")
Back to Journals » Journal of Asthma and Allergy » Volume 14
Dysregulated Metabolism in the Pathophysiology of Non-Allergic Obese Asthma
Authors McCravy M, Ingram JL , Que LG
Received 25 September 2020
Accepted for publication 1 January 2021
Published 4 March 2021 Volume 2021:14 Pages 179—186
DOI https://doi.org/10.2147/JAA.S282284
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Amrita Dosanjh
Matthew McCravy, Jennifer L Ingram, Loretta G Que
Division of Pulmonary, Allergy and Critical Care Medicine, Department of Medicine, Duke University Medical Center, Durham, NC, 27710, USA
Correspondence: Matthew McCravy
Division of Pulmonary, Allergy and Critical Care Medicine, Department of Medicine, Duke University Medical Center, Box 102349, Durham, NC, 27710, USA
Tel +1 731-571-4816
Email [email protected]
Abstract: Asthma is an obstructive airway disease that is characterized by reversible airway obstruction and is classically associated with atopic, TH 2 driven inflammation. Landmark studies in the second half of the twentieth century identified eosinophils as a key mediator of inflammation and steroids, both inhaled and systemic, as a cornerstone of therapy. However, more recently other phenotypes of asthma have emerged that do not respond as well to traditional therapies. In particular, obese patients who develop asthma as adults are less likely to have eosinophilic airway inflammation and do not respond to traditional therapies. Obese patients often have metabolic comorbidities such as impaired glucose tolerance and dyslipidemias, also known as metabolic syndrome (MetS). The unified pathophysiology of metabolic syndrome is not known, however, several signaling pathways, such as the neuropeptide glucagon-like peptide-1 (GLP-1) and nitric oxide (NO) signaling have been shown to be dysregulated in MetS. These pathways are targeted by commercially available medications. This review discusses the potential roles that dysregulation of the GLP-1 and NO signaling pathways, along with arginine metabolism, play in the development of asthma in obese patients. GLP-1 receptors are found in high density in the lung and are also detectable in bronchoalveolar lavage fluid. NO has long been associated with asthma. We hypothesize that these derangements in metabolic signaling pathways underpin the asthmatic phenotype seen in obese patients with non-eosinophilic airway inflammation and poor response to established therapies. While still an active area of research, novel interventions are needed for this subset of patient who respond poorly to available asthma therapies.
Keywords: asthma, obesity, metabolic syndrome, glucagon-like peptide-1, nitric oxide
Asthma is defined as an obstructive airway disease with a significant bronchodilator response. While bronchial hyperreactivity was historically thought to be the primary feature of asthma, more recent evidence demonstrated a heterogeneous, inflammatory process as a key aspect of asthma pathobiology.1 Classical asthma is atopic, characterized by a high peripheral eosinophil count and steroid responsiveness.2,3 However, subphenotypes of asthma with different inflammatory fingerprints and variable responses to treatment exist.4 In particular, obese patients with asthma are more likely to have a neutrophilic phenotype and are less likely to respond to steroids.5 The metabolic underpinnings of this finding are poorly understood, though evidence suggests that airway disease may be associated with metabolic derangements accompanying obesity rather than the mechanical effects of obesity (airway compression and increased effort to breathe).6
A clinical entity likely to be asthma has been recognized in Western medicine at least since the Code of Hammurabi. Remarkably, it was not until the 1980s that physicians and researchers began to understand asthma as an inflammatory disorder associated with eosinophils.7 Since that time, research has revealed that asthma is far more heterogenous than initially recognized. On a cellular level, the inflammatory milieu in the airways of asthmatic patients can be broadly classified into eosinophilic and non-eosinophilic inflammation.8 A neutrophilic pattern of inflammation appears to be more steroid resistant, either inhaled or systemic.5
Another phenotype of asthma that does not conform to the classical pattern is obese asthma. Observational data has shown that obese patients are more likely to have an airway neutrophilic inflammatory pattern and are resistant to inhaled corticosteroids.9 Further research is needed to definitively identify the cellular and molecular mechanisms contributing to the unique phenotype observed in patients with obesity and asthma. Data suggest that the metabolic derangements seen in obese patients, manifested as metabolic syndrome, play a role in modulating airway responses in asthma.6
Metabolic syndrome is defined as having five components: abdominal obesity, hypertriglyceridemia, reduced high-density lipoprotein (HDL) levels, hypertension, and insulin resistance/hyperglycemia.10 The etiology of metabolic syndrome is not fully understood; yet, we hypothesize that neuropeptides involved in its development are at least partly responsible for lung disease in these patients. An analysis of the National Health and Nutrition Examination Survey (NHANES) database demonstrated that metabolic syndrome is associated with worse lung function regardless of the patient’s body mass index (BMI).11 This analysis was replicated in a Korean population, and these authors also found a significant association between metabolic syndrome and impaired lung function. This association was not seen with simple obesity.6
An important neuropeptide that has gained much clinical interest in recent years is glucagon-like peptide-1 (GLP-1). GLP-1 has important functions in modulating glucose homeostasis and insulin sensitivity in patients with obesity and type 2 diabetes.12 Beyond glucose homeostasis, trials of medications acting on the GLP-1 signaling pathway result in weight loss in obese patients, indicating a role for GLP-1 in the development of obesity.13 Furthermore, GLP-1 is known to be dysregulated in patients with metabolic syndrome.14 With the previously mentioned association between worsening asthma and metabolic syndrome, targeting the GLP-1 pathway in the treatment of obese patients with asthma is a plausible potential therapeutic strategy.
GLP-1 Physiology and Lung Function
The notion of incretins, insulin secretagogues, first arose in the 1960s when researchers observed that oral administration of glucose increases plasma insulin concentration more than an equivalent dose of intravenous glucose. This first incretin to be discovered was gastric inhibitory polypeptide (GIP). GLP-1 and GLP-2 were discovered in the 1980s. GLP-1 is produced by intestinal L-cells, members of the neuroendocrine system, in response to a meal. These cells are diffusely distributed throughout the intestine and produce several neuropeptides in addition to GLP-1.15 The canonical function of GLP-1 is to increase insulin secretion in pancreatic β cells via a cyclic adenosine monophosphate (cAMP)-dependent mechanism after.16 This signaling occurs via the G protein-coupled receptor, GLP-1 receptor (GLP-1R).
GLP-1R is found on pancreatic β cells, as expected, but also on a host of other cell types. In addition to stimulating insulin secretion, GLP-1R signaling is also associated with improved β cell survival.17 One of the organ systems with an unexpectedly high concentration of GLP-1R is the respiratory system. The receptors are localized throughout the lung and are found in the pulmonary vasculature, airway smooth muscle cells, airway epithelial cells and type II alveolar cells.18 While the function of these receptors is poorly characterized, GLP-1 signaling on type II alveolar cells acts in a cAMP-dependent manner. This mechanism is consistent with signaling pathways seen in pancreatic islet cells and mediates production of surfactant in a mouse mode.19 Functionally, GLP-1R agonism improved outcomes in a mouse model of acute respiratory distress syndrome (ARDS).20 Additionally, GLP-1R agonism leads to inhibition of airway smooth muscle cell contraction.19
While these observations suggest a role for GLP-1 in the regulation of some endogenous pulmonary functions, the degree to which this signaling is involved in the pathophysiology of lung disease is still unknown. In addition to high pulmonary concentrations of GLP-1R, GLP-1, as well as other neuropeptides, are secreted in the airway as evidenced by their identification in the bronchoalveolar lavage (BAL) fluid of healthy adult volunteers.21 The cellular source of these peptides is not known. Additionally, it is not clear if these peptides have an endogenous signaling function or if they are merely bystanders in the alveolar space.
In a mouse model of respiratory syncytial virus (RSV) infection, the GLP-1R agonist, liraglutide, decreased mucus production via alterations in the interleukin-13 (IL-13) and interleukin-33 (IL-33) signaling pathways.22 In mice with ovalbumin-induced allergic airways disease, administration of liraglutide prior to allergen exposure reduced airway resistance after disease onset.23 Similarly, the authors demonstrated improvements in murine airway resistance following liraglutide administration in combination with lipopolysaccharide (LPS), as a model of chronic obstructive pulmonary disease (COPD). The authors propose that the mechanism of improved pulmonary function is increased surfactant production, as surfactant protein D expression was increased in the mice treated with liraglutide. These effects on interleukin signaling and surfactant production have similarly been observed in mouse models of house dust mite-induced allergic airways diseases and LPS-induced ARDS.20,24
Evidence of a pulmonary clinical benefit of GLP-1R agonism in patients with obstructive airway diseases remains sparse. In a study of patients with COPD, peripheral blood mononuclear cells were found to have decreased levels of GLP-1R in comparison with control subjects. These reduced levels of GLP-1R were associated with deranged interferon gamma signaling. Treatment with liraglutide corrected these abnormalities.25 Finally, in a small pilot study of overweight patients with type 2 diabetes and asthma, patients who were receiving liraglutide had significantly fewer asthma exacerbations than patients receiving placebo.26
Pulmonary Neuroendocrine Cells
Thus, data suggest that GLP-1 signaling may have a significant regulatory effect on pulmonary function. However, a major gap in understanding is the mechanism whereby GLP-1 arrives in the lung – is GLP-1 transported from the gut to the airway through the systemic vascular circulation or do pulmonary cells secrete GLP-1 in situ? The airway epithelium features intrinsic neuroendocrine cells that produce neuropeptides, including calcitonin gene-related peptide (CGRP) and serotonin.27 GLP-1 is known to be produced by neuroendocrine cells in the intestine.15 Human BAL samples contain GLP-1.22 The combination of these three findings suggest a possible role for pulmonary neuroendocrine cells (pNECs) in linking GLP-1 signaling with pulmonary function either by local paracrine signaling or through secretion into the systemic blood supply. Of particular interest is the unanswered question of whether alterations in pNEC function and distribution occurs in patients with obesity. Intriguingly, Mathews et al recently reported, in a genetically obese mouse model of ozone-induced airways disease, an increased expression of interleukin-23-driven CGRP expression, suggesting that airway irritants stimulate increased neuropeptide expression in obese subjects.28
Mice that are genetically deficient in pNECs demonstrate less mucus production and fewer inflammatory cells after ovalbumin challenge.29 These effects are linked to the absence of CGRP and gamma-aminobutyric acid (GABA), which sensitize pulmonary type 2 innate lymphoid cells. In adults with asthma, sputum concentrations of substance P, another product of pNECs, correlates with asthma symptoms.30 Furthermore, in mice infected with RSV, airway hyperresponsiveness and mucus production is abrogated by prophylactic treatment with an inhibitor of substance P signaling.31
Pulmonary neuroendocrine cells are thought to function as “sensors” for air particles entering the lower airway, as they express olfactory receptors, and they release neuropeptides after exposure to inhaled volatile irritants.32 Furthermore, obese mice demonstrate greater increases in airway resistance and more inflammatory cells in BAL fluid following exposure to ozone than lean mice in a mechanism requiring increased pulmonary CGRP expression.28 As CGRP is primarily produced by neuroendocrine cells, this observation provides a potential mechanistic link between obesity and airway hyperreactivity.
Vagal nerve efferents terminate on pNECs suggesting a possible neurologic component to disease processes involving pNECs. In mice exposed to allergenic stimuli early in life, these vagal terminations are aberrant and resulted in mucus hyperproduction via a GABA-mediated mechanism.33 This effect was also observed in rhesus monkeys that were exposed to ozone as infants.34 Thus, both allergic and non-allergic stimuli may modulate pNEC functions and signaling to promote airway inflammation and remodeling.
Finally, pNECs function as stem cells that drive airway healing after injury.35 Several disease states are associated with an abnormal proliferation of neuroendocrine cells. Infants with persistent tachypnea of the newborn have increased density of neuroendocrine cells in both the pulmonary lobules and airway epithelium compared with healthy controls.36 Also, pNECs are hyperplastic in the bronchioles of patients with asthma compared with samples obtained from healthy volunteers.29
GLP-1 and Metabolic Syndrome
In addition to the potential direct action of GLP-1 within the lung, another possible mechanistic link between GLP-1 signaling and asthma is as a downstream effect of glucose resistance in the setting of the metabolic syndrome. GLP-1R agonists improve insulin sensitivity in a mouse model of diet-induced obesity and metabolic syndrome.37 At a molecular level, interleukin-6 (IL-6) secreted from adipocytes stimulates the production of GLP-1 from intestinal L-cells.38 While far from conclusive evidence, these data suggest that GLP-1 signaling is involved in a regulatory feedback loop with adipocytes. In humans, continuous infusion of GLP-1 via a pump resulted in improved insulin sensitivity over a period of 6 weeks.19
Beyond the epidemiologic link between metabolic syndrome and insulin resistance with poor lung function, insulin resistance modulates molecular mediators of pulmonary disease. In a study of mice with diet-induced obesity and metabolic syndrome, impaired glucose metabolism was associated with airway hyperreactivity and peribronchial and perivascular lung fibrosis.39 These effects were mediated though the transforming growth factor-beta (TGF-β) signaling pathway. Hyperinsulinemia reduces the sensitivity of airway smooth muscle cells to β2-adrenoreceptor mediated relaxation.40 This effect was observed in both airway smooth muscle cell culture and a mouse model of diet-induced obesity and provides a potential explanation for the poor response to bronchodilators in obese patients with asthma.41
Metabolic Syndrome, Nitric Oxide and Obese Asthma
Nitric oxide (NO) was first discovered to have physiologic function in 1987 when it was identified as the smooth muscle relaxant, endothelium-derived relaxing factor. While initially associated with cardiovascular physiology, NO is now known to function in several organ systems, including the lung. In healthy pulmonary physiology, NO is endogenously released in the airways by nitric oxide synthase (NOS) and relaxes vascular and airway smooth muscle.42 In pathologic conditions such as asthma, concentrations of NO in exhaled breath are increased compared to that of non-asthmatic control subjects. Furthermore, elevated levels of exhaled NO are associated with refractory symptoms and poor response to corticosteroids, though not with objective markers of lung function.43 These findings suggest that abnormal NO signaling may be the final common pathway of asthma pathophysiology in patients with metabolic derangements. However, these findings are not specific to patients with asthma; exhaled NO is elevated in other syndromes of pulmonary inflammation, such as allergic rhinitis and possibly even exposure to air pollution.44 The association between obesity, asthma, and exhaled NO is less clear. Exhaled NO is negatively correlated with BMI in atopic children, but not in children without evidence of allergic inflammation.45 In obese adult patients with asthma, a lower exhaled NO concentration is directly associated with more difficult-to-treat disease and indirectly associated with increasing BMI.46
Metabolic Syndrome and Dysregulated Arginine Metabolism
Several lines of evidence in laboratory models suggest a relationship between metabolic syndrome and dysregulated arginine metabolism.47 Arginine is derived from dietary protein intake, protein breakdown, and de novo arginine production. The latter may be linked to the availability of citrulline, which is the immediate precursor of arginine and limiting factor for de novo arginine production. The bioavailability and fate of arginine is highly dependent on its metabolism by various organs and enzymes. In addition to arginases I and II, arginine enters the NOS pathway, where it is metabolized to form NO. NO is one of the major downstream effectors of arginine metabolism. The fraction of exhaled NO is physiologic manifestation of airway NO production.48 Dysregulated L-arginine metabolism and decreased NO production cause increased airway hyperreactivity and decreased exhaled NO in mice with diet-induced obesity.49
Serum levels of asymmetric dimethylarginine (ADMA), a competitive inhibitor of NOS, are increased in patients with metabolic syndrome compared with age matched controls without metabolic syndrome. ADMA levels also correlate with insulin resistance in a cohort of children and adolescents with diabetes and metabolic syndrome.50 In obese patients with asthma, increased ADMA levels correlate with decreased fraction of exhaled nitric oxide (FeNO) measurements in the Severe Asthma Research Program (SARP) study cohort.51 Taken together, these data suggest a link between arginine metabolism and NOS function and metabolic phenotype. This association may reflect a disturbed balance between L-arginine and ADMA.
Supplementation with L-arginine can improve glucose tolerance as well as other markers of metabolic syndrome in animal models of diet-induced obesity. In genetically obese rats, supplementation with L-arginine reduced fat mass and improved biochemical markers of dysregulated lipid metabolism.52 When the authors replicated the experiment in Glp1r-deficient mice, they observed no effect on glucose tolerance. In contrast, the opposite effect, GLP-1 acting to alter arginine metabolism, is less well established. The long-acting GLP-1R agonist, exendin-4, decreases the production of ADMA in renal tubular cells of rats with streptozotocin-induced diabetes.53 In humans, observational data suggest a possible benefit from higher L-arginine consumption on incidence of metabolic syndrome, though a differential effect may exist between plant- and animal-derived L-arginine.54 The authors attributed this effect to enhanced gut secretion of GLP-1 after a meal enriched in L-arginine as other data demonstrated a postprandial rise in serum GLP-1 in humans after eating a meal enriched in L-arginine. In a recent open-labeled clinical trial testing supplementation with L-citrulline, a precursor to L-arginine, as add-on therapy to obese asthmatics with low or low-normal FeNO, L-citrulline increased FeNO and serum L-arginine levels and improved asthma control.55 These data suggest that manipulating the NO signaling axis in obese patients with asthma may provide clinical benefit and warrant further study. A schematic representation of a postulated mechanism for GLP-1-modulated L-arginine metabolism and airway responses in obesity and metabolic syndrome is shown in Figure 1.
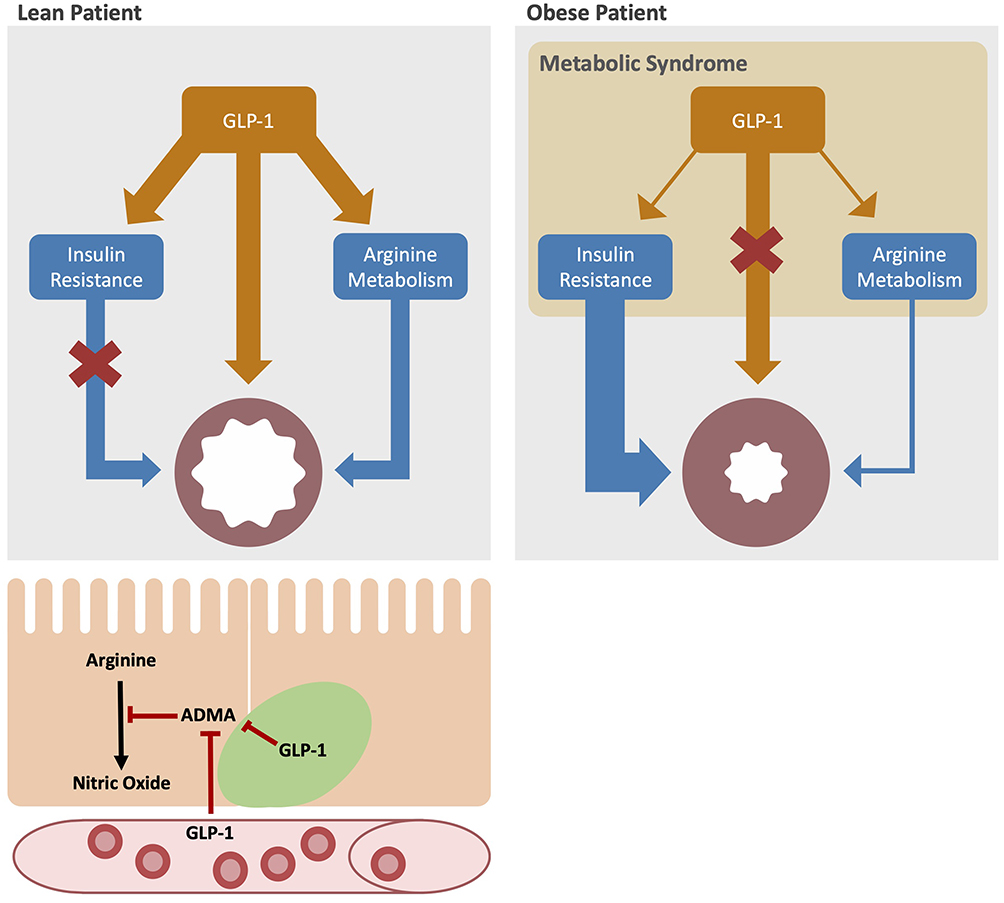 |
Figure 1 Metabolic pathways in obese patients with asthma: bottom left: GLP-1 delivered in the blood stream and, we hypothesize, secreted from pulmonary neuroendocrine cells (green) modulates arginine metabolism by decreasing ADMA production leading to an increase in nitric oxide (NO) production in airway epithelial cells (orange). Top left: in lean, healthy patients, GLP-1 acts to decrease insulin resistance, relax airway smooth muscle and increase airway NO production. Top right: in asthma patients with obesity and metabolic syndrome, GLP-1 production is decreased, leading to increased insulin resistance, decreased NO production, and increased smooth muscle contractility. All of these features contribute to bronchoconstriction. |
Researchers are investigating the potential modulatory effects of L-arginine supplementation on NO-mediated pathways for its effectiveness as a strategy for prevention and treatment of metabolic syndrome and its individual features and comorbidities, including abdominal obesity, type 2 diabetes and dyslipidemia and asthma. The effectiveness of L-arginine supplementation in obese patients with asthma and metabolic syndrome has not yet been determined; however, a small pilot study showed no difference in clinical asthma outcomes of patients who were supplemented with L-arginine and those who were not despite increasing levels of plasma L-arginine.56
Conclusions and Future Directions
In summary, asthma is a heterogeneous disease with multiple sub-phenotypes. Obese patients with asthma represent a phenotype that is unlikely to benefit from the cornerstone therapies currently employed in the treatment of allergic asthma. Despite the observation that these patients have a distinct airway inflammatory cell profile, the pathophysiology of this syndrome is not fully understood. Consequently, no good treatment options are available that are designed to mitigate the underlying process.
Several potentially intervenable pathways exist which are deranged in obese patients with asthma. These pathways include NO and GLP-1 signaling, which function abnormally in patients with metabolic syndrome. The degree to which endogenous GLP-1 production regulates lung function is unknown, and this lack of knowledge demonstrates a clear need for more work to be done to tease out the sources of pulmonary GLP-1 and its signaling effects.
Acknowledgment
Drs. Que and Ingram are supported by the NIH grants 1R01HL153641-01 and 1R01HL148542-01 (Que) and Ingram 5R01-HL130234 (Ingram).
Disclosure
Dr Loretta G Que reports a patent (reference number: T-006106) pending. The authors report no other conflicts of interest in this work.
References
1. Atkins PC, Norman ME, Zweiman B. Antigen-induced neutrophil chemotactic activity in man. Correlation with bronchospasm and inhibition by disodium cromoglycate. J Allergy Clin Immunol. 1978;62(3):149–155. doi:10.1016/0091-6749(78)90099-4
2. Bousquet J, Chanez P, Lacoste JY, et al. Eosinophilic inflammation in asthma. N Engl J Med. 1990;323(15):1033–1039. doi:10.1056/NEJM199010113231505
3. Haahtela T, Järvinen M, Kava T, et al. Comparison of a β2-agonist, terbutaline, with an inhaled corticosteroid, budesonide, in newly detected asthma. N Engl J Med. 1991;325(6):388–392. doi:10.1056/NEJM199108083250603
4. Halonen M, Stern D, Lohman C, et al. Two subphenotypes of childhood asthma that differ in maternal and paternal influences on asthma risk. Am J Respir Crit Care Med. 1999;160(2):564–570. doi:10.1164/ajrccm.160.2.9809038
5. Haldar P, Pavord ID, Shaw DE, et al. Cluster analysis and clinical asthma phenotypes. Am J Respir Crit Care Med. 2008;178(3):218–224. doi:10.1164/rccm.200711-1754OC
6. Kim M, Choi S, Choi S-H, et al. Metabolic syndrome and lung function in Korean children and adolescents: a cross-sectional study. Sci Rep. 2019;9(1):15646. doi:10.1038/s41598-019-51968-2
7. Durham SR, Carroll M, Walsh GM, Kay AB. Leukocyte activation in allergen-induced late-phase asthmatic reactions. N Engl J Med. 1984;311(22):1398–1402. doi:10.1056/NEJM198411293112202
8. Brasier AR, Victor S, Ju H, et al. Predicting intermediate phenotypes in asthma using bronchoalveolar lavage-derived cytokines. Clin Transl Sci. 2010;3(4):147–157. doi:10.1111/j.1752-8062.2010.00204.x
9. Telenga ED, Tideman SW, Kerstjens HAM, et al. Obesity in asthma: more neutrophilic inflammation as a possible explanation for a reduced treatment response. Allergy. 2012;67(8):1060–1068. doi:10.1111/j.1398-9995.2012.02855.x
10. Alberti KG, Eckel RH, Grundy SM, et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation. 2009;120(16):1640–1645. doi:10.1161/CIRCULATIONAHA.109.192644
11. Forno E, Han -Y-Y, Muzumdar RH, et al. Insulin resistance, metabolic syndrome, and lung function in US adolescents with and without asthma. J Allergy Clin Immunol. 2015;136(2):304–11.e8. doi:10.1016/j.jaci.2015.01.010
12. Zander M, Madsbad S, Madsen JL, et al. Effect of 6-week course of glucagon-like peptide 1 on glycaemic control, insulin sensitivity, and beta-cell function in type 2 diabetes: a parallel-group study. Lancet. 2002;359(9309):824–830. doi:10.1016/S0140-6736(02)07952-7
13. Husain M, Birkenfeld AL, Donsmark M, et al. Oral semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. 2019;381(9):841–851. doi:10.1056/NEJMoa1901118
14. Dineen SL, McKenney ML, Bell LN, et al. Metabolic syndrome abolishes glucagon-like peptide 1 receptor agonist stimulation of SERCA in coronary smooth muscle. Diabetes. 2015;64(9):3321–3327. doi:10.2337/db14-1790
15. Eissele R, GÖKE R, Willemer S, et al. Glucagon-like peptide-1 cells in the gastrointestinal tract and pancreas of rat, pig and man. Eur J Clin Invest. 1992;22(4):283–291. doi:10.1111/j.1365-2362.1992.tb01464.x
16. Drucker DJ, Philippe J, Mojsov S, et al. Glucagon-like peptide I stimulates insulin gene expression and increases cyclic AMP levels in a rat islet cell line. Proc Natl Acad Sci U S A. 1987;84(10):3434–3438. doi:10.1073/pnas.84.10.3434
17. Yusta B, Baggio LL, Estall JL, et al. GLP-1 receptor activation improves β cell function and survival following induction of endoplasmic reticulum stress. Cell Metab. 2006;4(5):391–406. doi:10.1016/j.cmet.2006.10.001
18. Rogliani P, Calzetta L, Capuani B, et al. Glucagon-like peptide 1 receptor: a novel pharmacological target for treating human bronchial hyperresponsiveness. Am J Respir Cell Mol Biol. 2016;55(6):804–814. doi:10.1165/rcmb.2015-0311OC
19. Romaní-Pérez M, Outeiriño-Iglesias V, Gil-Lozano M, et al. Pulmonary GLP-1 receptor increases at birth and exogenous GLP-1 receptor agonists augmented surfactant-protein levels in litters from normal and nitrofen-treated pregnant rats. Endocrinology. 2013;154(3):1144–1155. doi:10.1210/en.2012-1786
20. Zhu T, Li C, Zhang X, et al. GLP-1 analogue liraglutide enhances SP-a expression in LPS-induced acute lung injury through the TTF-1 signaling pathway. Mediators Inflamm. 2018;2018:3601454. doi:10.1155/2018/3601454
21. Mendivil CO, Koziel H, Brain JD, Fehrenbach H. Metabolic hormones, apolipoproteins, adipokines, and cytokines in the alveolar lining fluid of healthy adults: compartmentalization and physiological correlates. PLoS One. 2015;10(4):e0123344. doi:10.1371/journal.pone.0123344
22. Bloodworth MH, Rusznak M, Pfister CC, et al. Glucagon-like peptide 1 receptor signaling attenuates respiratory syncytial virus-induced type 2 responses and immunopathology. J Allergy Clin Immunol. 2018;142(2):683–687.e12. doi:10.1016/j.jaci.2018.01.053
23. Viby NE, Isidor MS, Buggeskov KB, et al. Glucagon-like peptide-1 (GLP-1) reduces mortality and improves lung function in a model of experimental obstructive lung disease in female mice. Endocrinology. 2013;154(12):4503–4511. doi:10.1210/en.2013-1666
24. Toki S, Goleniewska K, Reiss S, et al. Glucagon-like peptide 1 signaling inhibits allergen-induced lung IL-33 release and reduces group 2 innate lymphoid cell cytokine production in vivo. J Allergy Clin Immunol. 2018;142(5):1515–1528.e8. doi:10.1016/j.jaci.2017.11.043
25. Huang J, Yi H, Zhao C, et al. Glucagon-like peptide-1 receptor (GLP-1R) signaling ameliorates dysfunctional immunity in COPD patients. Int J Chron Obstruct Pulmon Dis. 2018;13(p):3191–3202. doi:10.2147/COPD.S175145
26. Khan F, Mat A, Hogan AE, et al. Preliminary asthma-related outcomes following glucagon-like peptide 1 agonist therapy. QJM. 2017;110(12):853–854. doi:10.1093/qjmed/hcx125
27. Stahlman MT, Jones M, Gray ME, et al. Ontogeny of neuroendocrine cells in human fetal lung. III. An electron microscopic immunohistochemical study. Labor Investig. 1987;56(6):629–641.
28. Mathews JA, Krishnamoorthy N, Kasahara DI, et al. Augmented responses to ozone in obese mice require IL-17A and gastrin-releasing peptide. Am J Respir Cell Mol Biol. 2018;58(3):341–351. doi:10.1165/rcmb.2017-0071OC
29. Sui P, Wiesner DL, Xu J, et al. Pulmonary neuroendocrine cells amplify allergic asthma responses. Science. 2018;360:6393. doi:10.1126/science.aan8546
30. Tomaki M, Ichinose M, Miura M, et al. Elevated substance P content in induced sputum from patients with asthma and patients with chronic bronchitis. Am J Respir Crit Care Med. 1995;151(3 Pt 1):613–617. doi:10.1164/ajrccm/151.3_Pt_1.613
31. Chu HW, Kraft M, Krause JE, et al. Substance P and its receptor neurokinin 1 expression in asthmatic airways. J Allergy Clin Immunol. 2000;106(4):713–722. doi:10.1067/mai.2000.109829
32. Gu X, Karp PH, Brody SL, et al. Chemosensory functions for pulmonary neuroendocrine cells. Am J Respir Cell Mol Biol. 2014;50(3):637–646. doi:10.1165/rcmb.2013-0199OC
33. Barrios J, Patel KR, Aven L, et al. Early life allergen-induced mucus overproduction requires augmented neural stimulation of pulmonary neuroendocrine cell secretion. FASEB J. 2017;31(9):4117–4128. doi:10.1096/fj.201700115R
34. Barrios J, Kho AT, Aven L, et al. Pulmonary neuroendocrine cells secrete γ-aminobutyric acid to induce goblet cell hyperplasia in primate models. Am J Respir Cell Mol Biol. 2019;60(6):687–694. doi:10.1165/rcmb.2018-0179OC
35. Stevens TP, McBride JT, Peake JL, et al. Cell proliferation contributes to PNEC hyperplasia after acute airway injury. Am J Physiol. 1997;272(3 Pt 1):L486–93. doi:10.1152/ajplung.1997.272.3.L486
36. Deterding RR, Pye C, Fan LL, et al. Persistent tachypnea of infancy is associated with neuroendocrine cell hyperplasia. Pediatr Pulmonol. 2005;40(2):157–165. doi:10.1002/ppul.20243
37. Parlevliet ET, Elst JPS-VD, Corssmit EPM, et al. CNTO736, a novel glucagon-like peptide-1 receptor agonist, ameliorates insulin resistance and inhibits very low-density lipoprotein production in high-fat-fed mice. J Pharmacol Exp Ther. 2009;328(1):240. doi:10.1124/jpet.108.144154
38. Wueest S, Laesser CI, Böni-Schnetzler M, et al. IL-6-type cytokine signaling in adipocytes induces intestinal GLP-1 secretion. Diabetes. 2018;67(1):36–45. doi:10.2337/db17-0637
39. Park YH, Oh EY, Han H, et al. Insulin resistance mediates high-fat diet-induced pulmonary fibrosis and airway hyperresponsiveness through the TGF-β1 pathway. Exp Mol Med. 2019;51(5):1–12.
40. Xu R, Gopireddy RR, Wu Y, et al. Hyperinsulinemia promotes heterologous desensitization of β2 adrenergic receptor in airway smooth muscle in obesity. FASEB J. 2020;34(3):3996–4008. doi:10.1096/fj.201800688RR
41. McGarry ME, Castellanos E, Thakur N, et al. Obesity and bronchodilator response in black and Hispanic children and adolescents with asthma. Chest. 2015;147(6):1591–1598. doi:10.1378/chest.14-2689
42. Gaston B, Drazen JM, Jansen A, et al. Relaxation of human bronchial smooth muscle by S-nitrosothiols in vitro. J Pharmacol Exp Ther. 1994;268(2):978–984.
43. Stirling RG, Kharitonov SA, Campbell D, et al. Increase in exhaled nitric oxide levels in patients with difficult asthma and correlation with symptoms and disease severity despite treatment with oral and inhaled corticosteroids. Asthma and Allergy Group. Thorax. 1998;53(12):1030–1034. doi:10.1136/thx.53.12.1030
44. De Prins S, Marcucci F, Sensi L, et al. Exhaled nitric oxide and nasal tryptase are associated with wheeze, rhinitis and nasal allergy in primary school children. Biomarkers. 2014;19(6):481–487. doi:10.3109/1354750X.2014.937362
45. Yao TC, Tsai H-J, Chang S-W, et al. Obesity disproportionately impacts lung volumes, airflow and exhaled nitric oxide in children. PLoS One. 2017;12(4):e0174691. doi:10.1371/journal.pone.0174691
46. Komakula S, Khatri S, Mermis J, et al. Body mass index is associated with reduced exhaled nitric oxide and higher exhaled 8-isoprostanes in asthmatics. Respir Res. 2007;8(1):32. doi:10.1186/1465-9921-8-32
47. De A, Rastogi D. Association of pediatric obesity and asthma, pulmonary physiology, metabolic dysregulation, and atopy; and the role of weight management. Expert Rev Endocrinol Metab. 2019;14(5):335–349. doi:10.1080/17446651.2019.1635007
48. Xu W, Comhair SAA, Janocha AJ, et al. Arginine metabolic endotypes related to asthma severity. PLoS One. 2017;12(8):e0183066. doi:10.1371/journal.pone.0183066
49. Singh VP, Aggarwal R, Singh S, et al. Metabolic syndrome is associated with increased oxo-nitrative stress and asthma-like changes in lungs. PLoS One. 2015;10(6):e0129850. doi:10.1371/journal.pone.0129850
50. Lee W, Lee HJ, Jang HB, et al. Asymmetric dimethylarginine (ADMA) is identified as a potential biomarker of insulin resistance in skeletal muscle. Sci Rep. 2018;8(1):2133. doi:10.1038/s41598-018-20549-0
51. Holguin F, Comhair SAA, Hazen SL, et al. An association between l-arginine/asymmetric dimethyl arginine balance, obesity, and the age of asthma onset phenotype. Am J Respir Crit Care Med. 2013;187(2):153–159. doi:10.1164/rccm.201207-1270OC
52. Fu WJ, Haynes TE, Kohli R, et al. Dietary L-arginine supplementation reduces fat mass in Zucker diabetic fatty rats. J Nutr. 2005;135(4):714–721. doi:10.1093/jn/135.4.714
53. Ojima A, Ishibashi Y, Matsui T, et al. Glucagon-like peptide-1 receptor agonist inhibits asymmetric dimethylarginine generation in the kidney of streptozotocin-induced diabetic rats by blocking advanced glycation end product-induced protein arginine methyltranferase-1 expression. Am J Pathol. 2013;182(1):132–141. doi:10.1016/j.ajpath.2012.09.016
54. Mirmiran P, Moghadam SK, Bahadoran Z, Ghasemi A, Azizi F. Dietary L-arginine intakes and the risk of metabolic syndrome: a 6-year follow-up in tehran lipid and glucose study. Prev Nutr Food Sci. 2017;22(4):263–270. doi:10.3746/pnf.2017.22.4.263
55. Holguin F, Grasemann H, Sharma S, et al. L-Citrulline increases nitric oxide and improves control in obese asthmatics. JCI Insight. 2019;4(24). doi:10.1172/jci.insight.131733.
56. Kenyon NJ, Last M, Bratt JM, et al. l-arginine supplementation and metabolism in asthma. Pharmaceuticals (Basel). 2011;4(1):187–201. doi:10.3390/ph4010187
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.