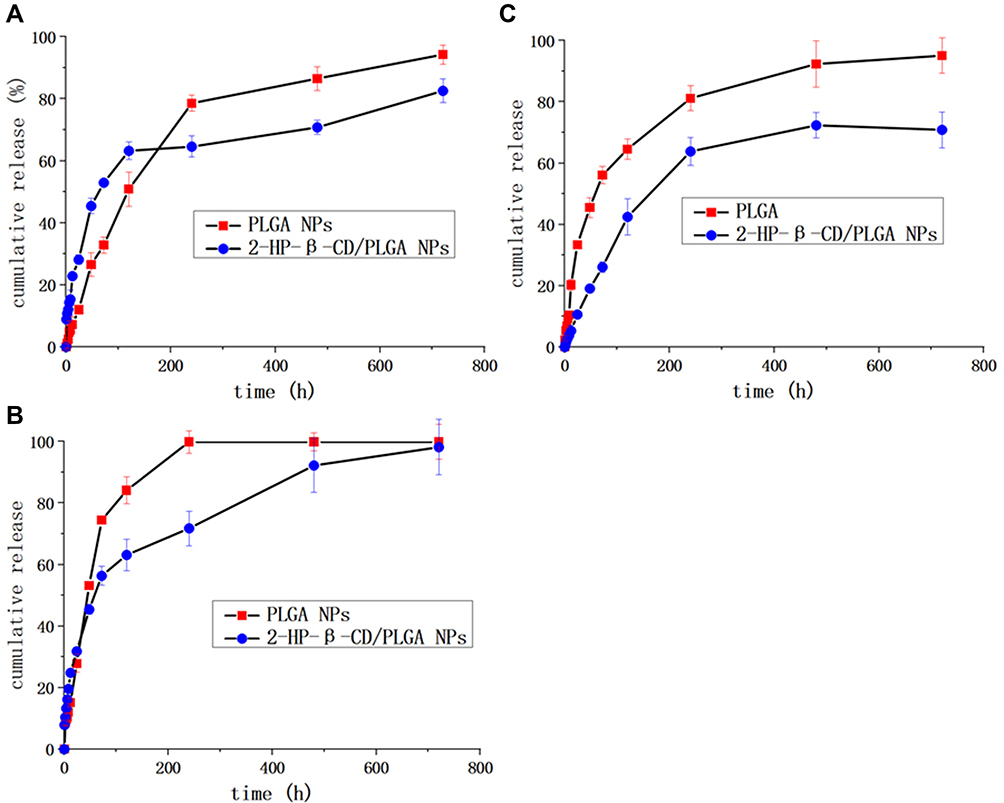 |
Figure 6 In vitro drug release profiles of 2-HP-β-CD modified NPs and naked PLGA NPs. (A) pH=7.4; (B) pH=6.8; (C) pH=5.0. Data are expressed as the means ± standard deviations (n=3). Abbreviations: PLGA NPs, poly(lactide-co-glycolide) nanoparticles; 2-HP-β-CD/PLGA NPs, 2-hydroxypropyl-beta-cyclodextrin modified nanoparticles. |
In vitro Cytotoxicity
To evaluate the in vitro antitumor activity of the NPs, MTT assays were performed through the treatment of A549 cells with free PTX, PLGA NP and 2-HP-β-CD/PLGA NP suspensions at a series of concentrations (Figure 7). All tested preparations exhibited an obvious dose-dependent trend at both 24 h and 72 h post-treatment. Cell growth was more significantly suppressed at 72 h treatment than that of 24 h. Moreover, free PTX groups exhibited the strongest toxicity at 24 h, which may have been due to the direct contact between PTX and A549 cells as opposed to the slow release from NPs. Interestingly, although both plain and modified PLGA NPs exhibited similar cytotoxicity after 24 h of treatment, cell growth was more extensively suppressed by the modified NPs as opposed to free PTX at relatively higher concentrations (≥1 μg/mL) in 72 h treatment. The cytotoxic mechanisms of the drugs are complex, and are related to drug release from the NPs and the cellular uptake efficiency of NPs or released drugs. In this study, the prepared NPs showed a relatively sustained drug-release rate. At lower doses (≤0.5 μg/mL), the NPs exhibited lower cytotoxicity than free PTX. However, the cytotoxicity disparity between free PTX and NPs was narrow at higher doses, with the cytotoxicity of 2-HP-β-CD modified NPs surpassing that of free PTX after 72 h of treatment. It is likely that the 2-HP-β-CD embedded nanostructure and the appropriate particle size of 2-HP-β-CD/PLGA NPs increased their cellular uptake efficiency, resulting in higher levels of cytotoxicity after 72 h of treatment.23 These findings were further assessed in cellular uptake studies.
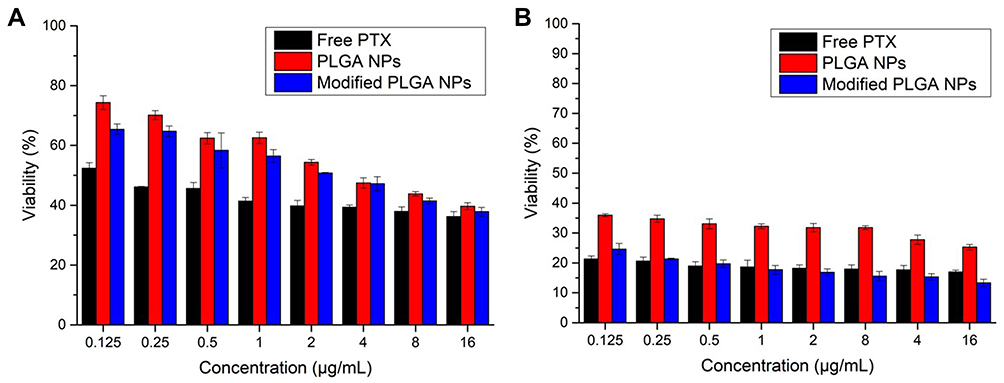 |
Figure 7 Viability of A549 cells cultured with free-PTX, PLGA NPs and 2-HP-β-CD/PLGA NPs for 24 h (A) and 72h (B). Data are expressed as the means ± standard deviations (n=3). Abbreviations: PTX, paclitaxel; PLGA NPs, poly(lactide-co-glycolide) nanoparticles; 2-HP-β-CD/PLGA NPs, 2-hydroxypropyl-beta-cyclodextrin modified poly(lactide-co-glycolide) nanoparticles. |
Cellular Uptake Study
To examine the effects of 2-HP-β-CD coating on the cellular uptake of NPs, fluorescently labeled PLGA and 2-HP-β-CD/PLGA NPs were incubated with A549 cells for 2 h and imaged on a fluorescence inversion microscope. Since both plain and modified NPs were based on PLGA cores, the initial levels of fluorescence were comparable across the NPs. As shown in Figure 8A, 2-HP-β-CD/PLGA NPs showed higher levels of cellular uptake than plain NPs, which was likely due to the latent reactivity of 2-HP-β-CD with the plasma membrane or the smaller size of the modified NPs.
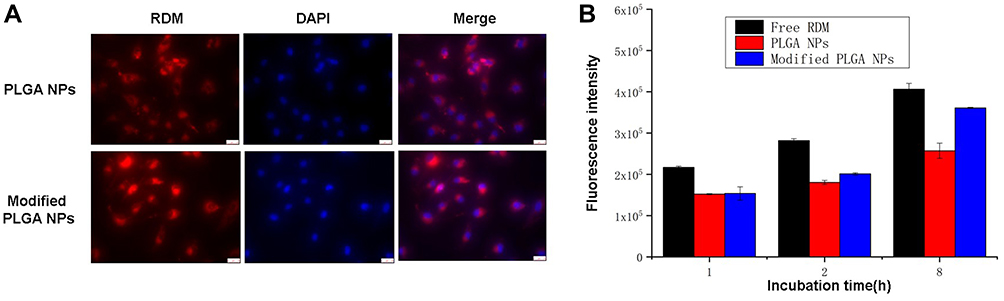 |
Figure 8 Biodistribution profiles of Rhodamine-B loaded NPs. (A)Fluorescence inversion microscope system images of A549 cells after incubation with naked PLGA NPs and 2-HP-β-CD modified PLGA NPs for 2 h. (B) Flow cytometry fluorescence intensity of free Rhodamine-B (free-RDM) and Rhodamine-B loaded NPs incubated with A549 cells after 1h, 2h and 8h incubation. Data are expressed as the means ± standard deviations (n=3). Abbreviations: RDM, Rhodamine-B; DAPI, 4ʹ,6-diamidino-2-phenylindole; PLGA NPs, fluorescence-labeled poly(lactide-co-glycolide) nanoparticles; Modified PLGA NPs, fluorescence-labeled 2-hydroxypropyl-beta-cyclodextrin modified poly(lactide-co-glycolide) nanoparticles. |
These observations were further confirmed by quantitative analysis using flow cytometry. Considering the sustained release profile of 2-HP-β-CD/PLGA NPs which may lead to a latent time-dependent cellular uptake effect, cells were treated with free-RDM, plain PLGA NPs, and modified NPs for 1, 2 and 8 h. As shown in Figure 8B, cells incubated with free-RDM showed the highest fluorescence intensity after 1, 2 and 8 h of incubation, whilst plain and modified PLGA NPs exhibited similar fluorescence intensities after 1 and 2 h of incubation. However, after 8 h, cells treated with 2-HP-β-CD/PLGA NPs showed the highest fluorescence intensity (p<0.05), confirming the additional contribution of 2-HP-β-CD. This corroborated the cytotoxicity data described above.
Pharmacokinetic Studies in Rats
The pharmacokinetic profiles of the NPs are shown in Figure 9, and relevant parameters are listed in Table 3. As predicted, both plain and modified NPs exhibited higher AUC values (1.4 and 2.4-fold higher, respectively) and greater circulating effects than free paclitaxel (free PTX). As shown in Table 3, free PTX showed the highest t1/2α values (0.41 ± 0.04 h) whilst plain and modified NPs were similar (0.21± 0.03 and 0.29 ± 0.01 h, respectively). However, free PTX was rapidly eliminated and decreased to 0.055 μg/mL within 6 h of injection, leading to relatively lower t1/2β values. These results may be due to the fact that free PTX can be rapidly transferred by glycoproteins and eventually metabolized by CYP3A enzymes, resulting in a fast decrease in blood concentrations.24 In addition, due to the EPR effect and hydrophilic surface characteristics, the NPs could more easily accumulate in various tissues or organs.25,26 Consequently, the in vivo circulating time of the NPs were significantly prolonged than that of free PTX, resulting in higher t1/2β and AUC values. Moreover, these profiles (t1/2β and AUC value) of 2-HP-β-CD/PLGA NPs were significantly higher than those of the plain NPs (3.4 and 1.7-fold increase, respectively, p<0.05). As shown in Figure 9, the blood concentrations of the PLGA NPs rapidly decreased 3 h post-injection, and declined to 0.058 ± 0.002 μg/mL within 8 h of injection. In comparison, 2-HP-β-CD/PLGA NPs maintained significantly higher blood concentrations (≥0.252 ± 0.032 μg/mL, p<0.05) at 12 h post-injection. It is likely that the hydrophilic surface modifications of 2-HP-β-CD could effectively reduce the adsorption of serum proteins and further enhance the circulating effects of 2-HP-β-CD/PLGA NPs, resulting in an improved bioavailability.
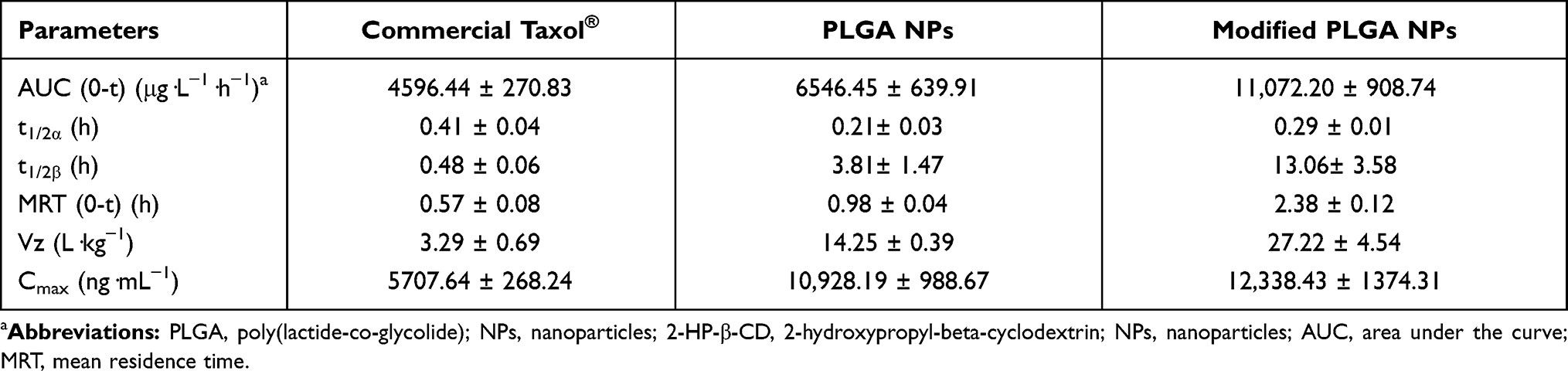 |
Table 3 Pharmacokinetic Profiles Following Intravenous Injection into Sprague–Dawley Rats at Doses of 20 Mg/Kg |
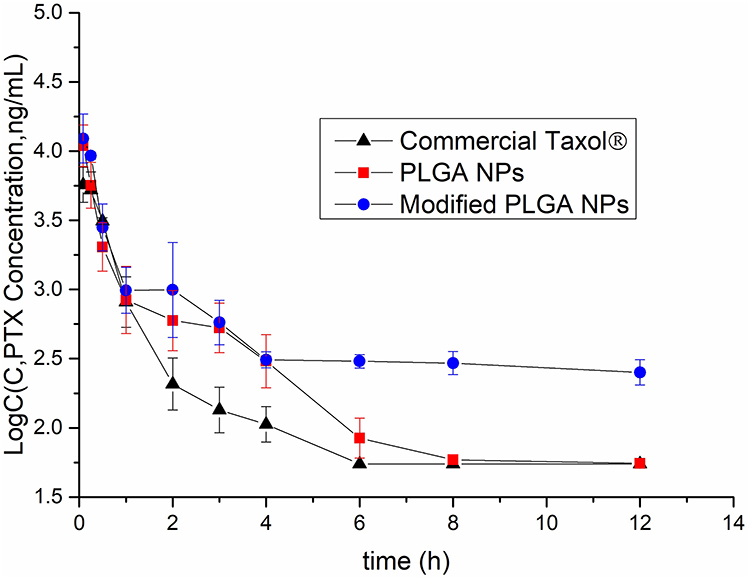 |
Figure 9 Pharmacokinetic profiles after an intravenous injection to Sprague–Dawley rats at the dose of 20 mg·kg−1. Data are expressed as the means ± standard deviations (n=6). Abbreviations: PTX, paclitaxel; Commercial Taxol®, commercial paclitaxel injection; PLGA NPs, poly(lactide-co-glycolide) nanoparticles; 2-HP-β-CD/PLGA NPs, 2-hydroxypropyl-beta-cyclodextrin modified poly(lactide-co-glycolide) nanoparticles. |
In vivo Biodistribution
As shown in Figure 10A, the location of the fluorescence signal during the early stages of drug metabolism for both NPs occurred in the liver and was determined by particle size. At 4 h post-injection, the NPs mainly distributed to the kidneys and bladder, and were presumably excreted by the kidneys. Both fluorescence labeled NPs rapidly distributed throughout the whole mice bodies of the mice, with 2-HP-β-CD/PLGA NPs showing higher fluorescence intensities than plain NPs within 2 h of injection, indicating that the modified NPs have a higher Cmax profile than plain NPs. Moreover, the fluorescence intensity of the PLGA NPs significantly decreased after 4 h of injection, whilst those of 2-HP-β-CD/PLGA-NPs were maintained at a relatively higher level, in agreement with the significantly prolonged t1/2β profiles of 2-HP-β-CD/PLGA NPs in pharmacokinetic studies. This notably longer circulating effect of the modified NPs was attributed to the 2-HP-β-CD present in the surface of PLGA. That is, the higher proportion of hydroxyl groups on the surface of 2-HP-β-CD further enhanced the surface hydrophilicity of PLGA, thus increasing its ability to be recognized by RES, resulting in higher AUC values and excellent in vivo circulating times of the 2-HP-β-CD/PLGA NPs.
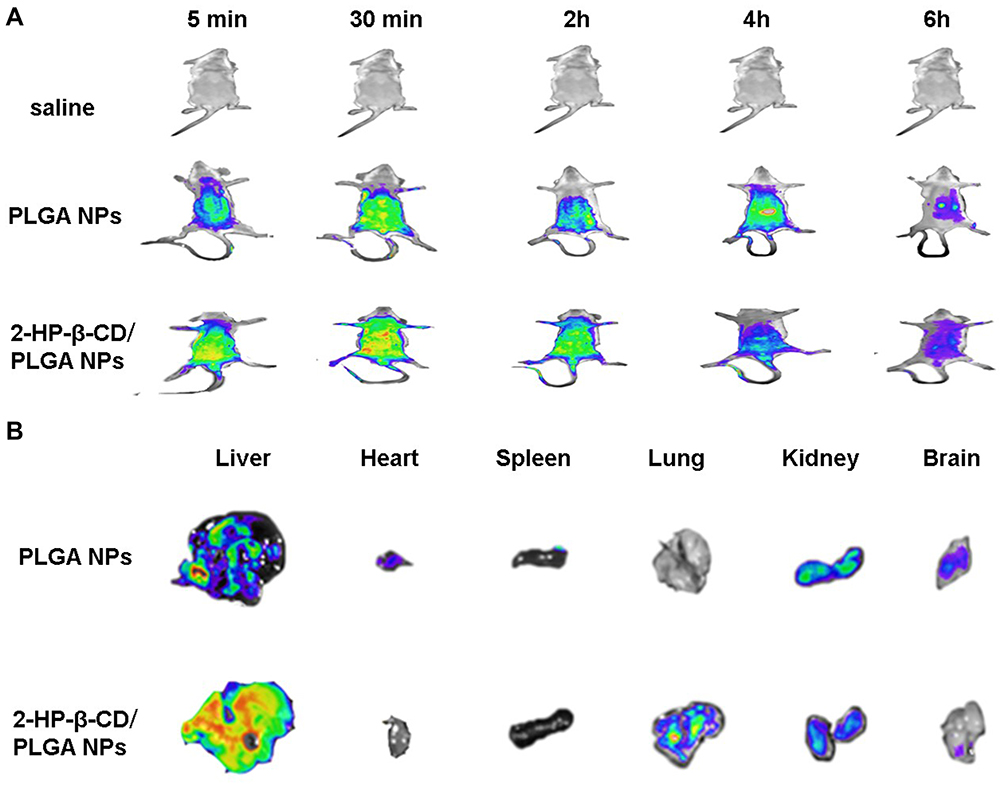 |
Figure 10 In vivo imaging and biodistribution analysis after tail vein injection of RDM-loaded NPs. (A) Real-time biodistribution imaging in mice. (B) In vitro visceral fluorescence imaging of mice organs after 6h post-injection. Abbreviations: RDM-loaded NPs, Rhodamine-B-loaded nanoparticles; PLGA NPs, fluorescence-labeled poly(lactide-co-glycolide) nanoparticles; Modified PLGA NPs, fluorescence-labeled 2-hydroxypropyl-beta-cyclodextrin modified poly(lactide-co-glycolide) nanoparticles. |
In vitro visceral imaging of the mouse organs is shown in Figure 10B. The NPs concentrated in the liver after 6 h of injection, which may have been due to their recognized by the RES and their passive targeting to the liver. The fluorescence intensity of the 2-HP-β-CD/PLGA-NPs in liver tissue was weaker than that of plain PLGA NPs. It is likely that the modified NPs achieved “invisibility” by utilizing the hydrophilic coating of 2-HP-β-CD, avoiding the scavenging effects of the immune system, prolonging in vivo circulation times. Interestingly, at 6 h post-injection, the NPs showed fluorescent signals in the mouse lungs only when surface-modified with 2-HP-β-CD, suggesting that the 2-HP-β-CD/PLGA-NPs can act as drug carriers the treatment of lung cancer.
Conclusions
In this study, a novel PTX-loaded biodegradable material was established using a simple and feasible method that optimized the particle profiles, drug release processes and biological behavior of the NPs. Firstly, we investigated the effects of 2-HP-β-CD coating and the MW of PLGA on the particle size, zeta potential, and drug load efficiency of the NPs. The results showed that the PLGA MW exhibited influenced the nano-properties of the NPs, and that the 2-HP-β-CD-NPs differed according to MW due to steric effects. In addition, 2-HP-β-CD modifications significantly decreased the particle size and enhanced the encapsulation efficiency (EE%). The EE% of 2-HP-β-CD/PLGA NPs were 49.12% higher than that of plain PLGA NPs, indicating that 2-HP-β-CD encapsulates certain levels of PTX, resulting in higher EE% values. As the plain and modified NPs with an MW of 10 k showed the most significant differences in particle size, zeta potential and EE%, they were selected for further assessments. The NP morphologies were confirmed by XRD and FT-IR, and the 2-HP-β-CD modification led to superior drug release profiles and higher cancer cell-specific toxicity. 2-HP-β-CD/PLGA NPs showed notable sustained-release properties at pH 7.4 with drug release rates that were accelerated at pH 6.8 and 5.0, considered an appropriate pH-responsive profile for tumor treatment. MTT assays showed that the 2-HP-β-CD surface modifications enhanced the cytotoxicity of NPs to A549 cells, likely due to their smaller particle size. These results were further confirmed in cellular uptake studies. Furthermore, pharmacokinetic analysis showed that the 2-HP-β-CD/PLGA NPs exhibited prolonged in vivo circulatory times and improved pharmacokinetic profiles than the PLGA NPs. At 6 h post-injection, only the modified NPs were targeted to mouse lungs, highlighting 2-HP-β-CD/PLGA-NPs as potential carriers for lung cancer treatment. On the basis of the drug release profiles reported above, we hypothesized that the combination of 2-HP-β-CD and PLGA was as follows: after ultrasonication, the hydrophobic segments of PLGA in the organic phase were captured by 2-HP-β-CD through supramolecular effects. Owing to their hydrophilic surface coating, these formerly hydrophobic segments of PLGA became hydrophilic and partly or entirely embedded in the outer hydrophilic nanostructure upon NP self-assembly. PTX was then encapsulated in the inner nanostructure of the NP composed of hydrophobic segments.
In summary, we developed a simple and efficient surface modification strategy that can alter the particle size, drug release profiles, cellular toxicity and biological fate of NPs, improving their potential as drug delivery systems for cancer therapeutics and diagnostics.
Abbreviations
PTX, paclitaxel; PLGA, poly(lactide-co-glycolide); 2-HP-β-CD, 2-hydroxypropyl-β-cyclodextrin; NPs, nanoparticles; MTT, methyl thiazolyl tetrazolium assay; EE%, encapsulation efficiency; TEM, transmission electron microscopy; FT-IR, Fourier transform infrared spectroscopy; SD rats, Sprague–Dawley rats; AUC, area under the curve; MRT, mean residence time.
Acknowledgments
Kangyu Zheng, Xiangmei Liu and Junfeng Ban are co-first authors for this study.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agreed to be accountable for all aspects of the work.
Funding
We acknowledge the financial support from the Natural Science Foundation of Guangdong Province(Grant Number 2019A1515011018); The planned Science and Technology Project of Guangdong Province(2017B020231001); Guangdong Graduate Education Innovation Program: Project Funded by Guangdong Province Joint Training Graduate Demonstration Base (Guangzhou General Pharmaceutical Research Institute Co., Ltd.) Guangdong Pharmaceutical University “Innovation and Enhancing Project” and Guangdong “Climbing” Program for Undergraduates (2021).
Disclosure
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1. Sun Q, Zhou Z, Qiu N, Shen Y. Rational design of cancer nanomedicine: nanoproperty integration and synchronization. Adv Mater. 2017;29(14).
2. Ma Z, Li J, Lin K, et al. Pharmacophore hybridisation and nanoscale assembly to discover self-delivering lysosomotropic new-chemical entities for cancer therapy. Nat Commun. 2020;11(1):4615. doi:10.1038/s41467-020-18399-4
3. Kobayashi H, Choyke PL. Super enhanced permeability and retention (SUPR) effects in tumors following near infrared photoimmunotherapy. Nanoscale. 2016;8(25):12504–12509. doi:10.1039/C5NR05552K
4. Luk BT, Zhang L. Current advances in polymer-based nanotheranostics for cancer treatment and diagnosis. ACS Appl Mater Interfaces. 2014;6(24):21859–21873. doi:10.1021/am5036225
5. Lan H, Zhang W, Jin K, Liu Y, Wang Z. Modulating barriers of tumor microenvironment through nanocarrier systems for improved cancer immunotherapy: a review of current status and future perspective. Drug Deliv. 2020;27(1):1248–1262. doi:10.1080/10717544.2020.1809559
6. Jain RK. Determinants of tumor blood flow: a review. Cancer Res. 1988;48(10):2641–2658.
7. Jacobetz MA, Chan DS, Neesse A, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut. 2013;62(1):112–120. doi:10.1136/gutjnl-2012-302529
8. Konerding MA, Miodonski AJ, Lametschwandtner A. Microvascular corrosion casting in the study of tumor vascularity: a review. Scanning Microsc. 1995;9(4):
9. Kolacna L, Bakesova J, Varga F, et al. Biochemical and biophysical aspects of collagen nanostructure in the extracellular matrix. Physiol Res. 2007;56(Suppl 1):S51–S60.
10. Levental KR, Yu H, Kass L, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139(5):891–906. doi:10.1016/j.cell.2009.10.027
11. Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010;7(11):653–664. doi:10.1038/nrclinonc.2010.139
12. Bouzin C, Feron O. Targeting tumor stroma and exploiting mature tumor vasculature to improve anti-cancer drug delivery. Drug Resist Updat. 2007;10(3):109–120. doi:10.1016/j.drup.2007.03.001
13. Boucher Y, Jain RK. Microvascular pressure is the principal driving force for interstitial hypertension in solid tumors: implications for vascular collapse. Cancer Res. 1992;52(18):5110–5114.
14. Schudel A, Chapman AP, Yau MK, et al. Programmable multistage drug delivery to lymph nodes. Nat Nanotechnol. 2020;15(6):491–499. doi:10.1038/s41565-020-0679-4
15. Stylianopoulos T, Martin JD, Chauhan VP, et al. Causes, consequences, and remedies for growth-induced solid stress in murine and human tumors. Proc Natl Acad Sci U S A. 2012;109(38):15101–15108. doi:10.1073/pnas.1213353109
16. Chen H, Zhang W, Zhu G, Xie J, Chen X. Rethinking cancer nanotheranostics. Nat Rev Mater. 2017;2(7). doi:10.1038/natrevmats.2017.24
17. Lin J, Yin Q, Chen B, et al. A magnetism/laser-auxiliary cascaded drug delivery to pulmonary carcinoma. Acta Pharm Sin B. 2020;10(8):1549–1562. doi:10.1016/j.apsb.2019.12.017
18. Fonseca C, Simoes S, Gaspar R. Paclitaxel-loaded PLGA nanoparticles: preparation, physicochemical characterization and in vitro anti-tumoral activity. J Control Release. 2002;83(2):273–286. doi:10.1016/S0168-3659(02)00212-2
19. Hill LE, Taylor TM, Gomes C. Antimicrobial efficacy of poly (DL-lactide-co-glycolide) (PLGA) nanoparticles with entrapped cinnamon bark extract against Listeria monocytogenes and Salmonella typhimurium. J Food Sci. 2013;78(4):N626–N632. doi:10.1111/1750-3841.12069
20. Cirpanli Y, Bilensoy E, Lale DA, Calis S. Comparative evaluation of polymeric and amphiphilic cyclodextrin nanoparticles for effective camptothecin delivery. Eur J Pharm Biopharm. 2009;73(1):82–89. doi:10.1016/j.ejpb.2009.04.013
21. Trapani G, Lopedota A, Boghetich G, et al. Encapsulation and release of the hypnotic agent zolpidem from biodegradable polymer microparticles containing hydroxypropyl-beta-cyclodextrin. Int J Pharm. 2003;268(1–2):47–57. doi:10.1016/j.ijpharm.2003.08.016
22. Ho BN, Pfeffer CM, Singh A. Update on nanotechnology-based drug delivery systems in cancer treatment. Anticancer Res. 2017;37(11):5975–5981. doi:10.21873/anticanres.12044
23. Win KY, Feng SS. Effects of particle size and surface coating on cellular uptake of polymeric nanoparticles for oral delivery of anticancer drugs. Biomaterials. 2005;26(15):2713–2722. doi:10.1016/j.biomaterials.2004.07.050
24. Kirby SM, Anna SL, Walker LM. Effect of surfactant tail length and ionic strength on the interfacial properties of nanoparticle-surfactant complexes. Soft Matter. 2017;14(1):112–123. doi:10.1039/C7SM01806A
25. Maeda H, Bharate GY, Daruwalla J. Polymeric drugs for efficient tumor-targeted drug delivery based on EPR-effect. Eur J Pharm Biopharm. 2009;71(3):409–419. doi:10.1016/j.ejpb.2008.11.010
26. Maeda H, Nakamura H, Fang J. The EPR effect for macromolecular drug delivery to solid tumors: improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv Drug Deliv Rev. 2013;65(1):71–79. doi:10.1016/j.addr.2012.10.002
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.