Back to Journals » OncoTargets and Therapy » Volume 13
DUSP9 Suppresses Proliferation and Migration of Clear Cell Renal Cell Carcinoma via the mTOR Pathway
Authors Luo J ![]() , Luo X, Liu X, Fang Z, Xu J
, Luo X, Liu X, Fang Z, Xu J ![]() , Li L
, Li L
Received 20 November 2019
Accepted for publication 4 February 2020
Published 13 February 2020 Volume 2020:13 Pages 1321—1330
DOI https://doi.org/10.2147/OTT.S239407
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Arseniy Yuzhalin
Jing Luo, Xing Luo, Xin Liu, Zhenqiang Fang, Jie Xu, Longkun Li
Department of Urology, Second Affiliated Hospital, Army Medical University, Chongqing, People’s Republic of China
Correspondence: Longkun Li; Jie Xu
Department of Urology, Second Affiliated Hospital, Army Medical University (Third Military Medical University), Chongqing, People’s Republic of China
Tel +86 13983615565
; +86 15736136290
Fax +86 023-68763217
Email [email protected]; [email protected]
Background: Clear cell renal cell carcinoma (ccRCC) is one of the most common urologic tumors. However, the carcinogenic mechanism of ccRCC remains unclear. This study aimed to investigate the effects of dual specificity phosphatase 9 (DUSP9) in ccRCC.
Methods: Cell proliferation and migration abilities were detected by Cell Counting kit-8, wound-healing (scratch) assay and transwell assay. The expression of mRNA in ccRCC was measured by qPCR. Western blot and immunohistochemical staining were used for protein expression. In addition, nude mouse xenograft experiment establishes an in vivo model to detect the inhibitory effect of DUSP9 on tumor proliferation.
Results: DUSP9 was significantly down-regulated in both ccRCC cell lines and ccRCC tissues compared to that in non-cancer cell lines and normal tissues. Besides, DUSP9 suppressed proliferation and migration of ccRCC cell lines in vitro. Importantly, the inhibition of tumor growth by DUSP9 was confirmed by xenograft tumor studies. And DUSP9 could inhibit both phosphorylation of mTOR and expression of its pathway-associated proteins Sox2, c-Myc, and HIF-1α, which are involved in cell proliferation and migration.
Conclusion: Taken together, our results uncovered DUSP9 as a tumor suppressor in ccRCC, acting by regulating cell proliferation and migration via the mTOR pathway.
Keywords: dual specificity phosphatase 9, clear cell renal cell carcinoma, proliferation, migration, mTOR
Introduction
Clear cell renal cell carcinoma (ccRCC) is the most commonly reported malignant renal tumor, and its incidence is increasing every year.1 According to the global cancer statistics released in 2018, 403,262 new cases of renal cell carcinoma and 175,098 deaths have been reported, showing its trend of growth.2 ccRCC comprises of 75% of all RCCs.3 Radical nephrectomy is the main treatment for patients with ccRCC; however, quite a few patients miss the timing of surgery during treatment,4 and the poor effect of chemotherapy and radiotherapy often leads to poor prognosis. Although targeted drugs have been commonly used to treat ccRCC, resistance to the drug results in dissatisfactory long-term effects.5 Therefore, a clarification of the molecular mechanism of ccRCC and development of new therapeutic targets are of utmost significance for further understanding the proliferation, metastasis, and targeted drug resistance, as well as guiding patient treatment.
Dual specificity phosphatases (DUSPs) family belongs to the mitogen-activated protein kinase phosphatases (MKPs), and includes ten catalytically active enzymes.6 DUSPs are considered to be important regulators of key signaling pathways in many diseases,7 and signaling pathway abnormalities are essential for the development and progression of cancers.8 Different kinds of DUSPs can specifically target different MAPKs. DUSP9, also known as MKP4, was first reported in 1997 by Muda et al.9 It is generally considered to be dephosphorylated by MAP kinases ERK, p38, and JNK.8–13 Recent studies have shown that DUSP9 is down-regulated in gastric cancer,10 hepatocellular carcinoma,14 colorectal cancer,12 and squamous cell carcinoma.15 In ccRCC, Luv et al16 had shown DUSP9 to act as a biomarker of diagnosis and prognosis. Wu et al17 had found the ratio of DUSP9 reduction was higher in high-stage and high-grade ccRCC, and the reduction of DUSP9 expression was associated with poor prognosis. Till date, the functional role and regulatory mechanism of DUSP9 in ccRCC remain unclear, and would require further investigation.
In this study, we performed various experiments, both in vitro and in vivo, and found DUSP9 expression to be consistently low in ccRCC tissues and cell lines. Overexpression of DUSP9 remarkably inhibited cell proliferation and migration. In addition, xenograft tumors with DUSP9 overexpression were also smaller in size. Mechanistically, we found DUSP9 to inhibit the activation of mTOR and expression of its downstream proteins. Together, these results indicated DUSP9 as a key tumor suppressor in ccRCC, and provided a new molecular mechanism for the development of ccRCC.
Materials and Methods
Cell Culture
Cell lines of human ccRCC, including 769-P, OS-RC-2, 786-O, and Caki-1, were obtained from the American Type Culture Collection (Rockville, MD, USA), and routinely tested in order to rule out mycoplasma contamination. The 769-P, OS-RC-2, and 786-O cell lines were cultured in RPMI-1640 medium (Gibco, Carlsbad, CA, USA) with 10% fetal bovine serum (FBS) (Gibco-Life Technologies, CA, USA). Caki-1 cell line was cultured in McCoy’s 5A (Modified) Medium (Gibco-Life Technologies, CA, USA). Line HK-2 was grown in K-SFM (Gibco-Life Technologies, CA, USA) serum free medium. All cell lines were incubated at 37 °C, 5% CO2, and 95% air.
Human Tumor Specimens
Tumor samples of patients with ccRCC were obtained from the Department of Urology, Second Affiliated Hospital, Army Medical University (Third Military Medical University), Chongqing, P. R. China. Based on histopathological examination, the samples were confirmed as ccRCC. The study was conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the Ethics Committee of the Army Medical University (Third Military Medical University) of China. Each patient had provided written informed consent prior to surgery. Part of the DUSP9 expression data was derived from the TCGA database (http://ualcan.path.uab.edu/).18
Lentiviral Transfection
Lentiviral vector construction of DUSP9 overexpression and negative control (NC) was commercially performed by Sangon, China. DUSP9 lentiviral overexpression system contained the green fluorescent protein gene and HA-tag for tracking the transfection efficiencies. All cells were cultured in 6-well culture dishes at 50% density, and 769-P, 786-O, and OS-RC-2 cells were infected with DUSP9 overexpression lentivirus, along with NC. The MOI (multiplicity of infection) value was set to 10 based on the results of our previous experiments. According to the instructions, all transfections were supplemented with 5 μg/mL Polybrene. Screening was conducted with 2 μg/mL puromycin (Sigma) for 2 weeks to obtain stably transfected cells.
Cell Proliferation Assay
Pretreated cells were planted into 96-well plates, and 2 × 103 cells per well were seeded and cultured for 1, 2, 3, 4 and 5 days. At the end of the culture, 10 μL of Cell Counting Kit-8 reagent (CCK-8, Kumamoto, Japan) was added to each well. After 2 h of incubation at 37 °C, 5% CO2, and 95% air, absorbance was recorded at 450 nm using a microplate reader. Cell proliferation could be assessed by absorbance values, in accordance with the protocol.
Transwell Assay
Transwell chambers (Corning Costar, Lowell, MA, USA) were used to investigate cell migration. Pretreated suspension cells (1 × 105) in 200 µL of 2% serum medium were seeded into the upper chamber; 600 μL of medium with 10% serum was added to the lower chamber. Cells were fixed and stained after incubation for 24 h, according to the manufacturer’s protocol, and each experiment was repeated at least thrice. The number of cells, from the upper chamber to the lower chamber, was calculated in six random fields for analysis.
RNA Isolation and q-PCR
Total RNA was extracted from cell lines and clinical specimens using the RNAiso PLUS reagent (Takara, Japan), and cDNA was synthesized using PrimeScriptTM RT reagent Kit (Takara, Japan), according to the manufacturer’s protocol. All q-PCR mRNA assays were performed with SYBR Green PCR Kit reagent (Qiagen, Valencia, USA) using a StepOnePlus Real-Time PCR system (Applied Biosystems, USA). Specific primers for mRNA were purchased from Sangon Biotech (Shanghai, China). GAPDH was used as endogenous control for mRNA. Calculation of fold change for the expression of mRNA was done by 2−ΔΔCt method. All primer sequences are provided in Table 1.
 |
Table 1 Nucleotide Sequences of Primers Used for RT-PCR Reactions |
Western Blot Assay
Total protein was harvested using RIPA lysis buffer (Beyotime, China). Protease inhibitor PMSF (Beyotime, China) was added to the lysis buffer prior to use. Protein concentration was determined using the BCA method. Approximately 40 μg of cells or tumor specimen proteins were transferred to nitrocellulose membranes after SDS-PAGE. The membrane was blocked with 5% skim milk for 2 h, followed by incubation with primary antibody against DUSP9 (Abcam, ab194355, 1:1000), GAPDH (Proteintech, 60004-1-Ig, 1:3000), α-Tubulin (Proteintech, 11224-1-AP, 1:1000), HA-Tag (CST, 3724S, 1:1000), mTOR (CST, 2972, 1:1000), phospho(p)-mTOR (Ser2448)(CST, 5536, 1:1000), HIF-1α (Proteintech, 20960-1-AP, 1:1000), c-Myc (Proteintech, 10828-1-AP, 1:1000), and Sox2 (CST, 3579, 1:1000) overnight at 4 °C, and then with a secondary antibody at 37 °C for 1 h. Related proteins were detected using the PierceTM ECL Western Blotting Substrate kit (Thermo Scientific, USA). Protein levels were normalized against GAPDH or α-Tubulin.
Nude Mouse Xenograft Experiment
Animal experiments were conducted following the guidelines by the Xinqiao Hospital and the Animal Care and Use Committee of the Army Medical University (Third Military Medical University). And the experiment protocol was approved by the Ethics Committee of the Army Medical University (Third Military Medical University) of China. Four-week-old female nude mice were purchased from the Laboratory Animal Center of the Army Medical University. All nude mice were raised in a standard SPF environment and fed on autoclave laboratory rodent feed. Ten nude mice were randomly divided into 2 groups (control group and experimental group, n = 5 each group). They were injected subcutaneously into the flank region (both sides/each nude mouse) with 2 × 106 stably transduced OS-RC-2 cells mixed in matrix gel (1:1 volume). Tumor size was monitored every 3 days with a caliper, and the volume was calculated using the following formula: V = length × (width2)/2. Three weeks after implantation, the mice were humanely sacrificed by CO2 inhalation and their tumor weights were measured.
Immunohistochemical Staining
Tumors were excised from nude mice, fixed in 4% paraformaldehyde, embedded in paraffin, and cut into 4 μm slices. The rehydrated paraffin sections were first treated with 3% H2O2 for 30 min and then incubated at room temperature with ImmunoBlock reagent for 30 min. The sections were then incubated with primary antibodies against Ki-67 (Proteintech, 27309-1-AP, 1:5000) and DUSP9 (Abcam, ab194355, 1:500), overnight at 4 °C. The sections were finally treated with secondary antibodies for 30 min and developed using a developer for 10 min. An IHC-based scoring standard was used to semi-quantitatively analyze the percentage of positive cells in Ki-67 and DUSP9 (positive cell score: 0 = percentage of negative staining; 1 = < 15%; 2 = 15–25%; 3 = 25–50%; 4 = 50–75%; 5 = 75–100%, each high power field (40 ×)). Five randomly selected fields of view were analyzed (40 ×) for quantification. The proportion of positively stained cells per slice in the NC and DUSP9 overexpression groups was determined subsequently.
Wound Healing (Scratch) Assay
Wound healing test was designed to investigate the migratory ability of cells. Approximately 70 μL of stable transfected cell suspension, at a concentration of approximately 5 × 105 cells/mL, was added to each well of the culture-insert (ibidi, Germany) and placed in an incubator. After 24-h culture, the culture insert was removed using sterile forceps and culture continued in serum-free medium. Photographs were taken under a microscope every 6 h and results analyzed accordingly.
Statistical Analysis
Statistical analysis was conducted using SPSS 17.0. All data were expressed as the mean ± SD. A two-tailed unpaired t-test or one-way analysis of variance (ANOVA) was performed to compare the differences between the groups. Repeated measures analysis of variance was performed for analyzing the growth curve of subcutaneous xenografts in nude mice and in CCK8 experiment. P value < 0.05 was regarded as statistically significant.
Results
DUSP9 Expression Was Significantly Down-Regulated in ccRCC Cell Lines and Tissues
We analyzed the HPA RNA-seq (DUSP9 gene) in normal tissues with NCBI gene sequence ID: 1852, and found DUSP9 to be only enriched in the kidney and placenta (Figure 1A). We compared the DUSP9 expression levels in ccRCC tissues with that in normal tissues using the TCGA database, and found DUSP9 expression to be significantly decreased in the tumors (Figure 1B). We also examined DUSP9 expression in ccRCC cell lines and tumor tissues. Western blot and q-PCR results suggested the expression of DUSP9 to be significantly reduced in four ccRCC cell lines (PCR results showed the expression of DUSP9 was reduced in 786-O, 769-P, Caki-1 and OS-RC-2 cells by approximately 89.56%, 81.48%, 74.68% and 78.94%, respectively.) compared to that in HK-2 cell line (normal renal tubular epithelial cells) (Figure 1C and E). Results from patient tissues were consistent with that from cell lines (PCR results showed the expression of DUSP9 was reduced in ccRCC tissues by approximately 81.07% on average.) (Figure 1D and F). Taken together, DUSP9 was found to be significantly down-regulated in ccRCC, and was closely related to tumorigenesis.
 |
Figure 1 DUSP9 expression is down-regulated in ccRCC. (A) RNA-Seq was performed on tissue samples from 95 human individuals, representing 27 different tissues, in order to determine tissue-specificity of all protein-coding genes; the data were obtained from NCBI gene sequence ID: 1852. (B) Expression of DUSP9 in ccRCC tissues was significantly down-regulated compared to that in normal tissues, using data obtained from TCGA dataset. (C, D) Western blot and (E, F) q-PCR analysis showed the DUSP9 levels to be significantly down-regulated in ccRCC cell lines and tumor tissues compared to that in normal cell lines and tissues (T denotes tumor tissues, N denotes tumor adjacent to normal tissues); *p < 0.05, **p < 0.01, ***p < 0.001. All data were representatives of three independent experiments. Abbreviations: DUSP9, dual specificity phosphatase 9; ccRCC, clear cell renal cell carcinoma; RNA-Seq, RNA sequencing. |
Overexpression of DUSP9 Inhibited Cell Proliferation and Migration in ccRCC in vitro
To investigate the effects of DUSP9 in ccRCC, we constructed two DUSP9 overexpressing cell lines (769-P and 786-O) using lentiviruses. We detected transfection efficiency by fluorescence microscopy and Western blotting (Figure 2A and B). Cell proliferation being closely related to tumorigenesis, we first examined the association of DUSP9 with ccRCC proliferation. Results of CCK-8 experiments showed the activity of cells overexpressing DUSP9, in both cell lines, to be lower than that in the negative control (NC) group (769-P and 786-O cells viability decreased by approximately 41.69% and 37.18% on the fifth day), thereby revealing that DUSP9 has the effect of inhibiting proliferation of ccRCC (Figure 2C).
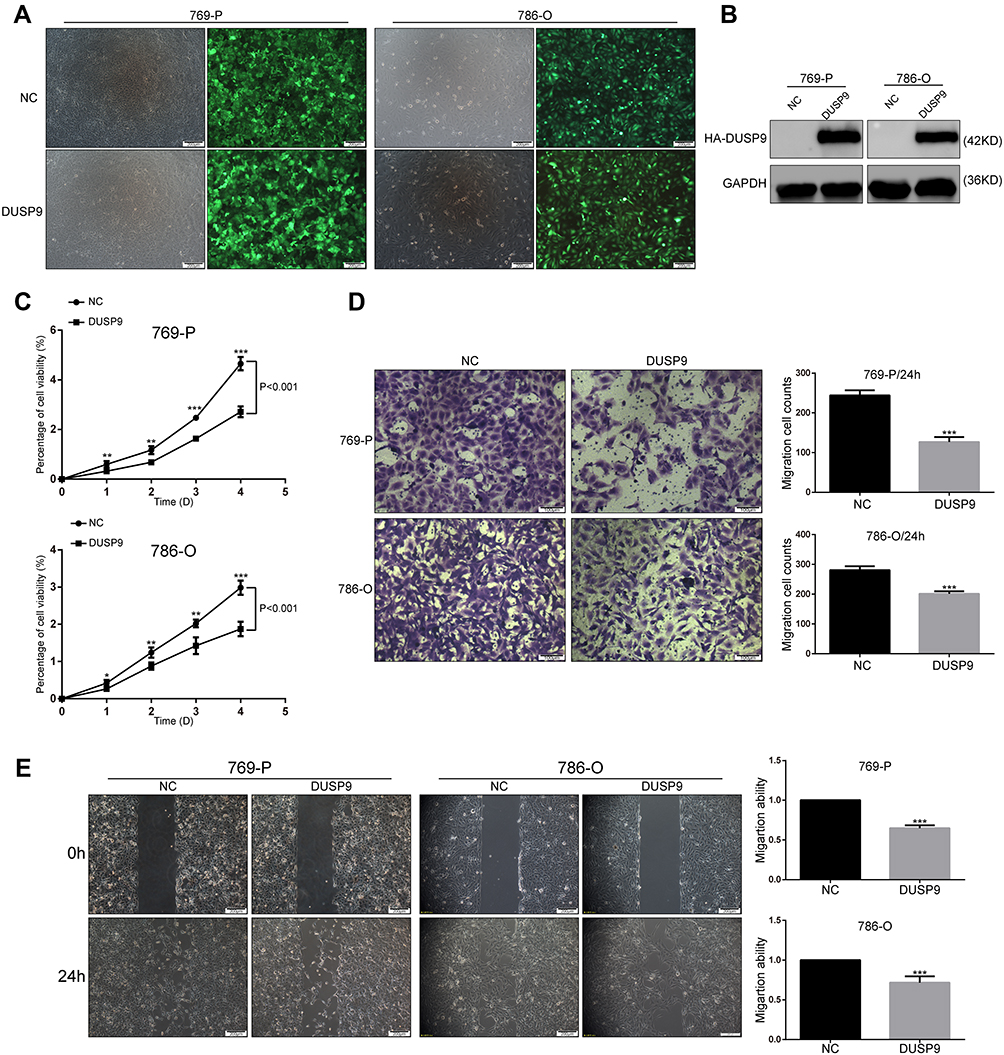 |
Figure 2 DUSP9 inhibits ccRCC cell proliferation and migration in vitro. (A, B) (A) Fluorescence labeling and (B) Western blot analysis confirmed the successful overexpression of DUSP9 in 769-P and 786-O cells. (C) CCK-8 viability assay demonstrated the overexpression of DUSP9 to significantly reduce the proliferation of 786-O and 769-P cells (Repeated-measures analysis of variance, P < 0.001). (D) Transwell assay and (E) wound-healing assay confirmed the overexpression of DUSP9 to inhibit migration in 769-P and 786-O cells; *p < 0.05, **p < 0.01, ***p < 0.001. All data were representatives of three independent experiments. Abbreviations: DUSP9, dual specificity phosphatase 9; NC, negative control; HA, HA protein tag; CCK-8, Cell Counting Kit-8. |
ccRCC is considered to be a tumor with high metastatic characteristics.19,20 In order to determine whether DUSP9 has an effect on the migration of ccRCC cells, we performed transwell and wound-healing (scratch) assays. As shown in Figure 2D and E, transwell results showed that 769-P and 786-O cells migration decreased by approximately 48.08% and 28.28% after overexpression of DUSP9, respectively. The same wound-healing (scratch) assays showed a reduction of migration of 769-P and 786-O cells approximately 35.19% and 28.32%, respectively. Therefore, the data suggested that DUSP9 can inhibit ccRCC cell proliferation and migration.
Overexpression of DUSP9 Inhibited Tumor Growth in ccRCC in vivo
To investigate whether DUSP9 inhibited ccRCC in vivo, we conducted nude mouse xenograft experiments. Previous studies had confirmed that OS-RC-2 cells have high tumor-forming efficiency in nude mice,21 and DUSP9 was also found to be less expressed in OS-RC-2 cells (Figure 1C and E). Therefore, OS-RC-2 cells were selected for in-vivo experiments. Results showed the volume (Figure 3A–C and E) and weight (Figure 3D) of xenograft tumors in DUSP9 overexpression group to be significantly less than that of tumors in NC group, where the volume was reduced by approximately 62.94% compared to NC, and the weight was reduced by approximately 62.81%. Immunohistochemical staining analysis of subcutaneous tumors in nude mice showed the overexpression of DUSP9 to reduce the expression of cell proliferation marker Ki-67 (Figure 3F). The results were consistent with those from in-vitro proliferation assays, thus suggesting that DUSP9 can inhibit ccRCC proliferation.
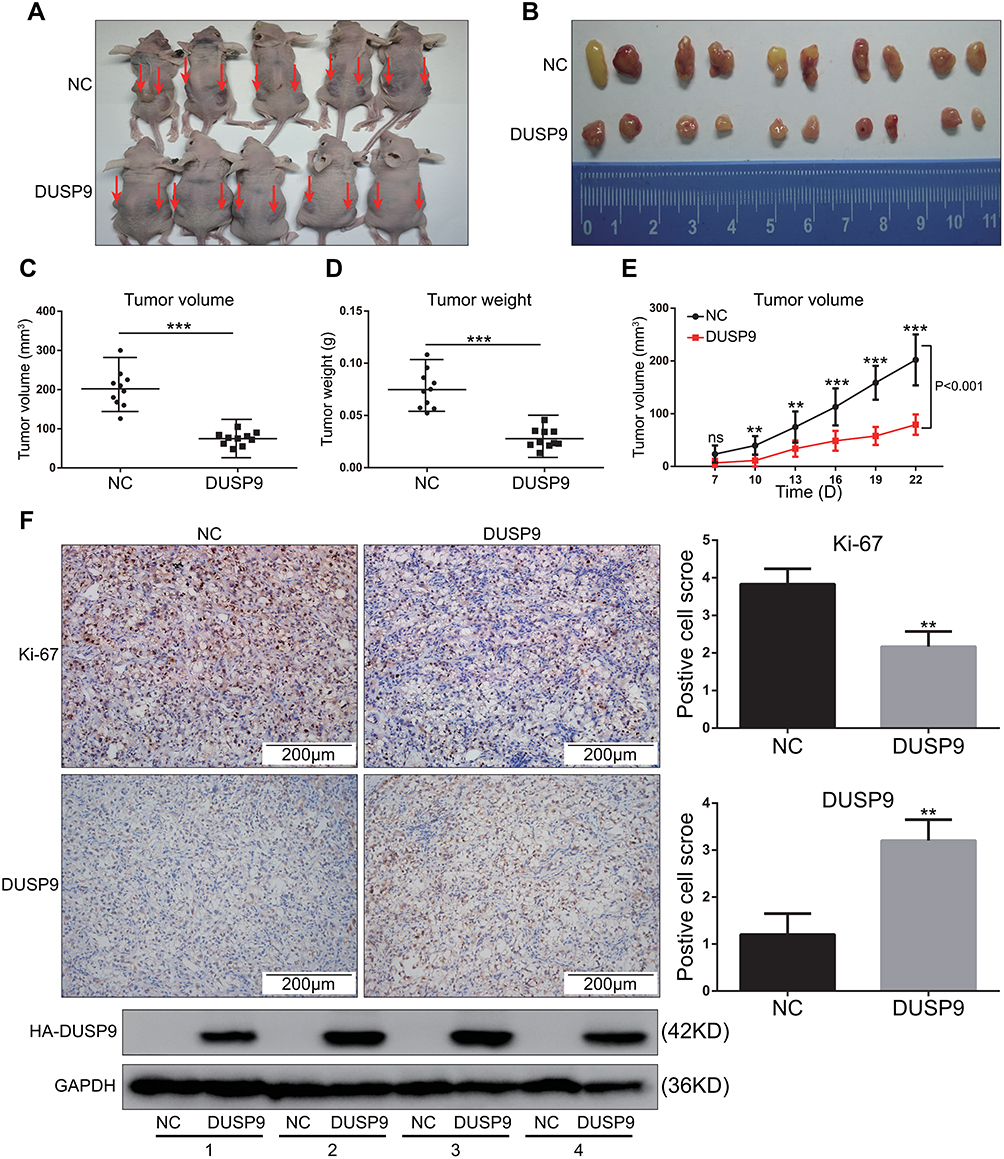 |
Figure 3 DUSP9 inhibits ccRCC tumor growth in vivo. (A) Two groups (NC and DUSP9 overexpression) of stably transfected OS-RC-2 cell suspensions (2 × 106 cells) in mixed matrix gel (1:1 volume) were injected subcutaneously into nude mice (n = 5 in each group, injections on both sides/each nude mouse, indicated by arrows). (B) Images of the xenograft tumors from the two groups (NC and DUSP9 overexpression) of stably transfected OS-RC-2 cells injected into the nude mice after 22 days. (C, D) Mean tumor volumes and tumor weights of DUSP9 overexpressing OS-RC-2 group were significantly smaller than those of OS-RC-2 NC group (n = 10 in each group). Xenograft tumors of the DUSP9 overexpressing group and NC group were dissected to determine their volumes and weights, and then analyzed comparatively. (E) The growth curve of subcutaneous xenograft tumor in nude mice. Tumor volumes were calculated every three days after one week of injection. (F) Histopathology of xenograft tumors in nude mice. IHC staining of DUSP9 and Ki-67 in DUSP9 overexpression or NC group xenografts are shown; Western blot analysis of eight xenograft tumor samples from two groups (n = 4 in each group); **p < 0.01, ***p < 0.001. Abbreviations: DUSP9, dual specificity phosphatase 9; NC, negative control; HA, HA protein tag. |
Overexpression of DUSP9 Inhibited mTOR Pathway Activation
mTOR pathway plays a crucial role in the development and treatment of ccRCC.22 To investigate whether DUSP9 could be associated with mTOR, we examined the changes in mTOR and p-mTOR (Ser2448) after overexpression of DUSP9 in ccRCC cell lines using Western blotting. Grayscale analysis showed that overexpression of DUSP9 reduced the expression of p-mTOR protein in 769-P and 786-O cells by approximately 79.50% and 49.90%, respectively (Figure 4A). At the same time, the expression of Sox2, c-Myc and HIF-1α downstream of the mTOR pathway was also suppressed. PCR results showed that the Sox2 expression of 769-P and 786-O cells was reduced by approximately 67.04% and 48.49%, c-Myc expression was decreased by approximately 30.66% and 44.75%, and HIF-1α expression was decreased by approximately 30.42% and 33.94%. (Figure 4B–D). These data together suggested DUSP9 as a negative regulator of the mTOR pathway in ccRCC.
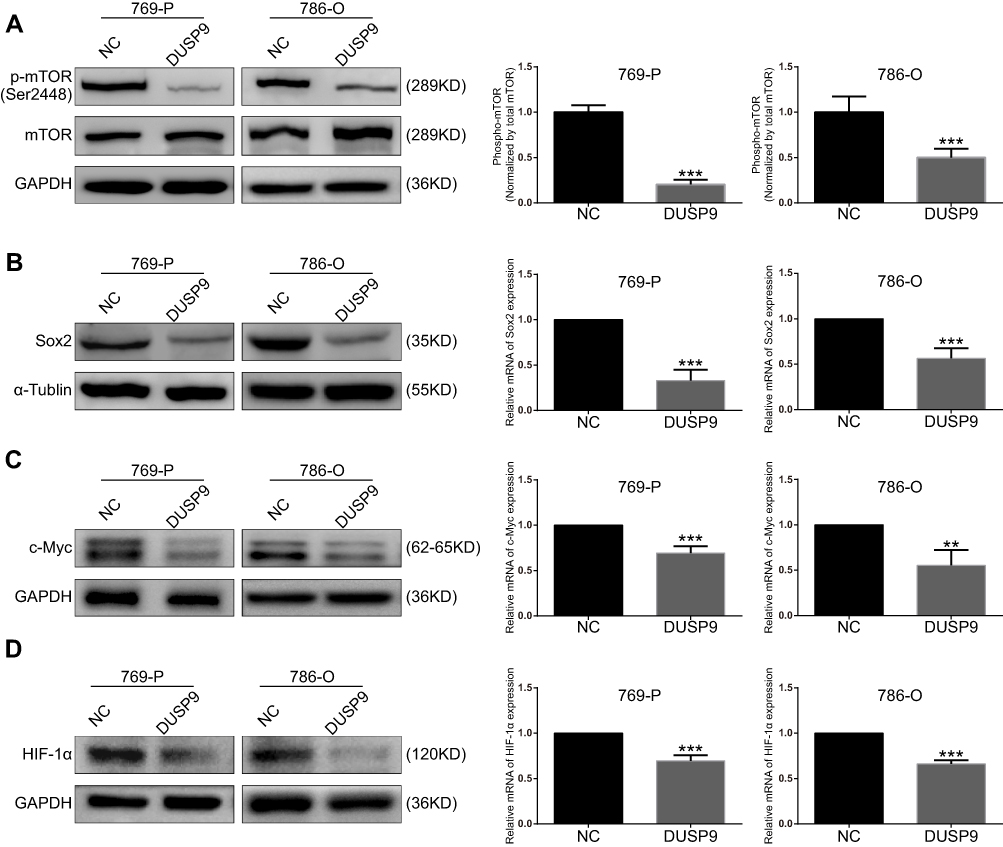 |
Figure 4 DUSP9 inhibits mTOR pathway activation. (A) Western blot analysis demonstrated inhibition of mTOR phosphorylation by the overexpression of DUSP9 in 769-P and 786-O cells at Ser2448. (B, C and D) Western blot and q-PCR analysis indicated overexpression of DUSP9 to inhibit Sox2, c-Myc, and HIF-1α protein and mRNA expression in 769-P and 786-O cells; **p < 0.01, ***p < 0.001. All data were representatives of three independent experiments. Abbreviations: DUSP9, dual specificity phosphatase 9; NC, negative control. |
Discussion
DUSP deregulation has been reported in a variety of cancers.23 DUSP9 is a DUSP with the characteristic of differential expression, being remarkably high only in the kidneys of adults. Many studies have proven DUSP9 to act as a tumor suppressor in cancer. For example, Liu et al15 had demonstrated DUSP9 reconstitution in squamous cell carcinoma to lead to G2-M phase-related cell death and microtubule disruption. Liu et al14 found DUSP9 to be down-regulated in hepatocarcinoma tissues and proliferating hepatoma cells, and to be associated with overall survival and recurrence time. Jenner et al12 had reported the establishment of a methylation screening method for DUSP9 in colorectal cancer, indicating the aberrant methylation of DUSP9 promoter as a phenotype of CPG island methylation. In ccRCC, Zhou et al24 had found DUSP9 mRNA expression to be down-regulated, by deep sequencing technology, and Wu et al17 had found the low expression of DUSP9 to result in poor prognosis. However, the functional information and regulatory mechanisms of DUSP9 in ccRCC remained unclear. In this study, we provided new evidence for the significant low expression of DUSP9 in ccRCC, and also demonstrated that overexpression of DUSP9 reduced proliferation and migration (Figures 1 and 2). Finally, we explored the downstream pathway of DUSP9 and found it to inhibit the activation of proliferation-related mTOR pathway (Figure 4).
Phosphatases are closely linked to cancer, and their abnormal expression and function characterize cancer cells.25 Here, we demonstrated DUSP9 to be only enriched in the kidney of adults, using HPA RNA-Seq analysis of normal tissue of the Gene series Gene ID: 1852, thus indicating that DUSP9 may be tissue-specific for the kidney (Figure 1A). By comparing the expression of DUSP9 in ccRCC tumors and paracancerous tissues, and normal cell lines and tumor cell lines, we found DUSP9 to be significantly down-regulated in tumors (Figure 1B–F). Experiments in ccRCC cell lines have indicated the overexpression of DUSP9 to inhibit cell proliferation and migration (Figure 2). Further, nude mice xenograft experiments have confirmed the overexpression of DUSP9 to inhibit tumor growth in vivo (Figure 3). All the above results collectively suggested DUSP9 to play an important role in carcinogenesis and ccRCC progression as a tumor suppressor.
mTOR is a highly conserved serine/threonine kinase of the PI3K-related kinase family, and plays a key role in the regulation of cell growth, proliferation, and survival.26 mTOR activation is closely linked to the activation of mTORC1,27 and the available mTOR inhibitors mainly act on mTORC1.28 Studies have found the conventional activation of mTORC1 to be primarily due to phosphorylation of Ser2448.29 Previous studies had shown mTOR to be closely related to carcinogenesis and recurrence mechanism of ccRCC.30 Kruck et al31 found phosphorylated-mTOR (p-mTOR) levels to be higher in renal cell carcinoma specimens than in benign renal parenchyma. One of the two most targeted drugs currently used in the treatment of ccRCC is an inhibitor of mTOR signaling pathway (everolimus and temsirolimus).32 However, the outcome of almost all patients taking the drugs generate resistance.33 Therefore, finding a method to combat ccRCC resistance or develop a new therapeutic drug is urgently required. Recently, many researchers have made new progress, such as Elie et al has reported on the good therapeutic effect of ccRCC new chemotherapy drug RANCE-1.34 In this study, we found the possibility of a connection between DUSP9 and mTOR. Overexpression of DUSP9 can, at least, inhibit mTOR phosphorylation at the Ser2448 site (Figure 4A), thereby reducing activation of the mTOR pathway. At the same time, overexpression of DUSP9 reduced the mRNA and protein expression of Sox2, c-Myc and HIF-1α downstream of the mTOR pathway (Figure 4B–D). These results together indicated that DUSP9 could act as a negative regulator in the mTOR pathway.
There are, however, certain limitations of the study. Although we demonstrated decreased phosphorylation of mTOR (Ser2448) in ccRCC cells after DUSP9 overexpression, there could probably be other targets of DUSP9, which may also affect cancer cell growth and migration. Therefore, further research is recommended in future in this regard.
Conclusion
In conclusion, our study demonstrates, for the first time, the possible inhibition of proliferation and migration in ccRCC by DUSP9 via the mTOR pathway (Figure 5). Hence, our findings provide new clues for elucidating the carcinogenic mechanism of ccRCC, and might provide new possibilities for clinical treatment of patients.
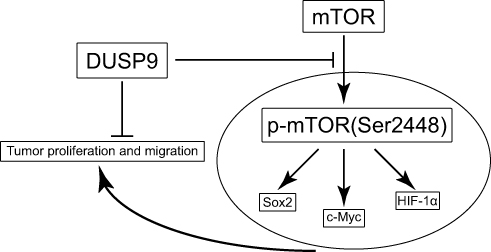 |
Figure 5 Schematic of the model DUSP9 might inhibit ccRCC proliferation and migration through the mTOR pathway. Abbreviations: DUSP9, dual specificity phosphatase 9; ccRCC, clear cell renal cell carcinoma. |
Acknowledgments
This work was supported by the National Natural Science Foundation of China [grant/award number 81772702] and partly by the Clinical Medical Research Talents Training Program of Army Medical University [grant/award number 2018XLC2017].
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Ljungberga B, Han YC, Jacqmin D, Lee JE, Weikert S, Kiemeney LA. The epidemiology of renal cell carcinoma. Eur Urol. 2011;60(4):615–621.
2. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi:10.3322/caac.v68.6
3. Inamura K. Renal cell tumors: understanding their molecular pathological epidemiology and the 2016 WHO classification. Int J Mol Sci. 2017;18(10):2195. doi:10.3390/ijms18102195
4. van der Veldt AAM, Meijerink MR, van den Eertwegh AJM, Boven E. Targeted therapies in renal cell cancer: recent developments in imaging. Target Oncol. 2010;5(2):95–112. doi:10.1007/s11523-010-0146-5
5. Hsieh JJ, Purdue MP, Signoretti S, et al. Renal cell carcinoma. Nat Rev Dis Primers. 2017;3:17009. doi:10.1038/nrdp.2017.9
6. Theodosiou A, Ashworth A. MAP kinase phosphatases. Genome Biol. 2002;3(7):
7. Patterson Kate I, Brummer T, O’Brien Philippa M, Daly Roger J. Dual-specificity phosphatases: critical regulators with diverse cellular targets. Biochem J. 2009;418(3):475–489. doi:10.1042/BJ20082234
8. Keyse SM. Dual-specificity MAP kinase phosphatases (MKPs) and cancer. Cancer Metastasis Rev. 2008;27(2):253–261. doi:10.1007/s10555-008-9123-1
9. Muda M, Boschert U, Smith A, et al. Molecular cloning and functional characterization of a novel mitogen-activated protein kinase phosphatase, MKP-4. J Biol Chem. 1997;272(8):5141–5151. doi:10.1074/jbc.272.8.5141
10. Wu F, Lv T, Chen G, et al. Epigenetic silencing of DUSP9 induces the proliferation of human gastric cancer by activating JNK signaling. Oncol Rep. 2015;34(1):121–128.
11. Lu H, Tran L, Park Y, et al. Reciprocal regulation of DUSP9 and DUSP16 expression by HIF1 controls ERK and p38 MAP kinase activity and mediates chemotherapy-induced breast cancer stem cell enrichment. Cancer Res. 2018;78(15):4191–4202. doi:10.1158/0008-5472.CAN-18-0270
12. Jenner S, Wiedorn KH, Techel D. Development of a DUSP9 methylation screening assay. Pathol Oncol Res. 2015;21(1):123–130. doi:10.1007/s12253-014-9797-3
13. Kidger AM, Keyse SM. The regulation of oncogenic Ras/ERK signalling by dual-specificity mitogen activated protein kinase phosphatases (MKPs). Semin Cell Dev Biol. 2016;50:125–132. doi:10.1016/j.semcdb.2016.01.009
14. Liu J, Ni W, Xiao M, Jiang F, Ni R. Decreased expression and prognostic role of mitogen-activated protein kinase phosphatase 4 in hepatocellular carcinoma. J Gastrointest Surg. 2013;17(4):756–765. doi:10.1007/s11605-013-2138-0
15. Liu Y, Lagowski J, Sundholm A, Sundberg A, Kulesz-Martin M. Microtubule disruption and tumor suppression by mitogen-activated protein kinase phosphatase 4. Cancer Res. 2007;67(22):10711–10719. doi:10.1158/0008-5472.CAN-07-1968
16. Iuv C, Kniazeva TG, Peter S, et al. [Molecular portrait of human kidney carcinomas: the gene expression profiling of protein-tyrosine kinases and tyrosine phosphatases which controlled regulatory signals in the cells]. Mol Biol (Mosk). 2002;36(3):480–490. doi:10.1023/A:1016059313254
17. Wu S, Wang Y, Sun L, et al. Decreased expression of dual-specificity phosphatase 9 is associated with poor prognosis in clear cell renal cell carcinoma. BMC Cancer. 2011;11:413. doi:10.1186/1471-2407-11-413
18. Chandrashekar DS, Bashel B, Balasubramanya SAH, et al. UALCAN: a portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia. 2017;19(8):649–658. doi:10.1016/j.neo.2017.05.002
19. Moch H, Cubilla AL, Humphrey PA, Reuter VE, Ulbright TM. The 2016 WHO classification of tumours of the urinary system and male genital organs-Part A: renal, penile, and testicular tumours. Eur Urol. 2016;70(1):106–119. doi:10.1016/j.eururo.2016.02.028
20. Hes O. [International Society of Urological Pathology (ISUP) Vancouver Classification of Renal Neoplasia 2012]. Cesk Patol. 2014;50(4):137–141. Czech.
21. Brodaczewska KK, Szczylik C, Fiedorowicz M, Porta C, Czarnecka AM. Choosing the right cell line for renal cell cancer research. Mol Cancer. 2016;15(1):83. doi:10.1186/s12943-016-0565-8
22. Hudes GR. Targeting mTOR in renal cell carcinoma. Cancer. 2010;115(S10):2313–2320. doi:10.1002/cncr.24239
23. Low HB, Zhang Y. Regulatory roles of MAPK phosphatases in cancer. Immune Netw. 2016;16(2):85–98. doi:10.4110/in.2016.16.2.85
24. Zhou L, Chen J, Li Z, et al. Integrated profiling of microRNAs and mRNAs: microRNAs located on Xq27.3 associate with clear cell renal cell carcinoma. PLoS One. 2010;5(12):e15224.
25. Dedobbeleer M, Willems E, Freeman S, Lombard A, Goffart N, Rogister B. Phosphatases and solid tumors: focus on glioblastoma initiation, progression and recurrences. Biochem J. 2017;474(17):2903–2924. doi:10.1042/BCJ20170112
26. Guo H, German P, Bai S, et al. The PI3K/AKT pathway and renal cell carcinoma. J Genet Genomics. 2015;42(7):343–353. doi:10.1016/j.jgg.2015.03.003
27. Perl A. mTOR activation is a biomarker and a central pathway to autoimmune disorders, cancer, obesity, and aging. Ann N Y Acad Sci. 2015;1346(1):33–44. doi:10.1111/nyas.12756
28. Mollica V, Di Nunno V, Gatto L, et al. Resistance to systemic agents in renal cell carcinoma predict and overcome genomic strategies adopted by tumor. Cancers (Basel). 2019;11(6):830. doi:10.3390/cancers11060830
29. Copp J, Manning G, Hunter TJ. TORC-specific phosphorylation of mTOR: phospho-Ser2481 is a marker for intact mTORC2. Cancer Res. 2009;69(5):1821.
30. Daniel S, Singer EA, Ramaprasad S. Molecular pathways in renal cell carcinoma: recent advances in genetics and molecular biology. Curr Opin Oncol. 2015;27(3):217–223.
31. Kruck S, Bedke J, Hennenlotter J, et al. Activation of mTOR in renal cell carcinoma is due to increased phosphorylation rather than protein overexpression. Oncol Rep. 2010;23(1):159–163.
32. Capitanio U, Montorsi F. Renal cancer. Lancet (London, England). 2016;387(10021):894–906. doi:10.1016/S0140-6736(15)00046-X
33. Duran I, Lambea J, Maroto P, et al. Resistance to targeted therapies in renal cancer: the importance of changing the mechanism of action. Target Oncol. 2016;12(1):19–35.
34. Elie BT, Hubbard K, Pechenyy Y, Layek B, Prabha S, Contel M. Preclinical evaluation of an unconventional ruthenium-gold-based chemotherapeutic: RANCE-1, in clear cell renal cell carcinoma. Cancer Med. 2019. doi:10.1002/cam4.2322
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.