Back to Journals » Vascular Health and Risk Management » Volume 17
Early Detection of Negative Smoking Impacts: Vascular Adaptation Deviation Based on Quantification of Circulated Endothelial Activation Markers
Authors Kumboyono K ![]() , Nurwidyaningtyas W
, Nurwidyaningtyas W ![]() , Chomsy IN
, Chomsy IN ![]() , Wihastuti TA
, Wihastuti TA ![]()
Received 8 December 2020
Accepted for publication 3 March 2021
Published 23 March 2021 Volume 2021:17 Pages 103—109
DOI https://doi.org/10.2147/VHRM.S296293
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Harry Struijker-Boudier
Kumboyono Kumboyono,1 Wiwit Nurwidyaningtyas,2 Indah Nur Chomsy,2 Titin Andri Wihastuti3
1Nursing Department, Faculty of Medicine, University of Brawijaya, Malang, 65145, Indonesia; 2Doctoral Program of Medical Science, Faculty of Medicine, University of Brawijaya, Malang, 65145, Indonesia; 3Department of Biomedicine, Faculty of Medicine, University of Brawijaya, Malang, 65145, Indonesia
Correspondence: Kumboyono Kumboyono Email [email protected]
Introduction: Smoking can cause vascular damage in the form of an inflammatory reaction characterized by endothelial activation. Endothelial activation forms a pathological adaptation pattern so that it can induce the atherogenesis process. Several markers, such as E-selectin, platelet-derived micro particles (PMPs) and hematopoietic stem cell (HSC) can identify the activation of endothelial in circulating blood. Therefore, the deviation of vascular adaptation due to smoking can be detected early through the feedback mechanism between E-selectin, PMPs, and HSC.
Purpose: This study aims to analyze the initial picture of the negative impact of smoking on vascular adaptation by measuring E-selectin, PMPs, and HSC in the peripheral blood circulation. Participant criteria and methods: Peripheral blood samples (5 mL) were taken from each participant, both the smoking group (n = 30) and the non-smoker group (n = 31) to obtain peripheral blood mononuclear cells (PBMNC). PBMNC was isolated using ficoll-based gradient centrifugation. The flow cytometry assay method used to measure the E-selectin, PMPs and hematopoietic stem cells.
Results: The mean of circulating E-selectin in smokers was higher than that of non-smokers. On the other hand, the average number of PMPs and HSCs in smokers was lower than non-smokers.
Conclusion: Smoking increases the risk of accelerated vascular block formation, as indicated by an increase in the amount of circulating E-selectin. The increase in E-selectin in the blood vessels mediates the increased adhesion of PMPs in the vascular area so that the number of circulating PMPs in smokers decreases. The decrease in circulating PMPs decreases the signal of vascular repair, which is characterized by a decline in the number of HSCs.
Keywords: endothelial injury, cardiovascular disease, atherosclerosis, peripheral blood mononuclear cells, smoker
Introduction
Smoking is one of the risky behaviours that can increase cardiovascular disease incidence. The long manifestation of smoking’s adverse effect has to lead the perception among smokers that smoking does not harm health. Therefore, it is necessary to conduct studies that can detect the progression of cardiovascular disorders before the actual manifestation of the disease.
The endothelium has an essential role in regulating the coagulation system and vascular tone.1 Changes in the structure and function of the endothelium are a description of the response to vascular damage.2–4 Long-term smoking habits can interfere with endothelial function through the oxidative stress pathway.5,6 Nicotine is the most toxic component of cigarettes that can induce endothelial cell death.7–10 The increasing the number of circulating endothelial cells which is the final stage of the endothelial damage process is a sign for the endothelial cell death.11 Endothelial damage can be preceded by endothelial activation, which is indicated by an increase in the amount of E-selectin.1 E-Selectin or the so-called CD62 antigen-like family member E (CD62E) is an adhesion molecule that expressed when endothelial cells are activated by inflammation.12 E-selectin is also present in activated platelets as a consequence of endothelial injury.13 E-Selectin mediates leucocyte adherence to injury areas and stimulates vascular inflammatory responses.13–15 E-Selectin is also considered a component involved in the self-renewal regulation of endothelial injury by Hematopoietic Stem Cell (HSC). Because of this dual role, E-selectin referred to as a regulator of cell-trafficking to areas of inflammation in humans.16 Soluble E-selectin can be identified in blood circulation by the ELISA method.15,17
Endothelial damage causes loss of the primary vascular protective barrier so that circulating platelets adhere to collagen easily beneath the endothelial cells. Attachment of platelets to the area of endothelial injury causes platelets to become activated and produce higher amounts of Platelet-Derived Micro Particles (PMPs). The coagulation effect in the injured area by releasing thrombin, thereby increasing platelet adhesion to endothelial cells. At this stage thrombus formation in the injured area has begun.13,18,19
The presence of PMPs in the blood circulation, both in pathological and physiological conditions, can be identified by identifying the surface marker with the CD41 marker.20 A high circulating CD41 count can also be found in healthy individuals, not accompanied by endothelial/platelet activation. This condition is because megakaryocytes, as platelet precursors, also express CD41. So, the increase of CD41 levels in the circulation has two meanings, as a marker of platelet activation in pathological conditions and an increase in the number of megakaryocytes in physiological conditions.20
Increased circulating CD41 will be detected by bone marrow (BM) as a signal that causes platelet activation to signal increased vascular repair needs. BM will produce high amounts of HSC when an extensive vascular endothelial injury occurs. The presence of HSC in the circulation can be determined using CD133.21–23 Circulating HSCs will undergo maturation to become Progenitor Endothelial Cells (EPC). The presence of PMPs plays a role in facilitating the planting of progenitor cells in endothelial areas that experience injury by increasing the attachment of EPC to fibrinogen.24,25 Endothelial damage is known to cause various forms of cellular responses. To maintain vascular homeostasis, as a form of vascular adaptation, can be identified by quantifying the circulating soluble marker as a marker of endothelial activation. This study does not seem to present anything new because the markers used have been established. Still, the study of vascular status in heavy smokers (> 10 cigarettes per day) for five years at the molecular level is a research that has not been widely carried out. This study aims to analyze the differences in the initial view of vascular adaptation in the smoker and non-smoker groups by measuring the amount of E-selectin, PMPs, and HSC in the peripheral blood circulation.
Materials and Methods
Research Design and Baseline Characteristic of Participants
This study used a cross-sectional design—recruitment of participants through filling in the willingness to take part in research via a google form. The google form link is distributed through social media for two weeks to get two groups of participants, namely the smokers and non-smokers. The Google form contains statements about participants’ objectives, benefits, rights and obligations, as well as research procedures. Prospective participants are given the opportunity to learn the information, if the prospective participant has understood and agreed, they are asked to sign the informed consent electronically to follow the research.
The smoking group is consisted of people who had a smoking history of no more than five years at the time of the study. Meanwhile, the non-smoking group were people who had never smoked at all until the study ends. All participants in both the smoking and non-smoking groups had no history of cardiovascular and metabolic diseases such as diabetes mellitus, hypertension, infection, obesity, or cancer. The history of the disease was retrieved from the medical records of each participant. After two weeks of recruitment, there were 30 and 31 participants in the smoker and non-smoker groups, respectively.
Data Collection Procedure
Participants were confirmed not to be taking drugs before blood collection was carried out. Then vital signs (blood pressure, pulse, respiration, and body temperature) are examined. Blood collection is carried out in the Central Laboratory of Biomedical, Faculty of Medicine, Brawijaya University. Laboratory analysts did not know the smoking status of each participant during the process of collecting and analyzing the studied blood samples.
A total of 5 mL of peripheral blood which has been taken using the venous punction procedure, then accommodated in an Ethylene Diamine Tetra Acetic (EDTA) tube. Blood was diluted with Axisxhield’s Lymphoperm in the same volume ratio in a 15 mL tube, then centrifuged for 30 minutes at 6000 rpm. Buffy coat, which is the result of PBMNC centrifugation, was taken with a clean pipette and accommodated in a new tube and added up to 10 mL of phosphate-buffered saline (PBS), centrifuged again for 10 min at 1200 rpm. The PBS washing procedure repeated for two times. The pellets are ready for staining.
Blood samples for measurement of E-selectin, CD41, and CD133 were obtained from the participants’ peripheral blood from both smokers and non-smokers. None of the participants took antiplatelet drugs, anti-inflammatory drugs, or hypolipidemic agents. This screening excludes any conditions thought to influence the number of platelets microparticles as a parameter in the present study.
Flow Cytometry Analysis to Evaluate the Number of Circulating E-Selectin, PMPs and HSC
The detection of E-selectin was confirmed by staining Phycoerythrin (PE)-conjugated monoclonal anti-human CD62E Biolegend Cat No. 322606 on the obtained PBMNC. Platelet microparticles carried out by adding 5 µL of Peridinin Chlorophyll (PerCP)-conjugated anti-human von Willebrand Factor (vWF/CD41) from (Biolegend, UK; catalogue number: 303720). Meanwhile, measurement of HSC counts was confirmed by adding 5µL of anti-human CD133 Allophycocyanine (APC mouse-conjugated monoclonal antibody IgG2A clone # 170411 [FAB cat no. 11331A]. The flowcytometric analyzed using the BD FACSMelody ™ Cell Sorter, and BD FACSChorusTM software simplifies sorting by using a streamlined workflow.
Data Analysis
All data were presented as mean value ± standard deviation. The comparison between the two groups was analyzed by independent t-test. The statistical significance was assumed if the null hypothesis failed to reject based on p-value <0.05.
Ethical Clearance
This research has been approved by the Research Ethics Committee of Brawijaya University with Reference Number 1206-KEP-UB-2019. All of the patients participate in this study provided informed consent that has been in accordance with the Declaration of Helsinki.
Results
Characteristics of smokers and non-smokers: height, weight, and pulse can be seen in Table 1. Each of these characteristics was not significantly different in the two groups. The analysis then continued by identifying the differences in the number of E-Selectin, PMPs, and HSC between the smokers and non-smokers (Figure 1 and Table 2).
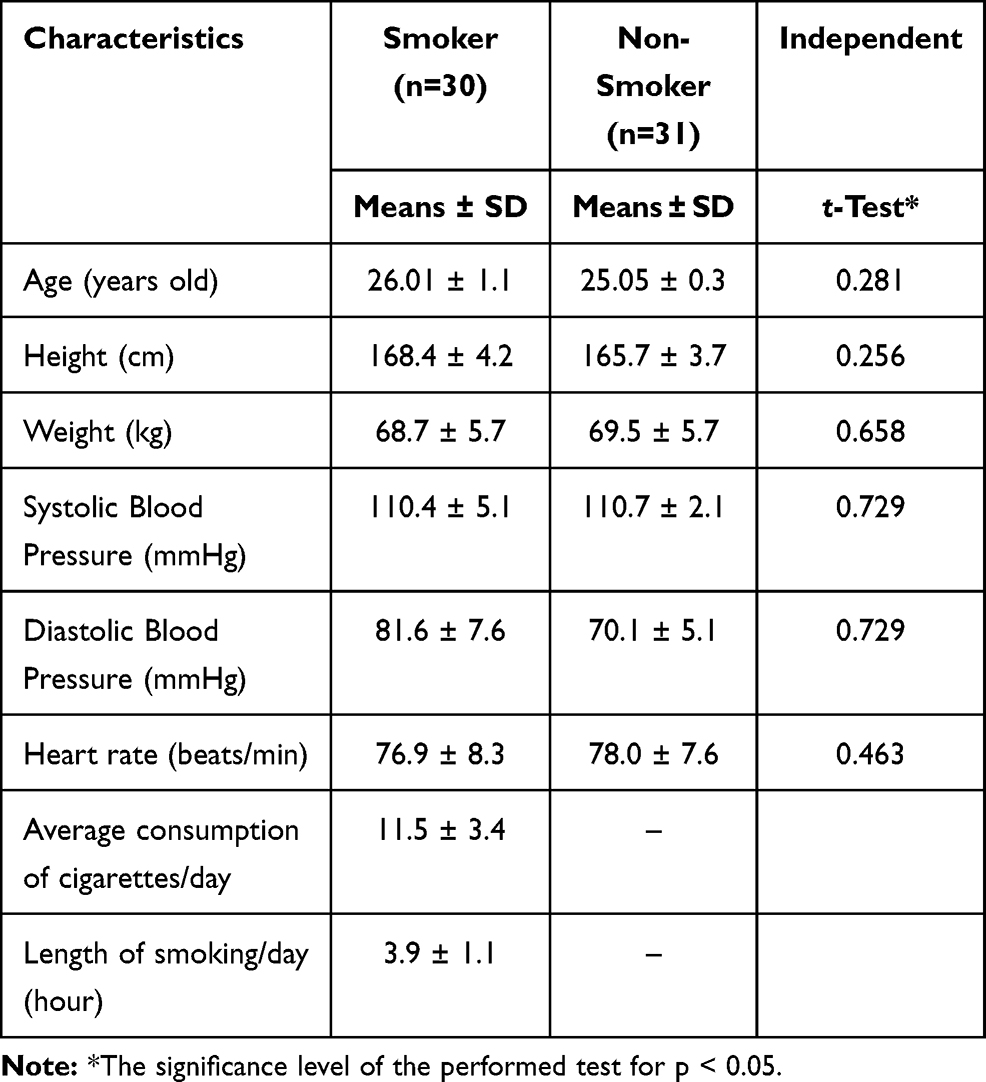 |
Table 1 The Baseline Characteristics of Smoker and Non-Smoker Group |
 |
Table 2 The Quantification of the Mean E-Selectin, PMPs, and HSC |
 |
Figure 1 Scatter plot diagram of the results of the analysis of E-selectin (CD62E), PMPS (CD41), and HSC (CD133) with flow cytometry. (A) Gate view of E-selectin analysis in smoker group. (B) Gate view of PMPs analysis in smokers group. (C) Gate view of HSC analysis in smokers group. (D) Gate view of E-selectin analysis in non-smokers analysis of PMPs in the non-smoker group. (E) Gate view of PMPs analysis in non-smokers group. (F) Gate view of HSCs analysis in non-smokers group. |
Based on Figure 1, it is known that the mean number of E-selectin (measured by the CD62E marker) in the smoker group was higher than in the non-smoker group. Meanwhile, the mean number of PMPs (measured through the CD41 marker) in the smoker group was lower than that of non-smokers. Meanwhile, the mean number of HSCs (measured using the CD133 marker) in the smoker group was lower in the non-smoker group. This condition indicates endothelial activation, which indicates vascular inflammation in the smoker group.
Table 2 shows that there is a significant difference between the mean E-selectin, PMPs, and HSC between the smoker and non-smoker groups. The mean of circulating E-selectin in smokers was higher than that of non-smokers. On the other hand, the mean of PMPs and HSCs in smokers was lower than that of non-smokers.
Discussion
Atherosclerotic disease is associated with endothelium function changes that occur in some CVD risk groups such as smokers, diabetes, hypertension and hyperlipidemia. Various previous studies have suggested that smoking can cause damage endothelium function.26 Damage to endothelium function due to long-term smoking habits is caused by increased production of free radicals that cause endothelial cell degradation resulting in decreased nitric oxide production.5,6 Some endothelial activation markers become the initial stage of vascular lesions’ development.27
The endothelial cells activate several Endothelial Microparticles (EMPs) released into circulation. The EMPs can be measure through surface makers such as E-selectin (CD62E), vascular cell adhesion molecules-1 (VCAM-1), and intercellular adhesion molecules-1 (ICAM-1).28 The platelets will increase rapidly to respond to endothelial injuries and secrete granule content into circulation called PMPs. Some platelet activation markers in circulation include P-selectin, CD40L, PF4, and GP IIb/IIIa (CD41)29,30 So, it is known that PMPs reflect platelet activation while EMPs markers become a representation of activation or endothelial dysfunction.31 The interaction of leukocytes and platelets in vascular endothelium induces inflammatory pathways, thrombosis and atherogenesis.32,33
This study showed that the number of mononuclear cells expressing E-selectin in the blood edge of smokers was higher than the non-smoker group. The increase in E-selectin in smokers in this study can be attributed to the results of previous studies that mentioned that smoking habits cause dysfunction and endothelial activation.34,35 Detection of endothelial activation can be measured through circulating markers including one of them is by measuring soluble E-selectin.36–38 In this study, the circulating of cells that expressed E-selectin in the smoker group showed the same pattern as the soluble E-selectin measurement in another study describing the endothelial injury. The number of E-selectin circulated results from an increase in active proteolytic cleavage from the surface of endothelial cells that have been activated. Still, there is no explanation of E-selectin’s biological activity and clearance.39 In addition to showing endothelial activation, E-selectin’s increase may provide an overview of vascular inflammatory alteration.17
Active smoking of one cigarette caused an immediate and significant increase in the numbers of circulating PMPs (CD41), CD45 and VE-cadherin (CD144).19 Platelet-derived MPs (PMPs) are also called platelet dust, are ultramicro-membranous vesicles that are 0.1–1.0 μm in diameter. PMPs are involved in various physiological functions, including endothelial repair.40 To evaluate PMPs’ role in the restoration of endothelial injury in the smoker group, quantification of circulating PMPs positive cell using flow cytometry in PBMNC smokers. This study’s results are different from previous studies, the number of mononuclear cells that express markers of PMPs in smokers in this study tends to be lower compared to the non-smoker group. Some study mentioned that PMPs can improve adherence on the walls of the damaged vascular endothelium to stimulate the expression of adhesion receptors gene, such as epithelial membrane protein-1 and increased ICAM-1 expression, thereby mediating platelet recruitment and leukocytes to repair endothelial damage.4,41,42
The decrease in the number of PMPs in smokers this time can be attributed to previous study statements regarding the possibility of increasing the number of PMPs in vascular endothelial dysfunction due to smoking habits. Platelets become the largest source of microparticle production in circulation. The number of PMPs in circulation can reach 70% to 90% of the total existing microparticles; this indicates the vital role of platelet function in shaping the homeostasis response of vascular systems.4,20
According to the present study, smoking is a potentially reversible risk factor and seems to be causally associated with the hematopoietic system.43,44 The CD133, also known as prominin or AC133, is a highly conserved antigen expressed on hematopoietic stem cells.45 This protein is absent on mature endothelial cells and monocytic cells and expressed in EPCs.21,46 Evaluation of the effects of smoking on circulating human hematopoietic stem cells using CD133 markers remains unclear. This study showed that the number of CD133 positive cell circulating in the group of smokers tended to be lower compared to the non-smoker group.
Tobacco components in cigarettes can directly affect various organs’ functioning because they contain pro-inflammatory effects and cytotoxic effects. Inhalation of oxidant molecules in cigarettes causes various damage that can activate inflammatory responses (increased TNF-α, IL-1, IL-6 and IL-8).47 When systemic inflammation occurs, HSCs located in bone marrow will undergo activation, expansion and proliferation.48 The mobilization of bone marrow HSCs intended for tissue repair mechanisms that occur dynamically and are self-regulated by stem cells.
The mechanism of proliferation and differentiation of HSCs due to inflammatory stimulus is part of the acute immune response. The accumulation of exposure to inflammatory cytokines causes disruption and loss of function of HSCs.49
Another study mentioned that pro-inflammatory cytokines are associated with decreased of stem cell factor (SCF), which are molecules that play a role in the mobilization of HSCs from bone marrow to circulation.50,51 So, it is likely that the mechanisms involved in decreasing the number of CD133 positive cells in smokers’ circulation are related to previous studies that stated that the accumulation of oxidants in smokers leads to increased production of inflammatory cytokines that can inhibit the signalling in the bone marrow.
Research Limitations
Measurements of vascular endothelial damage and inflammatory cytokines in smokers not measured in this study may continue the next research process. The correlation of research parameters in this study is associated with several previous studies that discussed the effects of cigarettes on endothelial damage and the impact of cigarette components on inflammatory induction of oxidative stress related to bone marrow failure.
Conclusion
The increase in circulating E-selectin positive cells in smokers indicates activation and endothelial injury as an indicator of the early stages of vascular lesions’ formation. Platelets respond to the presence of injury in the endothelium through an increase in the number of positive PMPs cells, followed by adhesion of positive PMPs cells in the endothelial area experiencing an injury. Thus, the number of positive PMPs cells in the circulation tends to decrease. The low number of PMPs in the circulation cannot stimulate the bone marrow to release HSCs into the circulation. When the number of PMPs in the circulation decreases, it cannot provide sufficient signalling for the bone marrow to release adequate circulating HSCs to mediate endothelial injury repair.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Roldan V, Marin F, Lip G, Blann A. Soluble E-selectin in cardiovascular disease and its risk factor. Thromb Haemost. 2003;90:1007–1020.
2. Woywodt A, Bahlmann FH, De Groot K, Haller H, Haubitz M. Circulating endothelial cells: life, death, detachment and repair of the endothelial cell layer. Nephrol Dial Transplant. 2002;17:1728–1730. doi:10.1093/ndt/17.10.1728
3. Goligorsky MS. Pathogenesis of endothelial cell dysfunction in chronic kidney disease: a retrospective and what the future may hold. Kidney Res Clin Pract. 2015;34(2):76–82. doi:10.1016/j.krcp.2015.05.003
4. Koupenova M, Clancy L, Heather A, et al. Circulating platelets as mediators of immunity, inflammation, and thrombosis. Circ Res. 2018;122:337–351. doi:10.1161/CIRCRESAHA.117.310795
5. Toda N, Toda H. Nitric oxide-mediated blood flow regulation as affect by smoking and nicotine. Eur J Pharmacol. 2020;649:1–3. doi:10.1016/j.ejphar.2010.09.042
6. Aicher A, Andreas MZ, Dimmeler S. Mobilizing endothelial progenitor cells. Hypertension. 2005;45:321–325. doi:10.1161/01.HYP.0000154789.28695.ea
7. Alamanda V, Singh S, Lawrence NJ, Chellappan SP. Nicotine-mediated induction of E-selectin in aortic endothelial cells requires Src kinase and E2F1 transcriptional activity. Biochem Biophys Res Commun. 2007;418(1):56–61. doi:10.1016/j.bbrc.2011.12.127
8. Versari D, Lerman LO, Lerman A. The importance of reendothelialization after arterial injury. Curr Pharm Des. 2007;13:1811–1824. doi:10.2174/138161207780831239
9. Pober JS, Min W, Bradley JR. Mechanisms of endothelial dysfunction, injury, and death. Annu Rev Pathol. 2009;4:71–95. doi:10.1146/annurev.pathol.4.110807.092155
10. Papathanaiou G, Mamali A, Papafloratos S, Zerva E. Effect of smoking and cardiovascular function: the role of nicotine and carbon monoxide. Health Sci J. 2014;8(2).
11. Blann AD, Woywodt A, Bertolini F, et al. Circulating endothelial cells. Biomarker of vascular disease. Thromb Haemost. 2005;93:228–235. doi:10.1160/TH04-09-0578
12. Collins T, Williams A, Johnston GI, et al. Structure and chromosomal location of the gene for endothelial-leukocyte adhesion molecule 1. J Biol Chem. 1991;266(4):2466–2473. doi:10.1016/S0021-9258(18)52267-5
13. Inami N, Nomura S, Kikuchi H, et al. P-selectin and Platelet-derived Microparticles associated with monocyte activation markers in patients with pulmonary embolism. Clin Appl Thromb Hemost. 2003;9(4):309–316. doi:10.1177/107602960300900406
14. Barbier V, Erbani J, Fiveash C, et al. Endothelial E-selectin inhibition improves acute myeloid leukaemia therapy by disrupting vascular niche-mediated chemoresistance. Nat Commun. 2020;11:2042. doi:10.1038/s41467-020-15817-5
15. Cherian P, Hankey G, Eikelboom J, et al. Endothelial and platelet activation in acute ischemic stroke and its etiological subtypes. Stroke. 2003;34:2132–2137. doi:10.1161/01.STR.0000086466.32421.F4
16. Silva M, Videira PA, Sackstein R. E-selectin ligands in the human mononuclear phagocyte system: implications for infection, inflammation, and immunotherapy. Front Immunol. 2018;8:1878.
17. Eikendal ML, Bots ML, Gohar A, et al. Circulating levels of P-selectin and E-selectin relate to cardiovascular magnetic resonance-derived aortic characteristics in young adults from the general population, a cross-sectional study. J Cardiovasc Magn Reson. 2018;20:54. doi:10.1186/s12968-018-0473-8
18. Freedman J, Loscalzo J, Barnard M, et al. Nitric oxide release from activated platelet inhibits platelet recruitment. J Clin Invest. 1997;100:350–356. doi:10.1172/JCI119540
19. Mobarrez F, Antoniewicz L, Bosson JA, et al. The effects of smoking on levels of endothelial progenitor cells and microparticles in the blood of healthy volunteers. PLoS One. 2014;9(2):e90314. doi:10.1371/journal.pone.0090314
20. Boilard E, Nigrovic PA, Larabee K, et al. Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science. 2010;327:580–583. doi:10.1126/science.1181928
21. Urbich C, Dimmeler S. Endothelial progenitor cells: characterization and role in vascular biology. Circ Res. 2004;95:343–353. doi:10.1161/01.RES.0000137877.89448.78
22. Kou F, Zhu C, Wan H, et al. Endothelial progenitor cells as the target for cardiovascular disease prediction, personalized prevention, and treatments: progressing beyond the state-of-the-art. EPMA J. 2020;11(4):629–643. doi:10.1007/s13167-020-00223-0
23. Gargett C, Schwab K, Zillwood R, et al. Isolation and culture of epithelial progenitors and mesenchymal stem cells from human endometrium. Biol Reprod. 2009;80(6):1136–1145. doi:10.1095/biolreprod.108.075226
24. Heiss C, Keymel S, Niesler U, et al. Impaired progenitor cell activity in age-related endothelial dysfunction. J Am Coll Cardiol. 2005;45:1441–1448. doi:10.1016/j.jacc.2004.12.074
25. Italiano J, Mairuhu A, Flaumenhaft R. Clinical relevance of microparticles from platelets and megakaryocytes. Curr Opin Hematol. 2010;17(6):578–584. doi:10.1097/MOH.0b013e32833e77ee
26. Wihastuti TA, Lestaari RD, Heriansyah T. The effect of darapladib therapy for the expression of Lp-PLA2 in dyslipidemia and type 2 diabetes mellitus atherosclerosis model. J Appl Pharm Sci. 2018;8(04):064–070.
27. Rabelink TJ, de Boer HC, van Zonneveld AJ. Endothelial activation and circulating markers of endothelial activation in kidney disease. Nat Rev Nephrol. 2010;6(7):404–414.
28. Daiber A, Steven S, Weber A, et al. Targeting vascular (endothelial) dysfunction. Br J Pharmacol. 2017;174(12):1591–1619.
29. Roldán V, Marín F, Lip GY, Blann AD. Soluble E-selectin in cardiovascular disease and its risk factors. A review of the literature. Thromb Haemost. 2003;90(12):1007–1020. doi:10.1160/TH02-09-0083
30. Yun SH, Sim EH, Goh RY, et al. Platelet activation: mechanisms and potential biomarker. Biomed Res Int. 2016;9060143:5.
31. Mörtberg J, Lundwall K, Mobarrez F, Wallén H, Jacobson SH, Spaak J. Increased concentrations of platelet- and endothelial-derived microparticles in patients with myocardial infarction and reduced renal function- a descriptive study. BMC Nephrol. 2019;20(1):71. doi:10.1186/s12882-019-1261-x
32. Thomas MR, Storey RF. The role of platelets in inflammation. Thromb Haemost. 2015;114(09):449–458. doi:10.1160/TH14-12-1067
33. Totani L, Evangelista V. Platelet-leukocyte interactions in cardiovascular disease and beyond. Arterioscler Thromb Vasc Biol. 2010;30(12):2357–2361. doi:10.1161/ATVBAHA.110.207480
34. Golbidi S, Laher I. Exercise and the aging endothelium. J Diabetes Res. 2013;2013:789607. doi:10.1155/2013/789607
35. Liao JK. Linking endothelial dysfunction with endothelial cell activation. J Clin Invest. 2013;123(2):540–541. doi:10.1172/JCI66843
36. Ross R. Atherosclerosis: an inflammatory disease. N Engl J Med. 1999;340:115–126. doi:10.1056/NEJM199901143400207
37. Price DT, Loscalzo J. Cellular adhesion molecules and atherogenesis. Am J Med. 1999;107:85–97. doi:10.1016/S0002-9343(99)00153-9
38. Leeuwenberg JF, Smeets EF, Neefjes JJ, et al. E-selectin and intercellular adhesion molecule-1 are released by activated human endothelial cells in vitro. Immunology. 1992;77:543–549.
39. Aleisa FA, Sakashita K, Lee JM, et al. Functional binding of E-selectin to its ligands is enhanced by structural features beyond its lectin domain. J Biol Chem. 2020;295(11):3719–3733. doi:10.1074/jbc.RA119.010910
40. Breen KA, Sanchez K, Kirkman N, et al. Endothelial and platelet microparticles in patients with antiphospholipid antibodies. Thromb Res. 2015;135(2):368–374. doi:10.1016/j.thromres.2014.11.027
41. Khlgatian M, Nassar H, Chou HH, Gibson FC, Genco CA. Fimbria-dependent activation of cell adhesion molecule expression in Porphyromonas gingivalis-infected endothelial cells. Infect Immun. 2002;70:257–267. doi:10.1128/IAI.70.1.257-267.2002
42. Lukasik M, Rozalski M, Luzak B, et al. Enhanced platelet-derived microparticle formation is associated with carotid atherosclerosis in convalescent stroke patients. Platelets. 2013;24(1):63–70. doi:10.3109/09537104.2011.654292
43. Pedersen K, Colak Y, Ellervik C, et al. Smoking and increased white and red blood cells: a mendelian randomization approach in the copenhagen general population study. Arterioscler Thromb Vasc Biol. 2019;39:965–977. doi:10.1161/ATVBAHA.118.312338
44. Siggins RW, Hossain F, Rehman T, Melvan JN, Zhang P, Welsh DA. Cigarette smoke alters the hematopoietic stem cell niche. Med Sci (Basel). 2014;2(1):37–50. doi:10.3390/medsci2010037
45. Takahashi M, Matsuoka Y, Sumide K, et al. CD133 is a positive marker for a distinct class of primitive human cord blood-derived CD34-negative hematopoietic stem cells. Leukemia. 2014;28(6):1308–1315. doi:10.1038/leu.2013.326
46. Zahran A, Abdel-Tahim M, Refaat A, et al. Circulating hematopoietic stem cells, endothelial progenitor cells and cancer stem cells in hepatocellular carcinoma patients: contribution to diagnosis and prognosis. Acta Oncologica. 2020;59:2020. doi:10.1080/0284186X.2019.1657940
47. Strzelak A, Ratajczak A, Adamiec A, Feleszko W. Tobacco smoke induces and alters immune responses in the lung triggering inflammation, allergy, asthma and other lung disease: a mechanistic review. Int J Environ Res Public Health. 2018;15(5):1033. doi:10.3390/ijerph15051033
48. Mitroulis I, Kalafati L, Bornhauser M, Hajishengallis G, Chavakis T. Regulation of the bone marrow niche by inflammation. Front Immunol. 2020;11:1540. doi:10.3389/fimmu.2020.01540
49. Trompouki E, Mullen L, Reyes D, Yodoi J, Kim S, Schuettplez L. Editorial: inflammatory signaling in bone marrow failure and hematopoietic malignancy. Front Immunol. 2017;8:660. doi:10.3389/fimmu.2017.00660
50. Balandran J, Purizaca J, Ensico J, et al. Pro-inflammatory-related loss of CXCL12 niche promote acute lymphoblastic leukemic progression at the expense of normal lymphopoiesis. Front Immunol. 2016;7:666. doi:10.3389/fimmu.2016.00666
51. Moll N, Ransohoff R. CXCL12 and XCR4 in bone marrow physiology. Expert Rev Hematol. 2010;3(3):315–322. doi:10.1586/ehm.10.16
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.