Back to Journals » Journal of Inflammation Research » Volume 18
Dynamic Regulation of Macrophage Polarization in Acute Myocardial Infarction and Its Therapeutic Potential
Authors Xu A ![]() , Xu S, Tan X, Sun Q, Song Y, Nong Y
, Xu S, Tan X, Sun Q, Song Y, Nong Y ![]() , Wang X, Zeng Y, Fan H
, Wang X, Zeng Y, Fan H ![]() , Zhou Y
, Zhou Y ![]()
Received 27 May 2025
Accepted for publication 17 November 2025
Published 13 December 2025 Volume 2025:18 Pages 17363—17385
DOI https://doi.org/10.2147/JIR.S543139
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Anh Ngo
Anchen Xu,1,2 Shuai Xu,1,2 Xin Tan,1,2 Qiaoyi Sun,1,2 Yahui Song,3 Yuxin Nong,1,2 Xiangyu Wang,1,2 Yiyao Zeng,1,2 Huimin Fan,1– 3 Yafeng Zhou1,2
1Department of Cardiology, The Fourth Affiliated Hospital of Soochow University, Suzhou Dushu Lake Hospital, Medical Center of Soochow University, Suzhou, 215000, People’s Republic of China; 2Department of Hypertension, The Fourth Affiliated Hospital of Soochow University, Institute for Hypertension, Soochow University, Suzhou, 215000, People’s Republic of China; 3Center of Translational Medicine and Clinical Laboratory, The Fourth Affiliated Hospital to Soochow University, Suzhou Dushu Lake Hospital, Suzhou, 215028, People’s Republic of China
Correspondence: Yafeng Zhou, Email [email protected] Huimin Fan, Email [email protected]
Abstract: Acute myocardial infarction (AMI) remains one of the leading causes of mortality and disability worldwide, involving complex immune and inflammatory responses. Among these, macrophages play a pivotal role as key immune cells. The polarization state of macrophages determines their function in both myocardial injury and repair. In the early phase of AMI, M1 macrophages promote inflammation and facilitate the clearance of necrotic tissue by releasing pro-inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β), and interleukin-6 (IL-6). However, excessive or prolonged M1 polarization may contribute to myocardial fibrosis and further deterioration of cardiac function. In contrast, M2 macrophages promote tissue repair and anti-inflammatory responses in the later phase by secreting anti-inflammatory cytokines such as interleukin-10 (IL-10) and transforming growth factor-beta (TGF-β), thereby reducing fibrosis and facilitating myocardial remodeling. This review summarizes the dynamic changes in macrophage polarization during AMI and elaborates on their roles in myocardial injury, inflammation, and tissue repair. Furthermore, it highlights recent advances in therapeutic strategies aimed at modulating macrophage polarization to improve AMI outcomes, including mTOR inhibitors, sodium-glucose co-transporter 2 (SGLT2) inhibitors, glucagon-like peptide-1 (GLP-1) receptor agonists, and gene-editing technologies such as CRISPR/Cas9. Overall, this review underscores the importance of regulating macrophage polarization, particularly the transition from M1 to M2, as a promising therapeutic target for AMI. Modulating macrophage function may provide novel insights into enhancing myocardial repair and preventing adverse cardiac events.
Keywords: acute myocardial infarction, myocardial remodeling, macrophage polarization, M1/M2 macrophages, macrophage-based therapeutic strategies
Introduction
Acute myocardial infarction (AMI), also known as myocardial infarction, remains one of the leading causes of mortality and disability worldwide, posing a severe threat to human health.1 AMI is characterized by acute occlusion of the coronary arteries, leading to insufficient blood supply to the affected myocardial regions, resulting in myocardial necrosis and life-threatening consequences. The primary causes of AMI include coronary atherosclerosis, arrhythmias, and respiratory failure, all of which contribute to inadequate blood supply, myocardial hypoxia, and excessive oxygen consumption. The underlying pathology of AMI is complex and multifactorial, involving atherosclerosis, endothelial dysfunction, inflammation, and immune responses.2
As a critical component of the innate immune system, macrophages play a pivotal role in both the progression and resolution of AMI.3 Their role in myocardial infarction extends beyond traditional immune functions, as they regulate cardiac inflammation and repair through distinct polarization states. M1 macrophages primarily exert pro-inflammatory effects in the early phase, whereas M2 macrophages play a key role in tissue repair and anti-inflammatory responses. The ability of the heart to recover following AMI largely depends on immune regulation, particularly the dynamic changes in macrophages. As highly plastic immune cells, macrophages can polarize into either pro-inflammatory M1 or anti-inflammatory M2 phenotypes, significantly influencing the outcome of myocardial injury.4
In recent years, increasing evidence has demonstrated the diverse roles of macrophages in AMI, ranging from exacerbating inflammation and tissue damage to promoting repair and tissue remodeling. Understanding the dynamics of macrophage polarization in the context of AMI is crucial for developing therapeutic strategies aimed at modulating inflammation to enhance cardiac repair and reduce adverse outcomes such as heart failure. This review summarizes the mechanisms of macrophage polarization and its role in AMI progression, discusses the functional differences between polarized macrophages in myocardial injury and repair, and explores potential therapeutic strategies targeting macrophage polarization to mitigate post-infarction inflammation.
Macrophage Subtypes and Functions
Macrophages primarily originate from monocytes, which are differentiated from hematopoietic stem cells in the bone marrow. After entering the peripheral blood, monocytes migrate to various tissues where they further differentiate into tissue-specific macrophages. In addition to monocytes derived from the bone marrow, some macrophages may already exist during embryonic development and gradually distribute to different tissues during the organism’s development.5,6 Macrophages of distinct developmental origins exert non-redundant roles in both homeostasis and disease contexts. The main functions of macrophages include phagocytosing pathogens, clearing necrotic cells, presenting antigens, and regulating immune responses. In the heart, macrophages can respond rapidly following myocardial infarction (MI), exerting their immune regulatory and repair functions.7
Also, macrophages exhibit remarkable plasticity, enabling them to adapt to different microenvironmental stimuli and differentiate into various functional phenotypes, a process known as macrophage polarization.8 Generally, macrophages can be classified based on their functions and activation states into two major subtypes: classically activated M1 macrophages and alternatively activated M2 macrophages. M1 macrophages primarily function in innate and adaptive immunity, playing roles in Th1 cell recruitment, pathogen resistance, and tumor control. They also secrete numerous pro-inflammatory cytokines, such as TNF-α, IL-1, IL-6, IL-12, type I interferon (IFN), CXCL1-3, CXCL-5, and CXCL8-10, and exhibit strong antigen-presenting activity.9 The pro-inflammatory cytokines secreted by M1 macrophages are harmful to myocardial cells, potentially leading to or exacerbating myocardial damage. In contrast, M2 macrophages, which arise through selective or alternative activation pathways, can be induced by parasitic or fungal infections, immune complexes, or macrophage colony-stimulating factor (M-CSF) through Th2 cell activation. Unlike M1 macrophages, M2 macrophages significantly downregulate the expression of pro-inflammatory cytokines such as IL-12 and IL-23, while upregulating the expression of anti-inflammatory factors like IL-10 and IL-1RA. However, M2 macrophages themselves can also produce low levels of pro-inflammatory cytokines such as IL-1, IL-6, and TNF-α.9 There is accumulating evidence that M2 macrophages play significant roles in pathogen clearance, anti-inflammatory responses, metabolism, wound healing, tissue remodeling and repair, immune regulation, and tumor progression, including in malignancy.10
However, several studies have shown that macrophage polarization is not a simple dichotomy of M1/M2 types, but rather a complex, continuous spectrum.11 Following AMI, the local inflammatory microenvironment of the heart undergoes dynamic changes, resulting in highly heterogeneous macrophage phenotypes. These different phenotypes of macrophages play distinct roles in inflammation and tissue repair, and may influence the severity of myocardial damage and the quality of repair. Based on their phenotype and function, M1 and M2 macrophages can further be subdivided into multiple subtypes. Although a few studies suggest that M1 macrophages (pro-inflammatory) can be divided into M1a and M1b, this has not become a widely accepted view. M1 macrophages are primarily driven by IFN-γ and LPS stimulation, activating the NF-κB and STAT1 signaling pathways, and producing large amounts of pro-inflammatory cytokines such as TNF-α, IL-6, and IL-1β.12 Moreover, TLR activation significantly enhances the pro-inflammatory response of M1 macrophages and promotes the release of ROS and NO, exacerbating tissue damage.13 In contrast, M2 macrophages (anti-inflammatory) can be divided into M2a, M2b, M2c, and M2d subtypes. Since M2d is primarily studied in the context of tumor microenvironments, where it promotes angiogenesis, it is seldom mentioned in cardiovascular research, and thus will not be discussed here. M2a is induced by IL-4 and IL-13 and mainly promotes tissue repair and fibroblast activation.14 M2b macrophages possess immune-regulatory functions, secreting IL-10 while retaining the expression of some pro-inflammatory cytokines, thereby playing a balancing role in the inflammatory response.15 M2c, induced by IL-10 and TGF-β, plays a major role in anti-inflammatory responses and the clearance of apoptotic cells, making it an important subtype in myocardial repair.6 M1/M2 macrophages are not static entities nor are they strictly opposed to one another; rather, they undergo dynamic transitions following acute myocardial infarction. During the acute phase (1–3 days), M1 macrophages dominate and promote inflammation; in the subacute phase (3–7 days), M2a and M2b gradually increase, promoting angiogenesis and fibroblast activation; in the chronic phase (>7 days), M2c predominates, leading to anti-inflammatory repair (Table 1).16–18
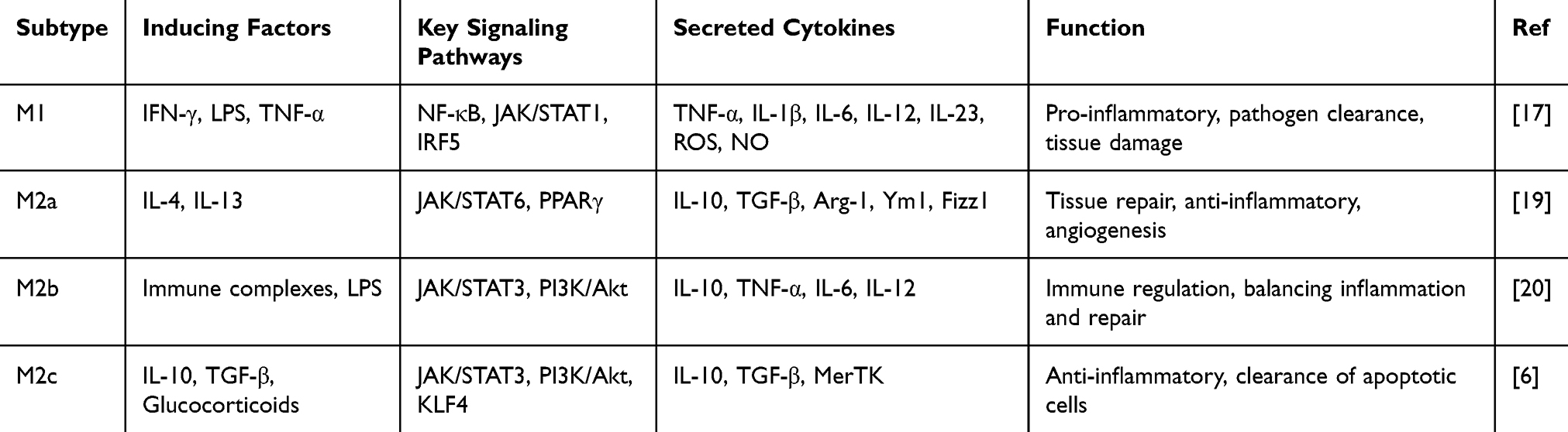 |
Table 1 Characteristics of Macrophage Polarization |
Notably, recent studies based on single-cell RNA sequencing (scRNA-seq) have revealed that macrophage polarization is far more dynamic and complex than the classical M1/M2 dichotomy. For example, scRNA-seq and lineage tracing have demonstrated that cardiac macrophages following MI comprise multiple transcriptionally distinct subsets with divergent functional trajectories. Embryonically derived macrophages largely retained reparative features, while recruited monocyte-derived macrophages exhibited temporal heterogeneity, initially adopting pro-inflammatory states before transitioning toward reparative phenotypes. These findings underscore that macrophage polarization exists on a continuous spectrum, driven by cues from the evolving myocardial microenvironment.21 In addition, spatial transcriptomics has enabled the mapping of macrophage subtypes within specific cardiac regions after MI, highlighting how their phenotypic state is shaped by spatial proximity to the infarct core, border zone, or remote myocardium. These spatial approaches have uncovered transitional macrophage states and revealed regionally specialized roles in inflammation resolution, angiogenesis, and fibrotic remodeling.19 Collectively, these findings suggest that future research directions should move beyond the simplistic M1-to-M2 paradigm and instead target specific macrophage subsets based on their spatiotemporal dynamics to optimize cardiac repair and limit adverse remodeling.
The Origin of Macrophages
In addition to their functional and phenotypic differences, is also a key factor in determining their role in AMI. Studies have shown that cardiac macrophages primarily originate from two distinct sources: embryonically derived macrophages and bone marrow-derived monocyte-derived macrophages.20
Embryonically Derived Cardiac Macrophages
Embryonically derived cardiac macrophages originate from the yolk sac and fetal liver, migrating to the heart during embryonic development and maintaining a stable microenvironment in cardiac tissue under homeostatic conditions.22 These macrophages have the ability for long-term self-renewal and do not rely on the replenishment of bone marrow-derived monocytes. Following myocardial infarction, embryonically derived macrophages predominantly exhibit an M2-like phenotype, secreting anti-inflammatory cytokines such as IL-10 and TGF-β, thereby suppressing inflammation and promoting tissue repair.
However, it is important to note that recent studies have demonstrated that embryonically derived macrophages are not exclusively anti-inflammatory. In the early stages following myocardial infarction, these macrophages can transiently adopt a pro-inflammatory phenotype, expressing cytokines such as TNF-α and IL-1β and activating NF-κB signaling. This phenotypic plasticity is highly context-dependent and shaped by injury-induced cues in the cardiac microenvironment.23,24 These findings highlight that the reparative role of embryonic macrophages is temporally regulated and not fixed, and caution should be taken when attributing strict M1/M2 classifications based solely on cellular origin.
Bone Marrow-Derived Monocyte-Derived Macrophages
After the onset of AMI, bone marrow-derived monocytes are extensively recruited to the injury site and rapidly differentiate into macrophages. During the acute phase, these macrophages predominantly exhibit an M1 phenotype, releasing large amounts of pro-inflammatory cytokines such as TNF-α, IL-6, and IL-1β, thereby enhancing the inflammatory response to clear necrotic cells and pathogens.25 However, as inflammation resolves, a subset of these macrophages can transition into the M2 phenotype, contributing to tissue repair. If this transition is impaired, it may lead to persistent inflammation, ultimately exacerbating cardiac damage and fibrosis.
Relationship Between Tissue Origin and Repair of Myocardial Infarction
Embryonically derived macrophages and bone marrow-derived macrophages exhibit distinct roles following AMI. Embryonically derived macrophages primarily regulate the repair process, whereas bone marrow-derived monocyte-derived macrophages predominantly drive the inflammatory response.21 Studies have shown that the survival of embryonically derived macrophages is crucial for myocardial injury recovery, whereas prolonged maintenance of the M1 phenotype in bone marrow-derived macrophages may lead to chronic inflammation, thereby impairing cardiac functional recovery.26
These studies suggest that the regulation of macrophage polarization after AMI is influenced not only by inflammatory signaling but also by temporal dynamics, cellular origin, and the local microenvironment. Therefore, future research should further explore strategies for precisely modulating macrophage polarization, facilitating the transition from M1 to M2 to mitigate inflammatory damage and promote myocardial repair (Table 2).27
 |
Table 2 Comparison of Embryonic-Derived and Bone Marrow-Derived Macrophages in AMI Repair |
Dynamics of Macrophage Polarization State in the Context of AMI
In the context of AMI, macrophages not only participate in the early inflammatory response but also play a critical role in tissue healing and cardiac remodeling. The polarization state of macrophages, characterized by the dynamic transition between M1 and M2 subtypes, directly influences myocardial tissue repair and long-term prognosis. Studies have shown that these temporal and spatial dynamics are crucial determinants of AMI outcomes.28,29 As previously discussed, M1 and M2 macrophages do not exist as singular entities but are further subdivided into multiple subtypes during different phases of inflammation to accommodate the needs of cardiac repair (Figure 1).
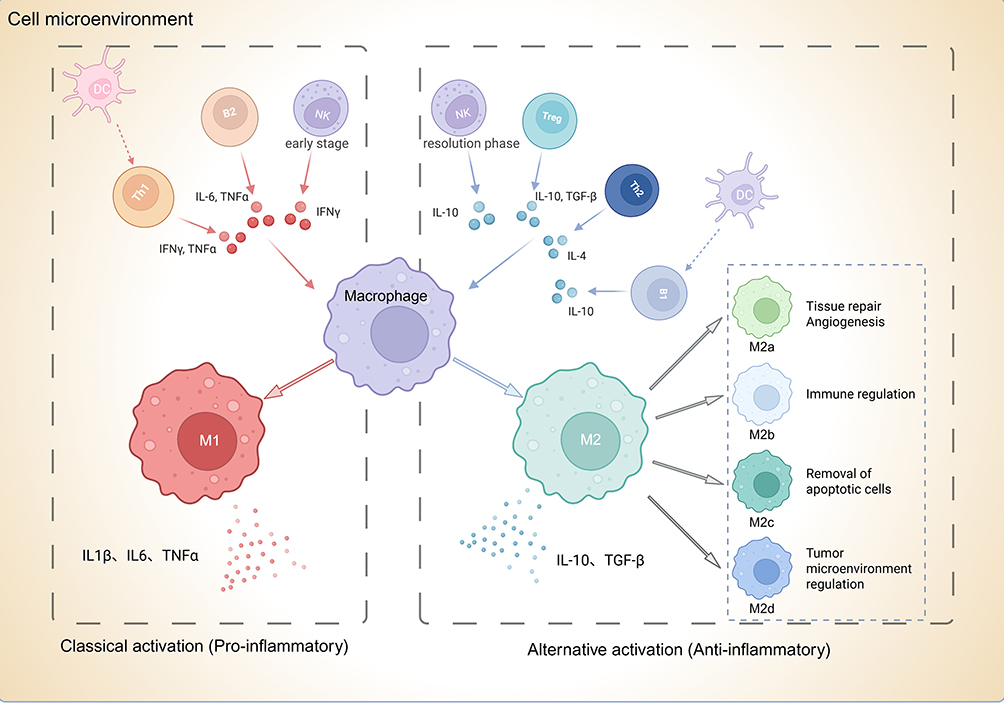 |
Figure 1 Polarization classification of macrophages and their interaction with other immune cells. Classical activation (M1, red) is driven by pro-inflammatory signals such as IFN-γ, TNFα, and IL-6 from Th1, B2, NK, and dendritic cells, leading to secretion of IL-1β, IL-6, and TNFα. Alternatively activated macrophages (M2, green) arise in response to anti-inflammatory cytokines including IL-4, IL-10, and TGF-β, produced by Treg, B1, and dendritic cells. M2 macrophages include distinct subtypes: M2a (IL-4/IL-13-induced, tissue repair and angiogenesis), M2b (immune regulation), M2c (removal of apoptotic cells), and M2d (tumor microenvironment regulation). Colored text indicators (M1, M2, M2a–M2d) correspond to macrophage phenotypes and their primary functions as illustrated in the figure. |
The Role of Immune Cells in Macrophage Polarization
After AMI, damaged tissue activates both innate and adaptive immune responses, with neutrophils, T cells, B cells, dendritic cells, and NK cells regulating macrophage polarization through cytokine secretion and direct cell-cell interactions. These immune cells influence the duration of the inflammatory response and the quality of cardiac repair.
Interaction of Neutrophils with Macrophages
Within hours after AMI, neutrophils are rapidly recruited to the site of injury, where they clear necrotic cells and influence macrophage polarization through the release of neutrophil extracellular traps (NETs).30 Elastase and myeloperoxidase (MPO) released by NETs can stimulate M1 macrophage polarization, thereby enhancing the pro-inflammatory response. However, excessive NETs release may exacerbate inflammation and worsen myocardial injury. As neutrophils undergo apoptosis, macrophages engulf apoptotic neutrophils, promoting the transition to the M2 macrophage phenotype and releasing anti-inflammatory cytokines such as IL-10 and TGF-β, which contribute to inflammation resolution and myocardial tissue repair.31
T Cells Regulate Macrophage Polarization
T cells play a crucial role in immune regulation following AMI. Th1 cells secrete IFN-γ, which promotes M1 macrophage polarization, enhances the inflammatory response, and may contribute to increased myocardial fibrosis. In contrast, Th2 cells primarily secrete IL-4 and IL-13, facilitating M2 macrophage polarization, suppressing inflammation, and accelerating myocardial repair.32 Regulatory T cells (Tregs) modulate macrophage function by releasing IL-10, thereby inhibiting excessive inflammatory responses and promoting myocardial repair.33
B Cells Regulate Macrophage Polarization
B cells play a crucial role in adaptive immunity, and their subtypes also influence macrophage polarization. B1 cells primarily secrete IL-10, promoting M2 macrophage activation, which helps reduce myocardial fibrosis and chronic inflammation.34 In contrast, B2 cells may facilitate M1 macrophage polarization by secreting pro-inflammatory cytokines such as TNF-α and IL-6, thereby prolonging the inflammatory response and potentially impairing cardiac functional recovery.35
Role of Dendritic Cells (DCs) in Macrophage Polarization
Following AMI, dendritic cells (DCs) function as antigen-presenting cells (APCs) and cooperate with macrophages to regulate immune responses. By presenting antigens, DCs activate T cells, thereby indirectly influencing macrophage polarization.36
Effects of Natural Killer Cells (NK Cells) on Macrophages
NK cells can secrete IFN-γ during the early stages of AMI, promoting M1 macrophage polarization and exacerbating early inflammation. However, during the resolution phase of inflammation, a subset of NK cells can also produce IL-10, facilitating M2 macrophage polarization and supporting tissue repair.37
Macrophage Status Prior to Infarction
Before myocardial infarction occurs, the state and functional changes of macrophages in the heart are often closely related to underlying pathological conditions such as atherosclerosis and chronic inflammation. Macrophages play a critical role in the formation of atherosclerotic plaques. Monocytes recruited from the bloodstream infiltrate the arterial intima, where they engulf large amounts of low-density lipoprotein (LDL), forming foam cells and promoting local inflammation.38,39 Chronic inflammation within the arteries activates M1 macrophages, leading to the secretion of large quantities of cytokines such as TNF-α and IL-1β, as well as matrix metalloproteinases (MMPs), which further contribute to plaque instability.40,41 The persistence of this chronic inflammatory state increases the risk of plaque rupture, making it one of the primary triggers of myocardial infarction.42,43
Macrophage Response in Early Acute Myocardial Infarction
In the early phase of AMI (acute phase, 0–3 days), M1 macrophages dominate the inflammatory response and serve as the primary infiltrating cells in the arterial intima.44,45 Ischemia-reperfusion injury in the local tissue induces the release of large amounts of pro-inflammatory cytokines, leading to the rapid activation of M1 macrophages. During this stage, M1 macrophages exert diverse effects on the injured myocardium. On one hand, they are induced by IFN-γ and LPS to secrete large amounts of TNF-α, IL-1β, and IL-6, amplifying inflammatory signaling and promoting the recruitment of neutrophils and monocytes.46 On the other hand, M1 macrophages are activated via TLR signaling to release reactive oxygen species (ROS) and nitric oxide (NO), thereby enhancing oxidative stress and further exacerbating myocardial injury.13 These cells drive neutrophil recruitment by secreting large quantities of pro-inflammatory cytokines such as IL-1β, TNF-α, and IL-6, which intensify the local inflammatory response and facilitate the clearance of necrotic tissue.47,48 The infiltration of macrophages at this stage is crucial for initiating the early immune response, clearing necrotic cardiomyocytes, and triggering the healing process.25
However, excessive or prolonged M1 macrophage activation can lead to sustained inflammation, resulting in more severe tissue damage and ultimately contributing to adverse cardiac remodeling, such as myocardial fibrosis and scar formation.47,49 Persistent activation of M1 macrophages has been closely associated with impaired cardiac function and an increased risk of long-term adverse cardiac events in AMI patients. Chronic inflammation may disrupt the local immune balance in cardiac tissue, exacerbating fibrosis and promoting the onset of heart failure.50 Studies have shown that excessive activation of M1 macrophages following myocardial infarction is strongly linked to the maintenance of a chronic inflammatory state, which reduces the heart’s compensatory capacity and further compromises cardiac function.51
Studies have found that excessive activation of M1 macrophages after myocardial infarction is closely associated with the maintenance of a chronic inflammatory state, which leads to a decline in cardiac compensatory capacity and further impairs cardiac function. Chronic inflammation not only promotes increased cardiomyocyte apoptosis but also contributes to cardiac fibrosis, weakening both systolic and diastolic function. Pro-inflammatory cytokines persistently secreted by M1 macrophages, such as IL-6 and TNF-α, further stimulate fibroblast activation and excessive collagen deposition, resulting in myocardial fibrosis. This process reduces myocardial elasticity, progressively deteriorates cardiac function, and increases the risk of heart failure. Therefore, regulating M1 macrophage activity, particularly in the early stages following acute myocardial infarction, is crucial for improving long-term patient outcomes.
Role of M2-Type Macrophages in Cardiac Repair
As the inflammation resolution phase (3–7 days) begins, cardiac tissue clears necrotic cells and transitions into the repair stage. During this process, the proportion of M1 macrophages gradually decreases, while M2 macrophages, particularly the M2a and M2b subtypes, increase and become dominant. M2 macrophages primarily promote tissue repair and exert anti-inflammatory effects.52 M2a macrophages, induced by IL-4 and IL-13, facilitate fibroblast proliferation and collagen deposition, contributing to angiogenesis and myocardial tissue repair. However, excessive activation of M2a macrophages may lead to pathological fibrosis, ultimately impairing cardiac function.17 Meanwhile, M2b macrophages are induced by IL-10 and TLR activation, exhibiting both pro-inflammatory and anti-inflammatory properties. They help balance inflammation and the repair process, preventing premature resolution of inflammation to ensure adequate clearance of necrotic tissue.53 The dynamic balance between M2a and M2b macrophages determines the trajectory of the repair process, where excessive M2a activity may result in excessive fibrosis, whereas M2b macrophages help maintain immune homeostasis.
As the disease progresses into the tissue remodeling phase (>7 days), M2c macrophages become the predominant subtype. Induced by IL-10 and TGF-β, M2c macrophages primarily function in anti-inflammatory responses, immunosuppression, and the clearance of apoptotic cells. Additionally, M2c macrophages play a crucial role in regulating the extent of cardiac fibrosis, preventing excessive scar formation, and promoting long-term stable myocardial remodeling. If M2c activation is impaired, it may result in persistent chronic inflammation or excessive fibrosis, ultimately compromising cardiac function.10 Therefore, precisely modulating M2c activity in clinical interventions may be a key strategy for improving AMI prognosis.
In summary, M2 macrophages contribute to reducing inflammatory damage and promoting tissue repair by secreting anti-inflammatory cytokines such as IL-10 and TGF-β. They facilitate fibroblast activation and proliferation, as well as angiogenesis, thereby accelerating the tissue healing process. The role of M2 macrophages in tissue regeneration, scar formation, and vascularization is particularly crucial for the long-term remodeling of cardiac function.54 The transition from M1 to M2 macrophages is a critical stage in post-myocardial infarction cardiac repair. M2 macrophages help to limit excessive inflammatory responses while simultaneously promoting cellular regeneration and tissue remodeling, which aids in preventing myocardial fibrosis and excessive scar formation.55 This process not only influences myocardial tissue recovery but also has long-term effects on overall cardiac function and morphology.
Effect of Macrophage Polarization Imbalance on Cardiac Remodeling
Although the transition from M1 to M2 macrophages is generally considered a crucial step in the healthy repair process, any disruption in this transition may lead to maladaptive cardiac remodeling, increasing the risk of heart failure.56 For instance, if the pro-inflammatory response of M1 macrophages persists or if the anti-inflammatory function of M2 macrophages fails to take effect in a timely manner, it can exacerbate myocardial fibrosis and reduce the amount of functional cardiac tissue.57 In such cases, the heart’s compensatory capacity declines further, increasing the risk of heart failure and other cardiac complications. Moreover, macrophage polarization may be influenced by metabolic disorders. Patients with metabolic diseases such as diabetes and obesity often exhibit an imbalance in macrophage polarization, making them more susceptible to prolonged inflammation, delayed cardiac repair, and adverse post-MI outcomes.58 The hyperglycemic and hyperlipidemic environment in these patients affects macrophage function, driving them toward an M1 phenotype and further intensifying local inflammation. In diabetic patients, the inflammatory response is often more persistent and difficult to resolve, significantly increasing the risk of myocardial fibrosis and functional impairment. Therefore, managing underlying metabolic disorders is essential for restoring macrophage function and maintaining a balanced M1/M2 polarization following myocardial infarction.
Based on the dynamic regulation of macrophage polarization, recent studies have developed various therapeutic strategies targeting M1/M2 polarization. Some research has focused on modulating macrophage polarization to reduce inflammation and promote myocardial repair. For example, certain drugs, such as everolimus and sirolimus, have been shown to facilitate the transition of macrophages from M1 to M2 by influencing cytokine signaling pathways.59 These drugs regulate macrophage function by inhibiting the mTOR signaling pathway, thereby suppressing the pro-inflammatory response of M1 macrophages while enhancing the anti-inflammatory and reparative capacity of M2 macrophages. Meanwhile, nanomedicine-based drug delivery systems and gene-editing technologies have also been explored for modulating macrophage polarization to improve post-infarction tissue repair.60 Targeted macrophage polarization-modulating therapies have demonstrated significant potential in both preclinical and clinical studies. For instance, by inhibiting the sustained activation of M1 macrophages or enhancing M2 macrophage function, it may be possible to effectively control the inflammatory response, reduce fibrosis following myocardial infarction, and ultimately improve long-term cardiac function.61
Macrophages play a complex and multifaceted role in AMI, with their polarization state—the dynamic transition between M1 and M2 subtypes—being a key determinant of myocardial repair and long-term cardiac prognosis. The pro-inflammatory response of M1 macrophages during the acute phase is essential for clearing necrotic cells; however, their prolonged activation may lead to tissue damage and worsening cardiac function. In contrast, M2 macrophages contribute significantly to tissue repair and cardiac remodeling, promoting the reduction of myocardial fibrosis and functional recovery. Understanding the spatiotemporal dynamics of macrophages in AMI and the mechanisms regulating their polarization provides a critical theoretical foundation for developing novel therapeutic strategies (Figure 2).54,62
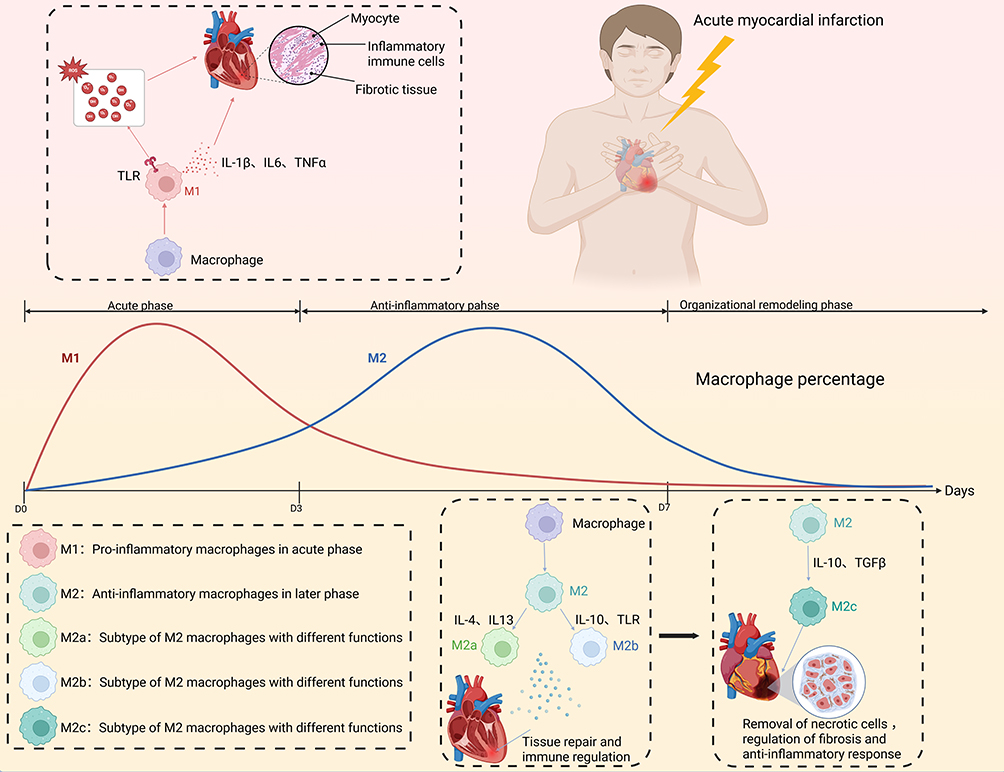 |
Figure 2 Progression of macrophage polarization and its function during acute myocardial infarction (AMI) progression. After AMI onset, M1 macrophages (pro-inflammatory; red) dominate in the acute phase (D0–D3), releasing cytokines (IL-1β, IL-6, TNFα) that amplify inflammation. During the anti-inflammatory phase (D3–D7), M2 macrophages (blue) become predominant, including subtypes M2a (IL-4/IL-13-induced, tissue repair), M2b (immune-regulatory, IL-10), and M2c (IL-10/TGFβ-induced, anti-inflammatory and fibrosis regulation). The organizational remodeling phase (>D7) is characterized by resolution of inflammation and tissue healing. Colored text indicators (M1, M2, M2a, M2b, M2c) correspond to macrophage phenotypes and their functions, as defined in the legend box within the figure. |
Mechanisms of Polarization Regulation in Macrophages
The polarization state of macrophages plays a crucial role in the onset and progression of AMI. Both the acute-phase inflammatory response and the subsequent tissue repair process are closely linked to macrophage polarization. The mechanisms regulating this process can be divided into intrinsic and extrinsic regulatory mechanisms.
Intrinsic Regulatory Mechanisms
Intrinsic regulatory mechanisms primarily refer to the internal processes by which macrophages regulate their polarization state through signaling pathways, transcription factors, and metabolic processes. These internal mechanisms directly influence how macrophages respond to external signals and determine their polarization direction.
Regulation of Signaling Pathways
Macrophage polarization is regulated by several classic signaling pathways, with the most important being the NF-κB, JAK/STAT, and PI3K/Akt pathways. The NF-κB pathway is the core regulatory pathway for M1 macrophage polarization. NF-κB (nuclear factor kappa B) is a key transcription factor that regulates the expression of genes associated with inflammation, immune responses, and cell survival. Under normal conditions, NF-κB binds to its inhibitor IκB and remains in an inactive state. When the cell is stimulated by pathogen-associated molecular patterns (PAMPs), such as lipopolysaccharides (LPS), or damage-associated molecular patterns (DAMPs), such as high-mobility group box 1 (HMGB1), IκB is phosphorylated and degraded, releasing NF-κB. NF-κB then translocates to the nucleus and activates the expression of pro-inflammatory genes.63,64 Following the onset of myocardial infarction (MI), the necrosis and hypoxia of myocardial cells stimulate the release of large amounts of DAMPs, such as HMGB1 and heat shock proteins. These molecules activate Toll-like receptors (TLRs) or NOD-like receptors (NLRs), initiating the NF-κB signaling pathway and promoting the secretion of pro-inflammatory cytokines, such as TNF-α, IL-1β, and IL-6, driving M1 macrophage polarization.65 These pro-inflammatory cytokines further recruit neutrophils and exacerbate the local inflammatory response. This strong inflammatory reaction is essential for the clearance of necrotic cells and the initiation of the early repair process, but excessive activation of the NF-κB pathway may result in prolonged inflammation, aggravated myocardial fibrosis, and ultimately hinder cardiac functional recovery.66
Similarly, M2 polarization involves multiple signaling pathways, with the JAK/STAT and PI3K/Akt pathways being two well-established mechanisms recognized in the academic community. Notably, the JAK/STAT pathway plays a role not only in M1 polarization but also in M2 polarization. The JAK/STAT signaling pathway is an important pathway for cells to respond to cytokine signals. This pathway is composed of the tyrosine kinase JAK and the signal transducers and activators of transcription (STAT). When cytokines such as IL-4, IL-10, or IFN-γ bind to their receptors, JAK kinases are activated and phosphorylate downstream STAT proteins. The phosphorylated STAT proteins dimerize and translocate to the nucleus, initiating the transcription of specific genes.67 In the early stages following MI, IFN-γ activates JAK1 and JAK2, promoting the phosphorylation of STAT1 and driving M1 macrophage polarization. These polarized macrophages exhibit strong pro-inflammatory activity, secreting large amounts of TNF-α and IL-1β. However, during the repair phase of MI, anti-inflammatory cytokines such as IL-4 and IL-10 activate JAK3 and STAT6 signaling pathways, promoting M2 macrophage polarization and enhancing tissue repair and angiogenesis.68 Thus, the JAK/STAT signaling pathway demonstrates dual functions in the post-MI phases, with pro-inflammatory effects in the early phase and anti-inflammatory repair in the later phase.
The PI3K/Akt signaling pathway plays a crucial role in cell survival, proliferation, and metabolic regulation. PI3K is activated by stimulation through cell surface receptors, producing PIP3, which then activates the downstream Akt. Activated Akt further regulates a series of downstream targets, promoting cell survival and anti-apoptotic functions.69 Following MI, the PI3K/Akt signaling pathway plays an important role in M2 macrophage polarization. Anti-inflammatory cytokines such as TGF-β, released by cardiomyocytes and fibroblasts during repair, can activate this pathway, promoting the conversion of macrophages from M1 to M2. Activation of Akt not only promotes the secretion of anti-inflammatory cytokines but also inhibits the expression of pro-inflammatory genes, thereby reducing M1 macrophage activity.70 Additionally, Akt can regulate macrophage metabolism through the mTOR signaling pathway, enhancing fatty acid oxidation, which promotes M2 polarization and accelerates tissue repair.71 However, the regulatory function of this pathway may not be strictly unidirectional. Studies have shown that the PI3K/Akt pathway exhibits context-dependent dual functionality in macrophage biology. Under pro-inflammatory conditions, such as stimulation by lipopolysaccharide (LPS) or interferon-γ (IFN-γ), Akt activation can paradoxically drive M1 polarization via engagement of the mTORC1–HIF-1α axis, thereby enhancing glycolytic flux and upregulating IL-1β expression.72,73 Mechanistically, Akt promotes the metabolic shift toward glycolysis—a hallmark of M1 macrophages—through mTOR signaling, while concurrently stabilizing the transcription factor HIF-1α, which plays a central role in pro-inflammatory gene expression. This functional duality underscores the complexity of PI3K/Akt-mediated regulation in macrophage polarization and indicates that its downstream effects are highly dependent on upstream stimuli and the surrounding microenvironment. Therefore, when designing therapeutic strategies targeting this pathway, careful consideration must be given to disease stage, tissue context, and inflammatory status to avoid unintended amplification of M1-driven inflammation.
Regulation of Transcription Factors
In addition to signaling pathways, transcription factors also play a critical role in macrophage polarization. For M1 polarization, the transcription factor IRF5 is considered a key regulator, as it directly promotes the expression of inflammation-related genes.74 In contrast, for M2 polarization, PPARγ and KLF4 are two essential transcription factors. PPARγ promotes M2 macrophage polarization by cooperating with STAT6. KLF4, on the other hand, suppresses inflammatory responses while promoting tissue repair, thereby driving M2 macrophage polarization.75
Metabolic Regulation
In addition to signal transduction, macrophage polarization is tightly regulated by cellular metabolism. Understanding these metabolic shifts is critical, as they not only drive macrophage polarization but also dictate the magnitude of myocardial injury, the extent of ventricular remodeling, and the patients’ risk of progressing to heart failure. After the onset of AMI, significant changes occur in the metabolic state of macrophages, directly influencing their polarization direction and function. Studies have shown that M1 macrophages primarily rely on glycolysis to maintain their pro-inflammatory state, while M2 macrophages depend on oxidative phosphorylation and fatty acid oxidation to promote tissue repair.76 Therefore, metabolic regulation plays a key role in the dynamic changes of macrophages after AMI and may become an important therapeutic target for intervention. These metabolic transitions not only determine macrophage phenotype but also influence the extent of myocardial injury, ventricular remodeling, and the risk of progression to heart failure, highlighting their relevance as therapeutic targets in clinical settings.
In the early stages of AMI, local hypoxia, pro-inflammatory cytokines (such as IFN-γ and TNF-α), and TLR signaling activate M1 macrophages, with their metabolic profile primarily relying on glycolysis rather than mitochondrial oxidative phosphorylation.77 This characteristic helps rapidly generate ATP to meet the demands of amplifying inflammatory signals, but it also leads to increased oxidative stress, worsening myocardial injury. M1 macrophages enhance glycolytic flux by upregulating the activity of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3), increasing glucose uptake and accelerating ATP production.78 Since glycolysis is independent of oxygen, it allows macrophages to survive in low-oxygen environments while supporting the rapid activation of inflammatory signals.79 Meanwhile, the TCA cycle in M1 macrophages is impaired, leading to the accumulation of succinate within the cells and inhibiting mitochondrial complex II (succinate dehydrogenase, SDH), which further strengthens the inflammatory state.80 Succinate, as an inflammation signaling molecule, promotes the stabilization of HIF-1α by inhibiting SDH, thereby enhancing IL-1β release and intensifying the pro-inflammatory response.81 Persistent dominance of glycolytic metabolism in M1 macrophages is associated with prolonged inflammation and adverse cardiac remodeling, which are major contributors to poor clinical outcomes such as reduced ejection fraction and increased heart failure risk. These metabolic changes enable M1 macrophages to rapidly adapt to the hypoxic and inflammatory microenvironment in the early phase of AMI. However, if inflammation persists for too long, it may lead to irreversible myocardial damage and promote fibrosis formation.
As AMI progresses into the inflammation resolution and repair phase (3–7 days post-infarction), the signaling of anti-inflammatory cytokines (such as IL-4 and IL-10) increases, driving the transition of M1 macrophages to M2 macrophages. This transition is accompanied by significant metabolic changes, shifting towards oxidative phosphorylation (OXPHOS) and fatty acid oxidation (FAO).82 During this phase, mitochondrial electron transport chain activity is enhanced, increasing ATP generation, while glycolysis dependency decreases.83 The restoration of SDH activity reduces succinate accumulation, thereby decreasing HIF-1α stability, limiting IL-1β release, and suppressing local inflammation.84 On the other hand, M2 macrophages highly express CD36 (fatty acid transporter), which promotes fatty acid uptake and generates ATP through FAO to support tissue repair and extracellular matrix (ECM) remodeling.85 Additionally, M2 macrophages exhibit strong arginine metabolism capacity, converting L-arginine into proline via arginase-1 (Arg-1), which stimulates fibroblast activation and promotes myocardial regeneration.86 These metabolic adaptive changes help M2 macrophages promote myocardial repair, reduce fibrosis, and restore cardiac function. Targeting these metabolic shifts offers a promising strategy to fine-tune the inflammatory-to-reparative balance, potentially reducing infarct size, limiting fibrosis, and improving long-term cardiac function in AMI patients.
Several clinical drugs have been developed to intervene in the inflammatory response of AMI by regulating metabolic levels. PFKFB3 inhibitors (such as PFK-158) can reduce M1 polarization and decrease the secretion of pro-inflammatory cytokines, thereby mitigating post-AMI inflammatory damage.85 Omega-3 fatty acids enhance M2 metabolic activity, promote tissue repair, and reduce excessive fibrosis.87 These findings suggest that metabolic interventions to regulate M1/M2 transition may become a potential strategy for improving AMI prognosis.
Regulation of microRNAs
Beyond metabolic and transcriptional regulation, post-transcriptional mechanisms—particularly those involving microRNAs (miRNAs)—have emerged as critical modulators of macrophage phenotype and function in the context of AMI. In recent years, the role of microRNAs (miRNAs) in macrophage polarization has garnered increasing attention. miR-155 has been shown to be a critical promoter of M1 macrophage polarization; it enhances the activity of the NF-κB pathway by inhibiting the SOCS1 signaling, thereby driving the inflammatory response.88,89 In contrast, miR-21 and miR-146a are considered promoters of M2 polarization, as they suppress the expression of pro-inflammatory cytokines and facilitate the conversion of macrophages to an anti-inflammatory phenotype.90 The regulatory roles of these miRNAs offer a novel perspective, suggesting that controlling macrophage polarization through non-coding RNAs could provide new therapeutic strategies for the treatment of acute myocardial infarction. By fine-tuning inflammatory gene expression and macrophage phenotype, miRNA-based modulation holds potential to reduce infarct size, limit adverse ventricular remodeling, and ultimately improve cardiac recovery in AMI patients.
External Regulatory Mechanisms
External regulatory mechanisms primarily refer to the factors from the microenvironment and external stimuli that influence macrophage polarization. Compared to intrinsic regulatory mechanisms, the influence of external microenvironmental factors on macrophage polarization is more indirect, often acting as an auxiliary mechanism to modulate intrinsic mechanisms. However, the impact of external microenvironmental factors on macrophages should not be overlooked. These microenvironmental factors include inflammatory cytokines, metabolic states, mechanical stress, and pharmacological interventions, all of which collectively affect macrophage function in AMI.
Regulation of Inflammatory Factors
After the onset of AMI, local tissues release a large number of inflammatory cytokines, such as TNF-α, IL-1β, and IFN-γ. These cytokines bind to receptors on the surface of macrophages, activating pro-inflammatory signaling pathways and promoting M1 macrophage polarization.91 Additionally, anti-inflammatory cytokines such as TGF-β and IL-10 play a role in the later stages, driving the transition of macrophages to the M2 phenotype and promoting tissue repair.32 The dynamic changes in these inflammatory cytokines play a crucial temporal regulatory role in the inflammatory response and repair process following myocardial infarction. As previously mentioned, these mechanisms are critical to both the initiation and resolution of AMI.
Impact of Metabolic Diseases
The metabolic state of macrophages directly influences or determines the process of macrophage polarization, acting as an intrinsic regulatory mechanism involved in the development of AMI. However, metabolic disorders associated with conditions such as obesity and diabetes often exacerbate the imbalance in macrophage polarization. Studies have confirmed that in patients with obesity and diabetes, M1 macrophage polarization is more pronounced, while the function of M2 macrophages is relatively limited. This polarization imbalance makes these patients more susceptible to chronic inflammation and fibrosis after myocardial infarction, ultimately impairing cardiac function and leading to adverse outcomes such as heart failure.92 Additionally, hyperglycemia and hyperlipidemia can further promote M1 polarization by upregulating inflammatory cytokines and free radical production, thereby exacerbating cardiac inflammation and remodeling.93
Regulation of Mechanical Stress
Mechanical stress in the heart following myocardial infarction, such as tension and shear forces, is one of the key factors influencing macrophage polarization. After myocardial injury, changes in the heart’s structure and function lead to increased mechanical stress, which in turn triggers cardiac remodeling and fibrosis. In the acute phase following myocardial infarction, hemodynamic abnormalities in the heart generate sustained mechanical pressure, especially in cases of heart failure. This pressure has a significant impact on the polarization of macrophages.
In the early stages of myocardial infarction, mechanical stress activates multiple signaling pathways, particularly the NF-κB and JAK/STAT pathways, promoting the release of pro-inflammatory cytokines such as IL-6 and TNF-α, which drive macrophages toward M1 polarization. During this phase, M1 macrophages enhance local inflammation by secreting large amounts of pro-inflammatory cytokines, further exacerbating myocardial injury.51 Additionally, mechanical stress directly affects infiltrating immune cells and cardiomyocytes through wall tension and shear forces, maintaining the inflammatory environment. This not only inhibits early tissue repair but may also accelerate the progression of cardiac fibrosis. During the recovery phase post-MI, sustained mechanical stress, especially during ventricular remodeling, may hinder tissue repair and induce chronic inflammation. Due to structural changes in the heart and increased mechanical pressure, M1 macrophages may remain in a pro-inflammatory state for an extended period, continuously secreting pro-inflammatory cytokines, leading to further myocardial fibrosis development.94 The prolonged upregulation of IL-6 and TNF-α exacerbates inflammation, resulting in increased cardiomyocyte apoptosis and suppressing normal myocardial repair.95
Overall, mechanical stress plays a crucial regulatory role in macrophage polarization following myocardial infarction. During the acute phase, mechanical stress enhances the release of pro-inflammatory cytokines, promoting M1 macrophage polarization and exacerbating the inflammatory response. In the recovery phase, sustained mechanical stress may hinder tissue repair, leading to myocardial fibrosis and worsening cardiac function. Therefore, regulating the impact of mechanical stress on macrophage polarization, especially at different stages post-MI, may offer new therapeutic approaches for cardiac repair.
Regulation of Drugs
Recent studies have confirmed that several drugs can alleviate post-MI inflammation and tissue damage by modulating macrophage polarization. For instance, mTOR inhibitors such as sirolimus and everolimus can promote the transition of macrophages from M1 to M2 by inhibiting the mTOR signaling pathway, thereby reducing the inflammatory response.96 Additionally, IL-1β inhibitors, such as canakinumab, have been used clinically. These inhibitors suppress the release of inflammatory cytokines, reduce the duration of M1 polarization, and promote myocardial repair.56 The application of nanomedicine-based drug delivery systems in regulating macrophage polarization has also gained significant attention. Specific nanoparticles can be designed to target macrophages and deliver anti-inflammatory drugs or miRNAs, directly modulating macrophage polarization. For example, certain nanoparticles can load miR-146a to suppress pro-inflammatory signaling, thus promoting M2 polarization of macrophages. This technology holds promise as a new therapeutic strategy for inflammation regulation following acute myocardial infarction.97,98
Emerging Mechanisms Regulating M1–M2 Transition
In addition to classical signaling pathways such as NF-κB and JAK/STAT, recent studies have identified a range of emerging regulatory mechanisms that govern the M1–to–M2 macrophage transition, offering novel targets for therapeutic intervention in AMI. One important mechanism involves metabolic reprogramming enzymes that actively shape macrophage phenotype. For instance, PFKFB3, a key regulator of glycolysis, is upregulated in M1 macrophages and enhances pro-inflammatory polarization by increasing fructose-2,6-bisphosphate production and glycolytic flux. Inhibition of PFKFB3 has been shown to suppress M1 activation and promote a shift toward reparative M2 phenotypes.99 Similarly, succinate dehydrogenase impairment in M1 macrophages leads to succinate accumulation, stabilizing HIF-1α and maintaining IL-1β expression, whereas restoration of SDH activity facilitates M2 transition.77 Beyond metabolic enzymes, epigenetic regulators such as histone deacetylases (HDACs), sirtuins, and bromodomain-containing proteins (such as BRD4) have also been implicated in macrophage polarization. For example, SIRT1 promotes M2 polarization by deacetylating and inhibiting NF-κB p65 subunits, reducing inflammatory gene expression.100 In contrast, HDAC3 is associated with M1 polarization, and its inhibition has been reported to enhance M2-associated gene programs.101 Furthermore, BRD4, a reader of histone acetylation, facilitates the transcription of inflammatory genes and sustains M1 polarization; BRD4 inhibition using BET inhibitors has shown promise in reprogramming macrophages toward an anti-inflammatory state.102 These emerging targets provide additional layers of control in macrophage plasticity and open new avenues for therapeutic modulation of post-infarction inflammation and tissue repair.
Macrophage-Related Therapeutic Strategies
Macrophages play a crucial role in both the inflammatory response and tissue repair following MI. In recent years, therapeutic strategies targeting macrophage polarization have become an important area of research aimed at improving the prognosis of AMI patients, reducing myocardial injury, and promoting cardiac function recovery. The polarization state of macrophages, which manifests as pro-inflammatory M1 and anti-inflammatory reparative M2 phenotypes, plays a key role in the balance between inflammation and repair. This balance is critical for cardiac remodeling after myocardial infarction. From this perspective, several macrophage-related therapeutic approaches for MI have been explored.
Drug Therapy Targeting Macrophage Polarization
As mentioned earlier, the mTOR signaling pathway plays a crucial regulatory role in macrophage polarization and metabolism, particularly in promoting the transition of M1 macrophages to M2 macrophages through mTOR inhibition, thereby alleviating inflammation after MI.72,103 Sirolimus and everolimus, as mTOR inhibitors, have been widely used to reduce post-MI inflammation and cardiac fibrosis. These drugs inhibit the mTOR signaling pathway, reducing the activity of M1 macrophages while promoting the activation of anti-inflammatory M2 macrophages, thereby decreasing myocardial injury and accelerating tissue repair.59 However, systemic mTOR inhibition can lead to off-target effects, including impaired endothelial cell proliferation, delayed wound healing, and increased susceptibility to infections due to broad immunosuppressive activity. Long-term use has also been associated with metabolic disturbances such as dyslipidemia and insulin resistance, which may offset cardiovascular benefits in certain patients. These adverse effects underscore the need for targeted delivery systems or selective mTORC1/2 modulation to minimize systemic toxicity while preserving therapeutic efficacy.104
IL-1β, a pro-inflammatory cytokine secreted by M1 macrophages, plays a central role in the inflammatory response following myocardial infarction. Canakinumab, a monoclonal antibody targeting IL-1β, effectively reduces chronic inflammation after MI by inhibiting IL-1β activity.56 Large-scale clinical studies have shown that canakinumab significantly reduces the incidence of recurrent cardiovascular events in MI patients and improves long-term prognosis.105,106 In addition to suppressing M1 macrophage activity, canakinumab also promotes M2 macrophage polarization, thus enhancing cardiac repair. Despite its anti-inflammatory efficacy, IL-1β blockade carries the risk of increased infections, particularly respiratory tract infections, as observed in large-scale trials. Moreover, the high cost and requirement for repeated subcutaneous injections may limit its accessibility. Patient selection is crucial, as not all individuals with post-MI inflammation exhibit IL-1β-driven pathology, and biomarker-guided stratification may optimize therapeutic benefit.107
Atherosclerosis is a major risk factor for AMI, and it is associated with dysregulation of lipoprotein metabolism. Clinically, oral drugs such as statins and injectable agents like PCSK9 inhibitors are commonly used for treatment. Recent studies and clinical trials have suggested that PCSK9 inhibitors also play a role in the prevention and treatment of AMI. Macrophages play an essential role in the formation and progression of atherosclerotic plaques, and cholesterol metabolism disruption is the primary driving force behind atherosclerosis. PCSK9 inhibitors (such as alirocumab and evolocumab) regulate cholesterol metabolism, reduce M1 macrophage polarization in plaques, and decrease plaque instability, thereby lowering the risk of MI.108 Translational considerations remain important for real-world adoption. While outcome benefits are robust for LDL-C lowering and secondary prevention, direct evidence that PCSK9 inhibitors modulate macrophage phenotypes in the acute MI setting is limited. High cost, injection burden, and variable long-term adherence constrain population-level impact. Response heterogeneity across clinical subgroups also argues for precision approaches.109 These drugs not only lower the levels of low-density lipoprotein cholesterol (LDL-C) in the blood but also influence macrophage polarization, promoting the cardiac repair process.
Gene Therapy and miRNA Regulation
In recent years, gene therapy technologies have made breakthrough progress in cardiovascular disease research, particularly in the regulation of macrophage polarization following AMI. Gene therapy can alter macrophage polarization by editing key signaling pathway genes or through miRNA-mediated transcriptional regulation, thereby influencing inflammation and tissue repair processes.110
The CRISPR/Cas9 gene editing technology has been used in recent years to directly regulate the polarization direction of macrophages. By knocking out or activating specific genes, it can improve the inflammatory and repair responses after myocardial infarction.111 For instance, the NF-κB signaling pathway is a key regulatory axis for the pro-inflammatory function of M1 macrophages. Research has shown that knocking out NF-κB key regulatory factors (such as p65/RelA) effectively reduces the pro-inflammatory response of M1 macrophages and promotes M2 polarization.112 CRISPR/Cas9-mediated NF-κB blockage has shown potential in reducing myocardial fibrosis after AMI in animal experiments.113 Furthermore, the JAK/STAT signaling pathway is also crucial in the M1/M2 transition. Targeting JAK1 reduces IFN-γ-induced M1 polarization, while activating STAT6 enhances IL-4-mediated M2 promotion, accelerating myocardial tissue repair.114 The optimization of gene delivery systems is also an important direction for CRISPR technology development. Current research primarily focuses on comparing viral vectors and non-viral delivery systems. Adeno-associated virus (AAV) vectors can express genes long-term to maintain M2 polarization, while non-viral delivery systems (such as lipid nanoparticles, LNP) have been used to deliver CRISPR components, showing excellent safety and efficacy in animal experiments.115 Although CRISPR-mediated macrophage polarization regulation is still in preclinical research, its high specificity and long-term regulatory effects offer new possibilities for AMI treatment.116
As non-coding RNAs, miRNAs can precisely regulate macrophage polarization by targeting mRNA for degradation or translation suppression. Several miRNAs have been identified as directly involved in the M1/M2 regulation related to AMI. miR-155, an inflammation-related miRNA, enhances NF-κB signaling by inhibiting suppressor of cytokine signaling 1 (SOCS1), thereby promoting the pro-inflammatory function of M1 macrophages. Knockout of miR-155 reduces inflammation after AMI and improves myocardial repair.117 miR-21 and miR-146a tend to promote M2 polarization; miR-21 enhances JAK/STAT3 signaling by inhibiting programmed cell death protein 4 (PDCD4), promoting M2 polarization, and showing reduced fibrosis and improved cardiac function in animal models of AMI.118 miRNA-based regulatory strategies have shown potential efficacy in animal experiments, but there is still a need to optimize delivery methods to improve stability and targeting.
Currently, miRNA delivery technologies mainly include nanoparticle systems (LNPs) and viral vectors. Nanoparticle systems can effectively protect miRNA and improve cellular uptake rates. Studies have shown that LNP-based miR-21 delivery promotes M2 polarization and reduces AMI fibrosis in mouse models. AAV-mediated overexpression of miR-146a has shown reduced inflammatory damage and improved myocardial function. However, miRNA delivery faces several challenges, including the short half-life of miRNAs, the need to develop more stable delivery methods, and avoiding non-specific targeting issues to reduce side effects. Future research will focus on optimizing miRNA delivery systems, improving their stability and targeting, and integrating metabolic regulation and immune intervention strategies for more precise AMI interventions.119
Immune-Modulating Therapy
Studies have shown that the infusion of stem cells or the inhibition of immune checkpoints can artificially alter the polarization direction of macrophages at the site of MI, thereby promoting cardiac repair. Stem cell therapy, particularly the infusion of bone marrow-derived monocytes or macrophages, has emerged as a novel therapeutic strategy for modulating post-myocardial infarction inflammation. These stem cells can regulate the local immune environment, promoting the polarization of macrophages toward the M2 phenotype, reducing cardiac inflammation and fibrosis. Clinical studies have demonstrated that stem cell infusion can accelerate tissue repair after myocardial infarction and improve cardiac function.120
Immune checkpoint inhibitors, which are typically used in cancer immunotherapy, have recently been found to have potential in regulating macrophage function. For example, blocking the PD-1/PD-L1 pathway enhances M2 macrophage polarization and reduces the pro-inflammatory activity of M1 macrophages, thus providing protective effects during the repair process after myocardial infarction.121
Macrophage Regulatory Strategies Incorporating Metabolic Disorders
Metabolic diseases such as diabetes and obesity are closely associated with the continuation of inflammatory responses and fibrosis after MI. In a hyperglycemic and hyperlipidemic environment, macrophages are more prone to polarize toward the M1 phenotype, leading to persistent pro-inflammatory responses and further exacerbating myocardial injury.92 For patients with metabolic diseases, strategies to regulate macrophage polarization should be combined with metabolic regulation. For instance, SGLT2 inhibitors (such as dapagliflozin) and GLP-1 receptor agonists (such as liraglutide) have been shown to reduce inflammation by modulating the metabolic state, thereby promoting the polarization of macrophages toward the M2 phenotype.122,123 The anti-inflammatory benefits of SGLT2 inhibitors and GLP-1 receptor agonists appear to be partly dependent on baseline metabolic status, with stronger effects observed in patients with type 2 diabetes or insulin resistance. Their efficacy in non-diabetic, lean, or elderly populations remains less certain. Additionally, gastrointestinal intolerance (for GLP-1 agonists) and genitourinary infections (for SGLT2 inhibitors) may limit tolerability in some patients (Table 3).124
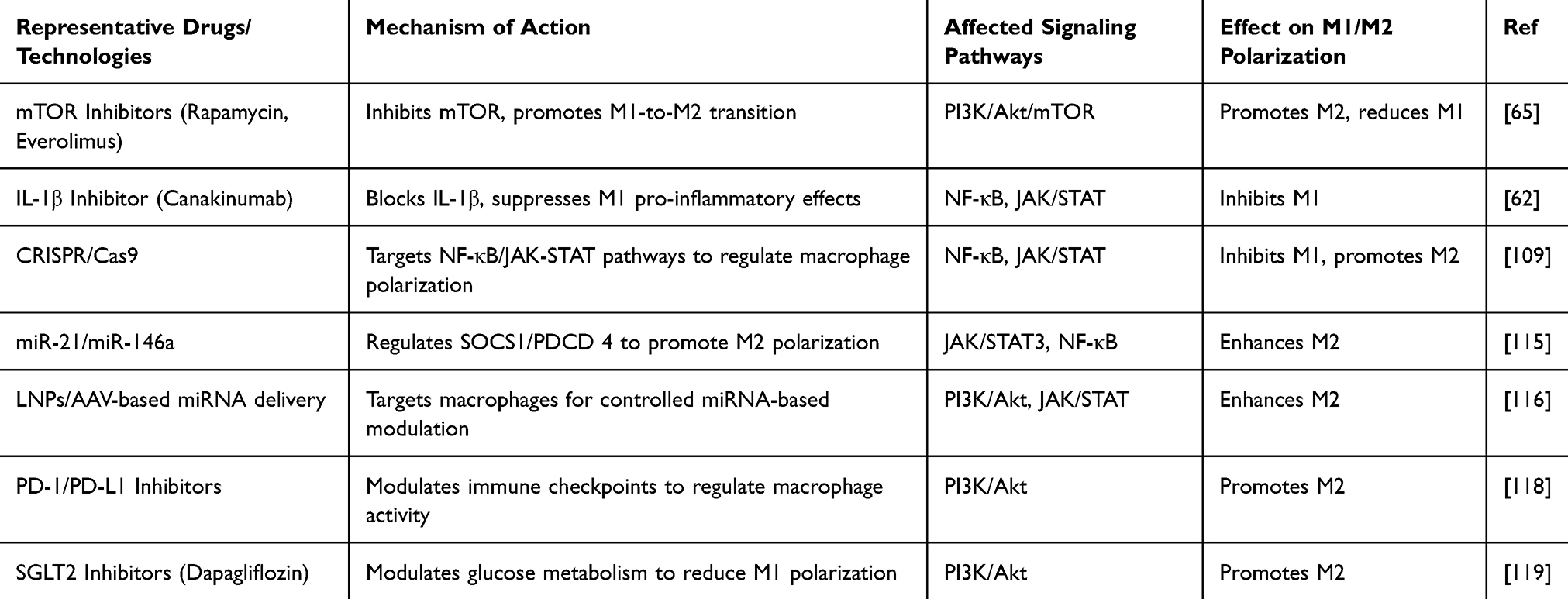 |
Table 3 Effects of Different Therapeutic Strategies on Macrophage Polarization |
Despite the growing interest in targeting macrophage polarization as a therapeutic approach for AMI, each strategy presents unique advantages and potential limitations. For instance, while mTOR inhibitors and IL-1β monoclonal antibodies have demonstrated promising anti-inflammatory effects in clinical studies, concerns remain regarding immunosuppression, infection risk, and high cost. Similarly, miRNA- and gene-editing–based interventions offer high mechanistic precision but face substantial challenges related to delivery efficiency, molecular stability, and long-term safety. Stem cell therapy and nanoparticle-based systems are emerging approaches with encouraging potential, though their reproducibility and clinical efficacy have yet to be fully established. A concise comparison of representative therapeutic approaches—including their molecular targets, translational stages, benefits, and limitations—provides insight into their respective strengths and challenges (Table 4).
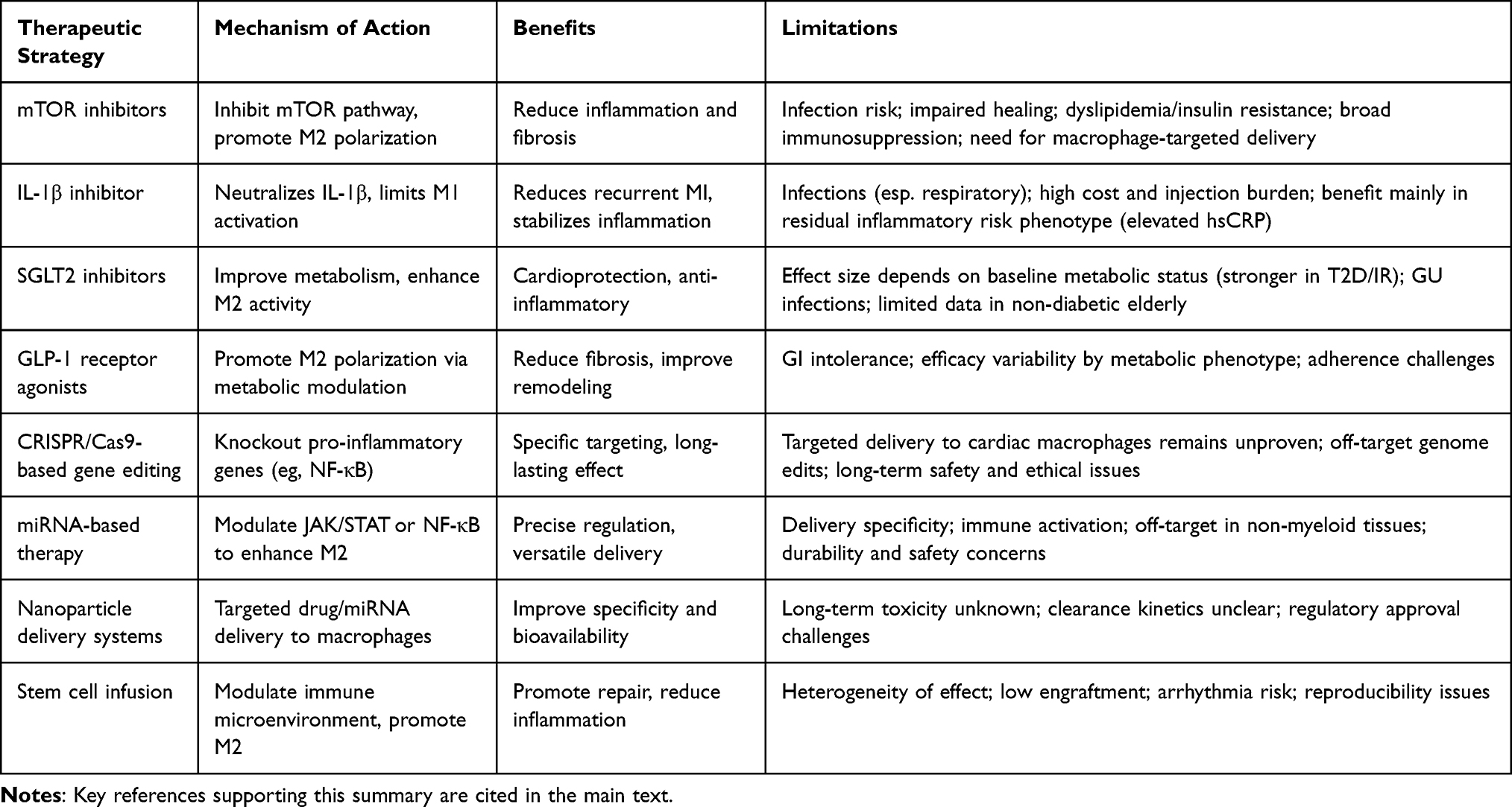 |
Table 4 Comparative Overview of Macrophage Polarization–Targeted Therapeutic Strategies in Acute Myocardial Infarction |
In parallel with these mechanistic advances, several macrophage-targeting strategies have entered clinical evaluation in AMI populations. The IL-1β monoclonal antibody canakinumab was assessed in the pivotal CANTOS trial (NCT01327846,https://clinicaltrials.gov/study/NCT01327846), which enrolled over 10,000 patients with prior myocardial infarction and elevated high-sensitivity C-reactive protein. Canakinumab significantly reduced recurrent cardiovascular events, thereby providing robust evidence for inflammation-targeted therapy in post-MI patients.56 Additionally, the ongoing DAPA-MI trial (NCT04564742,https://clinicaltrials.gov/study/NCT04564742) is evaluating the SGLT2 inhibitor dapagliflozin for its effects on left ventricular remodeling and inflammation following AMI. While mTOR inhibitors and GLP-1 receptor agonists have shown anti-inflammatory and anti-fibrotic effects in other cardiovascular contexts, their direct clinical application in AMI remains exploratory. Furthermore, miRNA- and CRISPR-based therapies are still in the preclinical stage, with translational potential contingent upon the development of efficient delivery systems and favorable safety profiles. Collectively, current clinical evidence provides preliminary support for macrophage polarization–targeted interventions in AMI, yet further large-scale validation remains necessary.
The Role of Macrophage Polarization in Other Cardiovascular Diseases
Although this review primarily focuses on the dynamic regulation and therapeutic implications of macrophage polarization in AMI, macrophages—due to their wide distribution and functional plasticity—are also critically involved in the pathogenesis of various other cardiovascular diseases.
In atherosclerosis (AS), M1 macrophages contribute to plaque formation and instability by secreting pro-inflammatory cytokines such as IL-1β and TNF-α, thereby increasing the risk of plaque rupture and thrombus formation. In contrast, M2 macrophages exert anti-inflammatory and reparative functions by releasing IL-10 and TGF-β, which help stabilize plaques and facilitate tissue remodeling.125 Modulating the M1/M2 balance has emerged as an important strategy for managing vascular inflammation, with metabolic agents such as SGLT2 inhibitors showing potential in shifting macrophage polarization toward a more reparative phenotype.126
In chronic heart failure (CHF), persistent low-grade inflammation plays a major role in adverse cardiac remodeling and functional decline. As resident immune cells in the heart, macrophages are deeply involved in regulating myocardial fibrosis, apoptosis, and impaired regeneration. Studies have demonstrated that distinct M2 subtypes perform specialized functions at different stages of HF progression—for example, M2a macrophages promote angiogenesis, while M2c subtypes contribute to inflammation resolution and tissue repair. Targeted modulation of specific M2 subtypes may thus represent a promising approach to attenuate fibrosis and improve clinical outcomes in CHF.127
Moreover, macrophage polarization is implicated in the progression of several other cardiovascular pathologies, including viral myocarditis, cardiac allograft rejection, hypertensive cardiomyopathy, and valvular heart disease. In viral myocarditis, M1 macrophages drive early myocardial injury, while M2 macrophages support inflammation resolution and scar formation in later stages.128 In hypertensive cardiomyopathy, chronic pressure overload promotes sustained pro-inflammatory macrophage activation, contributing to myocardial fibrosis and diastolic dysfunction.129 These findings underscore that macrophage polarization is not only temporally regulated but also shaped by disease-specific tissue microenvironments.
Therefore, advancing our understanding of macrophage polarization dynamics, metabolic programming, and cellular interactions across different cardiovascular contexts may help establish a broader immunotherapeutic framework. Future strategies focusing on temporal intervention windows, subtype-specific modulation, and targeted delivery systems are expected to foster the development of precision therapies not only for AMI but also for AS, CHF, and other immune-related cardiac diseases.
Conclusion and Future Perspectives
Macrophages play a dual role in the pathological process of AMI, acting as both major drivers of early inflammation and key regulators of later tissue repair. M1 macrophages dominate the inflammatory response during the acute phase of AMI by secreting pro-inflammatory cytokines (such as IL-1β, TNF-α, and IL-6). Through the NF-κB signaling pathway, M1 macrophages promote neutrophil recruitment, exacerbate local inflammation, and assist in the clearance of necrotic tissue. However, if M1 polarization persists for too long, it may lead to excessive inflammation, causing further myocardial damage and impairing cardiac function recovery. As inflammation transitions into the resolution phase, M2 macrophages begin to dominate the repair process. They secrete anti-inflammatory cytokines, such as IL-10 and TGF-β, and promote angiogenesis, fibroblast activation, and collagen remodeling via JAK/STAT and PI3K/Akt signaling pathways, thereby accelerating myocardial repair and cardiac function recovery.130,131 In addition to these classic immune pathways, recent evidence highlights the roles of metabolic reprogramming (such as glycolysis, fatty acid oxidation), epigenetic modifications, and non-coding RNAs (such as miRNAs) as highly actionable targets for modulating macrophage function.
The dynamic balance between M1 and M2 polarization is critical for the outcome of cardiac remodeling after AMI. Any disruption in this polarization process can lead to aggravated cardiac fibrosis, reduced myocardial function, and even increase the risk of heart failure. Therefore, strategies aimed at reducing M1 pro-inflammatory polarization and enhancing M2 reparative polarization have become an important focus in AMI treatment research. In recent years, multiple strategies have been proposed to optimize this transition process, including pharmacological interventions, gene editing, miRNA regulation, and immune modulation. For instance, mTOR inhibitors (such as everolimus) and IL-1β inhibitors (such as canakinumab) have shown potential in clinical settings to reduce inflammatory damage. Moreover, SGLT2 inhibitors (such as dapagliflozin) are currently under clinical investigation (DAPA-MI trial) and may exert macrophage-modulating effects beyond glycemic control. Gene editing technologies, such as CRISPR/Cas9, can precisely guide macrophages to switch to an anti-inflammatory repair phenotype by regulating the NF-κB or JAK/STAT signaling pathways. Furthermore, miRNAs (such as miR-21, miR-146a) are considered key regulators of macrophage polarization, and their delivery systems (such as LNP nanoparticles or AAV viral vectors) are continuously being optimized to improve therapeutic stability and targeting. However, these strategies still face challenges related to delivery efficiency, tissue targeting, and long-term safety, requiring further optimization before clinical application.132
In future AMI treatment strategies, the precise regulation of macrophage function remains a core challenge. Combining gene therapy, miRNA regulation, AI-assisted analysis, and targeted metabolic interventions may further optimize M1/M2 conversion, reduce fibrosis, and improve myocardial regeneration. Additionally, new biomaterials (such as smart nanoparticles) are being explored for more efficient delivery of intervention factors to enhance therapeutic stability and targeting. Despite these promising directions, several knowledge gaps remain. These include a lack of comprehensive single-cell transcriptomic data in human AMI, insufficient longitudinal studies tracking macrophage phenotypic transitions in vivo, and limited understanding of how systemic factors such as aging or comorbidities influence macrophage dynamics. With advancements in biomedical technologies, personalized, precision treatment strategies based on macrophage polarization regulation will become key strategies for improving AMI prognosis.
Abbreviations
AAV, Adeno-associated virus; APCs, Antigen-presenting cells; AMI, Acute myocardial infarction; Arg-1, Arginase-1; AS, Atherosclerosis; CHF, Chronic heart failure; DAMPs, Damage-associated molecular patterns; DCs, Dendritic cells; GLP-1, glucagon-like peptide-1; HMGB1, High-mobility group box 1; IFN, Type I interferon; IL-1β, Interleukin-1 beta; IL-6, Interleukin-6; IL-10, Interleukin-10; LDL, Low-density lipoprotein; LDL-C, Low-density lipoprotein cholesterol; LNP, Lipid nanoparticles; LPS, Lipopolysaccharides; M-CSF, Macrophage colony-stimulating factor; MI, Myocardial infarction; miRNAs, microRNAs; MMPs, Matrix metalloproteinases; MPO, Myeloperoxidase; NETs, Neutrophil extracellular traps; NF-κB, Nuclear factor kappa B; NK cells, Natural killer cells; NLRs, NOD-like receptors; NO, Nitric oxide; PAMPs, Pathogen-associated molecular patterns; PDCD4, Programmed cell death protein 4; PFKFB3, 6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3; ROS, Reactive oxygen species; SDH, Succinate dehydrogenase; SGLT2, Sodium-glucose co-transporter 2; SOCS1, suppressor of cytokine signaling 1; STAT, Signal transducers and activators of transcription; TGF-β, Transforming growth factor-beta; TLRs, Toll-like receptors; TNF-α, tumor necrosis factor-alpha; Tregs, Regulatory T cells.
Acknowledgments
We appreciate all authors whose publications could be included in our review.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by grants from National Natural Science Foundation of China (81873486, 82403624), the Science and Technology Development Program of Jiangsu Province-Clinical Frontier Technology (BE2022754), Clinical Medicine Expert Team (Class A) of Jinji Lake Health Talents Program of Suzhou Industrial Park (SZYQTD202102), Suzhou Key Discipline for Medicine (SZXK202129), Demonstration of Scientific and Technological Innovation Project (SKY2021002), Suzhou Dedicated Project on Diagnosis and Treatment Technology of Major Diseases (LCZX202132), Research on Collaborative Innovation of medical engineering combination (SZM2021014), Research on Collaborative Innovation of medical engineering combination (SZM2022003), Suzhou Key Laboratory of Diagnosis and Treatment of Panvascular Diseases (SZS2023021), Natural Science Foundation of Jiangsu Province (BK20240437), Suzhou Science and Education Youth Science and Technology Project (KJXW2023086).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Roth GA, Mensah GA, Johnson CO, et al. Global burden of cardiovascular diseases and risk factors, 1990-2019: update from the GBD 2019 study. J Am Coll Cardiol. 2020;76(25):2982–3021. doi:10.1016/j.jacc.2020.11.010
2. Visan I. Myocardial infarct inflammation. Nat Immunol. 2018;19(2):99. doi:10.1038/s41590-017-0037-3
3. Mouton AJ, DeLeon-Pennell KY, Rivera Gonzalez OJ, et al. Mapping macrophage polarization over the myocardial infarction time continuum. Basic Res Cardiol. 2018;113(4):26. doi:10.1007/s00395-018-0686-x
4. Alvarez-Argote S, O’Meara CC. The evolving roles of cardiac macrophages in homeostasis, regeneration, and repair. Int J Mol Sci. 2021;22(15). doi:10.3390/ijms22157923
5. Wang N, Liang H, Zen K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front Immunol. 2014;5:614. doi:10.3389/fimmu.2014.00614
6. Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496(7446):445–455. doi:10.1038/nature12034
7. Boutilier AJ, Elsawa SF. Macrophage polarization states in the tumor microenvironment. Int J Mol Sci. 2021;22(13). doi:10.3390/ijms22136995
8. Dijkgraaf EM, Heusinkveld M, Tummers B, et al. Chemotherapy alters monocyte differentiation to favor generation of cancer-supporting M2 macrophages in the tumor microenvironment. Cancer Res. 2013;73(8):2480–2492. doi:10.1158/0008-5472.Can-12-3542
9. Orecchioni M, Ghosheh Y, Pramod AB, Ley K. Macrophage polarization: different gene signatures in M1(LPS+) vs classically and M2(LPS-) vs. alternatively activated macrophages. Front Immunol. 2019;10:1084. doi:10.3389/fimmu.2019.01084
10. Zhou D, Huang C, Lin Z, et al. Macrophage polarization and function with emphasis on the evolving roles of coordinated regulation of cellular signaling pathways. Cell Signal. 2014;26(2):192–197. doi:10.1016/j.cellsig.2013.11.004
11. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13. doi:10.12703/p6-13
12. Italiani P, Boraschi D. From monocytes to M1/M2 macrophages: phenotypical vs functional differentiation. Front Immunol. 2014;5:514. doi:10.3389/fimmu.2014.00514
13. Murray PJ, Allen JE, Biswas SK, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41(1):14–20. doi:10.1016/j.immuni.2014.06.008
14. Gordon S, Plüddemann A, Martinez Estrada F. Macrophage heterogeneity in tissues: phenotypic diversity and functions. Immunol Rev. 2014;262(1):36–55. doi:10.1111/imr.12223
15. Wang LX, Zhang SX, Wu HJ, Rong XL, Guo J. M2b macrophage polarization and its roles in diseases. J Leukoc Biol. 2019;106(2):345–358. doi:10.1002/jlb.3ru1018-378rr
16. Nahrendorf M, Pittet MJ, Swirski FK. Monocytes: protagonists of infarct inflammation and repair after myocardial infarction. Circulation. 2010;121(22):2437–2445. doi:10.1161/circulationaha.109.916346
17. Shiraishi M, Shintani Y, Shintani Y, et al. Alternatively activated macrophages determine repair of the infarcted adult murine heart. J Clin Invest. 2016;126(6):2151–2166. doi:10.1172/jci85782
18. Pinto AR, Paolicelli R, Salimova E, et al. An abundant tissue macrophage population in the adult murine heart with a distinct alternatively-activated macrophage profile. PLoS One. 2012;7(5):e36814. doi:10.1371/journal.pone.0036814
19. Mantri M, Scuderi GJ, Abedini-Nassab R, et al. Spatiotemporal single-cell RNA sequencing of developing chicken hearts identifies interplay between cellular differentiation and morphogenesis. Nat Commun. 2021;12(1):1771. doi:10.1038/s41467-021-21892-z
20. Bajpai G, Schneider C, Wong N, et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat Med. 2018;24(8):1234–1245. doi:10.1038/s41591-018-0059-x
21. Dick SA, Macklin JA, Nejat S, et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat Immunol. 2019;20(1):29–39. doi:10.1038/s41590-018-0272-2
22. Epelman S, Lavine KJ, Beaudin AE, et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity. 2014;40(1):91–104. doi:10.1016/j.immuni.2013.11.019
23. Lavine KJ, Epelman S, Uchida K, et al. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc Natl Acad Sci U S A. 2014;111(45):16029–16034. doi:10.1073/pnas.1406508111
24. Leid J, Carrelha J, Boukarabila H, Epelman S, Jacobsen SE, Lavine KJ. Primitive embryonic macrophages are required for coronary development and maturation. Circ Res. 2016;118(10):1498–1511. doi:10.1161/circresaha.115.308270
25. Peet C, Ivetic A, Bromage DI, Shah AM. Cardiac monocytes and macrophages after myocardial infarction. Cardiovasc Res. 2020;116(6):1101–1112.
26. Heidt T, Courties G, Dutta P, et al. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ Res. 2014;115(2):284–295. doi:10.1161/circresaha.115.303567
27. Kang M, Jia H, Feng M, et al. Cardiac macrophages in maintaining heart homeostasis and regulating ventricular remodeling of heart diseases. Front Immunol. 2024;15:1467089. doi:10.3389/fimmu.2024.1467089
28. Sun X, Li Y, Deng Q, et al. Macrophage polarization, metabolic reprogramming, and inflammatory effects in ischemic heart disease. Front Immunol. 2022;13:934040.
29. Simões FC, Riley PR. Immune cells in cardiac repair and regeneration. Development. 2022;149(8):dev199906.
30. Swirski FK, Nahrendorf M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science. 2013;339(6116):161–166. doi:10.1126/science.1230719
31. Bateman RM, Sharpe MD, Jagger JE, et al. 36th International Symposium on Intensive Care and Emergency Medicine: Brussels, Belgium. 15-18 March 2016. Crit Care. 2016;20(Suppl 2):94. doi:10.1186/s13054-016-1208-6
32. Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol. 2014;11(5):255–265. doi:10.1038/nrcardio.2014.28
33. Weirather J, Hofmann UD, Beyersdorf N, et al. Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ Res. 2014;115(1):55–67. doi:10.1161/circresaha.115.303895
34. Comi G, Bar-Or A, Lassmann H, et al. Role of B cells in multiple sclerosis and related disorders. Ann Neurol. 2021;89(1):13–23. doi:10.1002/ana.25927
35. Zhao E, Xu H, Wang L, et al. Bone marrow and the control of immunity. Cell Mol Immunol. 2012;9(1):11–19. doi:10.1038/cmi.2011.47
36. Anzai A, Anzai T, Nagai S, et al. Regulatory role of dendritic cells in postinfarction healing and left ventricular remodeling. Circulation. 2012;125(10):1234–1245. doi:10.1161/circulationaha.111.052126
37. Yan X, Anzai A, Katsumata Y, et al. Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J Mol Cell Cardiol. 2013;62:24–35. doi:10.1016/j.yjmcc.2013.04.023
38. Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013;13(10):709–721. doi:10.1038/nri3520
39. Geovanini GR, Libby P. Atherosclerosis and inflammation: overview and updates. Clin Sci. 2018;132(12):1243–1252.
40. Libby P, Buring JE, Badimon L, et al. Atherosclerosis. Nat Rev Dis Primers. 2019;5(1):56. doi:10.1038/s41572-019-0106-z
41. Swirski FK, Nahrendorf M. Cardioimmunology: the immune system in cardiac homeostasis and disease. Nat Rev Immunol. 2018;18(12):733–744.
42. Khoury MK, Yang H, Liu B. Macrophage biology in cardiovascular diseases. Arterioscler Thromb Vasc Biol. 2021;41(2):e77–e81. doi:10.1161/atvbaha.120.313584
43. Libby P. Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32(9):2045–2051. doi:10.1161/atvbaha.108.179705
44. Ong SB, Hernández-Reséndiz S, Crespo-Avilan GE, et al. Inflammation following acute myocardial infarction: multiple players, dynamic roles, and novel therapeutic opportunities. Pharmacol Ther. 2018;186:73–87. doi:10.1016/j.pharmthera.2018.01.001
45. Prabhu SD, Frangogiannis NG. The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circ Res. 2016;119(1):91–112.
46. Mills CD, Lenz LL, Ley K. Macrophages at the fork in the road to health or disease. Front Immunol. 2015;6:59. doi:10.3389/fimmu.2015.00059
47. Nahrendorf M, Swirski FK. Monocyte and macrophage heterogeneity in the heart. Circ Res. 2013;112(12):1624–1633. doi:10.1161/circresaha.113.300890
48. Westman PC, Lipinski MJ, Luger D, et al. Inflammation as a driver of adverse left ventricular remodeling after acute myocardial infarction. J Am College Cardiol. 2016;67(17):2050–2060.
49. Epelman S, Liu PP, Mann DL. Role of innate and adaptive immune mechanisms in cardiac injury and repair. Nat Rev Immunol. 2015;15(2):117–129. doi:10.1038/nri3800
50. Weissman D, Maack C. Mitochondrial function in macrophages controls cardiac repair after myocardial infarction. J Clin Invest. 2023;133(4). doi:10.1172/jci167079
51. Sager HB, Hulsmans M, Lavine KJ, et al. Proliferation and recruitment contribute to myocardial macrophage expansion in chronic heart failure. Circ Res. 2016;119(7):853–864. doi:10.1161/circresaha.116.309001
52. Li L, Cao J, Li S, et al. M2 macrophage-derived sEV regulate pro-inflammatory CCR2(+) macrophage subpopulations to favor post-AMI cardiac repair. Adv Sci. 2023;10(14):e2202964. doi:10.1002/advs.202202964
53. Lavin Y, Winter D, Blecher-Gonen R, et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell. 2014;159(6):1312–1326. doi:10.1016/j.cell.2014.11.018
54. Kim SH, Lee KY, Chang K. The protective role of TREM2 in the heterogenous population of macrophages during post-myocardial infarction inflammation. Int J Mol Sci. 2023;24(6). doi:10.3390/ijms24065556
55. DeBerge M, Shah SJ, Wilsbacher L, Thorp EB. Macrophages in heart failure with reduced versus preserved ejection fraction. Trends Mol Med. 2019;25(4):328–340. doi:10.1016/j.molmed.2019.01.002
56. Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377(12):1119–1131. doi:10.1056/NEJMoa1707914
57. Chen C, Wang J, Liu C, Hu J. Cardiac resident macrophages: key regulatory mediators in the aftermath of myocardial infarction. Front Immunol. 2023;14:1207100. doi:10.3389/fimmu.2023.1207100
58. Choi C, Jeong YL, Park KM, et al. TM4SF19-mediated control of lysosomal activity in macrophages contributes to obesity-induced inflammation and metabolic dysfunction. Nat Commun. 2024;15(1):2779. doi:10.1038/s41467-024-47108-8
59. Weichhart T, Hengstschläger M, Linke M. Regulation of innate immune cell function by mTOR. Nat Rev Immunol. 2015;15(10):599–614. doi:10.1038/nri3901
60. Benjamin EJ, Muntner P, Alonso A, et al. Heart disease and stroke statistics-2019 update: a report from the American Heart Association. Circulation. 2019;139(10):e56–e528. doi:10.1161/cir.0000000000000659
61. Mantovani A, Garlanda C, Locati M. Macrophage diversity and polarization in atherosclerosis: a question of balance. Arterioscler Thromb Vasc Biol. 2009;29(10):1419–1423. doi:10.1161/atvbaha.108.180497
62. Rizzo G, Gropper J, Piollet M, et al. Dynamics of monocyte-derived macrophage diversity in experimental myocardial infarction. Cardiovasc Res. 2023;119(3):772–785. doi:10.1093/cvr/cvac113
63. Shin JJ, Park J, Shin HS, Arab I, Suk K, Lee WH. Roles of lncRNAs in NF-κB-mediated macrophage inflammation and their implications in the pathogenesis of human diseases. Int J Mol Sci. 2024;25(5). doi:10.3390/ijms25052670
64. Mussbacher M, Derler M, Basílio J, Schmid JA. NF-κB in monocytes and macrophages - an inflammatory master regulator in multitalented immune cells. Front Immunol. 2023;14:1134661. doi:10.3389/fimmu.2023.1134661
65. Williams JW, Giannarelli C, Rahman A, Randolph GJ, Biology KJCM. Classification, and phenotype in cardiovascular disease: JACC macrophage in CVD series (Part 1). J Am Coll Cardiol. 2018;72(18):2166–2180. doi:10.1016/j.jacc.2018.08.2148
66. Deng YF, Xu QQ, Chen TQ, et al. Kinsenoside alleviates inflammation and fibrosis in experimental NASH mice by suppressing the NF-κB/NLRP3 signaling pathway. Phytomedicine. 2022;104:154241. doi:10.1016/j.phymed.2022.154241
67. Fortelny N, Farlik M, Fife V, et al. JAK-STAT signaling maintains homeostasis in T cells and macrophages. Nat Immunol. 2024;25(5):847–859. doi:10.1038/s41590-024-01804-1
68. Hilgendorf I, Gerhardt LM, Tan TC, et al. Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ Res. 2014;114(10):1611–1622. doi:10.1161/circresaha.114.303204
69. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168(6):960–976. doi:10.1016/j.cell.2017.02.004
70. Vergadi E, Ieronymaki E, Lyroni K, Vaporidi K, Tsatsanis C. Akt signaling pathway in macrophage activation and M1/M2 polarization. J Immunol. 2017;198(3):1006–1014. doi:10.4049/jimmunol.1601515
71. Tian T, Chen L, Wang Z, Zhu M, Xu W, Wu B. Sema3A drives alternative macrophage activation in the resolution of periodontitis via PI3K/AKT/mTOR signaling. Inflammation. 2023;46(3):876–891. doi:10.1007/s10753-022-01777-z
72. Byles V, Covarrubias AJ, Ben-Sahra I, et al. The TSC-mTOR pathway regulates macrophage polarization. Nat Commun. 2013;4:2834. doi:10.1038/ncomms3834
73. Ip WKE, Hoshi N, Shouval DS, Snapper S, Medzhitov R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science. 2017;356(6337):513–519. doi:10.1126/science.aal3535
74. Ni Z, Zhao X, Dai X, Zhao L, Xia J. The role of interferon regulatory factor 5 in macrophage inflammation during osteoarthritis. Inflammation. 2019;42(5):1821–1829. doi:10.1007/s10753-019-01044-8
75. Odegaard JI, Chawla A. The immune system as a sensor of the metabolic state. Immunity. 2013;38(4):644–654. doi:10.1016/j.immuni.2013.04.001
76. O’Neill LA, Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med. 2016;213(1):15–23. doi:10.1084/jem.20151570
77. Tannahill GM, Curtis AM, Adamik J, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 2013;496(7444):238–242. doi:10.1038/nature11986
78. Mills EL, Kelly B, Logan A, et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell. 2016;167(2):457–470.e13. doi:10.1016/j.cell.2016.08.064
79. Ghesquière B, Wong BW, Kuchnio A, Carmeliet P. Metabolism of stromal and immune cells in health and disease. Nature. 2014;511(7508):167–176. doi:10.1038/nature13312
80. Diskin C, Pålsson-McDermott EM. Metabolic Modulation in Macrophage Effector Function. Front Immunol. 2018;9:270. doi:10.3389/fimmu.2018.00270
81. Huang SC, Everts B, Ivanova Y, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol. 2014;15(9):846–855. doi:10.1038/ni.2956
82. Van den Bossche J, Baardman J, de Winther MP. Metabolic characterization of polarized M1 and M2 bone marrow-derived macrophages using real-time extracellular flux analysis. J Vis Exp. 2015;(105). doi:10.3791/53424
83. Zhang D, Tang Z, Huang H, et al. Metabolic regulation of gene expression by histone lactylation. Nature. 2019;574(7779):575–580. doi:10.1038/s41586-019-1678-1
84. Nomura M, Liu J, Rovira II, et al. Fatty acid oxidation in macrophage polarization. Nat Immunol. 2016;17(3):216–217. doi:10.1038/ni.3366
85. Van den Bossche J, LA O, Menon D. Macrophage immunometabolism: where are we (going)? Trends Immunol. 2017;38(6):395–406. doi:10.1016/j.it.2017.03.001
86. Liu PS, Wang H, Li X, et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol. 2017;18(9):985–994. doi:10.1038/ni.3796
87. Endo J, Arita M. Cardioprotective mechanism of omega-3 polyunsaturated fatty acids. J Cardiol. 2016;67(1):22–27. doi:10.1016/j.jjcc.2015.08.002
88. Zhang Z, Pan X, Yang S, et al. miR-155 promotes ox-LDL-induced autophagy in human umbilical vein endothelial cells. Mediators Inflamm. 2017;2017:9174801. doi:10.1155/2017/9174801
89. Nazari-Jahantigh M, Wei Y, Noels H, et al. MicroRNA-155 promotes atherosclerosis by repressing Bcl6 in macrophages. J Clin Invest. 2012;122(11):4190–4202. doi:10.1172/jci61716
90. Curtale G, Mirolo M, Renzi TA, Rossato M, Bazzoni F, Locati M. Negative regulation of Toll-like receptor 4 signaling by IL-10-dependent microRNA-146b. Proc Natl Acad Sci U S A. 2013;110(28):11499–11504. doi:10.1073/pnas.1219852110
91. Handari SD, Rohman MS, Sargowo D, et al. Novel impact of colchicine on interleukin-10 expression in acute myocardial infarction: an integrative approach. J Clin Med. 2024;13(16). doi:10.3390/jcm13164619
92. Zhang S, Zhu X, Chen Y, Wen Z, Shi P, Ni Q. The role and therapeutic potential of macrophages in the pathogenesis of diabetic cardiomyopathy. Front Immunol. 2024;15:1393392. doi:10.3389/fimmu.2024.1393392
93. Komal S, Han SN, Cui LG, et al. Epigenetic regulation of macrophage polarization in cardiovascular diseases. Pharmaceuticals. 2023;16(2). doi:10.3390/ph16020141
94. Duncan SE, Gao S, Sarhene M, et al. Macrophage activities in myocardial infarction and heart failure. Cardiol Res Pract. 2020;2020:4375127. doi:10.1155/2020/4375127
95. Ma Y, Mouton AJ, Lindsey ML. Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl Res. 2018;191:15–28. doi:10.1016/j.trsl.2017.10.001
96. Han R, Gao J, Zhai H, Xiao J, Ding Y, Hao J. RAD001 (everolimus) attenuates experimental autoimmune neuritis by inhibiting the mTOR pathway, elevating Akt activity and polarizing M2 macrophages. Exp Neurol. 2016;280:106–114. doi:10.1016/j.expneurol.2016.04.005
97. Phạm TL, Yin Y, Kwon HH, et al. miRNA 146a-5p-loaded poly(d,l-lactic-co-glycolic acid) nanoparticles impair pain behaviors by inhibiting multiple inflammatory pathways in microglia. Nanomedicine. 2020;15(11):1113–1126. doi:10.2217/nnm-2019-0462
98. Yang J, Shuai J, Siow L, et al. MicroRNA-146a-loaded magnesium silicate nanospheres promote bone regeneration in an inflammatory microenvironment. Bone Res. 2024;12(1):2. doi:10.1038/s41413-023-00299-0
99. Wang D, Bi X, Zhao L, et al. Targeting SphK1/S1PR3 axis ameliorates sepsis-induced multiple organ injury via orchestration of macrophage polarization and glycolysis. Biochim Biophys Acta Mol Cell Res. 2025;1872(1):119877. doi:10.1016/j.bbamcr.2024.119877
100. Kauppinen A, Suuronen T, Ojala J, Kaarniranta K, Salminen A. Antagonistic crosstalk between NF-κB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell Signal. 2013;25(10):1939–1948. doi:10.1016/j.cellsig.2013.06.007
101. Chen X, Barozzi I, Termanini A, et al. Requirement for the histone deacetylase Hdac3 for the inflammatory gene expression program in macrophages. Proc Natl Acad Sci U S A. 2012;109(42):E2865–74. doi:10.1073/pnas.1121131109
102. Belkina AC, Nikolajczyk BS, Denis GV. BET protein function is required for inflammation: brd2 genetic disruption and BET inhibitor JQ1 impair mouse macrophage inflammatory responses. J Immunol. 2013;190(7):3670–3678. doi:10.4049/jimmunol.1202838
103. Zhang X, Kapoor D, Jeong SJ, et al. Identification of a leucine-mediated threshold effect governing macrophage mTOR signalling and cardiovascular risk. Nat Metab. 2024;6(2):359–377. doi:10.1038/s42255-024-00984-2
104. Paplomata E, O’Regan R. The PI3K/AKT/mTOR pathway in breast cancer: targets, trials and biomarkers. Ther Adv Med Oncol. 2014;6(4):154–166. doi:10.1177/1758834014530023
105. Woo J, Lu D, Lewandowski A, Ridker PM, Ebert BL, Steensma D. Canakinumab effects on erythropoiesis, cardiovascular risk, and clonal hematopoiesis: proteogenomic analysis of the cantos randomized clinical trial. Blood. 2022;140(Supplement 1):2236–2237. doi:10.1182/blood-2022-167275
106. Ridker PM, MacFadyen JG, Everett BM, Libby P, Thuren T, Glynn RJ. Relationship of C-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: a secondary analysis from the CANTOS randomised controlled trial. Lancet. 2018;391(10118):319–328. doi:10.1016/s0140-6736(17)32814-3
107. Everett BM, Cornel JH, Lainscak M, et al. Anti-inflammatory therapy with canakinumab for the prevention of hospitalization for heart failure. Circulation. 2019;139(10):1289–1299. doi:10.1161/circulationaha.118.038010
108. Hummelgaard S, Vilstrup JP, Gustafsen C, Glerup S, Weyer K. Targeting PCSK9 to tackle cardiovascular disease. Pharmacol Ther. 2023;249:108480. doi:10.1016/j.pharmthera.2023.108480
109. Hadjiphilippou S, Ray KK. Evolocumab and clinical outcomes in patients with cardiovascular disease. J R Coll Physicians Edinb. 2017;47(2):153–155. doi:10.4997/jrcpe.2017.212
110. O’Connell RM, Rao DS. Baltimore D. microRNA regulation of inflammatory responses. Annu Rev Immunol. 2012;30:295–312. doi:10.1146/annurev-immunol-020711-075013
111. Hulsmans M, Sam F, Nahrendorf M. Monocyte and macrophage contributions to cardiac remodeling. J Mol Cell Cardiol. 2016;93:149–155. doi:10.1016/j.yjmcc.2015.11.015
112. Zhang Y, Wang B, Chen J, Li T. Role of exosomal miRNAs and macrophage polarization in gastric cancer: a novel therapeutic strategy. Eur J Pharmacol. 2025;990:177268. doi:10.1016/j.ejphar.2025.177268
113. Shapouri-Moghaddam A, Mohammadian S, Vazini H, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. 2018;233(9):6425–6440. doi:10.1002/jcp.26429
114. Liu H, Li Q, Chen Y, et al. Suberosin attenuates rheumatoid arthritis by repolarizing macrophages and inhibiting synovitis via the JAK/STAT signaling pathway. Arthritis Res Ther. 2025;27(1):12. doi:10.1186/s13075-025-03481-3
115. Kenjo E, Hozumi H, Makita Y, et al. Low immunogenicity of LNP allows repeated administrations of CRISPR-Cas9 mRNA into skeletal muscle in mice. Nat Commun. 2021;12(1):7101. doi:10.1038/s41467-021-26714-w
116. Ferraresso F, Leung J, Kastrup CJ. RNA therapeutics to control fibrinolysis: review on applications in biology and medicine. J Thromb Haemost. 2024;22(8):2103–2114. doi:10.1016/j.jtha.2024.04.006
117. Wang J, Wu Q, Wang X, et al. Targeting macrophage phenotypes and metabolism as novel therapeutic approaches in atherosclerosis and related cardiovascular diseases. Curr Atheroscler Rep. 2024;26(10):573–588. doi:10.1007/s11883-024-01229-z
118. Li M, Zhang T, Li P, et al. IL-4-primed human umbilical cord mesenchymal stem cells-derived extracellular vesicles facilitate recovery in spinal cord injury via the miR-21-5p/PDCD4-mediated shifting of macrophage M1/M2 polarization. Life Sci. 2025;364:123441. doi:10.1016/j.lfs.2025.123441
119. Fei Y, Yu X, Liu P, Ren H, Wei T, Cheng Q. Simplified lipid nanoparticles for tissue- and cell-targeted mRNA delivery facilitate precision tumor therapy in a lung metastasis mouse model. Adv Mater. 2024;36(48):e2409812. doi:10.1002/adma.202409812
121. Thorp EB. Cardiac macrophages and emerging roles for their metabolism after myocardial infarction. J Clin Invest. 2023;133(18). doi:10.1172/jci171953
120. Cortés-Morales VA, Vázquez-González WG, Montesinos JJ, et al. Human bone marrow mesenchymal stem cells promote the M2 phenotype in macrophages derived from STEMI patients. Int J Mol Sci. 2023;24(22):doi:10.3390/ijms242216257.
122. Lin XF, Cui XN, Yang J, et al. SGLT2 inhibitors ameliorate NAFLD in mice via downregulating PFKFB3, suppressing glycolysis and modulating macrophage polarization. Acta Pharmacol Sin. 2024. doi:10.1038/s41401-024-01389-3
123. Yang L, Chen L, Li D, et al. Effect of GLP-1/GLP-1R on the polarization of macrophages in the occurrence and development of atherosclerosis. Mediators Inflamm. 2021;2021:5568159. doi:10.1155/2021/5568159
124. Zelniker TA, Wiviott SD, Raz I, et al. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials. Lancet. 2019;393(10166):31–39. doi:10.1016/s0140-6736(18)32590-x
125. Huang J, Ding X, Dong Y, Zhu H. Growth differentiation factor-15 orchestrates inflammation-related diseases via macrophage polarization. Discov Med. 2024;36(181):248–255. doi:10.24976/Discov.Med.202436181.23
126. Zhang H, Gong X, Ni S, Wang Y, Zhu L, Ji N. C1q/TNF-related protein-9 attenuates atherosclerosis through AMPK-NLRP3 inflammasome singling pathway. Int Immunopharmacol. 2019;77:105934. doi:10.1016/j.intimp.2019.105934
127. Dick SA, Macklin JA, Nejat S, et al. Publisher correction: self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat Immunol. 2019;20(5):664. doi:10.1038/s41590-019-0363-8
128. Fairweather D, Beetler DJ, Musigk N, et al. Sex and gender differences in myocarditis and dilated cardiomyopathy: an update. Front Cardiovasc Med. 2023;10:1129348. doi:10.3389/fcvm.2023.1129348
129. Adamo L, Rocha-Resende C, Prabhu SD, Mann DL. Reappraising the role of inflammation in heart failure. Nat Rev Cardiol. 2020;17(5):269–285. doi:10.1038/s41569-019-0315-x
130. Artyomov MN, Van den Bossche J. Immunometabolism in the single-cell era. Cell Metab. 2020;32(5):710–725. doi:10.1016/j.cmet.2020.09.013
131. Mahjoubin-Tehran M, Kovanen PT, Xu S, Jamialahmadi T, Sahebkar A. Cyclodextrins: potential therapeutics against atherosclerosis. Pharmacol Ther. 2020;214:107620. doi:10.1016/j.pharmthera.2020.107620
132. Ganini C, Amelio I, Bertolo R, et al. Global mapping of cancers: the cancer genome Atlas and beyond. Mol Oncol. 2021;15(11):2823–2840. doi:10.1002/1878-0261.13056
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.