Back to Journals » Drug Design, Development and Therapy » Volume 15
Edaravone Alleviated Propofol-Induced Neurotoxicity in Developing Hippocampus by mBDNF/TrkB/PI3K Pathway
Authors Yang Y, Yi J, Pan M, Hu B, Duan H
Received 28 November 2020
Accepted for publication 9 March 2021
Published 30 March 2021 Volume 2021:15 Pages 1409—1422
DOI https://doi.org/10.2147/DDDT.S294557
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Manfred Ogris
Yangliang Yang, Jing Yi, Mengzhi Pan, Baoji Hu, Hongwei Duan
Department of Anesthesiology, Shanghai Pudong Hospital, Fudan University Pudong Medical Center, Shanghai, People’s Republic of China
Correspondence: Hongwei Duan
Department of Anesthesiology, Shanghai Pudong Hospital, Fudan University Pudong Medical Center, No. 2800 Gongwei Road, Shanghai, 201399, People’s Republic of China
Email [email protected]
Background: To investigate the neuroprotective effect of edaravone on excessive-dose propofol-induced neurotoxicity in the hippocampus of newborn rats and HT22 cells.
Methods: Cell proliferation was investigated by assessing ki67 expression in the neural stem of the hippocampus of newborn rats and by cell counting kit-8 (CCK8) assay in HT22 cells. Cell apoptosis was assessed in vivo by caspase 3 detection in Western blots and measurement of apoptosis in neurons and glial cells by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay. Apoptosis was analyzed by flow cytometry in HT22 cells. The Morris water maze was used to evaluate the long-term learning and memory ability of rats. Inflammatory factors were detected by enzyme-linked immunosorbent assay (ELISA). The expression of mBDNF/TrkB/PI3K pathway-related proteins was detected by Western blot and quantitative reverse transcription-polymerase chain reaction (q-RT PCR).
Results: In neonatal rat hippocampus and HT22 cells, edaravone increased cell proliferation and decreased cell apoptosis after excessive propofol-induced neurotoxicity. In addition, the levels of proinflammatory factors interleukin (IL)-6 and tumor necrosis factor (TNF)-α were reduced by edaravone pretreatment. The use of the tropomyosin receptor kinase B (TrkB) antagonist ANA-12 and TrkB agonist 7,8DHF with propofol groups showed that edaravone mitigated excessive propofol-induced neurotoxicity through the mature brain-derived neurotrophic factor (mBDNF)/TrkB/phosphoinositide 3-kinase (PI3K) pathway. However, the current dose of propofol did not significantly affect long-term learning and memory in rats.
Conclusion: Edaravone pretreatment ameliorated propofol-induced proliferation inhibition, neuroapoptosis, and neural inflammation by activating the mBDNF/TrkB/PI3K pathway.
Keywords: edaravone, propofol, hippocampus, the brain-derived neurotrophic factor, BDNF, the tyrosine kinase receptor B, TrkB, 7,8-dihydroxyflavone, 7,8-DHF, ANA-12
Introduction
Early-life exposure to general anesthetics may lead to neurotoxic consequences.1 While many animal models and some clinical studies have demonstrated the high sensitivity and vulnerability of the developing brain of the newborns to anesthetic neurotoxicity, the mechanism of this effect remains unclear.2 As the use of anesthesia is inevitable in the clinical environment, anesthesiologists strive to find ways to assure the safety of children and provide comfort during painful procedures.3 To minimize risks, the effects of anesthetics on the neurological system need to be understood.4 The development of the neurological system requires a series of elaborately complicated processes. In these processes, neural stem cells (NSCs) proliferate, migrate a considerable distance from the germination center to their destination, differentiate into neurons and glial, and ultimately mature and integrate into the existing neuron circuit.5 An increased risk of neurodegeneration associated with the use of anesthesia would have considerable social significance.4
Since the late 1980s, propofol, also known as 2,6-diisopropylphenol, has been used as an intravenous anesthetic for the induction and maintenance of general anesthesia. Propofol is widely used during invasive diagnostic and surgical procedures and for sedation, including in newborns and young children.1,4,6–8 Compared to other intravenous anesthetics, propofol-induced anesthesia is associated with fewer side effects and faster recovery. The benefits of propofol include the rapid onset of anesthesia and short recovery time,4 as well as neuroprotective effects in pathogenic situations.6,9,10 However, increasing evidence suggests that propofol has neurotoxic effects in children, causing concerns regarding brain development by inhibition of the neuronal activation of hippocampal neurons,9,11 potentially causing decreased neurocognitive function.9 Animal studies have confirmed that propofol induces apoptosis and cognitive deficits in the central nervous system during the peak synaptic administration during the first 2 weeks of life.7 Previous studies have tested whether general anesthesia with propofol shortly after learning interferes with memory consolidation and whether these effects are time dependent. Additionally, the systemic administration of propofol immediately after learning of a location in a water maze was shown to affect the consolidation of spatial memory in rats. Moreover, propofol-induced cytotoxicity is not only dose- but also time-dependent, consistent with the toxic effects of other inhalational general anesthesia (GA) agents.10 Previous studies have shown that propofol is the most effective agent to activate the tonic γ-aminobutyric acid type A (GABA-A) current in the immature hypothalamus. The upregulation of tonic GABA-A conductance is related to epilepsy and neurodevelopmental disorders and may also contribute to memory disorders after GA.3
Brain-derived neurotrophic factor (BDNF) is an important member of the neuron growth factor family that is widely expressed in the central nervous hippocampal system.12,13 Research on the effects of neurotrophins in animals was useful to understanding the role of BDNF in branching, synaptic differentiation, pruning, and structural changes.14 Elevated levels of endogenous BDNF promote the maturation of dendritic spines.5 BDNF is synthesized into glycosylated precursor protein (pro) BDNF (30–32kDa) and converted into the mature form (mBDNF) after translation, a process involving multiple subcellular pathways, which is essential for the development of the central nervous system (CNS).15 Evidence supports the “yin-yang effect”; that is, proBDNF and mBDNF trigger opposite biological effects by activating two different receptor systems.14 In other words, as a precursor protein, proBDNF may be further cleaved by the tissue plasminogen activator (tPA)/plasminogen system to produce mBDNF and the protein portion related to the pro-domain.16 In response to neuronal activity, mBDNF is highly expressed in many brain regions and promotes activity-dependent synaptic plasticity through tyrosine kinase receptor B (TrkB) receptors.5,6,17 Additionally, BDNF also plays an important role in spatial memory formation, retention, and recall.4
The findings of propofol-induced neurotoxicity raise questions regarding the potential neuroprotective effects of edaravone. Edaravone, a novel exogenous free radical scavenger, was first used clinically in 2001 to treat neural injury after acute cerebral ischemic stroke and has also been shown to provide neuroprotective effects against oxidative stress, neuronal apoptosis, and seizure-induced neuronal damage.18–21 A growing body of evidence suggests that edaravone exerts its neuroprotective effects by increasing mBDNF and Bcl-2 expression, decreasing caspase-3 activity, and promoting extracellular signal-regulated kinases 1/2 (ERK1/2) activation in cultured neurons.22 Meanwhile, the mBDNF/TrkB signaling pathway may play a significant role in propofol-induced learning and memory impairments.23 Therefore, our objective was to explore the protective effect of edaravone against propofol-induced hippocampal neurotoxicity in terms of neuron cell proliferation, cell apoptosis, and neuroinflammation and to demonstrate that this protective effect occurred through the mBDNF/TrkB/PI3K pathway.
Materials and Methods
Animals
120 Sprague Dawley (SD) rats (11–18g, 7 day of age) were purchased from Pudong Hospital Animal Experimental Center affiliated to Fudan University (Pudong Hospital, Shanghai, China). All animal experimental protocols used in the present work were approved by the Animal Experimental Ethics Committee of Fudan University and conducted according to the guidelines for the Care and Use of Laboratory Animals from the National Institutes of Health. All animals were kept in an air-conditioned room at 25◦C, with a 12-h light-dark cycle (light: 8:00 a.m.-8:00 p.m.). The standard laboratory food and water were available all the time. All efforts were made to minimize the animals’ suffering.
Cell Culture of HT22
The HT22 hippocampal cell line was provided by Cell Bank, Shanghai. Cells were cultured in DMEM containing 10% FBC (Invitrogen, Carlsbad, CA, USA), 100μ/mL penicillin and 100 ng/mL streptomycin (Invitrogen, Carlsbad, CA, USA) at 37°C in 5% CO2. When the cell density reached 60–70%, the cells were treated with edaravone (60μmol/L) for pretreating 30 min then different concentrations of propofol (10–400μmol/L) for 6h.
Drugs and Treatments
Propofol was purchased from GUORUI PHARMACEUTICAL (H20143252, Sichuan, China). Edaravone was purchased from BIDA PHARMACEUTICAL (H20070051, Jilin, China). ANA-12, TrkB antagonist, was purchased from MedChemExpres (MCE, Catalog HY-12497, USA). 7,8-dihydroxyflavone (7,8-DHF), TrkB agonist, was purchased from MedChemExpres (MCE, Catalog HY-W013372, USA). 20% Intralipid injection (Fresenius-kabi, Jiangsu, China).
For the first experiment, the SD rats were randomly divided into five groups (n = 3 in each group); namely, the control, intralipid, edaravone, ANA-12 (TrkB antagonist), and 7,8-dihydroflavone (7,8-DHF, TrkB agonist) groups. In the second experiment, the SD rats were randomly divided into five groups (n = 16 in each group); namely, the control, propofol (pro), edaravone + propofol (eda+pro), 7,8-DHF + propofol (7,8-DHF+pro), and ANA-12+propofol (ANA-12+pro) groups. The 7,8-DHF was dissolved in phosphate-buffered saline (PBS) containing 2% dimethyl sulfoxide (DMSO) administered at a dose of 5 mg/kg intraperitoneally.24 ANA-12 was dissolved in 98% corn oil containing 2% DMSO and administered at a dose of 0.5 mg/kg intraperitoneally. In experiment one, the rats were administrated 150 uL saline, 150 uL intralipid, 3 mg/kg edaravone, 0.5 mg/kg ANA-12, or 5 mg/kg 7,8-DHF by intraperitoneal injection. In experiment two, the rats were intraperitoneally initially administered 30 mg/kg propofol, followed by ad 15 mg/kg at 1-hour intervals to achieve propofol anesthesia (6-hour anesthesia). Edaravone (3 mg/kg), ANA-12 (0.5 mg/kg), or 7,8-DHF (5 mg/kg) were administered before propofol injection (Figure 1).
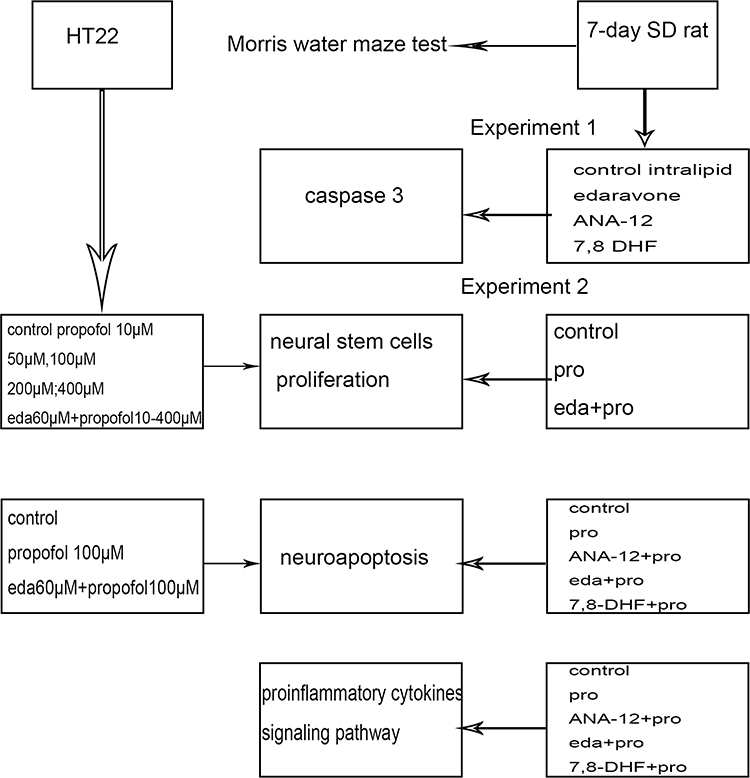 |
Figure 1 The experimental procedure. The two groups of textboxes under the left arrow were grouped into study HT22 cells; The four groups of textboxes under the right arrow were grouped into study 7-day SD rats; The four middle textboxes were methods. Abbreviations: 7,8 DHF, 7,8-dihydroxyflavone; pro, propofol; eda, edaravone. |
ELISA of Proinflammatory Factors (IL-6 and TNF-α)
Proinflammatory factors (IL-6 and TNF-α) in the hippocampus were measured using rat ELISA kit. The one of hippocampus tissues from same sample was ground and homogenized, then followed by centrifugation (5000 rpm) for 10 min at 4°C to suck out the supernatant. The IL-6 and TNF-α ELISA kits were detected at 450nm from instructions.
Immunofluorescence Assay
The prepared brain slices were washed with 50 mM PBS for 3 times for 10 min each, and then, the slices were incubated in 3% H2O2 for 30 min to block endogenous peroxidase activity, followed by immersion in blocking solution for 2h and then incubated overnight with primary antibodies: goat anti-Nestin (1:50, Abcam, UK), goat anti-ki67 (1:100, Abcam UK), goat anti-TrkB (1:100, Abcam UK), mouse anti–NeuN (1:200, Abcam UK), mouse anti–GFAP (1:200, Abcam UK), mouse anti–IBA1 (1:200, Abcam UK). Then, the sections were incubated for 2h at 25°C in the dark with the secondary antibodies: Alexa Fluor 488 goat anti-mouse (1:200, Beyotime, China) for TrkB and ki67; Alexa Fluor 594 IgG goat anti–rabbit (1:200, Beyotime, China) for Nestin, GFAP, IBA1 and NeuN. And then the sections were washed by PBS three times, next wiped the surrounding liquid and re-stained nucleus with DAPI. They were observed by the microscope and analyzed using Image-pro Plus 6.0 software (USA).
Double-Labeled Immunofluorescent Staining of TUNEL and NeuN or TUNEL and GFAP or TUNEL and IBA1
For TUNEL studies, P7 rat brains were paraffin-embedded and sectioned at 5 μm thickness. The anti-NeuN, anti-GFAP or anti-IBA1 antibody at 1:200 dilution in PBS was incubated with the sections at 4°C overnight. Tissue sections were then biotinylated with Alexa Fluor 594 IgG goat anti–rabbit (1:200, Beyotime, China). Then, TUNEL staining was performed according to the manufacture’s protocol (Roche In Situ Cell Death Detection Kit, USA. TUNEL labeling was conducted with a mixture of 45 μL equilibration buffer, 5 μL nucleotide mix and 1 μL recombinant terminal deoxynucleotidyl transferase (rTdT) enzyme in a humidified, lucifugal chamber for 1 h at 37°C, and then Hoechst 33258 was used to stain nuclei. The number of cells stained positively for TUNEL and NeuN or TUNEL and GFAP or TUNEL and IBA1 of hippocampus was analyzed using Image-pro Plus 6.0 software (USA). At least five brain tissue sections were chosen for analysis from each animal and the average value from these sections was taken as final value of this animal.
Quantitative-RT PCR
Homogenize the frozen hippocampus tissue in 1 mL of triazole reagent with an ultrasound system. The homogenate was incubated at room temperature for 5 minutes to separate the nucleoprotein complex. Then, add 0.2 mL chloroform, shake vigorously to homogenize, and incubate at room temperature for 5 minutes. The sample was then centrifuged at 10,000g for 15 minutes at 4°C. The RNA-containing aqueous phase was mixed with 0.5 mL isopropanol, incubated at room temperature for 10 minutes, and then centrifuged at 10,000 g at 4°C for 10 minutes. The RNA particles were resuspended in 75% ethanol, centrifuged at 3500g for 5 minutes at 4°C, dried and dissolved in 20μL 0.1% DEPC water. Add 30μL of RNase-free ddH2O for 2 minutes at room temperature and mix to completely dissolve the precipitate to obtain the total RNA of the sample.
Total RNA was extracted with the Trizol kit (Invitrogen). cDNA synthesis was performed according to the manufacturer’s instructions. Real-time PCR was performed using the Veriti DX (Life). The following primers were used: BDNF- forward primer (5ʹ-TGAGCCGAGCTCATCTTTGC-3ʹ), reverse primer (5ʹ-ATAGCGGGCGTTTCCTGAAG-3ʹ) and GAPDH forward primer (5ʹ-TGCCACTCAGAAGACTGTGG-3ʹ), reverse primer (5ʹ-TTCAGCTGGGATGACCTT-3ʹ). The products of the PCR were analysed by the Roche480II Real-Time PCR System (Roche), and GAPDH was used as an endogenous control. The results were calculated by 2^−⊿⊿Ct, where Ct is the threshold cycle for each transcript.
Cell Counting Kit-8 (CCK-8) Assay
HT22 cells suspended in 100 μL DMEM medium were seed into a 96-well plate. The cell viability was assessed by the CCK8 (Dojindo Molecular Technologies, Japan). 10μL CCK8 solution was given to each well of the plate after a different dose of propofol (10μM, 50μM, 100μM, 200μM, 400μM) and edaravone (60μM) at 37°C in dark for 2 h. Absorbance (A) at 450 nm was recorded after 2 h incubation.
Flow Cytometry
For the apoptosis assay, 2×105 HT22 cells were obtained and washed with PBS 3 times. Then, 5 samples were resuspended in 100μL of binding buffer, stained with 5μL of Annexin-V and propidium iodide (PI), and stored at room temperature in the dark for 30 minutes. After staining, added an additional 400 μL of binding buffer to the sample and resuspend. Analysis was performed using a flow cytometer (Becton–Dickinson, Bedford, MA, USA).
Morris Water Maze
On day 21 after modeling, Morris water maze test was used to evaluate spatial learning and memory of rats (n=5 for each). The experimental apparatus was a circular pool (150 cm in diameter and 50 cm in height, containing water at 28°C) with water colored by blue ink and divided into 4 equally quadrants (S1, S2, S3, S4). The pool was placed in a test room containing different prominent visual cues in each quadrant. Within 60 seconds, each rat was randomly placed in one of the four starting quadrants, facing the pool wall, looking for a hidden circular platform (diameter 11 cm, 1–2 cm underwater). Regardless of whether the rat found the platform or swam in the water for 60 seconds, we placed it on the platform and rested for 20 seconds. Four tests were performed from each of the four quadrants every day for 5 consecutive days. The escape latency (EL) was defined as the time from the start of the recording to the finding of the platform. The average time calculated from the 4 quadrants in 5 days represents the performance of the rats during this process. On the sixth day, the platform was removed. The rats were allowed to swim in the water for 60 s, and the swimming time in the platform quadrant was recorded as the space exploration time (SET). The swimming lane, space exploration time and swimming speed for calculating escape latency are recorded through the video track.
Western Blotting
Proteins were extracted from hippocampus tissue in lysis buffer with protease inhibitor. The protein concentration was detected with a BCA kit, and the same amount of protein was subjected to 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, electrotransferred onto a polyvinylidene fluoride membrane, and blocked with 5% skimmed milk for 2 h. Membranes were incubated with the primary antibodies against mBDNF, proBDNF, p-TrkB, TrkB, p-PI3K, PI3K, p-AKT, AKT, p-CREB, CREB, caspase 3, GAPDH (1:1000, Proteintech, USA) overnight at 4°C, followed by incubation with a secondary horseradish peroxidase-conjugated antibody (1:4000, Proteintech, USA) at room temperature for 1h. The bands were visualized using a chemiluminescence (ECL) Western blotting detection kit.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism 5 (Graphpad Software, Inc., San Diego, CA) and SPSS version 20.0 (SPSS Inc., Chicago, IL). Data were presented as mean ± Standard Deviation (SD). The differences between different groups analyzed by one-way analysis of variance followed by Tukey post hoc comparisons were considered as statistically significant when p value <0.05.
Results
Lipid, Edaravone, ANA-12, and 7,8-DHF Did Not Induce Caspase-3 Activation in Newborn Rats
We demonstrated that the administration of intralipid, edaravone, ANA-12, or 7,8-DHF did not significantly affect the hippocampal expression of cleaved caspase-3 compared to that in the control group at 6h (Figure 2A and B, F (4,10) = 0.212, p >0.05 vs control), thus confirming that intralipid, edaravone, ANA-12, and 7,8-DHF did not induce neural apoptosis.
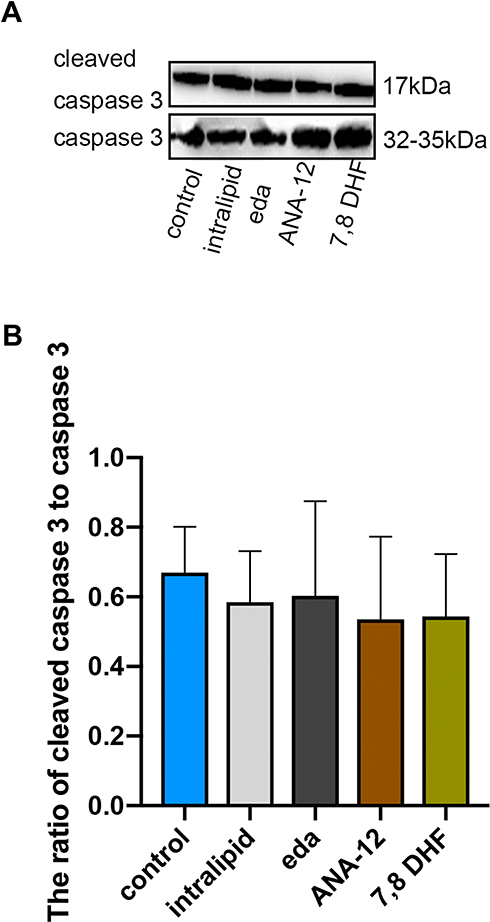 |
Figure 2 Intralipid, edaravone, ANA-12 and 7,8-DHF did not increase expression of cleaved caspase-3 in the hippocampus of P7 rats. (A) Representative Western blot of cleaved caspase-3; (B) quantitative analysis of cleaved caspase-3 in the hippocampus 6 hours. Results were the mean ±SD. Abbreviations: eda, edaravone; 7,8 DHF, 7,8-dihydroxyflavone. |
Edaravone Pretreatment Promoted Propofol-Induced Hippocampal Neural Stem Cells Proliferation in Newborn Rats and HT22 Cells
Neurogenesis is a vital procedure in brain development and includes neural stem cell proliferation.7 This proliferation was verified by measurement of ki67 positivity in the present study. The effects of propofol on neural stem cell proliferation were confirmed by the observation of a decreased number of ki67-positive cells. The pro group showed a reduced number of ki67-positive neural stem cells (Figure 3A and B, F (2,12) = 20.22, p <0.001 vs control group). Notably, edaravone pretreatment effectively increased neural stem cell proliferation compared to only propofol exposure (Figure 3A and B, p< 0.001 vs pro group). Pretreatment with edaravone significantly also markedly elevated cell viability compared to 100 μM propofol in HT22 cells (Figure 3C, p <0.001).
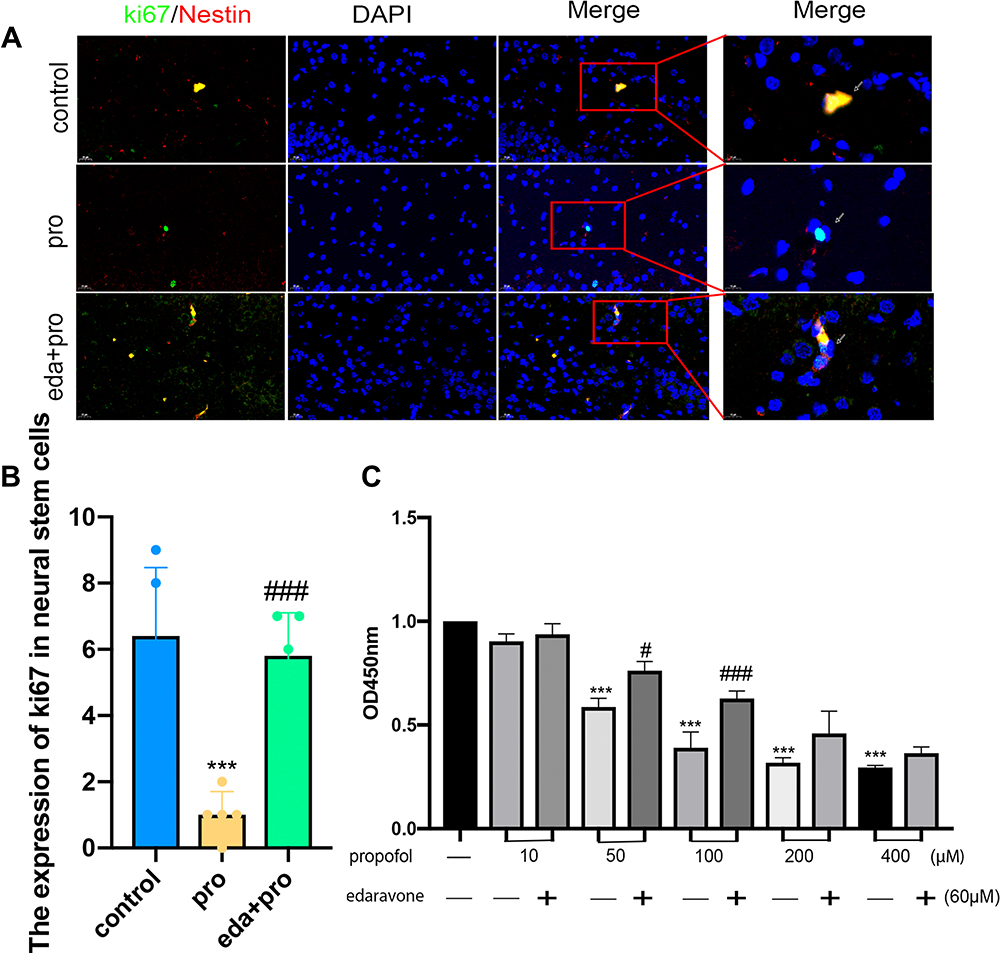 |
Figure 3 Pretreatment with edaravone attenuated propofol-induced reducing neural stem cells proliferation in the hippocampus of P7 rats and HT22 cells. (A) Representative ki67 images of neural stem cells. Green staining indicated ki67 positive cells and red staining indicated neural stem cells (Nestin). Scan bar = 20μm (40X); 10μm (100X). (B) Quantification of ki67 positive cells of neural stem cells in the hippocampus. (C) CCK8 detected that the proliferation of HT22 cells decreased with the increase of propofol dose, and edaravone increased cell proliferation in 50μM,100μM propofol. Results were the mean ±SD. ***p<0.001 as compared with control group. #p<0.05, ###p<0.001 as compared with corresponding to the concentration of propofol group. Abbreviations: pro, propofol; eda, edaravone. |
Edaravone Alleviated Propofol-Induced Hippocampal Cell Apoptosis and Reduced Astrocytes and Microglial Cells in Newborn Rats
We first evaluated the effect of edaravone pretreatment on 6 h of propofol exposure. As shown in Figure 4A and B, cleaved caspase 3 levels were significantly increased in both the pro and ANA-12+pro groups compared to that in the control group (F (4,10) =40.76, p<0.001). In contrast, the levels of cleaved caspase 3 were decreased in the eda+pro and 7,8- DHF+pro groups (p <0.001 vs, pro group), suggesting that apoptosis-related proteins cleaved caspase 3 supported the anti-apoptosis activity of edaravone. After propofol exposure, apoptosis was significantly increased in hippocampal neurons, astrocytes, and microglial cells (Figure 4C–F, F (4,60) =50.22, p <0.001 vs control group). However, edaravone pretreatment dramatically inhibited the apoptosis of neurons (p <0.01), astrocytes (p <0.001), and microglial cells (p <0.001) in the hippocampus compared to the pro group. Meanwhile, the numbers of astrocytes (Figure 4G, F (4,60) =10.55, p <0.05 vs control group) and microglial cells (p <0.001 vs control group) were increased in the propofol-induced and ANA-12 pretreatment groups but were restored with edaravone or 7.8-DHF pretreatment (p <0.05; p <0.01; p <0.001 vs pro group). Based on these findings, we hypothesized that edaravone alleviated propofol-induced hippocampal cell apoptosis and decreased astrocyte and microglial cell numbers.
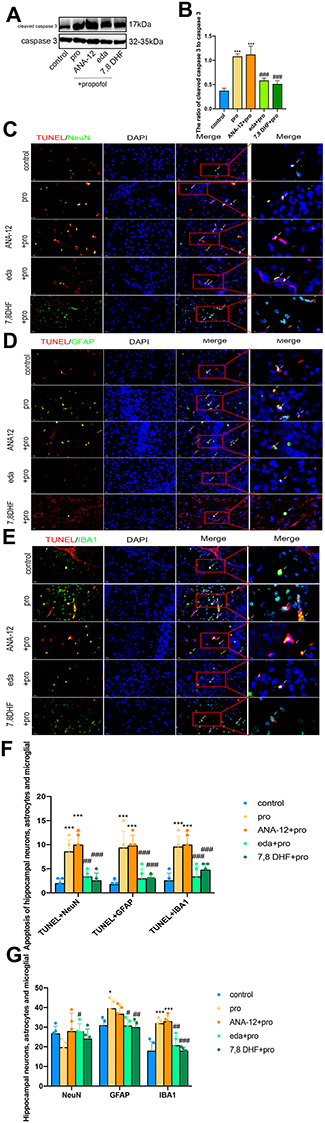 |
Figure 4 Pretreatment with edaravone attenuated propofol-induced neuroapoptosis in the hippocampus of P7 rats. (A) Representative Western blot of cleaved caspase-3 in the hippocampus 6 h in five groups. The quantitative analysis of cleaved caspase-3 at 6h (B). (C–E) Representative TUNEL images of neurons (C), astrocytes (D) and microglial cells (E) in the hippocampal areas. Green staining indicated TUNEL positive cells and red staining indicated neurons, astrocytes and microglial cells. Scan bar = 50μm (20X), 20μm (40X). (F) Quantification of TUNEL positive cells of neurons, astrocytes and microglial cells in the hippocampal areas. (G) Quantification of positive cells of neurons, astrocytes and microglial cells in the hippocampal areas. Results were the mean ±SD. *p <0.05, ***p<0.001 as compared with control group. #p<0.05, ##p<0.01, ###p<0.001 as compared with propofol group. Abbreviations: 7,8 DHF, 7,8-dihydroxyflavone; pro, propofol; eda, edaravone. |
Edaravone Reduced Propofol-Induced Cell Apoptosis in HT22 Cells and Propofol Did Not Affect Long-Term Learning and Memory
Flow cytometry analysis showed that edaravone significantly reduced propofol-induced apoptosis in HT22 cells (Figure 5A and B, F (2,6) =533.6, p <0.001 vs pro group). However, excessive propofol did not cause long-term learning and memory effects, as compared to the control group, the escape latency of the first five days was not statistically significant (Figure 5C, F (4,100) =2.76, p >0.05 vs control group). The learning and memory of the TrkB inhibitor group were worse than that of the control group on the sixth day of the target space exploration time (Figure 5D, F (4,20 =3.81, p <0.05 vs control group).
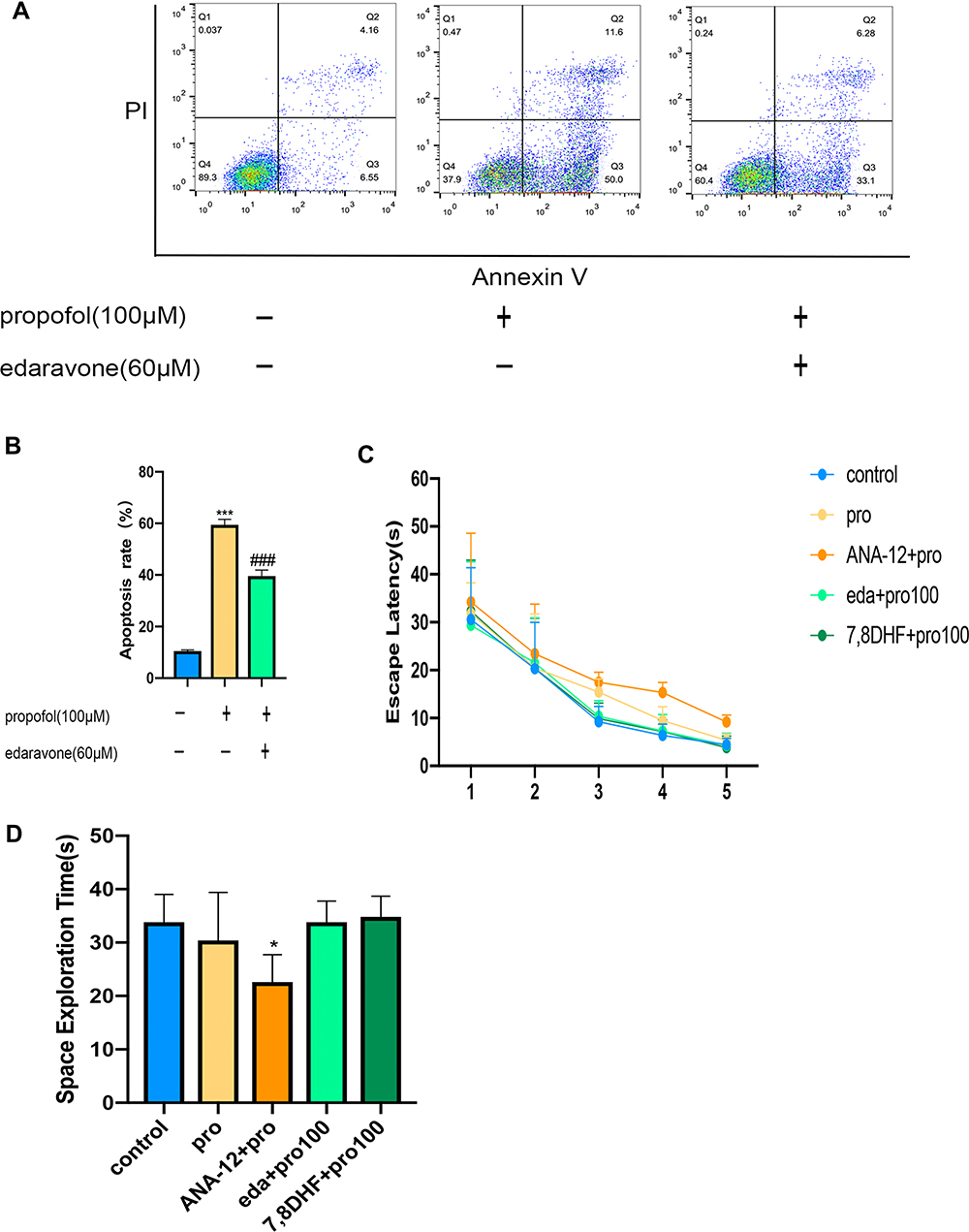 |
Figure 5 Edaravone reduced propofol-induced neuroapoptosis in HT22 cells and MWM detected long-term learning and memory. (A) Representative flow cytometry of cell apoptosis in three groups. (B) The quantitative analysis of apoptosis rate. (C) Escape latency proved rats in five groups were not statistically significant. (D) Rats in ANA-12+pro group reduced the space exploration. Results were the mean ±SD. *p <0.05, ***p<0.001 as compared with control group. ###p<0.001 as compared with propofol group. Abbreviations: 7,8 DHF, 7,8-dihydroxyflavone; pro, propofol; eda, edaravone. |
Edaravone Inhibited the Production of Proinflammatory Cytokines After Treatment with Excessive Propofol
We assessed the role of edaravone in propofol-induced proinflammatory cytokines of the hippocampus. In our study, edaravone or 7,8-DHF pretreatment decreased the production of TNF-α and IL-6 which was elevated after propofol exposure (Figure 6A, F (4,20) =12.09, p <0.05 vs pro group). These results suggested that edaravone protected developing rats against propofol-induced neurotoxicity by regulating proinflammatory cytokines.
 |
Figure 6 Edaravone inhibited the production of proinflammatory cytokines after excessive propofol treatment and regulates the ratio of mBDNF to proBDNF. (A) IL-6 and TNF-α were measured with edaravone. ANA-12 and 7.8 DHF intervention after propofol-induced P7 rats by ELISA. (B) Expression of mRNA levels of BDNF. (C and D) Representative Western blot of proBNDF and mBDNF in the hippocampus 6 h in three groups. The quantitative analysis of proBNDF and mBDNF at 6h (D). Results were the mean ±SD. *p<0.05, ***p<0.001 as compared with control group. #p<0.05, ###p<0.001 as compared with propofol group. Abbreviations: pro, propofol; eda, edaravone; IL-6, interleukin 6; TNF-α, tumor necrosis factor α; mBDNF, mature brain-derived neurotrophic factor. |
Edaravone Pretreatment Increased the Level of Hippocampal mBDNF in Developing Rats Exposed to Propofol
A lack of mBDNF significantly reduces synaptic plasticity, leading to decreased learning and memory.25 To determine the relationship between mBDNF and proBDNF, we first explored the mRNA expression of BDNF by quantitative reverse transcription-polymerase chain reaction (q-RT PCR), which did not differ significantly between groups (Figure 6B, F (2.6) =1.074, p >0.05 vs control group). However, edaravone pretreatment ameliorated the propofol-induced decrease in hippocampal mBDNF (Figure 6C and D, F (2.12) =29.73, p <0.001). Meanwhile, edaravone pretreatment inhibited the BDNF precursor, proBDNF (p <0.001 vs pro group), suggesting that edaravone promoted BDNF expression after propofol exposure to activate the downstream TrkB/PI3K signaling pathway.
Edaravone Increased TrkB/PI3K Signaling Pathway Activity to Inhibit Propofol-Induced Neurotoxicity
Double immunofluorescence staining was used to detect TrkB in hippocampal neurons, astrocytes, and microglial cells. As expected, TrkB was activated in all three cell types in the eda+pro group (Figure 7A). The ratios of phosphorylated TrkB to TrkB (Figure 7B and C, F (4,10) = 8.27, p <0.01 vs control), phosphorylated PI3K to PI3K (Figure 7B and D, F (4,10) = 21.32, p <0.001 vs control), phosphorylated protein kinase B (AKT) to AKT (Figure 7B and E, F (4,10) = 61.28, p <0.001 vs control), and phosphorylated CREB to CREB (Figure 7B and F, F (4,10) =35.36, p <0.001 vs control) decreased significantly after propofol treatment, indicating decreased activity of the TrkB/PI3K signaling pathway. This effect was reversed by edaravone (p <0.01 or p <0.001 vs pro group). Thus, edaravone decreased neurotoxicity after propofol treatment by increasing TrkB/PI3K pathway activation.
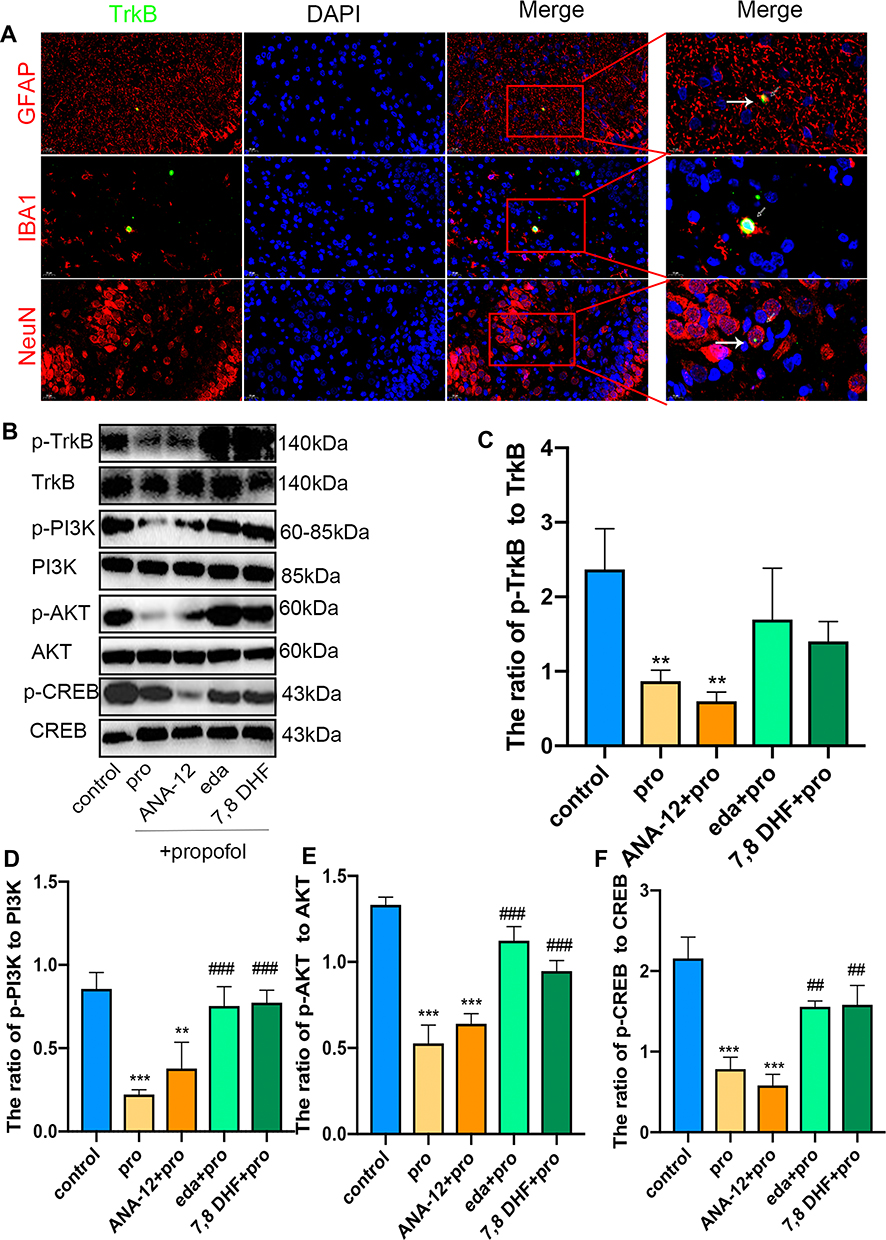 |
Figure 7 Edaravone increased the activity of the TrkB/PI3K/AKT signaling pathway to inhibit propofol-induced neurotoxicity. (A) Representative TrkB images of neurons, astrocytes and microglial cells in the hippocampal areas of eda+pro group. Green staining indicated TrkB positive cells and red staining indicated neurons, astrocytes and microglial cells. Scan bar = 20μm (40X), 10μm (100X). (B–F) Representative Western blot of p-TrkB, TrkB, p-PI3K, PI3K, p-AKT, AKT, p-CREB and CREB in the hippocampus 6 h in five groups. The quantitative analysis of p-TrkB and TrkB (C), p-PI3K and PI3K (D), p-AKT and AKT (E), p-CREB and CREB (F). Results were the mean ±SD. **p<0.01, ***p<0.001 as compared with control group. ##p<0.01, ###p<0.001 as compared with propofol group. Abbreviations: 7,8 DHF, 7,8-dihydroxyflavone; pro, propofol; eda, edaravone; TrkB, tropomyosin receptor kinase B; PI3K, phosphoinositide 3-kinase; AKT, phosphorylated protein kinase B; CREB, cyclic AMP response element-binding protein. |
Discussion
The mechanisms of anesthetic neurotoxicity include excessive and abnormal activation of the N-methyl-D-aspartate (NMDA) and GABA-A receptors,26 generation of free radicals,27 activation of P75 growth factor,28 inflammation, and excessive increase of Ca2,29 We considered using edaravone as a free radical scavenger to reduce anesthetic neurotoxicity. In cell proliferation, nestin is an intermediate neurofilament protein that can be used as a marker of neural stem cells for immunolocalization, indicating that neural stem cells are actively proliferating based on the incorporation of ki67. The down-regulated double-labeled immunofluorescent staining of ki67 and nestin in the propofol group in this study showed that propofol inhibited neural stem cell proliferation resulting in a relatively reduced transformation from neural stem cells to neurons and glial cells in the developing hippocampus. In the CCK8 detection of cell proliferation, propofol dose-dependently reduced HT22 cell proliferation. Edaravone pretreatment significantly increased cell viability in the reduction of cell proliferation mediated by propofol, which may be related to the elimination of free radicals produced by propofol and the maintenance of normal neuronal cell proliferation.
Regarding neuroapoptosis and neuroinflammation, excessive astrocytes affected normal cell development,12 while microglial activation caused the release of large amounts of pro-inflammatory factors.30 We found that edaravone reduced the numbers of astrocytes and microglia while reducing the expression of pro-inflammatory factors. This has also been demonstrated in previous studies. Edaravone protects astrocytes through the PI3K/Akt pathway against H2O2-induced oxidative stress and LPS-induced pro-inflammatory responses.31 Edaravone reduces oxidative stress by up-regulating the Nrf2/ARE pathway and also inhibiting inflammation by inhibiting the activation of NF-κB.32 The antioxidant capacity of edaravone has been confirmed in many disease models. For example, in cisplatin,32 excessive glutamate,33 and cyclophosphamide34 treatments, edaravone increased superoxide dismutase (SOD) and glutathione (GSH) levels, reduced malonaldehyde (MDA), levels, and showed an antioxidant effect. Edaravone combined with the lipid system enhanced the antioxidant function of edaravone.35 Caspase-3, as a member of the caspase family, is S-nitrosylated on its catalytic site Cys163, which inhibits its protease activity.18,36 The levels of cleaved caspase 3 were significantly increased in the propofol group, leading to cell death.36 Taken together, these results supported our hypothesis that excessive propofol can lead to neuroapoptosis and neuroinflammation in the developing hippocampus.
BDNF mRNA levels did not differ significantly among groups, suggesting that edaravone pretreatment or excessive propofol exposure affected the total expression of BDNF mRNA. However, the mBDNF/proBDNF ratio was decreased in the propofol group but was elevated in the edaravone pretreatment group. These findings indicated that the neuroprotective effect of edaravone was based on both antioxidant effects and enhanced synthesis of mBDNF.22 Thus, edaravone activated downstream TrkB expression by increasing mBDNF and decreasing proBDNF. To verify that edaravone exerted its neuroprotective effects by activating TrkB/PI3K signaling, we applied TrkB agonist 7, 8-DHF, and TrkB antagonist ANA-12 pretreatment to confirm the reliability of the pathway. Edaravone and 7,8-DHF pretreatment not only ameliorated neural stem cell proliferation but also reduced neural apoptosis and neuroinflammation.
Edaravone reversed nerve damage and inflammation caused by ANA-12 pretreatment. These findings suggest the neuroprotective effects of edaravone through the TrkB receptor. A study reported that TNF-α sustained neutrophil recruitment during inflammation through endothelial activation, while IL-6 regulated neutrophil trafficking during acute inflammation via STAT3.20 In the present study, we observed decreased secretion of TNF-α, IL-6, and IL-1β in developing rats after the administration of edaravone and TrkB agonist 7,8-DHF compared to excessive propofol-stimulated rats administered the TrkB antagonist ANA-12, suggesting that edaravone may provide anti-inflammatory effects through TrkB receptor activation. Recently, mBDNF was shown to bind to TrkB and exert a pro-survival effect by activating downstream signaling pathways, including the PI3K/AKT pathway, which could provide neuroprotective effects by reducing striatum apoptosis.12 The cyclic AMP response element-binding protein (CREB), which promotes continuous plastic changes and establishment of synaptic connections with other neurons in neurogenesis, is activated by the phosphorylation of AKT, while the translocation of CREB binding protein (CBP) to the promoter triggered the expression of protective proteins.12,36–38
In conclusion, edaravone pretreatment ameliorated propofol-induced proliferation inhibition, neuroapoptosis, and neural inflammatory through activation of the mBDNF/TrkB/PI3K pathway.
Data Sharing Statement
Data supporting the findings of our study are obtained from corresponding authors upon reasonable request.
Acknowledgment
This project was supported by the Key Specialty Construction Project of Pudong Health and Family Planning Commision of Shanghai (PWZzk2017-06).
Disclosure
The authors confirm that there are no conflicts of interest in this work.
References
1. Liu F, Liu S, Patterson TA, et al. Protective effects of xenon on propofol-induced neurotoxicity in human neural stem cell-derived models. Mol Neurobiol. 2020;57:200–207. doi:10.1007/s12035-019-01769-5
2. Ren G, Zhou Y, Liang G, et al. General anesthetics regulate autophagy via modulating the inositol 1,4,5-Trisphosphate receptor: implications for dual effects of cytoprotection and cytotoxicity. Sci Rep. 2017;7:12378. doi:10.1038/s41598-017-11607-0
3. Tesic V, Joksimovic SM, Quillinan N, et al. Neuroactive steroids alphaxalone and CDNC24 are effective hypnotics and potentiators of GABA (A) currents, but are not neurotoxic to the developing rat brain. Br J Anaesth. 2020;124:603–613. doi:10.1016/j.bja.2020.01.013
4. Upton DH, Popovic K, Fulton R, et al. Anaesthetic-dependent changes in gene expression following acute and chronic exposure in the rodent brain. Sci Rep. 2020;10:9366. doi:10.1038/s41598-020-66122-6
5. Ni GX, Liang C, Wang J, et al. Astragaloside IV improves neurobehavior and promotes hippocampal neurogenesis in MCAO rats through BDNF-TrkB signaling pathway. Biomed Pharmacother. 2020;130:110353. doi:10.1016/j.biopha.2020.110353
6. Xu F, Ma R, Zhang G, et al. Estrogen and propofol combination therapy inhibits endoplasmic reticulum stress and remarkably attenuates cerebral ischemia-reperfusion injury and OGD injury in hippocampus. Biomed Pharmacother. 2018;108:1596–1606. doi:10.1016/j.biopha.2018.09.167
7. Li J, Guo M, Liu Y, et al. Both GSK-3β/CRMP2 and CDK5/CRMP2 pathways participate in the protection of dexmedetomidine against propofol-induced learning and memory impairment in neonatal rats. Toxicol Sci. 2019;171:193–210. doi:10.1093/toxsci/kfz135
8. Moon DU, Esfahani-Bayerl N, Finke C, et al. Propofol modulates early memory consolidation in humans. eNeuro. 2020;7:7. doi:10.1523/ENEURO.0537-19.2020
9. Li Y, Jia C, Zhang D, et al. Propofol-induced neurotoxicity in hESCs involved in activation of miR-206/PUMA signal pathway. Cancer Biomarkers Section a Dis Markers. 2017;20:175–181. doi:10.3233/CBM-170167
10. Wang Y, Tian D, Wei C, et al. Propofol attenuates α-synuclein aggregation and neuronal damage in a mouse model of ischemic stroke. Neurosci Bull. 2020;36:289–298. doi:10.1007/s12264-019-00426-0
11. Yang L, Ton H, Zhao R, et al. Sevoflurane induces neuronal activation and behavioral hyperactivity in young mice. Sci Rep. 2020;10:11226. doi:10.1038/s41598-020-66959-x
12. Du L, Mei Z, Huang Y, et al. Protection of the Geum japonicum Thunb. var. chinense extracts against oxygen-glucose deprivation and re-oxygenation induced astrocytes injury via BDNF/PI3K/Akt/CREB pathway. Biomed Pharmacother. 2020;127:110123. doi:10.1016/j.biopha.2020.110123
13. Wang G, Su J, Li L, et al. Edaravone alleviates hypoxia-acidosis/reoxygenation-induced neuronal injury by activating ERK1/2. Neurosci Lett. 2013;543:72–77. doi:10.1016/j.neulet.2013.02.067
14. Miguez MJ, Bueno D, Espinoza L, et al. Among Adolescents, BDNF and Pro-BDNF lasting changes with alcohol use are stage specific. Neural Plast. 2020;2020:3937627. doi:10.1155/2020/3937627
15. Barreda Tomás FJ, Turko P, Heilmann H, et al. BDNF expression in cortical GABAergic interneurons. Int J Mol Sci. 2020;21:21. doi:10.3390/ijms21051567
16. Wu C, Lu J, Lu S, et al. Increased ratio of mature BDNF to precursor-BDNF in patients with major depressive disorder with severe anhedonia. J Psychiatr Res. 2020;126:92–97. doi:10.1016/j.jpsychires.2020.05.010
17. Kojima M, Otabi H, Kumanogoh H, et al. Reduction in BDNF from inefficient precursor conversion influences nest building and promotes depressive-like behavior in mice. Int J Mol Sci. 2020:21. doi: 10.3390/ijms21113984.
18. Hao LY, Dong L, Yu QX, et al. Edaravone inhibits procaspase-3 denitrosylation and activation through FasL-Trx2 pathway in KA-induced seizure. Fundam Clin Pharmacol. 2020;34:662–670. doi:10.1111/fcp.12556
19. Shi X, Ohta Y, Shang J, et al. Neuroprotective effects of SMTP-44D in mice stroke model in relation to neurovascular unit and trophic coupling. J Neurosci Res. 2018;96:1887–1899. doi:10.1002/jnr.24326
20. Zhao X, Zhang E, Ren X, et al. Edaravone alleviates cell apoptosis and mitochondrial injury in ischemia-reperfusion-induced kidney injury via the JAK/STAT pathway. Biol Res. 2020;53:28. doi:10.1186/s40659-020-00297-0
21. Alzoubi KH, Al Mosabih HS, Mahasneh AF. The protective effect of edaravone on memory impairment induced by chronic sleep deprivation. Psychiatry Res. 2019;281:112577. doi:10.1016/j.psychres.2019.112577
22. Okuyama S, Morita M, Sawamoto A, et al. Edaravone enhances brain-derived neurotrophic factor production in the ischemic mouse brain. Pharmaceuticals (Basel, Switzerland). 2015;8:176–185. doi:10.3390/ph8020176
23. Zhou J, Wang F, Zhang J, et al. The interplay of BDNF-TrkB with NMDA receptor in propofol-induced cognition dysfunction: mechanism for the effects of propofol on cognitive function. BMC Anesthesiol. 2018;18:35. doi:10.1186/s12871-018-0491-y
24. Pandey SN, Kwatra M, Dwivedi DK, et al. 7,8-Dihydroxyflavone alleviated the high-fat diet and alcohol-induced memory impairment: behavioral, biochemical and molecular evidence. Psychopharmacology. 2020;237:1827–1840. doi:10.1007/s00213-020-05502-2
25. Dong Y, Hong W, Tang Z, et al. Dexmedetomidine attenuates neurotoxicity in developing rats induced by sevoflurane through upregulating BDNF-TrkB-CREB and downregulating ProBDNF-P75NRT-RhoA signaling pathway. Mediators Inflamm. 2020;2020:5458061. doi:10.1155/2020/5458061
26. Ishiguro M, Kobayashi S, Matsuyama K, et al. Effects of propofol on IPSCs in CA1 and dentate gyrus cells of rat hippocampus: propofol effects on hippocampal cell’s IPSCs. Neurosci Res. 2019;143:13–19. doi:10.1016/j.neures.2018.05.003
27. Zhang C, Wang Y, Jin J, et al. Erythropoietin protects propofol induced neuronal injury in developing rats by regulating TLR4/NF-kappaB signaling pathway abstract. Neurosci Lett. 2019;712:134517. doi:10.1016/j.neulet.2019.134517
28. Schilling JM, Kassan A, Mandyam C, et al. Inhibition of p75 neurotrophin receptor does not rescue cognitive impairment in adulthood after isoflurane exposure in neonatal mice. Br J Anaesth. 2017;119:465–471. doi:10.1093/bja/aew299
29. Xing N, Xing F, Li Y, et al. Dexmedetomidine improves propofol-induced neuronal injury in rat hippocampus with the involvement of miR-34a and the PI3K/Akt signaling pathway. Life Sci. 2020;247:117359. doi:10.1016/j.lfs.2020.117359
30. Wang T, Zhu H, Hou Y, et al. Galantamine reversed early postoperative cognitive deficit via alleviating inflammation and enhancing synaptic transmission in mouse hippocampus. Eur J Pharmacol. 2019;846:63–72. doi:10.1016/j.ejphar.2018.12.034
31. Guo Z, Wu HT, Li XX, et al. Edaravone protects rat astrocytes from oxidative or neurotoxic inflammatory insults by restoring Akt/Bcl-2/Caspase-3 signaling axis. IBRO Rep. 2020;8:122–128. doi:10.1016/j.ibror.2020.04.003
32. Jangra A, Kwatra M, Singh T, et al. Edaravone alleviates cisplatin-induced neurobehavioral deficits via modulation of oxidative stress and inflammatory mediators in the rat hippocampus. Eur J Pharmacol. 2016;791:51–61. doi:10.1016/j.ejphar.2016.08.003
33. Bai X, Zhang C, Chen A, et al. Protective effect of edaravone on glutamate-induced neurotoxicity in spiral ganglion neurons. Neural Plast. 2016;2016:4034218. doi:10.1155/2016/4034218
34. Singh S, Kumar A. Protective effect of edaravone on cyclophosphamide induced oxidative stress and neurotoxicity in rats. Curr Drug Saf. 2019;14:209–216. doi:10.2174/1574886314666190506100717
35. Minnelli C, Laudadio E, Galeazzi R, et al. Synthesis, characterization and antioxidant properties of a new lipophilic derivative of edaravone. Antioxidants. 2019;8:258. doi:10.3390/antiox8080258
36. Niu HM, Ma DL, Wang MY, et al. Epimedium flavonoids protect neurons and synapses in the brain via activating NRG1/ErbB4 and BDNF/Fyn signaling pathways in a chronic cerebral hypoperfusion rat model. Brain Res Bull. 2020;162:132–140. doi:10.1016/j.brainresbull.2020.06.012
37. Lv J, Lu C, Jiang N, et al. Protective effect of ginsenoside Rh2 on scopolamine-induced memory deficits through regulation of cholinergic transmission, oxidative stress and the ERK-CREB-BDNF signaling pathway. Phytother Res. 2021;35:337–345. doi:10.1002/ptr.6804
38. Tao H, Chen X, Du Z, et al. Corn silk crude polysaccharide exerts anti-pancreatic cancer activity by blocking the EGFR/PI3K/AKT/CREB signaling pathway. Food Funct. 2020;11:6961–6970. doi:10.1039/d0fo00403k
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.