Back to Journals » Neuropsychiatric Disease and Treatment » Volume 14
Dysfunction of cortical synapse-specific mitochondria in developing rats exposed to lead and its amelioration by ascorbate supplementation
Authors Ahmad F, Salahuddin M, Alamoudi W ![]() , Acharya S
, Acharya S ![]()
Received 3 August 2017
Accepted for publication 15 January 2018
Published 21 March 2018 Volume 2018:14 Pages 813—824
DOI https://doi.org/10.2147/NDT.S148248
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Faraz Ahmad,1,2 Mohammad Salahuddin,3 Widyan Alamoudi,2 Sadananda Acharya1
1Department of Public Health, College of Public Health, Imam Abdulrahman Bin Faisal University, Dammam, Saudi Arabia; 2Neuroscience Department, Institute for Research and Medical Consultations, Imam Abdulrahman Bin Faisal University, Dammam, Saudi Arabia; 3Animal House Department, Institute for Research and Medical Consultations, Imam Abdulrahman Bin Faisal University, Dammam, Saudi Arabia
Background: Lead (Pb) is a widespread environmental neurotoxin and its exposure even in minute quantities can lead to compromised neuronal functions. A developing brain is particularly vulnerable to Pb mediated toxicity and early-life exposure leads to permanent alterations in brain development and neuronal signaling and plasticity, culminating into cognitive and behavioral dysfunctions and elevated risk of neuropsychiatric disorders later in life. Nevertheless, the underlying biochemical mechanisms have not been completely discerned.
Methods: Because of their ability to fulfill high energy needs and to act as calcium buffers in events of high intensity neuronal activity as well as their adaptive regulatory capability to match the requirements of the dynamicity of synaptic signaling, synapse-specific or synaptic mitochondria (SM) are critical for synaptic development, function and plasticity. Our aim for the present study hence was to characterize the effects of early-life Pb exposure on the functions of SM of prepubertal rats. For this purpose, employing a chronic model of Pb neurotoxicity, we exposed rat pups perinatally and postnatally to Pb and used a plethora of colorimetric and fluorometric assays for assessing redox and bioenergetic properties of SM. In addition, taking advantage of its ability as an antioxidant and as a metal chelator, we employed ascorbic acid (vitamin C) supplementation as an ameliorative therapeutic strategy against Pb-induced neurotoxicity and dysfunction of SM.
Results: Our results suggest that early-life exposure to Pb leads to elevated oxidative stress in cortical SM with consequent compromises in its energy metabolism activity. Ascorbate supplementation resulted in significant recovery of Pb-induced oxidative stress and functional compromise of SM.
Conclusion: Alterations in redox status and bioenergetic properties of SM could potentially contribute to the synaptic dysfunction observed in events of Pb neurotoxicity. Additionally, our study provides evidence for suitability of ascorbate as a significant ameliorative agent in tacking Pb neurotoxicity.
Keywords: synaptic, oxidative damage, heavy metal neurotoxicity, neuropsychiatric, mitochondrial bioenergetics, mitochondrial membrane potential
Introduction
Lead (Pb) is a naturally occurring toxic heavy metal, and its widespread use in recent decades has resulted in extensive environmental contamination and increased risk of exposure.1,2 Pb exposure can occur through contaminated air, water as well as food. Moreover, exposure to almost all forms of Pb (metallic, organic and inorganic) can lead to toxicity making it a high-risk environmental toxin of significant concern.2 Despite widespread efforts to contain its exposure, environmental Pb contamination is still a concern especially in urban areas of developing countries with emerging industrial production.1 Importantly, studies have shown that exposure to even low levels of Pb, previously thought to be permissible, can be neurotoxic and have deleterious cognitive and psychological outcomes.3,4 Thus, while blood Pb levels below 10 μg/dL was considered safe, recent studies have provided evidence suggesting that exposure to Pb resulting in blood Pb levels even below this limit can account for irreversible neurotoxicity and detrimental behavioral and cognitive outcomes.5–7
While Pb affects virtually all organs and organ systems in the body including cardiovascular, renal and reproductive systems,2 its ability to act as a potent and pervasive neurotoxin is well documented.8–10 Pb is permeable to the blood–brain barrier, possibly through the action of Ca2+-ATPase, and rapidly accumulates in neurons and astrocytes.11 In fact, the nervous system is regarded as among the most vulnerable target of Pb toxicity, which is evident by behavioral abnormalities, neuromuscular disabilities and cognitive deficits in events of Pb exposure.2,12 Along similar lines, several groups have shown impairment of brain functions upon Pb exposure in both animal and human studies.9 Proper maintenance, function and plasticity of synaptic signaling are critical factors for sensory-motor, cognitive and behavioral functions.13 Not surprisingly, Pb has been shown to induce alterations in both pre-synaptic and post-synaptic functions of neurons.5,9,14 While studies have implicated oxidative stress, interference with calcium signaling and gene expression defects in synaptic dysfunction induced by Pb,15 a complete understanding of molecular mechanisms and targets involved has remained elusive.
Immature nervous system is particularly susceptible to prenatal and postnatal lead exposure because of the presence of a still developing blood–brain barrier.16 Alterations in normal synaptic functions early in brain development can critically affect development, function and plasticity of the brain,5,16,17 in turn leading to permanent compromises in higher order brain functions. Not surprisingly then, developmental Pb neurotoxicity leads to permanent alterations in brain development culminating into irreparable deficits in sensory-motor, cognitive and behavioral functions as well as elevated risk of psychological disorders and stress.4,18–20 Hence, understanding the molecular and cellular factors involved in synaptic dysfunction induced by Pb in the developing nervous system and devising therapeutic strategies against it are of prime importance.
Synapse-specific mitochondria (or synaptic mitochondria [SM]) play essential regulatory roles in the development and physiology of synapses because of their ability to fulfill high energy demands and to regulate redox and calcium signaling.21–24 Additionally, SM fractions are capable of adaptive reconfiguration to suit with the dynamic changes associated with synaptic activity and plasticity.22,25–27 Because of these reasons, although sharing the same origin, SM are not identical to free (non-synaptic) mitochondria.21,23 These differences are in part governed by dissimilarities in their physical sizes, proteome, lipidome and enzyme profiles.21,24 Moreover, synaptic mitochondrial pool is more vulnerable to pathological insults and calcium overload and swelling, and its dysfunction is implicated in synaptic deficits commonly observed in pathologies like neurodegenerative conditions.21–23,26,28 Importantly, SM are important regulators of synaptic morphogenesis during brain development and neuronal plasticity not only because of their bioenergetic functions but also because they modulate synapse pruning and immunoinflammatory functions.17,21,26,27,29 In light of these evidences, alterations in proper functioning of cortical SM upon exposure to early-life stressors like Pb could potentially lead to critical disruptions in synaptic signaling, which could in turn have irreversible detrimental effect on high-order brain functions.
Ascorbic acid or vitamin C is a water-soluble vitamin with known neuromodulatory and neuroprotective properties in events of neurotoxicity and excitotoxicity30–33 as well as in neuropsychiatric diseases.34 Of note, neuroprotective effects of ascorbate supplementation in heavy metal toxicity is also well documented.35,36 Neuroprotective effects of ascorbate are thought to involve a multifaceted array of therapeutic mechanisms involving metal chelation, antioxidant and anti-apoptotic actions and modulation of neurotransmitter signaling.34,35 In fact, therapeutic effects of ascorbate in alleviation of lead toxicity have been documented by a number of groups.37–41
Our aims for this study were 1) to analyze deleterious effects of early-life (prenatal and postnatal) Pb exposure on SM of prepubertal rat cerebral cortices, and 2) to observe if oral supplementation of ascorbic acid could ameliorate these effects.
Materials and methods
Chemicals and reagents
All chemicals were of analytical grade and procured from EMD Millipore or Sigma-Aldrich Co.
Animals and experimental paradigms
All experiments involving animals were carried out in accordance with the institutional guidelines for animal care and use for scientific research and were approved by the Institutional Review Board of Imam Abdulrahman Bin Faisal University. Female Wistar rats were housed in cages with sexually mature males (2:1, male to female ratio) under a light-dark 12/12 regime in rooms with a controlled temperature of 25°C with free access to food chow and drinking water. After separation from the males, each pregnant female was housed in a separate cage. Pregnant females (gestation day GD15) were separated and divided into four groups: Ctrl (control), Pb (lead), Asc (ascorbic acid) and Pb+Asc (lead and ascorbic acid). While dams of the Ctrl and Asc groups received normal drinking water, Pb and Pb+Asc groups were provided with 0.2% lead acetate (2,000 ppm or 6.15 mM) in drinking water from GD15 until weaning of pups at postnatal day P21.42,43 Drinking water to all dams was provided ad libitum. Mothers of Asc and Pb+Asc dams were administered with 500 mg/kg body weight of ascorbic acid using an oral gavage from GD15 until weaning of pups at P21.37 After weaning (at P21), lead acetate and ascorbic acid treatments were stopped for all pups, and male and female pups of each group were separated and maintained with free access to food chow and normal drinking water. Only male pups were selected for experiments and were sacrificed on P30. This paradigm of oral route of Pb administration in drinking water has been used previously and was chosen because it emulates environmental exposure to this heavy metal.42–44 No significant difference in the volume of water consumed was observed between the groups.
Isolation of synapse-specific mitochondrial fraction
Rats were decapitated under anesthesia and brain cortices were dissected out. Synapse-specific mitochondria from cerebral cortices of P30 rats were isolated using Ficoll gradient-based ultracentrifugation as described previously.45 Briefly, cerebral cortices were homogenized in 10 times volume of isolation buffer (10 mM Tris, 1 mM EDTA(K), 320 mM sucrose, pH 7.4) using a Potter Elvehjem pestle and glass tube on ice. The homogenate was then centrifuged at 1,300× g for 10 min at 4°C and resultant supernatant (post-nuclear supernatant [PNS]) was further centrifuged at 17,000× g for 10 min at 4°C to obtain supernatant (post-mitochondrial supernatant [PMS]) and crude mitochondrial pellet (CMP). CMP was resuspended in isolation buffer and loaded onto a discontinuous 7.5%–10% Ficoll medium gradient (ratio of CMP suspension: 7.5% Ficoll: 10% Ficoll was 5:7:7) and centrifuged at 99,000× g for 30 min at 4°C in a Sorvall MX150 micro-ultracentrifuge (Thermo Fisher Scientific, Waltham, MA, USA). This separated CMP into myelin (at first interface), synaptosomes (at second interface) and free (non-synaptic) mitochondria as a pellet. Synaptosomal fraction was then collected and lysed by homogenization in 6 mM Tris pH 8.1 and the homogenate was centrifuged at 11,800× g for 10 min at 4°C. The pellet obtained was washed with 6 mM Tris pH 8.1 and resuspended in 3% Ficoll medium and layered on top of a discontinuous 4.5%–6% Ficoll medium gradient (ratio of 3% Ficoll suspension: 4.5% Ficoll: 6% Ficoll was 3.3:5:10). The gradient tube was centrifuged at 11,300× g for 30 min at 4°C and the pellet obtained was enriched in synapse-specific mitochondrial (SM) fraction and was washed twice with isolation buffer at 10,000× g for 10 min at 4°C. SM were resuspended in isolation buffer, aliquoted and stored at −80°C. Purity and integrity of SM fractions were assessed by enrichment of succinate dehydrogenase (SDH) activity and fumarase activity in presence and absence of Triton X-100, respectively (details are presented in following sections).
Mitochondrial enrichment assay
Enrichment of mitochondria was determined by assaying for the activity of SDH, a well-known mitochondrial marker protein. SDH activity in homogenate, PNS, PMS, CMP and enriched SM fractions was measured spectrophotometrically to verify enrichment at each stage of subcellular fractionation. Assay for SDH activity was performed essentially as described.46 Briefly, SM fraction or other subcellular fractions (0.1 mg protein each) were suspended in 50 mM (K) phosphate buffer, pH 7.4 containing 3 mM potassium ferricyanide (III) acting as an exogenous electron acceptor. Decrease in absorption (420 nm) upon addition of 50 mM succinate was followed to measure the rate of reduction of potassium ferricyanide to potassium ferrocyanide (II) at 30°C for 2 min (ε420 for potassium ferricyanide =1,040 M−1·cm−1). Reaction rate was calculated as nmol ferrocyanide formed per min per mg protein and is represented with respect to reaction rate from homogenate. SM fraction pretreated with 500 mM malonate, a competitive inhibitor of SDH, was used as a negative control. All spectroscopic measurements for this study were performed on an Infinite M200 PRO plate-reader (Tecan, Grodig, Austria).
Mitochondrial integrity assay
Integrity of inner membrane of SM pool isolated from rat cerebral cortices was measured using activity of fumarase, an enzyme localized in mitochondrial matrix as described previously.47 The assay is based upon formation of fumarate from L-malate (fumarase catalyzes the reversible reaction of conversion of malate to fumarate). The enzymatic reaction was monitored as increase in absorbance at 240 nm upon addition of 50 mM malate to SM fraction (0.1 mg protein) resuspended in isolation buffer in presence or absence of 0.2% Triton X-100. The linear part of the kinetic curve was used to calculate the slope of fumarase activity and expressed as μmol fumarate formed/min/mg protein (ε240 for fumarate =2.44 mM−1·cm−1). Integrity of SM pool was calculated as 1-(ν−TX/ν+TX), where ν−TX and ν+TX represent fumarase reaction rates in absence and presence of Triton X-100 and is expressed as a percentage.
Assays for reactive oxygen species
Reactive oxygen species (ROS) levels in SM samples isolated from cerebral cortical tissue of rat pups of the Ctrl, Pb and Pb+Asc groups were measured using a fluorometric method employing 2′,7′-dichlorofluorescein diacetate (DCFH-DA). In presence of ROS, non-fluorescent DCFH-DA is converted to fluorescent dichlorofluorescein (DCF). For the assay, mitochondrial sample (0.5 mg protein) was incubated with 10 μM DCFH-DA for 15 min at 30°C. Formation of DCF was monitored fluorometrically (excitation at 495 nm; emission at 530 nm). A standard curve of DCF was always used in all experiments, and ROS levels were calculated as μM DCF formed per mg protein and is represented with respect to Ctrl samples.
Assay for reactive nitrogen species
Nitric oxide (NO•) end products, total nitrites and nitrates (NOx), were measured using Griess reagent (0.2% N-(1-naphthyl) ethylenediamine dihydrochloride, 2% sulfanilamide and 10% phosphoric acid).48 SM samples (0.5 mg protein) were diluted to a final volume of 100 μL in isolation buffer and incubated with an equal volume of Griess reagent at room temperature in dark for 10 min. Absorbance was measured at 548 nm, and concentration of NOx levels was determined using a sodium nitrite standard curve and is represented as pmol/μg protein.
Oxidative damage assays
Oxidative damage to cortical SM pool was assessed by assays for lipid peroxidation, protein sulfhydryl oxidation and protein carbonylation using thiobarbituric acid (TBA), 5,5′-dithio-bis-(2-nitrobenzoic acid) (DTNB, Ellman reagent) and 2,4-dinitrophenylhydrazine (DNPH), respectively.48,49
Because sucrose present in isolation buffer can interfere with TBA assay,50 synaptic mitochondrial samples (1 mg protein) were washed twice with and resuspended in 100 μL of phosphate-buffered saline (PBS), pH 7.4. SM were then incubated with 200 μL of TBA-TCA-HCl reagent (15% trichloroacetic acid [TCA], 0.375% TBA and 0.25 M HCl) at 80°C for 20 min. A blank comprising the reaction mixture without mitochondria was always included. The samples were subsequently centrifuged at 17,000× g for 5 min and absorbance of the supernatant was measured at 535 nm. Lipid peroxides were determined as pmol thiobarbituric acid reactive substances (TBARS) per μg protein using an ε535 value of 1.56×105 M−1·cm−1 after subtraction of the absorbance of the blank sample.
Protein thiol oxidation was estimated indirectly using an assay that measures free protein thiols (reduced thiol groups).49 In brief, SM fractions (1 mg of protein) were treated with TCA (final concentration 10% w/v) for 10 min on ice, and the protein pellet was obtained after centrifugation at 17,000× g for 3 min. The pellets were then washed twice with 5% TCA and solubilized in 100 mM Tris, pH 8.8, containing 5% SDS. Re-solubilized protein pellet was incubated with 10 mM DTNB for 30 min in dark at 30°C. 2-Nitro-5-thiobenzoate (TNB) ion formed upon reaction of thiols with DTNB provided an estimate of free thiols and is estimated at 412 nm (ε412 for TNB =14,150 M−1·cm−1). Free (reduced) thiol groups in SM preparations were represented as μmol per μg protein after subtraction of the absorbance of the blank sample (lacking any mitochondria).
For quantification of protein carbonyls as another parameter of protein oxidation, SM fractions (1 mg protein; diluted in isolation buffer to a final volume of 100 μL) were incubated with 100 μL of 10 mM DNPH (prepared in 2N HCl) with intermittent shaking in dark at room temperature for 1 h. Buffer blanks without mitochondria were always used in all experiments. Proteins in the reaction mixture were precipitated by TCA (final concentration of 10% w/v) and pelleted by centrifugation at 17,000× g for 5 min. Protein pellets were washed thrice with acetone and dissolved in 8 M urea, 20 mM KH2PO4 (pH 2.3). Protein carbonyls formed were measured at 370 nm and expressed as pmol per μg protein using an ε370 of 2.2×10−4 M−1·cm−1.
Measurement of mitochondrial antioxidant capacity
Antioxidant power of SM preparations was analyzed following reduction (and consequent decolorization) of the radical cation form of ABTS (2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid)) (ABTS+•). The ABTS+• decolorization method has been used previously for analyzing the antioxidant power of biological samples including mitochondria.51,52 ABTS+• was produced by incubation of 7 mM ABTS with 2.45 mM potassium persulfate for 12–16 h at room temperature in dark. ABTS+• formed by this method is stable for at least 2 days in dark at room temperature and has absorption maxima at 420, 660 and 734 nm.51 Rate of ABTS+• decolorization at 734 nm upon addition of SM samples was measured for 20 min at 30°C. A blank without mitochondria was used in all experiments. Antioxidant capacities of SM samples are represented with respect to Ctrl group of animals.
Safranin uptake assay
Cationic lipophilic dyes like safranin O have been used as a tool for measurement of mitochondrial membrane potential (MMP, ΔΨm).47,53 The assay is based upon the principle that mitochondrial uptake of safranin O depends on its ΔΨm. For the assay, SM preparations (1 mg protein) were incubated in isolation buffer containing 10 μM safranin O for 15 min at 30°C in dark. Subsequently, the mitochondria were pelleted by centrifugation at 17,000× g for 5 min and fluorescence of mitochondrial pellet and supernatant was measured (excitation at 520 nm; emission at 570 nm). The ratio F(520 nm/570 nm) intramitochondrial/F(520 nm/570 nm) extramitochondrial was calculated to estimate relative mitochondrial uptake of safranin O and was used as an estimation of MMP.54 MMP of SM fractions is represented with respect to values of samples from Ctrl animals.
Mitochondrial MMP generation
MMP (ΔΨm) generation in presence of energizing substrates (malate and glutamate) was assessed using the principle of reduction of fluorescence (quenching) of safranin O in presence of energized mitochondria with a repolarized ΔΨm.53 Briefly, SM preparations (0.5 mg protein) were suspended in isolation buffer containing 10 mM KCl, 1 mM MgCl2 and 10 μM safranin O. ΔΨm generation was monitored by assessing the change in fluorescence of safranin O (excitation at 520 nm; emission at 570 nm) before and 15 min after addition of 50 mM malate and 50 mM glutamate at 30°C. Extent of decrease in safranin O fluorescence upon mitochondrial energization was calculated and used for estimation of MMP repolarization. ΔΨm generation of SM samples is represented with respect to values from Ctrl samples.
Mitochondrial complex assays
Complex I (NADH dehydrogenase) assay
Complex I activity assay was performed as described.55 Briefly, SM (0.1 mg protein) were suspended in isolation buffer containing 3 mM potassium ferricyanide (III). Reaction was initiated by addition of 60 mM NADH, and rate was monitored as decrease in absorption at 420 nm at 30°C. Reaction rate was calculated as nmol ferrocyanide formed per min per mg protein using the linear part of the curve. A negative control employing mitochondria pretreated with 1 mM rotenone was used for each sample to obtain the rotenone-sensitive NADH oxidation, which is considered to represent the actual complex I activity.56 Complex I activity is represented with respect to Ctrl values.
Complex II (succinate dehydrogenase) assay
Assay for complex II or SDH was performed essentially as described in the Mitochondrial enrichment assay section. Reaction rate of SM samples is represented with respect to rate of SM from Ctrl animals.
Complex III (coenzyme Q: cytochrome c oxidoreductase) assay
Assay for complex III activity was carried out as described elsewhere.56 SM samples (0.1 mg protein) suspended in isolation buffer were preincubated with 5 mM KCN to block cytochrome oxidase activity. Reaction was initiated by addition of 5 mM cytochrome c, and the rate of reaction (increase in amounts of reduced cytochrome c) was followed at 550 nm at 30°C (ε550 for reduced cytochrome c =21 mM−1·cm−1). Reaction rate was calculated as μmol cytochrome c reduced per min per mg protein and is represented with respect to rate of complex III activity of SM from Ctrl animals.
Complex IV (cytochrome c oxidase) assay
Reduction of cytochrome c and activity assay of complex IV (cytochrome c oxidase) was performed as described previously.56 Cytochrome c was reduced by addition of a few crystals of sodium dithionite, and reduction of cytochrome c was confirmed by change in color of the solution from blood red to pink. Purification of reduced cytochrome c was performed by desalting using a Sephadex G-25 column (Amersham, Uppsala, Sweden). For the activity assay of complex IV, SM samples (0.1 mg protein) were resuspended in isolation buffer and incubated with 0.3 mM reduced cytochrome c. The rate of oxidation of cytochrome c was followed at 30°C as decrease in absorption at 550 nm.46 Reaction rate was calculated as μmol cytochrome c oxidized per min per mg protein and is represented with respect to rate of SM from Ctrl animals.
ATP synthesis assay
ATP synthesis was measured indirectly by following consumption of inorganic phosphate (Pi) in presence of ADP and substrates malate and glutamate, employing a glucose-hexokinase trap method.46 Reaction mixture contained SM preparations (1 mg protein) resuspended in isolation buffer containing 50 mM glucose, 25 mM KH2PO4, 2 mM MgCl2, 5 units/mL of hexokinase (EC units, 1 μmol substrate/min), 2 mM ADP and 50 mM each of malate and glutamate. The reaction was allowed to proceed for 20 min at 30°C and terminated by addition of TCA (final concentration 10% w/v). The supernatant obtained after centrifugation at 10,000× g for 1 min was assayed for Pi using a phosphomolybdic acid-based protocol employing ascorbic acid as the reducing agent.57 A standard curve (1–50 mM KH2PO4) was used in all experiments, and consumption of Pi is represented with respect to Ctrl values.
Statistical analysis
All graphical representation of data and statistical analyses were performed using GraphPad Prism software. Data are represented as mean ± SEM as indicated in the figure legends. Statistical comparisons were made by using unpaired two-tailed Student’s t-test. One-way analyses of variance followed by post hoc tests with Newman–Keuls correction was used to compare multiple groups. Data were considered significant if p<0.05.
Results
Purified synaptic mitochondrial fraction obtained by Ficoll gradient-based ultracentrifugation is enriched in SDH activity and has high integrity
Activity of SDH, a mitochondrial marker enzyme, was measured in various cellular fractions to analyze the level of enrichment of the SM preparations isolated by the discontinuous gradient Ficoll medium-based ultracentrifugation protocol. SDH activity in synaptic mitochondrial preparation was found to be almost 7 times more than that in the starting homogenate material, indicating robust enrichment of mitochondria (Figure 1). Additionally, we assayed fumarase activity in presence or absence of detergent Triton X-100 to measure the integrity of SM fraction.47 Integrity of SM fraction was found to be 93.6%±3.2% (mean ± SD).
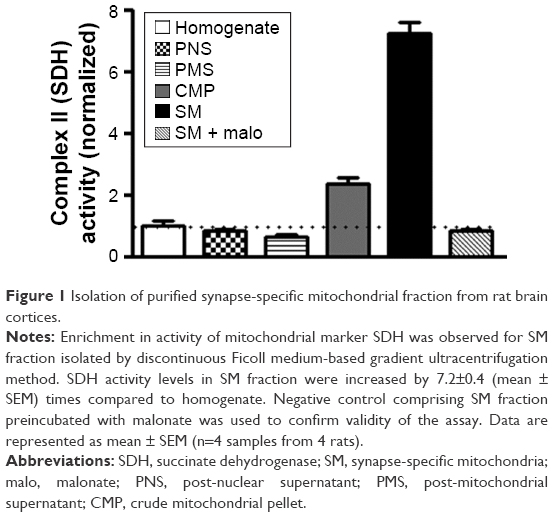 | Figure 1 Isolation of purified synapse-specific mitochondrial fraction from rat brain cortices. |
Ascorbic acid supplementation ameliorates oxidative damage and reduction in antioxidant capacity of SM induced by early-life Pb exposure
Oxidative stress in SM of rat cerebral cortices was measured using both DCFH-DA and Griess assays for the presence of ROS and reactive nitrogen species (RNS), respectively. Increase in ROS and NOx levels were observed in SM from rats of Pb group when compared to Ctrl group, indicating elevated oxidative stress. Pb-induced increase in oxidative stress was rescued by supplementation with ascorbate (Figure 2A and B).
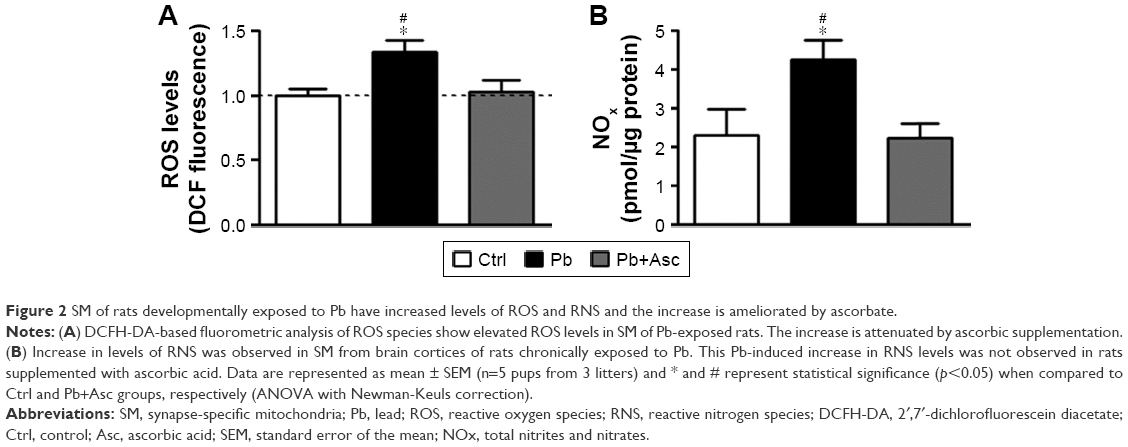 | Figure 2 SM of rats developmentally exposed to Pb have increased levels of ROS and RNS and the increase is ameliorated by ascorbate. |
Increase in oxidative stress in SM of rats of Pb group was accompanied by elevated levels of lipid and protein oxidation markers, as measured by the presence of TBARS, and protein carbonyls and oxidized protein thiols, respectively. Moreover, ascorbic acid treatment prevented the increase in lipid and protein oxidation in Pb-exposed rats (Figure 3A–C).
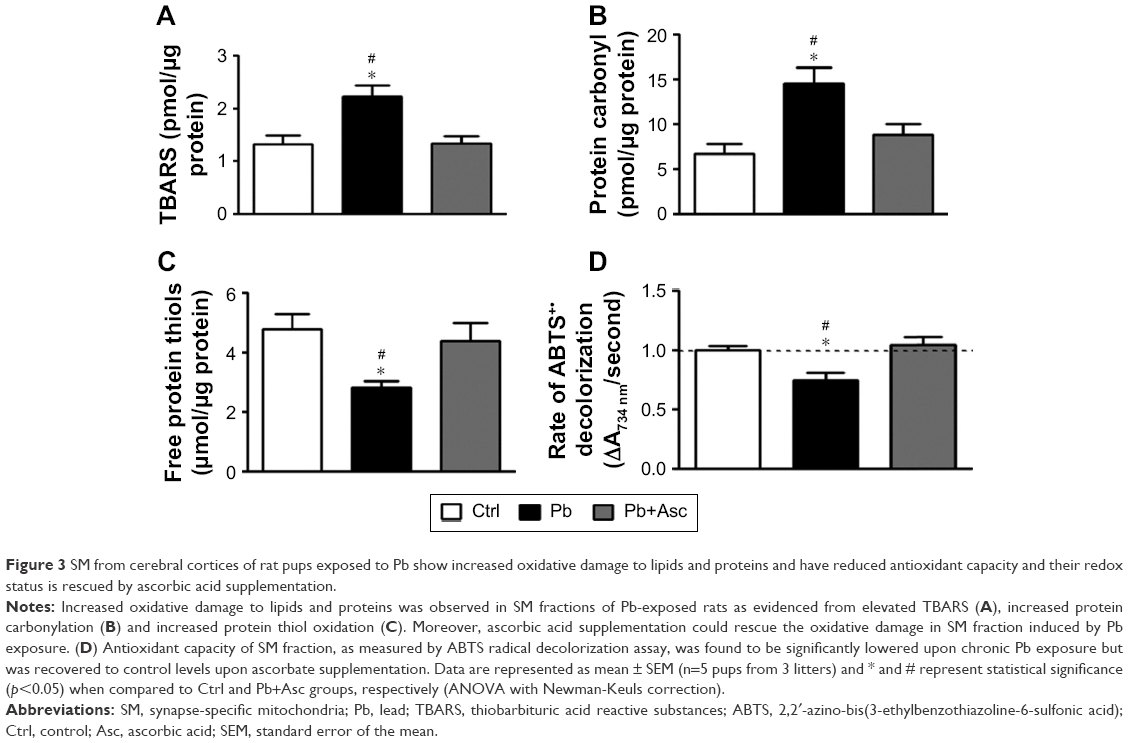 | Figure 3 SM from cerebral cortices of rat pups exposed to Pb show increased oxidative damage to lipids and proteins and have reduced antioxidant capacity and their redox status is rescued by ascorbic acid supplementation. |
Antioxidant capacity of SM fractions of rat cortices was measured by ABTS radical scavenging assay. Ability to scavenge ABTS+• was significantly reduced in SM fraction of rats from Pb group when compared to Ctrl rats. Antioxidant power of SM from prepubertal rats developmentally exposed to Pb was recovered in rats from Pb+Asc group, highlighting the rescue effects of ascorbate supplementation (Figure 3D).
The data suggest that increase in oxidative stress in SM fraction from prepubertal rats developmentally exposed to Pb is due to both increase in generation of ROS and RNS levels as well as reduction in the capacity to scavenge free radicals. Moreover, high levels of free radicals in SM of Pb-exposed rats resulted in increased protein and lipid damage. Interestingly, elevated levels of RNS/ROS, reduced radical scavenging and increased protein and lipid oxidation are prevented by supplementation with ascorbic acid.
Pb induced alterations in MMP of SM and its repolarization are rescued by ascorbate
Transmembrane potential of mitochondria (inside negative), also called as MMP (ΔΨm), is established by the proton pump of the mitochondrial respiratory chain and is a measure of functional state of mitochondria. ΔΨm is a critical regulator of several mitochondrial functions including bioenergetics (ATP production), mitochondrial dynamics (fusion and fission), redox homeostasis and apoptotic signaling.58 Using the cationic lipophilic probes, safranin O, we found that ΔΨm of SM isolated from Pb-exposed prepubertal rats was slightly but significantly reduced when compared to SM of aged matched controls (Figure 4A). Repolarization of energized SM (in presence of substrates glutamate and malate) was also assayed and found to be significantly reduced in SM isolated from rats developmentally exposed to Pb in comparison to Ctrl rats (Figure 4B), indicating compromises in mitochondrial bioenergetic enzyme functions. Deficits in both basal ΔΨm and repolarization of energized SM of Pb-exposed rats were recovered by supplementation with ascorbic acid (Figure 4A and B). However, ascorbic acid alone did not alter the basal or energized ΔΨm (Figure S1A and B).
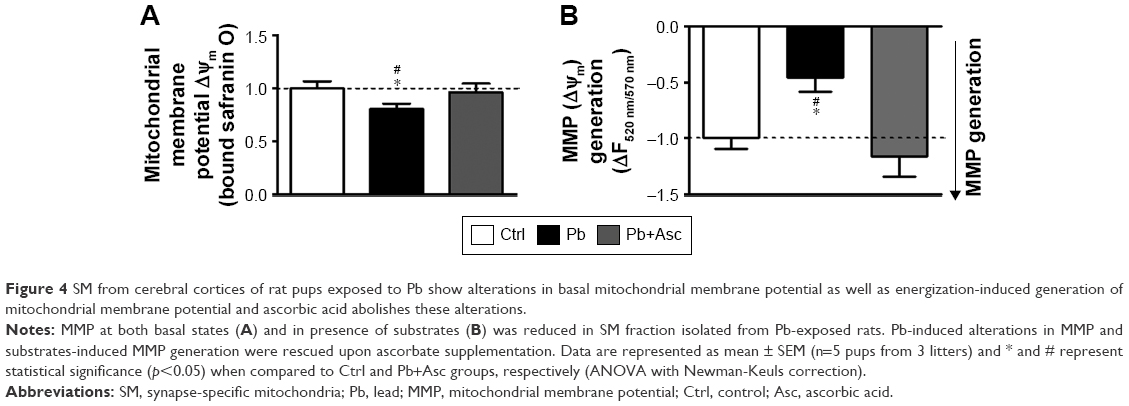 | Figure 4 SM from cerebral cortices of rat pups exposed to Pb show alterations in basal mitochondrial membrane potential as well as energization-induced generation of mitochondrial membrane potential and ascorbic acid abolishes these alterations. |
Ascorbic acid supplementation rescues compromised activities of electron transport chain oxidoreductases and ATP synthesis ability of cortical SM from rats developmentally exposed to Pb
To observe any dysregulation in the bioenergetic properties of SM of Pb-exposed prepubertal rats, we analyzed the activities of enzyme complexes I, II, III and IV of the electron transport chain. While activity levels of complex I and complex IV in SM fraction of Pb-exposed rats were similar to those from aged matched controls (Figure 5A and D), activities of complexes II and III were found to be significantly reduced in SM of Pb-exposed rats (Figure 5B and C). Moreover, while ascorbic acid supplementation rescued the Pb-mediated loss of complex II activity (Figure 5B), it was unable to change complex III activity levels (Figure 5C).
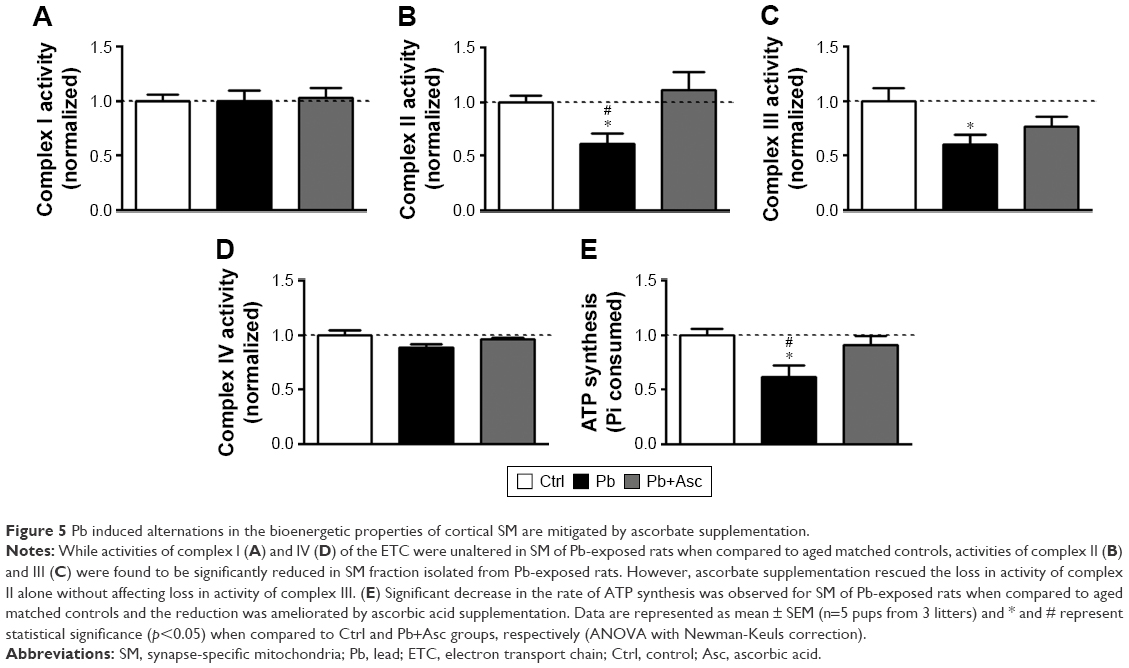 | Figure 5 Pb induced alternations in the bioenergetic properties of cortical SM are mitigated by ascorbate supplementation. |
We also observed a decrease in ATP synthesis in presence of substrates malate and glutamate in SM fractions isolated from Pb-exposed rats; however, complete recovery was observed in rats supplemented with ascorbic acid (Figure 5E). On the other hand, ascorbic acid alone did not have any effects on the bioenergetic properties of SM (Figure S1C–G).
The data suggest that reduction in bioenergetic activity of SM from prepubertal rats developmentally exposed to Pb is rescued with ascorbate supplementation.
Discussion
In the brain, Pb neurotoxicity is prominent in prefrontal cortex, hippocampus and cerebellum,10 where it results in morphological, structural and pathological changes in neuronal cells and their synaptic connections.8,59,60 Mechanisms of lead neurotoxicity are complex. Many of the toxic mechanisms associated with Pb neurotoxicity are because of its ability to substitute for divalent Ca2+ and Zn2+ cations,6 which critically regulate important cellular processes including cell signaling and gene expression. Among the primary targets of Pb neurotoxicity are synaptic signaling and its maintenance and plasticity.5,9,14 In fact, both electrophysiological and biochemical data point to a reduced efficiency of both pre-synaptic and post-synaptic machinery upon Pb exposure.8,14 While alterations in neurotransmitter (glutamate) release and N-methyl-D-aspartate receptor physiology are the best studied mechanisms of Pb-induced synaptic dysfunction,5,9 an interplay of several factors is thought to influence disruption of synaptic functions in Pb neurotoxicity. These include deleterious effects of Pb on redox and calcium homeostasis; cell signaling and death pathways; and membrane receptor trafficking and gene expression.15,16
Developing brain is particularly vulnerable to Pb neurotoxicity because of its immature blood–brain barrier.16 In fact, chronic early-life exposure to Pb is known to have permanent detrimental effects on development, maintenance and function of the synapses,16 leading to irreparable neurodevelopmental deficits in sensory-motor and cognitive skills.3,4,61 This is because neuronal signaling (both spontaneous and evoked) at the synapse in the developing brain critically affects synapse pruning and proper brain development and function,26,29 and any alterations in synapse functions early in the brain development would lead to permanent cognitive and behavioral effects.
Synapse-specific mitochondria (SM) form a specialized class of mitochondria different from free neuronal mitochondria because of their ability of acting as calcium buffers to prevent excitotoxicity and as energy providers for the compelling process of synaptic signaling.21–23,25 Indeed, SM are smaller in size compared to free non-SM and have an altered enzyme profile as well as a modified proteome and lipidome, and metabolome.21,24 Additionally, SM population has elevated vulnerability to pathological stresses and calcium overload and swelling.21–23 Alterations in proper functioning of cortical SM in prepubertal rats upon Pb exposure as observed in our study could potentially lead to critical disruptions in synaptic signaling, which in turn would affect higher order brain functions. Although, Pb-induced mitochondrial dysfunction has been observed earlier, most of the studies have been limited to in vitro cell culture systems62–65 or in vitro mitochondrial preparations.66,67 Many in vivo studies using rodent models have observed mitochondrial dysfunction in adult or aged rats.12,68 Few studies have reported the effects of early-life Pb exposure on brain mitochondria in prepubertal rats. For example, ultrastructural morphological changes in hippocampal44 and retinal69 mitochondria have been observed upon exposure of neonatal rats to Pb. Evidence for altered mitochondrial bioenergetic functions in primary cerebellar granular neurons of rats exposed to Pb70 and hippocampal and cerebellar neurons71 has also been provided. Other studies indicate that perinatal Pb exposure increases ROS levels in a calcium-dependent manner72 and reduces antioxidant system of brain mitochondria.42,73 However, none of these studies have specifically looked at the effects of Pb exposure on synapse-specific mitochondria of brain cerebral cortex, which is an important regulator of synapse function and plasticity. In this study, we sought to assess the deleterious effects of early-life (prenatal and postnatal) exposure to Pb on synapse-localized mitochondrial population in the developing rat brain.
Our results suggest that early-life exposure to Pb leads to disruptions in redox homeostasis of SM quite early during development and consequent compromises in its membrane potential and energy metabolism activity. Given the critical importance of SM in the synapse physiology, this potentially constitutes as a major pathogenic event in synaptic dysfunction leading to detrimental behavioral outcomes observed in events of Pb exposure. Because ascorbic acid (vitamin C) is capable of both reducing oxidative stress and chelating metal ions, it can potentially serve as a detoxifying agent for Pb toxicity. Use of ascorbic acid in amelioration of Pb poisoning has been elucidated previously.12,37–39 We used a dose of 500 mg ascorbic acid per kg body weight because it reduces the number of degenerating neurons upon Pb exposure to levels comparable to control untreated rats.37 In comparison, a dose of 100 mg/kg body weight could only slightly reduce the number of degenerating neurons, which still remained significantly higher than control rats.38,39 While supplementation of ascorbic acid alone did not affect bioenergetic properties of SM, we observed appreciable recovery of Pb-induced oxidative stress and functional compromise of SM by ascorbic acid supplementation. Our results suggest that rescue of Pb-mediated disruptions in SM functions by ascorbate could potentially be used as a therapeutic strategy against early-life Pb exposure.
Acknowledgments
The study was partly funded by Deanship of Scientific Research, Imam Abdulrahman Bin Faisal University, Saudi Arabia (Project No. 2016-087-IRMC). The authors thank Dr Khaldoon Alsamman, Dr Hatem K Herzallah and Dr Sultan T Al-Otaibi for assistance in experiments and in the analysis and review of the results.
Disclosure
The authors report no conflicts of interest in this work.
References
Meyer PA, Brown MJ, Falk H. Global approach to reducing lead exposure and poisoning. Mutat Res. 2008;659(1–2):166–175. | ||
Assi MA, Hezmee MN, Haron AW, Sabri MY, Rajion MA. The detrimental effects of lead on human and animal health. Vet World. 2016;9(6):660–671. | ||
Chiodo LM, Jacobson SW, Jacobson JL. Neurodevelopmental effects of postnatal lead exposure at very low levels. Neurotoxicol Teratol. 2004;26(3):359–371. | ||
Allen KA. Is prenatal lead exposure a concern in infancy? What is the evidence? Adv Neonatal Care. 2015;15(6):416–420. | ||
Neal AP, Guilarte TR. Molecular neurobiology of lead (Pb2+): effects on synaptic function. Mol Neurobiol. 2010;42(3):151–160. | ||
Garza A, Vega R, Soto E. Cellular mechanisms of lead neurotoxicity. Med Sci Monit. 2006;12(3):RA57–RA65. | ||
Bellinger DC, Bellinger AM. Childhood lead poisoning: the torturous path from science to policy. J Clin Invest. 2006;116(4):853–857. | ||
White LD, Cory-Slechta DA, Gilbert ME, et al. New and evolving concepts in the neurotoxicology of lead. Toxicol Appl Pharmacol. 2007;225(1):1–27. | ||
Toscano CD, Guilarte TR. Lead neurotoxicity: from exposure to molecular effects. Brain Res Brain Res Rev. 2005;49(3):529–554. | ||
Sharma P, Chambial S, Shukla KK. Lead and neurotoxicity. Indian J Clin Biochem. 2015;30(1):1–2. | ||
Marchetti C. Molecular targets of lead in brain neurotoxicity. Neurotox Res. 2003;5(3):221–236. | ||
Flora G, Gupta D, Tiwari A. Toxicity of lead: a review with recent updates. Interdiscip Toxicol. 2012;5(2):47–58. | ||
Sweatt JD. Neural plasticity and behavior – sixty years of conceptual advances. J Neurochem. 2016;139(Suppl 2):179–199. | ||
Sadiq S, Ghazala Z, Chowdhury A, Büsselberg D. Metal toxicity at the synapse: presynaptic, postsynaptic, and long-term effects. J Toxicol. 2012;2012:132671. | ||
Verstraeten SV, Aimo L, Oteiza PI. Aluminium and lead: molecular mechanisms of brain toxicity. Arch Toxicol. 2008;82(11):789–802. | ||
Neal AP, Guilarte TR. Mechanisms of lead and manganese neurotoxicity. Toxicol Res (Camb). 2013;2(2):99–114. | ||
Hagberg H, Mallard C, Rousset CI, Thornton C. Mitochondria: hub of injury responses in the developing brain. Lancet Neurol. 2014;13(2):217–232. | ||
Winneke G. Developmental aspects of environmental neurotoxicology: lessons from lead and polychlorinated biphenyls. J Neurol Sci. 2011;308(1–2):9–15. | ||
Grandjean P, Herz KT. Trace elements as paradigms of developmental neurotoxicants: lead, methylmercury and arsenic. J Trace Elem Med Biol. 2015;31:130–134. | ||
Hsiang J, Díaz E. Lead and developmental neurotoxicity of the central nervous system. Curr Neurobiol. 2011;2(1):35–42. | ||
Fedorovich SV, Waseem TV, Puchkova LV. Biogenetic and morphofunctional heterogeneity of mitochondria: the case of synaptic mitochondria. Rev Neurosci. 2017;28(4):363–373. | ||
Vos M, Lauwers E, Verstreken P. Synaptic mitochondria in synaptic transmission and organization of vesicle pools in health and disease. Front Synaptic Neurosci. 2010;2:139. | ||
Du H, Guo L, Yan S, Sosunov AA, McKhann GM, Yan SS. Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc Natl Acad Sci U S A. 2010;107(43):18670–18675. | ||
Dubinsky JM. Heterogeneity of nervous system mitochondria: location, location, location! Exp Neurol. 2009;218(2):293–307. | ||
Ly CV, Verstreken P. Mitochondria at the synapse. Neuroscientist. 2006;12(4):291–299. | ||
Raefsky SM, Mattson MP. Adaptive responses of neuronal mitochondria to bioenergetic challenges: roles in neuroplasticity and disease resistance. Free Radic Biol Med. 2017;102:203–216. | ||
Jeanneteau F, Arango-Lievano M. Linking mitochondria to synapses: new insights for stress-related neuropsychiatric disorders. Neural Plast. 2016;2016:3985063. | ||
Cavallucci V, Ferraina C, D’Amelio M. Key role of mitochondria in Alzheimer’s disease synaptic dysfunction. Curr Pharm Des. 2013;19(36):6440–6450. | ||
Li Z, Okamoto K, Hayashi Y, Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119(6):873–887. | ||
Ahmad A, Shah SA, Badshah H, et al. Neuroprotection by vitamin C against ethanol-induced neuroinflammation associated neurodegeneration in the developing rat brain. CNS Neurol Disord Drug Targets. 2016;15(3):360–370. | ||
Kim HJ, Song W, Jin EH, et al. Combined low-intensity exercise and ascorbic acid attenuates kainic acid-induced seizure and oxidative stress in mice. Neurochem Res. 2016;41(5):1035–1041. | ||
de Freitas P, Zanoni JN, Alves AM, de Miranda Neto MH. Neuroprotection and neurodegeneration in submucosal VIP-IR neurons in the jejunum of ascorbic acid supplemented aging Wistar rats. Nutr Neurosci. 2012;15(6):283–288. | ||
Rice ME. Ascorbate regulation and its neuroprotective role in the brain. Trends Neurosci. 2000;23(5):209–216. | ||
Moretti M, Fraga DB, Rodrigues ALS. Ascorbic acid to manage psychiatric disorders. CNS Drugs. 2017;31(7):571–583. | ||
Patrick L. Toxic metals and antioxidants: part II. The role of antioxidants in arsenic and cadmium toxicity. Altern Med Rev. 2003;8(2):106–128. | ||
Hsu PC, Guo YL. Antioxidant nutrients and lead toxicity. Toxicology. 2002;180(1):33–44. | ||
Sadeghi A, Ebrahimzadeh Bideskan A, Alipour F, Fazel A, Haghir H. The effect of ascorbic acid and garlic administration on lead-induced neural damage in rat offspring’s hippocampus. Iran J Basic Med Sci. 2013;16(2):157–164. | ||
Chang BJ, Jang BJ, Son TG, et al. Ascorbic acid ameliorates oxidative damage induced by maternal low-level lead exposure in the hippocampus of rat pups during gestation and lactation. Food Chem Toxicol. 2012;50(2):104–108. | ||
Han JM, Chang BJ, Li TZ, et al. Protective effects of ascorbic acid against lead-induced apoptotic neurodegeneration in the developing rat hippocampus in vivo. Brain Res. 2007;1185:68–74. | ||
Acharya UR, Rathore RM, Mishra M. Role of vitamin C on lead acetate induced spermatogenesis in swiss mice. Environ Toxicol Pharmacol. 2003;13(1):9–14. | ||
Patra RC, Swarup D, Dwivedi SK. Antioxidant effects of alpha tocopherol, ascorbic acid and L-methionine on lead induced oxidative stress to the liver, kidney and brain in rats. Toxicology. 2001;162(2):81–88. | ||
Gottipolu RR, Davuljigari CB. Perinatal exposure to lead: reduction in alterations of brain mitochondrial antioxidant system with calcium supplement. Biol Trace Elem Res. 2014;162(1–3):270–277. | ||
Harry GJ, Schmitt TJ, Gong Z, Brown H, Zawia N, Evans HL. Lead-induced alterations of glial fibrillary acidic protein (GFAP) in the developing rat brain. Toxicol Appl Pharmacol. 1996;139(1):84–93. | ||
Baranowska-Bosiacka I, Strużyńska L, Gutowska I, et al. Perinatal exposure to lead induces morphological, ultrastructural and molecular alterations in the hippocampus. Toxicology. 2013;303:187–200. | ||
Lai JC, Walsh JM, Dennis SC, Clark JB. Synaptic and non-synaptic mitochondria from rat brain: isolation and characterization. J Neurochem.1977;28(3):625–631. | ||
Dua R, Gill KD. Effect of aluminium phosphide exposure on kinetic properties of cytochrome oxidase and mitochondrial energy metabolism in rat brain. Biochim Biophys Acta. 2004;1674(1):4–11. | ||
Valenti D, Vacca RA, de Pinto MC, De Gara L, Marra E, Passarella S. In the early phase of programmed cell death in Tobacco Bright Yellow 2 cells the mitochondrial adenine nucleotide translocator, adenylate kinase and nucleoside diphosphate kinase are impaired in a reactive oxygen species-dependent manner. Biochim Biophys Acta. 2007;1767(1):66–78. | ||
Dutta M, Ghosh AK, Rangari V, et al. Silymarin protects against copper-ascorbate induced injury to goat cardiac mitochondria in vitro: involvement of antioxidant mechanisms(s). Int J Pharm Pharm Sci. 2014;6(8):422–429. | ||
El-Masry TA, Emara AM, El-Shitany NA. Possible protective effect of propolis against lead-induced neurotoxicity in animal model. J Evol Biol Res. 2011;3(1):4–11. | ||
Buege JA, Aust SD. Microsomal lipid peroxidation. Methods Enzymol. 1978;52:302–310. | ||
Katalinic V, Modun D, Music I, Boban M. Gender differences in antioxidant capacity of rat tissues determined by 2,2′-azinobis (3-ethylbenzothiazoline 6-sulfonate; ABTS) and ferric reducing antioxidant power (FRAP) assays. Comp Biochem Physiol C Toxicol Pharmacol. 2005;140(1):47–52. | ||
Cherian E, Sudheesh NP, Janardhanan KK, Patani G. Free-radical scavenging and mitochondrial antioxidant activities of Reishi-Ganoderma lucidum (Curt: Fr) P. Karst and Arogyapacha-Trichopus zeylanicus Gaertn extracts. J Basic Clin Physiol Pharmacol. 2009;20(4):289–307. | ||
Valenti D, de Bari L, De Filippis B, Ricceri L, Vacca RA. Preservation of mitochondrial functional integrity in mitochondria isolated from small cryopreserved mouse brain areas. Anal Biochem. 2014;444:25–31. | ||
Emaus RK, Grunwald R, Lemasters JJ. Rhodamine 123 as a probe of transmembrane potential in isolated rat-liver mitochondria: spectral and metabolic properties. Biochim Biophys Acta. 1986;850(3):436–448. | ||
Powell CS, Jackson RM. Mitochondrial complex I, aconitase, and succinate dehydrogenase during hypoxia-reoxygenation: modulation of enzyme activities by MnSOD. Am J Physiol Lung Cell Mol Physiol. 2003;285(1):L189–L198. | ||
Spinazzi M, Casarin A, Pertegato V, Salviati L, Angelini C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat Protoc. 2012;7(6):1235–1246. | ||
Katewa SD, Katyare SS. A simplified method for inorganic phosphate determination and its application for phosphate analysis in enzyme assays. Anal Biochem. 2003;323(2):180–187. | ||
Iannetti EF, Willems PH, Pellegrini M, et al. Towards high-content screening of mitochondrial morphology and membrane potential in living cells. Int J Biochem Cell Biol. 2015;63:66–70. | ||
Gąssowska M, Baranowska-Bosiacka I, Moczydłowska J, et al. Perinatal exposure to lead (Pb) induces ultrastructural and molecular alterations in synapses of rat offspring. Toxicology. 2016;373:13–29. | ||
Wilson MA, Johnston MV, Goldstein GW, Blue ME. Neonatal lead exposure impairs development of rodent barrel field cortex. Proc Natl Acad Sci U S A. 2000;97(10):5540–5545. | ||
Hu H, Téllez-Rojo MM, Bellinger D, et al. Fetal lead exposure at each stage of pregnancy as a predictor of infant mental development. Environ Health Perspect. 2006;114(11):1730–1735. | ||
Dabrowska A, Venero JL, Iwasawa R, et al. PGC-1α controls mitochondrial biogenesis and dynamics in lead-induced neurotoxicity. Aging (Albany NY). 2015;7(9):629–647. | ||
Geier DA, King PG, Geier MR. Mitochondrial dysfunction, impaired oxidative-reduction activity, degeneration, and death in human neuronal and fetal cells induced by low-level exposure to thimerosal and other metal compounds. Toxicol Environ Chem. 2009;91(3–4):735–749. | ||
Ye F, Li X, Li F, et al. Cyclosporin A protects against Lead neurotoxicity through inhibiting mitochondrial permeability transition pore opening in nerve cells. Neurotoxicology. 2016;57:203–213. | ||
Liu G, Wang ZK, Wang ZY, Yang DB, Liu ZP, Wang L. Mitochondrial permeability transition and its regulatory components are implicated in apoptosis of primary cultures of rat proximal tubular cells exposed to lead. Arch Toxicol. 2016;90(5):1193–1209. | ||
Devi CB, Jyotsna V, Kumari KK, Indravathi G, Thakur A. In vitro effect of lead on cholinergic and bioenergetic systems in synaptosomal and mitochondrial fractions of rat brain regions. Int J Adv Life Sci. 2014;7(1):1–10. | ||
Holtzman D, Shen Hsu J, Mortell P. In vitro effects of inorganic lead on isolated rat brain mitochondrial respiration. Neurochem Res. 1978;3(2):195–206. | ||
Flora SJ, Saxena G, Mehta A. Reversal of lead-induced neuronal apoptosis by chelation treatment in rats: role of reactive oxygen species and intracellular Ca(2+). J Pharmacol Exp Ther. 2007;322(1):108–116. | ||
Perkins GA, Scott R, Perez A, Ellisman MH, Johnson JE, Fox DA. Bcl-xL-mediated remodeling of rod and cone synaptic mitochondria after postnatal lead exposure: electron microscopy, tomography and oxygen consumption. Mol Vis. 2012;18:3029–3048. | ||
Baranowska-Bosiacka I, Gutowska I, Marchetti C, et al. Altered energy status of primary cerebellar granule neuronal cultures from rats exposed to lead in the pre- and neonatal period. Toxicology. 2011;280(1–2):24–32. | ||
Lalith Kumar V, Muralidhara. Ameliorative effects of ferulic Acid against lead acetate-induced oxidative stress, mitochondrial dysfunctions and toxicity in prepubertal rat brain. Neurochem Res. 2014;39(12):2501–2515. | ||
Yang X, Wang B, Zeng H, et al. Role of the mitochondrial Ca(2+) uniporter in Pb(2+)-induced oxidative stress in human neuroblastoma cells. Brain Res. 2014;1575:12–21. | ||
Sannadi S, Kadeyala PK, Gottipolu RR. Reversal effect of monoisoamyl dimercaptosuccinic acid (MiADMSA) for arsenic and lead induced perturbations in apoptosis and antioxidant enzymes in developing rat brain. Int J Dev Neurosci. 2013;31(7):586–597. |
Supplementary material
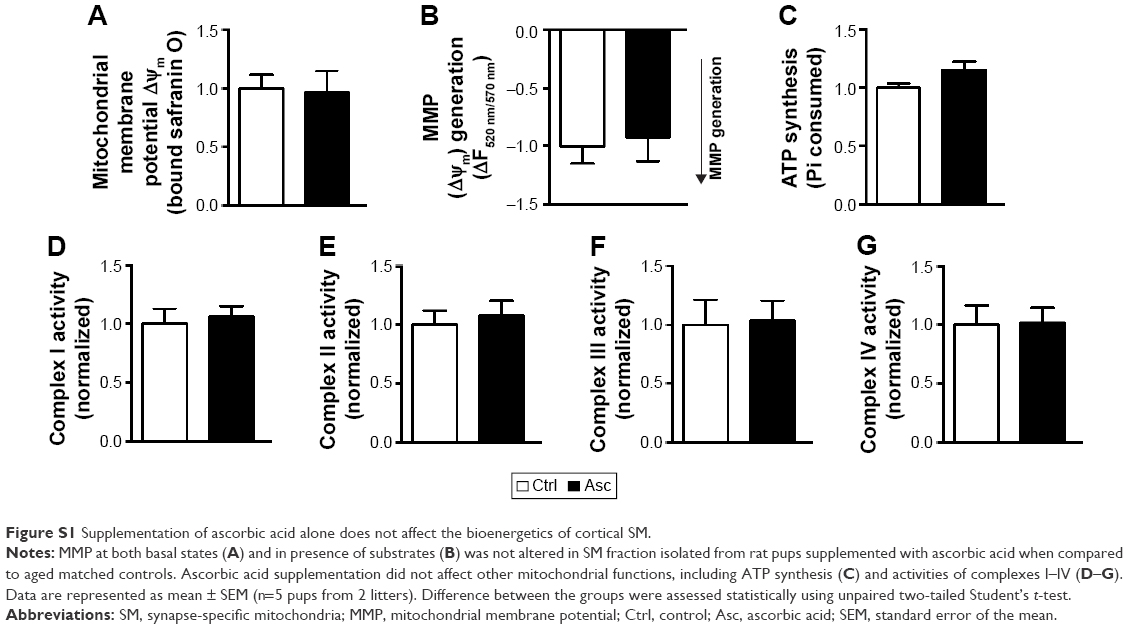 | Figure S1 Supplementation of ascorbic acid alone does not affect the bioenergetics of cortical SM. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.