Back to Journals » International Journal of General Medicine » Volume 19
Dual Regulation of Post-Translational Modification of Proteins: Bottlenecks and Breakthroughs in NK Cell Therapy for Glioblastoma
Authors Liu Y, Qiu Q, Deng H, Song P, Bu J, Zhang M
Received 21 November 2025
Accepted for publication 5 March 2026
Published 18 March 2026 Volume 2026:19 583369
DOI https://doi.org/10.2147/IJGM.S583369
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Kenneth Adler
Yushu Liu, Qingya Qiu, Hui Deng, Ping Song, Jiarui Bu, Mengxian Zhang
Department of Oncology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, 430030, People’s Republic of China
Correspondence: Mengxian Zhang, Email [email protected]
Abstract: Glioblastoma (GBM) is the most malignant primary central nervous system tumor in adults, with strong invasiveness, high recurrence, and poor prognosis. Natural killer (NK) cells, innate immune cells that eliminate glioma stem cells without MHC matching, show promise for GBM immunotherapy, but their efficacy is limited by GBM’s immunosuppressive tumor microenvironment (TME), especially via protein post-translational modifications (PTMs). This review summarizes seven key PTMs’ (phosphorylation, acetylation, glycosylation, methylation, ubiquitination, SUMOylation, lactylation) dual regulation on NK cell therapy: physiological PTMs enhance NK cytotoxicity, targeting, and persistence; aberrant PTMs block NK activation, induce exhaustion, and promote GBM immune escape. It also analyzes bottlenecks (insufficient NK activity/persistence, GBM’s PTM-mediated escape) and breakthroughs (PTM-targeted small molecules like TAK-981, CRISPR-edited NK cells, combination therapies). Future directions include BBB precision delivery, PTM-guided personalized therapy, and PTM crosstalk research, aiming to advance NK therapy’s clinical translation for GBM.
Keywords: glioblastoma, natural killer cells, post-translational modifications, immune escape, cancer immunotherapy, gene editing, combination therapy
Introduction
Glioblastoma (GBM) is the most common and highly malignant primary central nervous system tumor in adults, characterized by strong invasiveness, high recurrence, and extremely poor prognosis. Current standard treatment—surgical resection combined with temozolomide chemotherapy and radiotherapy—results in a median patient survival of <15 months and a 5-year survival rate of <5%.1–3 While high-dose ionizing radiation is the only well-established exogenous risk factor,4,5 other risks (eg, human cytomegalovirus infection, adolescent obesity, family cancer history) are still under investigation.6–8 Key biomarkers of GBM include EGFRvIII mutation (incidence ~30%), IDH1/2 mutation (~10%), and H3K27M mutation (incidence up to 80% in diffuse midline GBM), closely linked to aberrations in protein post-translational modifications (PTMs). IDH mutation leads to the accumulation of 2-hydroxyglutarate (2-HG), a metabolite associated with tumor epigenetic reprogramming; studies have shown that IDH-mutant glioma cells can downregulate NKG2D ligand expression on their surface, thereby evading NK cell immune surveillance.9 EGFRvIII overexpression, meanwhile, enables GBM to escape NK cell killing by regulating the expression of tumor cell surface immune molecules.10 GBM’s poor response to conventional therapies stems from its high heterogeneity, dense tumor structure, blood–brain barrier (BBB) obstruction, and immunosuppressive tumor microenvironment (TME),11 creating an urgent need for novel therapeutic strategies.
NK cells, as innate immune cells capable of recognizing and eliminating glioma stem cells (GSCs),12,13 hold substantial potential for GBM immunotherapy. Current immunotherapies such as chimeric antigen receptor (CAR)-T cell therapy encounter significant bottlenecks in GBM therapy, including high tumor heterogeneity, strong immunosuppression in the tumor microenvironment (TME), and limited blood–brain barrier (BBB) penetration. In contrast, NK cells do not require MHC matching and can kill infected or transformed cells via the perforin/granzyme pathway or death receptor pathway,14 and carry a low risk of inducing graft-versus-host disease (GVHD). These characteristics make NK cells uniquely suited to overcoming GBM immune tolerance. Moreover, GBM’s high invasiveness and stem-cell-like properties result in high recurrence rates after conventional radiotherapy and chemotherapy; the targeted cytotoxicity of NK cells against GSCs thus offers a novel strategy to tackle this issue.
Current NK cell-based core therapeutic strategies for GBM primarily encompass adoptive NK cell infusion, CAR-NK cell therapy, and cytokine-stimulated NK cell expansion: (1) Adoptive NK cell infusion: Autologous or allogeneic NK cells are expanded ex vivo with cytokines such as IL-2 and IL-18 to enhance their purity and cytotoxicity. Preclinical studies have shown that these expanded NK cells can cross the blood–brain barrier following intravenous infusion and significantly inhibit intracranial tumor growth in orthotopic GBM xenograft models.15 (2) CAR-NK cell therapy: NK cells are engineered to express chimeric antigen receptors targeting GBM-specific antigens (eg, EGFRvIII, HER2). Critical preclinical evidence demonstrates that EGFRvIII-CAR-NK cells specifically recognize and eliminate EGFRvIII-positive GBM cells, reduce tumor burden, and prolong survival in tumor-bearing mice.10 (3) Cytokine-stimulated NK cell expansion: Systemic administration of IL-2 or IL-15 boosts NK cell persistence and anti-tumor activity, but sustained stimulation may induce NK cell exhaustion—a constraint mitigable via PTM modulation. These strategies have been validated in preclinical research, underscoring the potential of NK cell therapy for GBM and the need for optimization through PTM targeting. Compared with CAR-T cell therapy, CAR-NK cell therapy holds broader clinical potential in solid tumors like GBM.10
Nevertheless, the therapeutic efficacy of NK cells is limited by multiple factors in the TME, among which PTMs act as a core regulatory mechanism.16,17 In contrast to strategies such as genetic modification (prone to off-target effects) and cytokine triggering (which may induce NK cell exhaustion), PTM modulation offers high precision, strong reversibility, and favorable safety. PTMs directly target the activity-regulating sites of key molecules involved in NK cell function, enhancing effector activity without altering the gene sequence; abnormal modifications can also be rapidly reversed using small-molecule inhibitors, thereby avoiding side effects associated with long-term interventions. Furthermore, PTM modulation exerts dual anti-tumor effects by acting simultaneously on NK cell activation and GBM immune escape, achieving superior synergy compared with single-target interventions. Through phosphorylation, acetylation, or glycosylation, PTMs dually regulate NK cell function and GBM immune evasion: physiological PTMs enhance NK cytotoxicity, targeting ability, and survival in the TME, while GBM-induced aberrant PTMs reduce GBM immunogenicity, block NK cell activation, or cause irreversible NK cell exhaustion—thus hindering therapeutic efficacy.
Despite progress in PTM-regulated NK therapy for GBM, challenges persist. In this review, we first elaborate on the biological implications of seven key PTMs, followed by an illustration of their dual regulatory mechanisms underlying NK cell-based therapy for GBM. We then analyze the current bottlenecks in therapeutic application and propose PTM-targeted strategies to surmount these limitations. Finally, we prospect three major future research directions (precise delivery, personalized therapy, PTM crosstalk) to provide a comprehensive reference for advancing clinical translation.
Biological Significance of PTMs of Proteins
PTMs are processes that alter protein structure and function via covalent attachment of chemical moieties.18 To date, over 450 PTM types have been identified. Different modifications modulate protein charge, hydrophobicity, conformation, and stability, thereby influencing cellular metabolism, signal transduction, and protein–protein interactions. This review focuses on seven of the most common PTMs (Figure 1).
 |
Figure 1 Schematic diagram of key molecules for seven common protein post-translational modifications regulating NK cell therapy for glioblastoma. This figure presents seven core types of protein post-translational modifications that regulate natural killer (NK) cell therapy for glioblastoma, along with their corresponding key regulatory/target molecules. The key molecules labeled for each modification are as follows: Phosphorylation: LCK, SHP-2; Acetylation: p50, HDAC1/2, ACAT1; Glycosylation: NKp30, CD16a, MGAT3/5; Methylation: METTL3, DNMT1; Ubiquitination: ITCH, CRLs, Rac1/RhoA; SUMOylation: UBC9, MEF, CRMP2; Lactylation: LDHA, GTPSCS, H3K18la. |
Phosphorylation
Phosphorylation is one of the most widely studied PTMs, and it affects protein activity and interactions by covalently adding phosphate group. It primarily occurs on serine, threonine, and tyrosine residues of target substrate proteins.19 The reversibility of phosphorylation is jointly regulated by kinases and phosphatases, such as LCK kinase mediating phosphorylation and SHP-2 phosphatase mediating dephosphorylation.20,21 Protein stability, interactions, subcellular localization, and enzymatic activity are determined by the specific substrate and phosphorylation site,22 while mutations in phosphoprotein sites can drive cancer initiation and progression by promoting tumor cell proliferation, invasion, metastasis, and inhibiting apoptosis.23,24
In summary, phosphorylation modulates the activity of key signaling molecules through site-specific modification, and its abnormal regulation is closely related to the dysfunction of NK cells and the immune escape of GBM. Targeting the kinases/phosphatases that regulate phosphorylation is an important direction to improve the efficacy of NK cell therapy.
Acetylation
Acetylation is the enzyme-catalyzed (eg, acetyltransferases) transfer of acetyl groups from acetyl-CoA to protein amino acid residues. Its main form—dynamic, reversible protein lysine acetylation—is regulated by lysine acetyltransferases (KATs) and lysine deacetylases (KDACs).25 KATs include the p300/CBP family, and KDACs are divided into classes I–IV;26 different subtypes have specific regulatory effects on NK cell function.27 Histone acetylation occurs on all core histones; H4K16ac (localized to gene promoters/enhancers), for instance, activates transcription. Reduced H4K16ac associates with cancers, cancer cell chemoresistance, and tumor progression.28,29 Non-histone lysine acetylation (eg, of oncoproteins) correlates with tumorigenesis.30,31
In summary, acetylation regulates gene transcription and protein activity through histone and non-histone modification, and the imbalance of acetylation homeostasis in the GBM microenvironment is a key factor inhibiting NK cell function. Regulating acetylation levels through KAT activators or HDAC inhibitors can effectively enhance the anti-tumor activity of NK cells.
Glycosylation
As another form of PTMs, glycosylation mainly affects protein stability and interactions. It is the process in which sugar chains attach to proteins and lipids under enzymatic control. Glycosyltransferases transfer sugar chains to proteins, forming glycosidic bonds with amino acid residues in proteins to produce glycoproteins.32 N-glycosylation mainly occurs in the Asn-X-Ser/Thr sequence, while O-glycosylation has no fixed sequence, and the two have different effects on the function of NK cell receptors.32 The role of glycosylation in tumor cells cannot be ignored; it participates in the progression of many malignant tumors, including proliferation, apoptosis, invasion, metastasis, and immune escape.33–36
In summary, glycosylation modulates the binding ability of NK cell receptors and the stability of effector molecules through sugar-chain modification, and abnormal glycosylation in the GBM microenvironment mediates the degradation of NK cell activation molecules and the escape of tumor cells. Targeting glycosylation modification enzymes or sugar-chain structures can effectively improve the recognition and killing ability of NK cells.
Methylation
Methylation is the methyltransferase-catalyzed transfer of active methyl groups to targets, without altering DNA sequence. It occurs in DNA,37 RNA,38 histones,39 and non-histones.40 In 1959, Richard P. Ambler found ε-N-methyllysine in flagellin hydrolysate—the first identification of protein methylation in living cells.41 Later, Kim and Paik showed methylated lysine cannot be coupled with tRNA, confirming histone methylation as a PTM.42 Lysine methylation includes monomethylation, dimethylation and trimethylation; histone H3K4me3 is associated with gene activation, and H3K27me3 is associated with gene silencing.43 Lysine methylation regulates gene expression (activation/inhibition) based on site/degree,44–47 affecting cancer. PRMT1 (catalyzing ~90% arginine methylation) activates oncogenic signaling via H4R3 dimethylation, promoting cancer.48
In summary, methylation regulates gene expression and protein function through DNA, RNA and protein modification, and the hypermethylation of NK cell activation-related genes in the GBM microenvironment leads to functional inhibition. Inhibiting methyltransferases or activating demethylases can reverse the epigenetic silencing of NK cells and restore their anti-tumor function.
Ubiquitination
Ubiquitination is the second most common PTM after phosphorylation, mainly involved in protein degradation.49 Ubiquitin (Ub) is a 76-amino-acid protein with seven lysine residues, each of which can be ubiquitinated to form unique polyubiquitin chains. It is catalyzed by three enzymes: E1 activating enzymes, E2 conjugating enzymes, and E3 ligases, with the latter determining substrate specificity. Polyubiquitin chains formed by Ub binding to target proteins are recognized by the 26S proteasome, leading to protein degradation.50 K48-linked ubiquitination mainly mediates protein degradation, and K63-linked ubiquitination is mainly involved in signal transduction.50 Ubiquitination plays a crucial regulatory role in tumors by affecting cell survival, proliferation, and differentiation. The regulation of protein degradation by ubiquitination provides a molecular basis for GBM to subsequently achieve immune evasion by ubiquitinating and degrading NKG2D ligands.
In summary, ubiquitination regulates the stability and signal transduction of proteins through different types of polyubiquitin chains, and abnormal ubiquitination in the GBM microenvironment leads to the degradation of NK cell functional molecules and the escape of tumor cells. Inhibiting the ubiquitin-proteasome pathway or specific E3 ligases can effectively maintain the function of NK cells and enhance their killing effect on GBM.
SUMOylation
SUMOylation is an enzymatic cascade where Small Ubiquitin-like Modifier (SUMO) proteins—structurally similar to ubiquitin—bind covalently/reversibly to target lysine residues, competing for substrates with ubiquitin. But SUMO differs in amino acid sequence/surface charge; hence, it has distinct functions:51 ubiquitination marks proteins for degradation, while SUMOylation stabilizes proteins and regulates their interactions/localization.52 It modulates processes like gene expression,53 DDR,54 nuclear-particle transport,55 cell cycle,56 and apoptosis.57 The mammalian SUMO family comprises four members: SUMO1, SUMO2, SUMO3, and SUMO4, with SUMO2 and SUMO3 being highly homologous. Their functions exhibit distinct preferences: SUMO1 typically mediates stable mono-SUMOylation, primarily regulating protein subcellular localization, interactions, and stability. In contrast, SUMO2/3 are more inclined to rapidly form poly-SUMO chains under cellular stress conditions such as DNA damage and heat shock, thereby modulating stress-responsive signaling pathways.58 Altered SUMO pathway components induce proliferation/resistance/metastasis via oncoproteins;59–63 dysregulated SUMOylation causes tumors, making SUMO a potential cancer target.64–68
In summary, SUMOylation regulates protein localization, stability and signal transduction through covalent modification, and abnormal SUMOylation in GBM inhibits the transcription of NK cell cytotoxic molecules and mediates the immune escape of tumor cells. Inhibiting the SUMOylation pathway can effectively restore the anti-tumor function of NK cells and is a potential target for GBM immunotherapy.
Lactylation
Lactylation regulates gene expression by adding lactyl groups to lysine residues (Kla), affecting cellular metabolism and functions. It modifies histones/non-histones and plays key roles in biological processes, especially tumorigenesis and progression.69 Lactylation is a key modification that links metabolic reprogramming in the tumor microenvironment to cellular functional regulation. Among these modifications, lactylation of histone H3 at lysine 18 (H3K18la) is one of the most well‑studied and central modifications.69 In tumors such as glioblastoma, lactate produced by enhanced glycolysis can be converted into lactyl‑CoA via enzymatic pathways involving nuclear GTPSCS, which in turn catalyzes H3K18la.70,71 This modification drives malignant processes such as metabolic reprogramming and immune evasion in tumors by regulating the expression of downstream genes.69,71 In the tumor microenvironment, lactate accumulates heavily, promoting tumorigenesis; local high lactate induces an acidic microenvironment that boosts immunosuppression and tumor invasion/metastasis.72 Its gene expression regulation here also impacts NK cell mitochondrial function, a core mechanism for GBM microenvironment inhibiting NK activity.73
In summary, lactylation is a metabolic PTM closely related to tumor glycolysis; the high lactate level in the GBM microenvironment induces abnormal lactylation of NK cells, leading to mitochondrial dysfunction and functional exhaustion. Inhibiting lactate production or lactylation modification can effectively restore the metabolic balance and anti-tumor function of NK cells.
Multi‑Omics Technologies for Identifying Clinically Relevant PTM Signatures in GBM
Recent advances in high‑throughput omics technologies have enabled systematic dissection of PTM landscapes in GBM and their association with clinical phenotypes, including prognosis, immune infiltration, and response to immunotherapy. Analogous to the multi‑omics framework applied to brain tumor‑related epilepsy,74 integrative omics approaches are now uncovering actionable PTM‑related biomarkers that can guide personalized NK cell‑based therapies.
Genomics and epigenomics have revealed that IDH1/2 mutations—present in ~10% of GBM—drive genome‑wide DNA methylation and histone methylation changes via the oncometabolite 2‑hydroxyglutarate (2‑HG).9,43 2‑HG accumulation correlates with downregulation of NKG2D ligands (ULBP1/3) and reduced NK cell cytotoxicity, providing a direct mechanistic link between a genomic alteration and PTM‑mediated immune evasion.9 Similarly, H3K27M mutations in diffuse midline gliomas (a GBM subtype) cause global loss of H3K27me3 and reciprocal gain of H3K27ac, which can be detected by ChIP‑seq and are associated with altered expression of immune ligands (eg, HLA‑E).73,75 DNA methylation profiling of GBM has identified MGMT promoter methylation as a standard predictive biomarker for temozolomide response; emerging data suggest that methylation status of NKG2D and PRF1 promoters in NK cells may influence their antitumor function, though this requires further validation in GBM cohorts.76,77
Transcriptomics (RNA‑seq) has been instrumental in defining GBM molecular subtypes (proneural, mesenchymal, classical) that exhibit distinct PTM enzyme expression profiles. For instance, mesenchymal GBM shows elevated HDAC1/2 and DNMT1 levels, which correlate with suppressed MICA/ULBP2 transcription and poorer survival (hazard ratio [HR] = 1.8, 95% CI 1.2–2.6).78,79 Single‑cell RNA‑seq of tumor‑infiltrating NK cells has revealed exhaustion trajectories characterized by downregulation of GZMB, IFNG, and KLF2 within 24 hours of TME entry, accompanied by upregulation of CISH and DUSP2.80,81 These transcriptional signatures can be used to construct NK cell dysfunction scores that predict response to adoptive NK therapy.81
Proteomics and PTM‑omics represent the most direct layer for quantifying PTM alterations. Mass spectrometry‑based phosphoproteomics has identified hyperphosphorylation of PLCγ and VAV1 in activated NK cells, while TGF‑β‑stimulated NK cells exhibit Smad2/3 phosphorylation correlating with reduced IFN‑γ production (r = –0.62, p < 0.01).13,82 Acetylome profiling of GBM tissues has shown global hypoacetylation of H3K9/27 in the tumor core, which is reversed by HDAC inhibitors.28,78 Lactylome analysis recently demonstrated that H3K18la enrichment at the promoters of IFNG and GZMB in NK cells is quantitatively correlated with lactate concentration in GBM interstitial fluid (R2 = 0.71) and with reduced intratumoral NK cell persistence.71,73 SUMOylome studies in glioma stem cells have identified UBC9‑mediated SUMO1 modification of PVR and CRMP2 as key events that impair NK cell recognition and cytotoxic granule transport.83–85
Metabolomics bridges metabolic reprogramming and PTM dynamics. Elevated lactate (10–20 mM) in the GBM microenvironment is not only a metabolic waste product but also a substrate for lysine lactylation, linking glycolytic activity (LDHA expression) to epigenetic silencing of NK effector genes.73,86 Oncometabolite 2‑HG, detected by LC‑MS, serves as a negative prognostic biomarker (median overall survival: 31 vs. 15 months for IDHwt) and simultaneously predicts resistance to NK cell killing via NKG2D ligand downregulation.9,79
Integration of these multi‑omics datasets enables the construction of PTM‑based risk scores and predictive models. For example, a four‑gene PTM enzyme signature (HDAC1, HDAC2, DNMT1, LDHA) stratifies GBM patients into high‑risk vs. low‑risk groups with significantly different overall survival (HR = 2.3, p < 0.001) and correlates with CD8+ T cell and NK cell infiltration scores derived from transcriptomic deconvolution algorithms.79 Such integrative approaches, as exemplified in brain tumor‑related epilepsy research,74 provide a roadmap for translating PTM discoveries into clinically actionable biomarkers for personalized NK cell immunotherapy.
The seven key PTMs form a “dual-regulation network” in GBM-NK cell crosstalk: physiological modifications (eg, phosphorylation of WASH, acetylation of p50) enhance NK cell cytotoxicity and metabolic adaptability, while aberrant modifications (eg, H3K18la, SUMOylation of MEF) drive GBM immune escape by silencing effector genes or inducing NK cell exhaustion. Targeting the regulatory enzymes of these PTMs (eg, HDAC, LDHA, SAE) is the core to breaking this imbalance. The following section will further elaborate on how this dual-regulation network specifically modulates NK cell therapy efficacy in GBM.
Dual Regulation of NK Cell Therapy for Glioblastoma by PTMs of Proteins
Promotive Effect
Phosphorylation
Phosphorylation modifications enhance NK cell activation signaling and effector molecule release. NK cells recognize MICA/B on the surface of GBM cells via NKG2D, activating signaling pathways and secreting perforin, granzyme B and other molecules.87 Specifically, LCK kinase mediates the phosphorylation of WASH protein at tyrosine 141 (Y141), and phosphorylated WASH activates the intracellular Arp2/3 complex, further promoting the rearrangement of cytoplasmic actin filaments (eg, the formation of actin-enriched regions at the immune synapse) to provide a “track” for lytic granule transport; the mutation of this site (Y141F) will completely block the activation of Arp2/3 complex, leading to the failure of NK cell lysosomal granule polarization.88 This process enhances NK cell cytoskeletal rearrangement and lysosomal granule polarization,88 facilitates effector molecule release, and improves the efficiency of GBM lysis.89 In ovarian and pancreatic cancer models, Luo et al90 found that Neo-2/15 exerts its function by activating the IL-2Rβγ signaling pathway: upon binding to its ligand, IL-2Rβγ activates intracellular JAK1/3 kinases, which induce the phosphorylation of STAT5 and Akt through phosphorylation modification, thereby promoting the expression of c-Myc and nuclear respiratory factor 1 (NRF1) in CAR-NK cells. Subsequently, c-Myc/NRF1 activates mitochondrial oxidative phosphorylation (OXPHOS) and the tricarboxylic acid (TCA) cycle, increasing ATP production, and ultimately enhancing the cytotoxicity and long-term survival of CAR-NK cells in the tumor microenvironment. This metabolic reprogramming mechanism provides a potential strategy for optimizing CAR-NK cells for GBM therapy (Figure 2).
 |
Figure 2 Key pathways of the promotive and inhibitory effects of phosphorylation modification on NK cell therapy for glioblastoma (Note: + indicates promotion, - indicates inhibition). This figure illustrates the core pathways of phosphorylation modification regulating natural killer (NK) cell therapy for glioblastoma (GBM): 1. Promotive effects: MICA/B→NKG2D→LCK→WASH→Arp2/3 complex→Lytic granule (+); Neo-2/15→IL-2Rβγ→JAK1/3→STAT5/Akt→c-Myc→OXPHOS/TCA cycle (+); 2. Inhibitory effects: GSCs→αv integrin→TGF-β→TGFBR2→Smad2/3→IFN-γ/perforin/granzyme B (-). |
In summary, physiological phosphorylation enhances the therapeutic effect by activating NK cell signaling pathways and enhancing metabolic adaptability, while abnormal phosphorylation inhibits NK cell function by blocking activation signals. Targeting phosphorylation regulatory enzymes (such as LCK kinase and TGF-β receptors) can become a key direction to enhance the efficacy of NK cell therapy.
Acetylation
Acetylation modification can maintain the functional homeostasis and targeted killing efficacy of NK cells. In terms of acetylation regulation, the acetylation homeostasis of NK cells themselves is critical for maintaining their GBM-killing function. In a colorectal cancer model, Wei et al91 found that under immune stimulation such as IL-18, ACAT1 in NK cells is phosphorylated at serine 60 (S60), and then ACAT1 translocates from mitochondria to the nucleus to exert acetyltransferase activity, directly acetylating NF-κB family member p50 at lysine 146 (K146). This modification impairs the DNA-binding ability and transcriptional repressive function of p50, thereby enhancing the cytotoxicity of NK cells. Exogenous acetate supplementation can rescue the acetylation deficiency caused by insufficient nuclear translocation of ACAT1 and further enhance NK cell cytotoxicity. Although this mechanism has not been directly verified in GBM, it suggests that regulating the acetylation homeostasis in NK cells may be a universal strategy to enhance their anti-tumor function, which warrants further investigation (Figure 3). In melanoma and leukemia models, Sohn et al92 demonstrated that exogenous acetate supplementation modulates NK cell histone acetylation via the ACLY pathway to enhance their effector function, and the role of this pathway in the regulation of GBM-associated NK cells remains to be experimentally verified. HDAC inhibitors can optimize therapeutic efficacy by blocking the deacetylation of key molecules: MS-275 (primarily developed for diffuse midline glioma (DMG) and equally effective for GBM-targeted NK cells) inhibits HDAC1/2, blocks the deacetylation of PLCγ downstream of NKG2D, enhances its binding ability with NKG2D downstream signal molecules, and thus amplifies the activation signal, ultimately enhancing the killing of H3K27M-mutant GSCs by NK cells;75 Entinostat maintains histone acetylation, stabilizes H3K27ac at the CAR gene promoter region, prevents CAR downregulation during CAR-NK cell expansion, and ensures targeting of EGFRvIII-positive GBM;93 combined use of pan-HDAC inhibitors with IL-2 inhibits HDAC1, relieves the suppression of the JAK2-STAT5B pathway, and promotes STAT5B acetylation to enhance NK cell proliferation.94
 |
Figure 3 Key pathways of the promotive and inhibitory effects of acetylation modification on NK cell therapy for glioblastoma (Note: + indicates promotion, - indicates inhibition). This figure illustrates the core molecular pathways of acetylation modification regulating natural killer (NK) cell therapy for glioblastoma (GBM): 1. Promotive effects: ACAT1→p50 (acetylation), HDAC1/2 inhibitors→PLCγ (acetylation maintenance), Entinostat→H3K27ac (stabilization at CAR gene promoter region), IL-2 + pan-HDAC inhibitors→JAK2-STAT5B (acetylation); 2. Inhibitory effects: TGF-β1→HDAC3→IFN-γ/perforin/granzyme B (transcriptional inhibition), acetyl-CoA deficiency→H3K9ac/H3K27ac (downregulation at NKG2D promoter region), OA→P300→c-Myc (acetylation inhibition), hypoxia→FASN→OA accumulation. |
In summary, physiological acetylation maintains the anti-tumor activity of NK cells by regulating gene transcription and protein activity, while abnormal deacetylation in the GBM microenvironment silences effector genes and inhibits NK cell function. Targeting HDAC with specific inhibitors can effectively restore the acetylation level of NK cells and enhance their killing effect on GBM.
Glycosylation
Glycosylation modification can optimize the function of NK cell receptors and the antibody-dependent cell-mediated cytotoxicity (ADCC) effect. Specific glycosylation modifications can enhance the targeted killing efficiency of NK cells against GBM cells by optimizing the function of NK cell activating receptors and the secretion of effector molecules. N-glycosylation of the NK cell activating receptor NKp30 is a core regulatory link: N-glycosylation at the Asn42 and Asn68 sites in its extracellular region mediates the formation of higher-order oligomers (eg, trimers, pentamers) of the receptor (only monomers remain after enzymatic deglycosylation). This enhances the binding affinity to B7-H6 on GBM cells; the glycan at the Asn42 site stabilizes the oligomeric state to strengthen activation signals, promoting the release of granzyme and perforin to kill GBM cells95 (Figure 4). Glycosylation of CD16a on the NK cell surface regulates ADCC activity: Kifunensine inhibits N-glycan processing, maintaining CD16a in the high-mannose type, which significantly enhances its binding affinity to therapeutic antibodies. The killing rate of primary NK cells against GBM cells increases, and ADCC activity is further enhanced by 33% when combined with afucosylated antibodies.96 In addition, fluorescein-labeled sialic acid (FL-SA) is integrated into NK cell glycans via the high sialylation pathway of GBM; the constructed anti-fluorescein CAR-NK cells enhance cytotoxicity against GBM and promote the secretion of IFN-γ and TNF-α.97 N-acetyl-D-glucosamine-calix[4]arene (GN4C) downregulates MGAT3/5 in NK cells to reduce abnormal glycans, thereby upregulating NKG2D expression and promoting IL-2 secretion to maintain proliferation—strengthening killing effects. This process depends on PI3K/ERK signaling.98
 |
Figure 4 Key pathways of the promotive and inhibitory effects of glycosylation modification on NK cell therapy for glioblastoma (Note: + indicates promotion, - indicates inhibition). This figure illustrates the core pathways of glycosylation modification regulating natural killer (NK) cell therapy for glioblastoma (GBM): 1. Promotive effects: NKp30→B7-H6→granzyme/perforin (+); 2. Inhibitory effects: SLAMF7→TM4SF5→Lysosome→Degradation (-). |
In summary, physiological glycosylation enhances the recognition and killing ability of NK cells by optimizing the structure and function of receptors and effector molecules, while abnormal glycosylation in the GBM microenvironment mediates the degradation of NK cell activation molecules and the escape of tumor cells. Regulating glycosylation modification through specific activators or inhibitors can effectively improve the anti-tumor efficacy of NK cells.
Methylation
Methylation modifications ensure NK cell survival, proliferation and ligand recognition efficiency. In terms of methylation regulation, multiple key mechanisms maintain NK cell function: extracellular IL-15 binds to the IL-15R (CD122/CD132) on the NK cell membrane to initiate downstream signaling; METTL3 mediates the m6A modification of Ptpn11 mRNA, promoting its translation into SHP-2 protein; enhanced SHP-2 activity further induces the phosphorylation and activation of AKT/MAPK, maintaining NK cell survival and proliferation, and laying a foundation for their anti-tumor function99 (Figure 5); Bugide et al found that the histone methyltransferase EZH2 inhibits the transcription of NKG2D ligands, and EZH2 inhibition upregulates ligand expression to enhance the clearance of cancer cells by NK cells;100 in H3K27M-mutant GBM, highly expressed LSD1 inhibits NKG2D ligands by maintaining low levels of H3K4me1/me2, and treatment with the LSD1 inhibitor 2-PCPA upregulates MICA expression, enhances the killing of GSCs by NK cells, and further prolongs the survival of xenograft mice when combined with NK cell infusion;101 studies have shown that the DNA damage response pathway (eg, cGAS-STING) can upregulate the expression of NKG2D ligands such as MICA on the surface of tumor cells,102 suggesting that radiotherapy may sensitize GBM cells to NK cell killing through this pathway, but the specific molecular mechanism and data on the improvement of clearance efficiency in GBM still need to be verified by original research.
 |
Figure 5 Key pathways of the promotive and inhibitory effects of methylation modification on NK cell therapy for glioblastoma (Note: + indicates promotion, - indicates inhibition). This figure illustrates the core pathways of methylation modification regulating natural killer (NK) cell therapy for glioblastoma (GBM): 1. Promotive effects: IL-15→CD122/CD132→METTL3→Ptpn11 mRNA→SHP-2→AKT/MAPK (+); 2. Inhibitory effects: 2-HG→DNMT→NKG2D promoter (-). |
In summary, physiological methylation maintains the survival, proliferation and ligand recognition ability of NK cells by regulating RNA and protein modification, while abnormal methylation in the GBM microenvironment silences NK cell activation-related genes and inhibits their function. Inhibiting methyltransferases or activating demethylases can reverse epigenetic silencing and restore the anti-tumor function of NK cells.
This section focuses on the promotive effects of PTMs on NK cell-based therapy for GBM. As PTMs that exert an inhibitory effect on therapy, the mechanisms of action of ubiquitination, SUMOylation, and lactylation will be elaborated in detail in subsequent relevant sections.
Inhibitory Effect
Phosphorylation and Ubiquitination Cooperatively Block Activation Pathways
Effective NK cell activation relies on a homeostatic balance where “activation signals outweigh inhibitory signals”, and abnormal PTMs can directly disrupt this balance. During phosphorylation modification, αv integrin of GSCs assists in the secretion of TGF-β into the extracellular space, which binds to TGFBR2 of NK cells, induces the phosphorylation of Smad2/3, thereby inhibiting the production of IFN-γ and the synthesis and release of perforin/granzyme B, and ultimately leading to NK cell functional inactivation13 (Figure 2). From the perspective of signal transduction rules, Smad2/3 phosphorylation is not only the initial step of the classical Smad pathway but also regulates NK cell IFN-γ production, cytotoxicity and tissue-resident marker expression through Smad4-dependent or independent pathways,82,103 indicating that dysregulated phosphorylation exerts multi-target inhibitory effects on activation signals.
At the level of ubiquitination regulation, clinical studies have shown that in patients with B-cell lymphoma, the level of CBLB ubiquitination in peripheral blood mononuclear cells (PBMCs) is negatively correlated with IFN-γ secretion and ADCC effect of NK cells.104 Although this study did not involve GBM, it suggests that CBLB ubiquitination may inhibit the activation of downstream pathways by regulating key activation signaling molecules such as PI3K and SOS1 in NK cells, and the role of this mechanism in the regulation of GBM-associated NK cell function warrants further investigation. In addition, ubiquitination can further enhance NK cell cytotoxicity by maintaining the stability of degranulation-related molecules such as granzyme B and perforin, providing a mechanistic basis for potentiating NK cell therapy for GBM.105 In a multiple myeloma model, Petillo et al105 found that inflammatory stress in the tumor microenvironment activates ubiquitin ligases such as Cullin-Ring ligases (CRLs), which degrade GTPases such as Rac1 and RhoA in NK cells through K48-linked ubiquitination—these molecules are crucial for the formation of immune synapses between NK cells and tumor cells and perforin polarization; the mutation of this ubiquitination site (K48R) can significantly improve the immune synapse formation disorder of NK cells. Gene expression profiling of intracranial tumors from orthotopic GBM xenografts revealed that NKC-treated tumors exhibit downregulated expression of CD70 (fold change: −5.64), a ligand of the NK cell activating receptor CD27, suggesting that GBM cells may evade NK cell surveillance by reducing the expression of activating receptor ligands.15 Conversely, the upregulation of CDH2 (fold change: 2.1), a ligand of the NK cell inhibitory receptor KLRG1, in NKC-treated tumors indicates an additional immune escape mechanism mediated by enhanced inhibitory signaling.15 CRLs-mediated degradation of Rac1 and RhoA leads to abnormal F-actin polymerization in NK cells, a more than 50% reduction in the localization rate of perforin to the immune synapse, and a significant decrease in degranulation efficiency, ultimately resulting in the loss of killing function. Whether this mechanism is applicable to GBM remains to be further verified. Ubiquitination mediated by inhibitory receptors can block NK cell activation signals: In T cells, the inhibitory receptor TIGIT recruits the E3 ubiquitin ligase ITCH upon ligand binding, and the classic inhibitory mechanism of suppressing calcium influx and the NF-κB pathway via K63-linked ubiquitination of key signaling molecules such as LAT/PLCγ has been confirmed;106 it is speculated that this pathway may exert a similar activation-inhibitory effect in NK cells, but the specific regulatory effect in the GBM-NK cell interaction remains to be experimentally elucidated (Figure 6).
 |
Figure 6 Key pathways of the inhibitory effects of ubiquitination, SUMOylation, and lactylation on NK cell therapy for glioblastoma (Note: - indicates inhibition). This figure illustrates the core inhibitory pathways of three modifications regulating natural killer (NK) cell therapy for glioblastoma (GBM): 1. Ubiquitination: TIGIT→ITCH→LAT/PLCγ→NF-κB/Ca2⁺ (-); 2. SUMOylation: MEF→Perforin (-); 3. Lactylation: LDHA→Lactate→H3K18la/Mitochondrial fragmentation (-). |
In summary, abnormal phosphorylation and ubiquitination in the GBM microenvironment cooperatively block the activation pathway of NK cells: phosphorylation inhibits the secretion of effector molecules by activating the TGF-β/Smad pathway, and ubiquitination mediates the degradation of key functional molecules of NK cells through K48-linked ubiquitination. Targeting the key molecules of these two modifications can effectively restore the activation signal of NK cells.
Acetylation and Methylation Cooperatively Silence Functional Genes
GBM cells escape NK cell killing through their own PTMs, and their tumor microenvironment (TME) disrupts NK cell acetylation homeostasis through a “nutrient deprivation-metabolic inhibition-immune interference” axis: the Warburg effect leads to glucose depletion and lactate production,92 reducing mitochondrial citrate levels and causing ACLY-mediated acetyl-CoA deficiency. Research in other cancer models (eg, melanoma) has shown that such ACLY-mediated acetyl-CoA deficiency can lead to a significant reduction in H3K9ac/H3K27ac levels at the NKG2D promoter (eg, by more than 50%),92 suggesting a potential conserved mechanism that may also impair NK cell activation in the GBM microenvironment; lactate oxidizes ACAT1, reducing nuclear p50 acetylation and 60% of CCL5/CXCL10, and impairing NK cell recruitment;91 M2-type tumor-associated macrophages secrete TGF-β1 to form an immunosuppressive microenvironment, and TGF-β1 binds to receptors on the NK cell surface, activates intracellular signals, upregulates HDAC3 expression and promotes its translocation to the nucleus. HDAC3 removes histone acetylation, inhibits the transcription of genes such as IFN-γ, reduces the synthesis of perforin/granzyme B, and decreases the GBM killing rate75 (Figure 3); GSCs enhance histone Kcr (with no change in Kac);107 oleic acid (OA) inhibits P300-mediated c-Myc acetylation and reduces IFN-γ;108 hypoxia upregulates FASN to promote OA accumulation, forming a “hypoxia-OA-PTMs disorder” cycle.102,108
Abnormal epigenetic signals in the GBM microenvironment can induce hypermethylation of NK cell activation-related genes, leading to their functional inhibition, which is an important mechanism of GBM immune escape. Existing studies have shown that IDH-mutant glioma cells can escape NK cell killing by downregulating their own NKG2D ligands (eg, ULBP3);9 meanwhile, in human CD8+ T cells and NK cells, the transcriptional activation of the NKG2D gene depends on the regulation of DNA demethylation and histone acetylation.76 It is speculated that in the GBM microenvironment, IDH mutations may indirectly affect the epigenetic state of NK cells through the metabolite 2-HG, thereby regulating NKG2D expression, but this causal relationship has not been directly verified by experiments and remains to be further investigated (Figure 5). In addition to NKG2D, abnormal methylation of genes encoding other activation receptors of NK cells (eg, DNAM1) may further inhibit their function. Although direct evidence is currently limited, referring to the regulatory mode of NKG2D, methylation signals in the GBM microenvironment may extensively silence NK cell activation-related genes, block their activation at the recognition stage, and ultimately significantly impair the killing ability of NK cells against GBM cells, thus supporting GBM immune escape.76,77
In summary, abnormal acetylation and methylation in the GBM microenvironment cooperatively silence the functional genes of NK cells: acetylation imbalance leads to the downregulation of effector gene transcription by reducing histone acetylation levels, and hypermethylation leads to the silencing of activation-related genes by modifying the promoter region of genes. Regulating the acetylation and methylation levels of NK cells can effectively restore the expression of functional genes and enhance their anti-tumor function.
Glycosylation and SUMOylation Interfere with Recognition and Effector Execution
Abnormal glycosylation in the GBM microenvironment inhibits NK cell function and promotes immune escape by degrading NK cell activation molecules and interfering with signal transduction. In a hepatocellular carcinoma model, Kim et al77 found that glycosylation at a specific site in the extracellular region of SLAMF7 on the NK cell surface enables its binding to TM4SF5 of tumor cells, thereby mediating the transport of SLAMF7 to lysosomes for degradation, leading to decreased expression of perforin and granzyme B in NK cells and a reduction in killing rate. Although this mechanism is derived from liver cancer research, it suggests that a similar glycosylation-mediated degradation mechanism of NK cell activation receptors may exist in GBM, which needs experimental verification (Figure 4). Methylglyoxal (MGO) and glyoxal (GO) in GBM induce non-enzymatic glycosylation of NK cell proteins to form advanced glycation end products (AGEs), among which MGO exerts a stronger inhibitory effect: 0.6 mM MGO reduces the binding rate of NK cells to target cells by 40%, and 1–2 mM MGO decreases the killing rate by more than 85% and metabolic activity by 40%, and AGEs may modify NK cell receptors or effector molecules.109 In addition, high fucosylation of the antibody Fc segment impairs the binding ability of NK cells to FcγRIIIa and reduces ADCC activity; the synergistic effect of afucosylated antibodies may be reversely inhibited by abnormal glycosylation of NK cells in the GBM microenvironment.110 Excessive glycosylation of NKp30 alters the residue environment near the N162 glycan chain, interferes with its binding to B7-H6 and impairs activation signals, and this abnormality is more likely to occur under inflammatory stress in GBM.95
Ubiquitin-like modifications can interfere with the efficiency of NK cell target recognition. In a multiple myeloma model, Zitti et al111 found that ubiquitin-like modifications retain PVR, the ligand of the NK cell activation receptor DNAM1, inside tumor cells, inhibit PVR surface expression, and reduce NK cell recognition. It is speculated that GBM cells may adopt a similar regulatory mode to affect the localization and exposure of PVR on the cell surface, thereby interfering with NK cell recognition, but direct evidence of this mechanism in GBM is lacking.112 On the other hand, ubiquitin-like modifications can directly inhibit the production of NK cell cytotoxic molecules: Basic research has shown that the transcription factor MEF can be inactivated by ubiquitin-like modifications,80 suggesting that ubiquitin-like modifications may indirectly affect the expression of cytotoxic molecules including perforin, but the specific role and regulatory mechanism of this pathway in NK cells remain to be confirmed (Figure 6).
In summary, abnormal glycosylation and SUMOylation in the GBM microenvironment interfere with the recognition and effector execution of NK cells: glycosylation mediates the degradation of NK cell activation receptors and the modification of effector molecules, and SUMOylation inhibits the surface expression of tumor cell ligands and the transcription of NK cell cytotoxic molecules. Targeting these two modifications can effectively improve the recognition and killing ability of NK cells against GBM.
Lactylation-Dominated Synergistic Inhibition by Multiple Metabolic Molecules
In addition, metabolic interference of PTMs such as lactylation exerts an auxiliary inhibitory effect on NK cell therapy. Metabolic abnormalities in GBM induce abnormal PTMs in NK cells and disrupt their energy balance, among which lactylation has the most significant impact: metabolic abnormalities in GBM enhance glycolysis, and lactate dehydrogenase A (LDHA) mediates lactate production and secretion into the extracellular space, and the increased extracellular lactate concentration forms a metabolically inhibitory microenvironment; multiple studies have confirmed that lactate can inhibit NK cell activity through a dual mechanism of “lactate accumulation + glucose deprivation”, downregulate NKp46/CD107a on the NK cell surface, and reduce the synthesis and release of perforin and granzyme B;86,113 meanwhile, lactate accumulation can disrupt the metabolic balance of NK cells and may impair their mitochondrial function, thereby further inhibiting NK cell activity86 (Figure 6). A recent study73 further clarified the key mechanism of lactylation-induced NK cell dysfunction: lactate-induced lysine lactylation (Kla) specifically modifies ROCK1 at the Lys13 site, promoting DRP1 Ser616 phosphorylation and subsequent mitochondrial fragmentation, which directly impairs NK cell cytotoxicity. Notably, the combination of nicotinamide riboside (NR, a NAD+ precursor) and honokiol (HKL, a SIRT3 activator) synergistically activates SIRT3-mediated delactylation of ROCK1, thereby inhibiting ROCK1-DRP1 signaling. In vitro experiments, this combination restored CD107a+ degranulation rate and granzyme B secretion in lactate-treated NK cells by ~60%, and significantly enhanced their killing efficiency against AML blast cells and GBM-related tumor cell lines (K562, HL60).73 In vivo leukemia xenograft models, adoptive transfer of NR+HKL-pretreated NK cells reduced tumor burden by ~45% and prolonged mouse survival by ~30% compared to untreated NK cells, validating the therapeutic potential of this lactylation-targeted strategy.73 Other metabolic abnormalities in GBM also interfere with NK cell function through PTMs: GBM cells highly express CD39/CD73, which can catalyze the production of a large amount of adenosine. Adenosine binds to ADORA2A on the NK cell surface to activate the cAMP-PKA pathway, thereby inhibiting IFN-γ secretion and perforin expression,114 and the negative correlation between adenosine levels and the expression of NK cell functional markers has been clinically confirmed in a variety of solid tumors; high expression of cyclooxygenase 2 (COX-2) produces prostaglandin E2 (PGE2), which downregulates NK cell NKG2D via EP2/EP4 and induces myeloid-derived suppressor cells (MDSCs) to produce TGF-β; COX-2 inhibitors (eg, celecoxib) can restore NKG2D expression and enhance killing efficiency;114 in addition to acetylation regulation, OA induces mitochondrial fragmentation in NK cells, impairs oxidative phosphorylation, and forms a metabolic PTMs inhibitory network with lactate.108
In summary, lactylation dominated by metabolic abnormalities in the GBM microenvironment and the synergistic inhibition of multiple metabolic molecules lead to NK cell metabolic imbalance and functional exhaustion: lactylation induces mitochondrial dysfunction and inhibits the secretion of effector molecules, and other metabolic molecules further aggravate the immunosuppressive microenvironment. Targeting lactate production or lactylation modification is an effective strategy to restore the anti-tumor function of NK cells.
Collectively, the dual regulation of PTMs on NK cell therapy is characterized by “synergistic promotion of physiological modifications and coordinated inhibition of aberrant modifications”: phosphorylation/acetylation/glycosylation/methylation jointly enhance NK cell activation and targeting, while ubiquitination/SUMOylation/lactylation cooperate to block killing function and promote GBM escape. Our insight is that this “multi-PTM crosstalk” is a key unmet target for improving therapy efficacy. Next, we will analyze the current bottlenecks of NK cell therapy in GBM, which are directly driven by abnormal PTMs (Table 1).
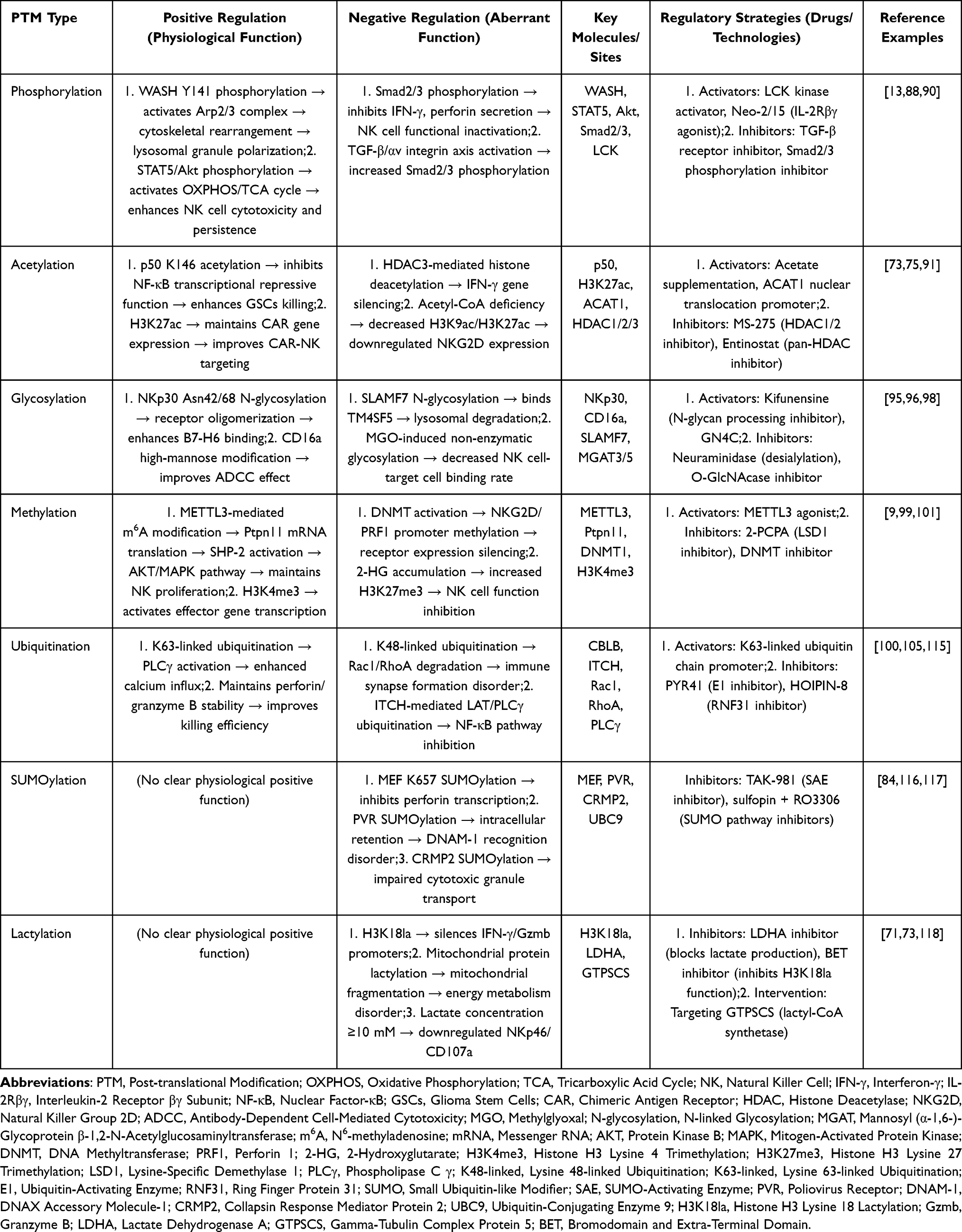 |
Table 1 PTM Dual Effects and Regulatory Strategies |
Current Bottlenecks in NK Cell Therapy
Challenges in NK Cell Activity and Persistence
Blockade of Activation Signals
In the dual regulation of GBM NK cell therapy by PTMs, low NK cell activation efficiency is the primary bottleneck. The GBM TME blocks the “resting-to-activated state” transition of NK cells through a variety of abnormal PTMs, making it difficult for in vitro prepared NK cells to initiate the killing program after infusion, resulting in the dilemma of “high infusion dose but low in vivo efficacy”. First, TGF-β-αv integrin axis-mediated phosphorylation dysregulation: GSCs contact NK cells through surface αv integrin, activate the TGF-β pathway, induce the phosphorylation of Smad2/3 in NK cells, block the synthesis of IFN-γ and the secretion signals of granzyme B and perforin, leading to NK cell “functional inactivation”.13 A study of 21 GBM patients showed that the mean fluorescence intensity (MFI) of phosphorylated Smad2/3 (p-Smad2/3) in tumor-infiltrating NK cells (TI-NK) was higher than that in peripheral blood NK cells (PB-NK), while the expression of activation markers such as NKG2D, CD16 and NKp30 was downregulated.13 Second, H3K27M mutation-related ubiquitination defects exacerbate the activation dilemma: in H3K27M-mutant GBM, homologous recombination repair defects may affect the expression of immune stimulatory molecules after damage, and although olaparib combined with radiotherapy can partially restore the expression of NK activation ligands (eg, MICA/B),13,119 its induction efficiency is still lower than that in wild-type GBM. In addition, the imbalance between inhibitory and activation receptor expression impairs activation potential: an analysis of tumor-infiltrating lymphocyte-derived NK cells (TIL-NK) from 60 patients showed that the expression rates of inhibitory receptors KIR2DL-1 and −2/3 were higher than those in healthy people, while the expression levels of activation receptors such as CD335, CD226 and CD159c were lower than those in normal people.120
Irreversible Functional Exhaustion
Even if a small number of NK cells overcome activation barriers to initiate the killing program, their function will decline rapidly and irreversibly in the GBM TME. This “irreversible exhaustion” directly limits the long-term efficacy of NK cell therapy, and the core mechanism is the dual regulation of “epigenetic silencing + microenvironmental inhibition” mediated by abnormal PTMs. Epigenetic silencing exhibits a synergistic effect of molecular networks: DNMT1 binds to BCL11B to maintain the DNA methylation of NK cell effector genes (Prf1, Gzmb, IFN-γ), while H3K27me3 directly inhibits the promoters of these genes, jointly silencing the transcription of cytotoxic molecules;77 GTPSCS localized in the GBM nucleus catalyzes the production of lactyl-CoA, promoting histone H3 lysine 18 lactylation (H3K18la); H3K18la enriches at the promoters of IFN-γ and Gzmb genes in NK cells, and forms a “DNA methylation-histone inhibition-histone lactylation” triple silencing network together with the DNMT1-H3K27me3 pathway.71 Time-resolved technology shows that NK cells exhibit downregulation of cytotoxic markers (NKG2D, CD107a) 6–12 hours after entering the TME, and fully enter the TGFB1-regulated functional dysfunction program within 24 hours, accompanied by a 2.8-fold increase in SMAD3 activity, a decrease in SMAD4 activity, and recruitment of HDACs to enhance the inhibition of NK cell activation.121 Notably, cytokine selection significantly impacts NK cell exhaustion: IL-15-engineered NK cells rapidly lose cytotoxicity after repeated GBM cell rechallenges, associated with upregulation of exhaustion-related genes (CISH, DUSP2);81 in contrast, IL-21-engineered NK cells maintain long-term function by avoiding exhaustion, which is mediated by CEBPD-induced metabolic reprogramming (enhanced oxidative phosphorylation) and epigenetic activation of effector gene expression.81 Different molecular subtypes of GBM can shape unique immunosuppressive microenvironments, and mesenchymal subtype GBM (MES GBM) has been confirmed to further impair the anti-tumor function of NK cells by regulating the polarization state of tumor-associated macrophages; GBM with high ALDH1A3 expression enhances glycolysis by binding to PKM2,118 which not only competes for glucose to cause NK cell metabolic imbalance but also induces lysine lactylation of mitochondrial respiratory chain proteins and glycolytic enzymes in NK cells, disrupting energy metabolism;118 in addition, lactate activates the GTPSCS-H3K18la pathway to drive high GDF15 expression, which inhibits NK cell degranulation and IFN-γ secretion through paracrine, and forms a triple inhibitory network together with tumor-associated macrophages and lactate metabolic stress, making it difficult for NK cells to recover their function even after leaving this microenvironment.71,121 In summary, irreversible exhaustion is the result of multi-dimensional synergistic effects, suggesting that targeting metabolic PTMs or blocking the TGFB1-SMAD3 signal may reverse the exhausted state.
Loss of Metabolic Adaptability
The extreme metabolic environment of the GBM TME causes concentration-dependent metabolic damage to NK cells through abnormal PTMs, making them unable to adapt to the glucose-deficient and high-lactate environment and shortening their intratumoral survival time; NK cells retain partial adaptability at low concentrations.122 Hypoxia is a key feature of the GBM microenvironment, and hypoxia and lactate accumulation can jointly disrupt the energy metabolic homeostasis of NK cells, inhibit the expression of mitochondrial respiratory chain-related genes through regulating the HIF1α-related signaling pathway, and simultaneously destroy the NAD+ homeostasis of NK cells, leading to mitochondrial dysfunction and impaired oxidative phosphorylation, which makes it difficult to meet the energy demand of NK cells even when glucose is sufficient.73,86 Epigenetic-metabolic synergy further blocks compensation: the expression of DNMT/HDAC is abnormally increased in GBM, and lactate-induced H3K18la may enhance their binding ability; DNMT1 binds to BCL11B to maintain the DNA methylation of NK cell glycolytic enzymes (HK2, PFK1), while HDAC inhibits their transcription, jointly blocking glycolytic compensation;123 DNMT/HDAC dual inhibitors can reverse metabolism-related abnormal PTMs, improve glycolysis, and prolong the survival time of NK cells in the simulated tumor microenvironment.124 The loss of metabolic adaptability occurs synchronously with functional inhibition: Zman-seq shows that NK cells transform from a cytotoxic state to a dysfunctional state within 24 hours after infiltrating the TME;121 in hypoxia experiments, the mitochondrial fragmentation rate of NK cells increases significantly within 24 hours, glycolysis efficiency decreases, the expression of the survival protein BCL-2 is reduced, and the intratumoral survival time is shortened.122
The Tumor Immune Escape Dilemma
GBM constructs a multi-dimensional regulatory network using PTMs to achieve immune escape through two core links: “impaired recognition of GBM by NK cells” and “blocked killing after recognition”, thereby weakening the anti-tumor effect of NK cells.
At the level of NK cell recognition, GBM reduces the recognition of immunogenic molecules through PTMs: abnormally activated TGF-β recruits HDAC1/2 and DNMT1 through the SMAD signal, and dual modification of the MICA/ULBP2 promoter inhibits its mRNA expression;125 TCGA data show that the expression of HDAC1/2 in GBM is higher than that in normal brain tissue (negatively correlated with MICA/ULBP2 transcription), and siRNA knockdown of HDAC1/2 can restore ligand expression;78 with the increase of GBM malignancy, TGF-β levels increase, while the surface positive rate of MICA/ULBP2 decreases;125 GBM highly expresses matrix metalloproteinases (MMP)/a disintegrin and metalloproteinase (ADAM), which cleave MICA/ULBP2, and the released soluble MICA (sMICA)/soluble ULBP2 (sULBP2) competes with NK cell NKG2D for binding, reducing the positive rate of NKG2D in patients;125 CD155 highly expressed in 80% of GBM has abnormal localization regulated by PTMs, impairing DNAM-1 recognition;83 HDAC also alters the expression of immune molecules through histone acetylation PTMs.116
At the level of NK cell killing inhibition, GBM is centered on ubiquitin-like modifications: the ubiquitin-like modification pathway is abnormally activated (miR-214 downregulation, PIAS3/UBA2 upregulation, USP34 stabilization of Pin1), resulting in a 40–60% increase in SUMO1 modification in GBM;84,126,127 GBM reduces its surface expression through self-PVR ubiquitin-like modification,84,111 and simultaneously targets NK cell effector molecules through ubiquitin-like modifications (CRMP2: impairing killing granule transport; ROCK1: inducing mitochondrial fragmentation in synergy with lactylation);73,85,117 TAK-981 and sulfopin + RO3306 can block ubiquitin-like modifications and restore killing function.84,117 PreOperative RT further blocks GBM immune escape at the killing stage by reducing TAM/microglia recruitment (lower Iba-1 expression) and inhibiting the senescence-associated secretory phenotype (SASP).128 PostOperative RT induces higher levels of tumor cell senescence (increased p21 expression) and SASP-related cytokines (eg, IL-6), which promote immune suppression and tumor recurrence.128 In contrast, PreOperative RT removes senescent cells during surgical resection, reducing SASP-mediated inhibition of NK cell killing function and enhancing the persistence of NK cell efficacy.128
The bottlenecks of NK cell therapy for GBM lie in NK cell functional defects and tumor immune escape: Abnormal PTMs in the TME impair NK cell activity and persistence by blocking activation signals, inducing irreversible exhaustion, and disrupting metabolic adaptability. Meanwhile, GBM reduces immunogenicity and interferes with recognition/killing via PTM regulation, forming an escape network. These multi-dimensional barriers interact synergistically to limit therapeutic efficacy, demanding targeted strategies for resolution.
Core Breakthrough Strategies Based on PTM Regulation
Enhancing NK Cell Activity and Persistence
Targeted Drugs
Small-molecule drugs targeting PTM regulatory enzymes can enhance NK cell activity and in vivo persistence through multiple pathways.117,129 The ubiquitin-like activating enzyme (SAE) inhibitor TAK-981 binds irreversibly to the SAE-SUMO complex, blocking the transfer of SUMO to the E2 enzyme UBC9: it relieves the ubiquitin-like modification-mediated inhibition of NK cell MEF protein at lysine 657, restoring the expression of perforin and granzyme B to 1.6 times the normal level; it also activates the type I interferon (IFN1) signal to upregulate NK cell IFNAR1. Preclinical studies have shown that TAK-981 can increase the number of NK cells, improve the CD107a positive rate and IFN-γ levels in colon cancer, breast cancer and lymphoma models, and prolong the median survival of tumor-bearing mice when combined with immune checkpoint inhibitors.117 Although there are currently no test data of TAK-981 in GBM models, its mechanism of action provides a new idea for overcoming GBM immunosuppression. Histone deacetylase (HDAC) inhibitors exhibit prominent potential in enhancing NK cell-mediated anti-GBM effects: class I HDAC inhibitor MS-275, which can cross the blood–brain barrier and is well-tolerated, not only upregulates the expression of NK cell-activating ligands (MICA/B, ULBP1-3) in diffuse midline glioma (DMG, a high-grade GBM subtype) cells but also induces the expression of the inhibitory ligand HLA-E.75 Mechanistically, MS-275 promotes STAT3 acetylation at lysine 685, which enhances the transcriptional activity of unphosphorylated STAT3 and directly drives HLA-E expression.75 Importantly, blocking the HLA-E-NKG2A inhibitory axis with monalizumab can further amplify the cytotoxicity of NK cells against MS-275-pretreated GBM/DMG cells; preclinical studies in orthotopic DMG mouse models have confirmed that the combination of MS-275, NK cell infusion, and NKG2A blockade achieves significant tumor growth inhibition and remarkably prolongs animal survival compared with single-agent or dual combination therapies.75 Notably, the cytokine IL-21, when genetically engineered into NK cells for autonomous secretion, demonstrates superior anti-GBM potential compared to IL-15, as it avoids neurotoxicity while enhancing NK cell polyfunctionality (simultaneous secretion of multiple effector cytokines) and metabolic fitness.81 IL-21-driven STAT3 activation is a key upstream signal for CEBPD-mediated NK cell functional enhancement, and small-molecule modulators targeting the IL-21/STAT3/CEBPD axis may provide a novel class of PTM-related drugs to improve NK cell therapy efficacy.81 In addition to MS-275, other HDAC inhibitors (eg, valproic acid) upregulate the expression of NKG2D ligands (MICA, ULBP2) in GBM cells and simultaneously enhance the transcription of IFNG and GZMB genes in NK cells; proteasome inhibitors (eg, bortezomib) reduce the degradation of GBM-derived immunosuppressive molecules (TGFβ, IL-10), induce the expression of stress antigens (HSP70), and prolong the intratumoral survival time of NK cells when combined with oncolytic viruses.129 These drugs provide strong preclinical evidence and practical support for the clinical translation of NK cell therapy for GBM.
Importantly, although the in vivo efficacy of HDAC inhibitor MS-275 combined with NK cells was established in an orthotopic diffuse midline glioma (DMG) model—a high-grade glioma sharing key molecular features with GBM (eg, H3K27M mutation) and classified as a GBM subtype under WHO 2021 classification—this provides the first preclinical proof-of-concept that PTM modulation can sensitize malignant gliomas to NK cell killing in the brain.75 Direct validation in classic IDH-wildtype GBM orthotopic models is urgently needed and represents a top priority for future research. Similarly, the SUMOylation inhibitor TAK-981 has demonstrated potent NK cell-enhancing effects in multiple solid tumor models, but its efficacy in GBM remains to be experimentally verified.117 These gaps highlight the critical need for dedicated GBM-specific preclinical studies to accelerate the clinical translation of PTM-targeted NK cell therapies.
Gene Editing
Unlike small-molecule drugs (which regulate PTMs indirectly through enzymes), gene editing directly modifies key molecules of NK cells, eliminates inhibitory signals and strengthens activation pathways, thereby improving the activity and persistence of NK cells in the GBM microenvironment. Using CRISPR/Cas9 technology, core strategies (knockout of inhibitory molecules, stabilization of activation receptors, optimization of cytokine pathways) have all shown efficacy in GBM models.130–132 CRISPR/Cas9 knockout blocks inhibitory signals: knockout of CIS optimizes the IL-2/IL-15 signal, enhances the phosphorylation of STAT3/STAT5, results in a 53.4–57.8% apoptosis rate of T98G GBM spheroids and 31.9–38.5% for U251MG GBM spheroids, and prolongs the intracranial survival time to 79.5 days;130 knockout of Siglec-7 (a key inhibitory receptor) enhances NK cell cytotoxicity against Siglec-7 ligand-positive GBM cells by 1.3–1.7-fold;132 knockout of CD96 or TIGIT (inhibitory receptors) improves NK cell degranulation and cytotoxicity against GBM cell lines (U-251 MG).131 Knock-in of functional genes at safe loci: introduction of HA tag or GFP reporter at the CD96 locus enables in vivo monitoring, with knock-in efficiency up to 16.5%;131 feeder-free cell expansion (2–4×108 NK cells can be obtained from 16 mL of peripheral blood with a knockout efficiency >80%) and optimized nucleofection technology (P3 buffer + DN-100 pulse code, survival rate >70%, negligible off-target effects) support clinical translation.130,131 Consistent with this, a recent study demonstrated that ex vivo expansion of human peripheral blood-derived NK cells using a chemically defined, feeder-free system supplemented with IL-2 and IL-18 yields highly purified (≥90%) functional NK cells, which maintain potent cytotoxicity against GBM cell lines and intracranial tumors in orthotopic xenograft models.15 Notably, intravenous administration of these expanded NK cells (either once or twice via retro-orbital sinus) prolonged the overall survival of NOG mice bearing U87MG-derived intracranial tumors, with no significant difference in efficacy between single and double infusion schedules, indicating that even a single dose of ex vivo-expanded NK cells can exert durable antitumor effects.15 A critical advancement is the engineering of NK cells to autonomously secrete IL-21, which outperforms IL-15-engineered NK cells in both safety and long-term anti-GBM activity.81 Intratumoral administration of IL-15 NK cells induces significant neurotoxicity (characterized by astrocytosis, microgliosis, and weight loss) without effective tumor control, while IL-21 NK cells mediate durable tumor eradication in multiple patient-derived GSC orthotopic models (eg, GSC20, GSC8–11, GSC267) without detectable toxicity.81 Mechanistically, IL-21 drives epigenetic reprogramming of NK cells via activation of STAT1/STAT3 signaling, which directly binds to the promoter of CEBPD (a key C/EBP family transcription factor) to enhance its expression.81 CEBPD further upregulates target genes such as KLF2 and BNIP3L, which are critical for maintaining NK cell metabolic fitness (enhanced oxidative phosphorylation, reduced glycolysis) and long-term cytotoxicity against recurrent GBM cells.81 Genetic deletion of CEBPD abrogates the superior anti-tumor activity of IL-21 NK cells, while CEBPD overexpression in unmodified NK cells mimics IL-21-mediated functional enhancement.81 Base editing (silencing TIGIT without off-target effects) improves safety, while multi-gene editing requires optimization of gRNA dosage and ratio to reduce the risk of chromosomal translocation.131 Gene editing has a synergistic effect with small-molecule drugs (microenvironment regulation), for example, TGF-β receptor-edited NK cells can effectively eliminate glioma stem cells; small-molecule drugs targeting PTM enzymes restore NK cell PTM balance/remodel the GBM microenvironment, jointly promoting clinical translation.
In summary, gene editing technology can accurately remodel the functional molecules of NK cells to overcome the PTM-mediated immunosuppressive barrier of GBM, and the combination with PTM-targeted drugs can exert a synergistic anti-tumor effect. The optimization of expansion and editing technology and the development of safe multi-gene editing strategies are the core to promote the clinical application of this technology in GBM therapy.
Blocking Tumor Immune Escape
Inhibiting Tumor Cell Escape Mediated by PTMs
Tumor cells escape the killing of NK cells and cytokine-induced killer (CIK) cells through abnormal PTMs, reshape immune recognition or inhibit NK cell function, and targeted PTM intervention has become a core strategy to overcome escape. In terms of glycosylation regulation, studies in multiple myeloma models have found that high sialylation of tumor cells can enrich Siglec-7/9 ligands, recruit SHP-1/2 phosphatases to block NK cell degranulation, cytokine secretion and ADCC effect;133 studies in neuroblastoma models have shown that the reduction of NK cell O-GlcNAc modification can damage receptor expression, reduce the release of effector molecules and killing efficiency.134 Although these studies do not involve GBM, they suggest that abnormal glycosylation may be a universal mechanism of tumor immune escape, and strategies such as neuraminidase desialylation or O-GlcNAcase inhibition may also be applicable to GBM, which needs experimental verification. Ubiquitination-mediated escape mechanisms have been reported in a variety of tumors: in melanoma and colorectal cancer, RNF31 drives linear ubiquitination of TNFR1, recruiting A20/TBK1 to block TNF-induced tumor cell death;135 in multiple myeloma, Nectin2 (CD112) degradation reduces NK cell activation, and this effect can be reversed by PYR41;115 in melanoma and colon cancer, PSME3 overexpression accelerates PTP degradation to inhibit MHC-I presentation, and PSME3 knockout can restore MHC-I expression and NK cell killing efficiency.136 These mechanisms provide a reference for exploring ubiquitination-related immune escape in GBM. Gene expression profiling of intracranial tumors from orthotopic GBM xenografts revealed that NKC-treated tumors exhibit downregulated expression of CD70 (fold change: −5.64), a ligand of the NK cell activating receptor CD27, suggesting that GBM cells may evade NK cell surveillance by reducing the expression of activating receptor ligands.15 Conversely, the upregulation of CDH2 (fold change: 2.1), a ligand of the NK cell inhibitory receptor KLRG1, in NKC-treated tumors indicates an additional immune escape mechanism mediated by enhanced inhibitory signaling.15 In terms of acetylation-dependent escape mechanisms, studies in multiple myeloma have shown that HDAC mediates the downregulation of NKG2D ligands and ADAM-induced MICA shedding, and panobinostat can reverse this effect, upregulate ligand expression, reduce soluble MICA release, and enhance NK cell killing.137 The application potential of this strategy in GBM warrants further investigation.
Combination Therapy Amplifies the Regulatory Effect of PTMs Modulation
Single PTM regulatory strategies are limited by the heterogeneity of the GBM microenvironment and the diversity of escape mechanisms, while combining PTM intervention with antibody therapy, gene-edited NK cell therapy or immune checkpoint blockade can effectively overcome GBM immune escape by multi-target synergy to amplify the immune activation effect. In terms of the combination of glycosylation-related PTM regulation and antibody therapy, in multiple myeloma models, the exposure of CD38 on tumor cells with desialylation by neuraminidase is increased, and NK cell-mediated ADCC is enhanced when combined with daratumumab;133 maintaining the O-GlcNAc modification of adoptive NK cells can preserve their killing efficiency.134 The ideas of these combination strategies can provide a reference for GBM-related research. In terms of the combination of ubiquitination-related PTM regulation and immune cell therapy, in melanoma and colorectal cancer models, the combined use of HOIPIN-8 with NK/CD8+ T cells can improve the tumor cell killing rate;135 in melanoma and colon cancer, the combination of PSME3 inhibition with anti-PD-1 can enhance the killing effect of NK/CD8+ T cells;136 TIPE2-knockout NK cells have increased infiltration ability and cytokine secretion, and double knockout with CISH can further improve the in vitro killing rate.138 Although these studies are mostly focused on other tumors, they provide a reference for the design of combination therapy strategies for GBM. In terms of the combination of acetylation-related PTM regulation and CIK cell therapy, in multiple myeloma models, the combined use of HDAC inhibitors with CIK cells can upregulate the expression of NKG2D ligands in tumor cells and IFN-γ in CIK cells, improving killing efficiency.137 Additionally, combining PreOperative RT with HDAC inhibitors may synergistically enhance NK cell efficacy by simultaneously regulating PTM-related transcriptional signatures (eg, G2M checkpoint activation) and acetylation homeostasis, further amplifying the anti-GBM effect.128 At present, direct evidence for the enhancement of NK efficacy by PTM modulator combination therapy in GBM models is still scarce, and most studies are still in the stage of other tumor models. Future research needs to strengthen the study of GBM-specific combination therapy.
Targeting the bottlenecks of NK cell therapy, core breakthrough strategies focus on PTM regulation: correcting abnormal modifications with targeted drugs, enhancing NK cell function via gene editing, and combining with antibody therapy, PreOperative RT, etc. These strategies synergistically address PTM-mediated immunosuppression by boosting NK activity and blocking tumor escape, offering a precise and efficient solution for GBM treatment.
Outlook for the Future Research Direction
To address current challenges in GBM NK cell therapy, future efforts should focus on precise breakthroughs in the following three directions to advance the translation of basic research to clinical practice.
Optimization of Precision Delivery Technology
The current delivery of CAR-NK cells and PTM modulators is limited by poor blood–brain barrier penetration, insufficient retention in the tumor microenvironment and off-target effects on normal tissues, and it is necessary to construct an efficient delivery system through carrier upgrading and cell adaptation. The “TfR + EGFRvIII” dual-targeting carrier can enhance blood–brain barrier penetration and tumor targeting, reduce retention in normal brain tissue (single-targeting carrier retains about 20% in normal brain tissue),139 and load HDAC inhibitors to upregulate GBM cell NKG2D ligands (increase by 1.8–2 times).135 Hypoxia/lactate-responsive carriers adapted to the tumor microenvironment are coupled with anti-lactylation antibodies to protect NK cell mitochondrial function; biocompatible materials reduce interference with NK cell receptors, or IL-15 modified fragments prolong the survival time of CAR-NK cells (currently 48–72 hours).138 Notably, intravenous administration of ex vivo-expanded NK cells via the retro-orbital sinus has been verified to effectively penetrate the blood–brain barrier and inhibit intracranial tumor growth in orthotopic GBM xenografts.15 This non-invasive delivery route avoids the risks associated with intracranial injection (eg, infection, bleeding) and is more suitable for patients ineligible for open surgery due to tumor location or systemic status.15 Additionally, immune cells can access the brain via the choroid plexus and circumventricular organs (fenestrated capillaries without BBB), further supporting the feasibility of intravenous delivery for GBM-targeted NK cell therapy.15 Importantly, PreOperative RT can synergistically improve the delivery efficiency and intratumoral retention of NK cells by remodeling the GBM microenvironment.128 Preclinical studies show that PreOperative RT reduces the density of immunosuppressive TAMs in the tumor core and border, reducing the physical and molecular barriers to NK cell infiltration.128 Moreover, PreOperative RT induces transcriptional changes in GBM cells that enhance their susceptibility to NK cell-mediated killing, improving the “functional delivery” efficiency of NK cells (ie, the proportion of delivered cells that exert anti-tumor effects).128 The combination of PreOperative RT with intravenous NK cell delivery has shown promising results in orthotopic models, with significantly prolonged survival compared to either therapy alone,128 providing a novel strategy to optimize precision delivery for GBM NK cell therapy.
PTM Marker-Guided Personalized Therapy
The molecular heterogeneity of GBM and individual differences in PTM modifications urgently require the establishment of a “PTM markers-TME-therapeutic response” regulatory system. Construct a multi-dimensional PTM marker panel to detect the expression of core PTM enzymes (HDAC high-expression subtypes have a higher response rate to inhibitors),79 key modification sites and TME molecules (adrenomedullin,140 sMICA/sULBP2);125 liquid biopsy enables dynamic monitoring and adjustment of adrenomedullin antagonist dosage (avoiding a 15% increase in vascular resistance in normal brain tissue).140 Marker-based therapeutic regimens include: the m6A high-risk group (high YTHDF2 expression) adopts Nutlin.3a + NK cell therapy (2.1 times higher sensitivity);141 cases with EGFR amplification/PLCG1-Y783 phosphorylation adopt EGFR inhibitor + CAR-NK cell therapy; hypoxia-TAM enriched cases adopt adrenomedullin antagonist preconditioning; cases with high TAM infiltration or high risk of early recurrence may benefit from PreOperative RT combined with NK cell infusion, as supported by interim results from the NeoGlioma Phase I/IIa clinical trial showing reduced TGF-β accumulation in TAMs and a favorable CD4:CD8 lymphocyte ratio in PreOperative SRS (stereotactic radiosurgery) treated patients.128
Deepening PTM Crosstalk Mechanisms
Current PTM research mostly focuses on a single pathway, and it is necessary to systematically analyze the crosstalk mechanisms such as “phosphorylation-acetylation” and “ubiquitin-like modification-lactylation”. Future research should explore the TGF-β-mediated “phosphorylation-epigenetic PTMs” crosstalk (inducing Smad2/3 phosphorylation,13 recruiting HDAC/DNMT to enhance H3K27me3)77 and the interaction between lactylation and H3K18la;71 another critical crosstalk axis is the “IL-21-induced phosphorylation-epigenetic reprogramming” pathway: IL-21 activates STAT1/STAT3 phosphorylation in NK cells,81 and phosphorylated STAT1/STAT3 binds to the CEBPD promoter to promote its transcription, further regulating the acetylation/methylation status of downstream target genes (eg, KLF2, BNIP3L).81 This “phosphorylation-transcription factor activation-epigenetic modification” crosstalk forms a positive regulatory loop to maintain NK cell long-term cytotoxicity and metabolic fitness in the GBM microenvironment.81 Design multi-target strategies: “HDAC-PRMT6” dual inhibitors,77,142 combined use of lactate dehydrogenase and BET inhibitors, or gene-edited NK cells (knockout of TGFBR2)13 + ubiquitin-like modification inhibitors (TAK-981117); additionally, strategies targeting the IL-21/STAT3/CEBPD crosstalk axis (eg, CEBPD overexpression combined with HDAC inhibitors) can synergistically enhance NK cell anti-GBM efficacy by simultaneously regulating phosphorylation and epigenetic PTMs;81 use GBM patient-derived organoids to simulate PTM crosstalk and verify the mechanism and scheme safety.
Future NK cell therapy for GBM should focus on three core directions: optimizing precision delivery to cross the BBB and enhance intratumoral retention; developing PTM marker-guided personalized regimens to adapt to patient heterogeneity; and deepening PTM crosstalk research to explore multi-target intervention potential. These three aspects synergize to form a comprehensive breakthrough system from delivery and adaptation to mechanism, facilitating clinical translation.
Conclusion
This review comprehensively analyzes the dual regulatory roles of seven key post-translational modifications (phosphorylation, acetylation, glycosylation, methylation, ubiquitination, SUMOylation, and lactylation) in NK cell-based immunotherapy for glioblastoma. PTMs not only synergistically enhance the cytotoxicity, targeting efficiency, and in vivo persistence of NK cells through physiological modification, but also suppress NK cell activation, trigger irreversible functional exhaustion, and promote immune escape of GBM via aberrant modification.
The unique value of this review lies in systematically delineating the dual-action network of PTMs in NK cell function regulation and GBM immune escape, and for the first time integrating the core mechanistic framework of “PTM abnormality – NK cell dysfunction – GBM therapeutic resistance”, offering a new perspective for deciphering the resistance mechanisms underlying GBM immunotherapy. Meanwhile, we highlight major clinical bottlenecks restricting NK cell therapy for GBM, including insufficient NK cell activity and persistence, inefficient precision delivery, and PTM-mediated GBM immune escape, and summarize corresponding PTM-targeted strategies such as small-molecule compounds, CRISPR/Cas9-engineered NK cells, and multi-target combination regimens.
This review further defines the core principle of PTM-mediated dual regulation in GBM-directed NK cell therapy: physiological phosphorylation, acetylation, glycosylation, and methylation potentiate therapeutic efficacy by activating NK cell signaling, sustaining metabolic adaptation, and improving ligand recognition, whereas abnormal ubiquitination, SUMOylation, and lactylation impair anti-tumor effects through functional gene silencing, mitochondrial injury, and immune escape facilitation.
Future investigations should focus on three priority directions: first, perform in vivo validation of PTM regulatory events, characterize modification profiles in clinical specimens, and develop highly specific small-molecule inhibitors targeting PTM enzymes; second, optimize precision delivery systems for NK cells and PTM modulators, explore intelligent BBB-penetrating carriers and biomaterial-assisted delivery platforms to substantially enhance in vivo delivery efficiency; third, elucidate PTM crosstalk networks and design multi-target intervention strategies to achieve precise manipulation of the complex PTM regulatory landscape.
The integration of PTM modulation, NK cell engineering, and precision delivery to establish a trinity therapeutic system of “modification regulation – cellular engineering – precise delivery” represents a critical step toward advancing clinical translation of NK cell therapy for GBM. These strategies are instrumental in overcoming therapeutic limitations and breaking through current bottlenecks of NK cell therapy in solid tumors such as GBM. They will not only facilitate translational development of GBM immunotherapy and improve patient prognosis, but also serve as a reference for developing PTM-targeted immunotherapies against other solid tumors. This review provides a conceptual framework for overcoming the immunosuppressive microenvironment of GBM through PTM modulation, and the proposed approaches are anticipated to accelerate clinical translation of NK cell therapy and improve patient outcomes.
Abbreviations
GBM, glioblastoma; NK, Natural killer; TME, tumor microenvironment; PTMs, post-translational modifications; BBB, blood–brain barrier; GSCs, glioma stem cells; MHC, major histocompatibility complex; CAR, chimeric antigen receptor; KATs, lysine acetyltransferases; KDACs, lysine deacetylases; Ub, Ubiquitin; SUMO, Small Ubiquitin-like Modifier; Kla, lysine residues; NRF1, nuclear respiratory factor 1; OXPHOS, oxidative phosphorylation; TCA, tricarboxylic acid; S60, serine 60; K146, lysine 146; DMG, diffuse midline glioma; ADCC, antibody-dependent cell-mediated cytotoxicity; FL-SA, fluorescein-labeled sialic acid; GN4C, N-acetyl-D-glucosamine-calix[4]arene; CR, complete remission; PD, disease progression; CRLs, Cullin-Ring ligases; 2-HG,2-hydroxyglutarate; DNMT, DNA methyltransferase; IDHwt, IDH-wild-type; MGO, Methylglyoxal; GO, glyoxal; AGEs, advanced glycation end products; LDHA, Lactate dehydrogenase A; PGE2, Prostaglandin E2; MFI, mean fluorescence intensity; TI-NK, tumor-infiltrating NK cells; PB-NK, peripheral blood NK cells; HRR, homologous recombination repair; TIL-NK, tumor-infiltrating lymphocyte-derived NK cells; H3K18la, histone H3K18 lactylation; MES GBM, Mesenchymal subtype GBM; SAE, SUMO-activating enzyme; IFN1, type I interferon; HDAC, Histone deacetylase.
Acknowledgments
We thank everyone in the team for their valuable discussions. We also acknowledge the figure resources provided by biogdp.com.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by the National Natural Science Foundation of China (No. 81772680).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Ostrom QT, Price M, Neff C, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2016–2020. Neuro Oncol. 2023;25(12 Suppl 2):iv1–27. PMID: 37793125; PMCID: PMC10550277. doi:10.1093/neuonc/noad149
2. van den Bent MJ, Geurts M, French PJ, et al. Primary brain tumours in adults. Lancet. 2023;402(10412):1564–1579. PMID: 37738997. doi:10.1016/S0140-6736(23)01054-1
3. Schaff LR, Mellinghoff IK. Glioblastoma and other primary brain malignancies in adults: a review. JAMA. 2023;329(7):574–587. PMID: 36809318; PMCID: PMC11445779. doi:10.1001/jama.2023.0023
4. Alexander BM, Cloughesy TF. Adult glioblastoma. J Clin Oncol. 2017;35(21):2402–2409. PMID: 28640706. doi:10.1200/JCO.2017.73.0119
5. Schwartzbaum JA, Fisher JL, Aldape KD, Wrensch M. Epidemiology and molecular pathology of glioma. Nat Clin Pract Neurol. 2006;2(9):494–503;quiz1pfollowing516. PMID: 16932614. doi:10.1038/ncpneuro0289
6. Joseph GP, McDermott R, Baryshnikova MA, Cobbs CS, Ulasov IV. Cytomegalovirus as an oncomodulatory agent in the progression of glioma. Cancer Lett. 2017;384:79–85. PMID: 27777041. doi:10.1016/j.canlet.2016.10.022
7. Moore SC, Rajaraman P, Dubrow R, et al. Height, body mass index, and physical activity in relation to glioma risk. Cancer Res. 2009;69(21):8349–8355. PMID: 19808953; PMCID: PMC2783605. doi:10.1158/0008-5472.CAN-09-1669
8. Rice T, Lachance DH, Molinaro AM, et al. Understanding inherited genetic risk of adult glioma - a review. Neurooncol Pract. 2016;3(1):10–16. PMID: 26941959; PMCID: PMC4774334. doi:10.1093/nop/npv026
9. Zhang X, Rao A, Sette P, et al. IDH mutant gliomas escape natural killer cell immune surveillance by downregulation of NKG2D ligand expression. Neuro Oncol. 2016;18(10):1402–1412. PMID: 27116977; PMCID: PMC5035522. doi:10.1093/neuonc/now061
10. Golán I, Rodríguez de la Fuente L, Costoya JA. NK Cell-Based Glioblastoma Immunotherapy. Cancers. 2018;10(12):522. PMID: 30567306; PMCID: PMC6315402. doi:10.3390/cancers10120522
11. Paw I, Carpenter RC, Watabe K, Debinski W, Lo HW. Mechanisms regulating glioma invasion. Cancer Lett. 2015;362(1):1–7. PMID: 25796440; PMCID: PMC4435977. doi:10.1016/j.canlet.2015.03.015
12. Gras Navarro A, Björklund AT, Chekenya M. Therapeutic potential and challenges of natural killer cells in treatment of solid tumors. Front Immunol. 2015;6:202. PMID: 25972872; PMCID: PMC4413815. doi:10.3389/fimmu.2015.00202
13. Shaim H, Shanley M, Basar R, et al. Targeting the αv integrin/TGF-β axis improves natural killer cell function against glioblastoma stem cells. J Clin Invest. 2021;131(14):e142116. PMID: 34138753; PMCID: PMC8279586. doi:10.1172/JCI142116
14. Chávez-Galán L, Arenas-Del Angel MC, Zenteno E, Chávez R, Lascurain R. Cell death mechanisms induced by cytotoxic lymphocytes. Cell Mol Immunol. 2009;6(1):15–25. PMID: 19254476; PMCID: PMC4002546. doi:10.1038/cmi.2009.3
15. Morimoto T, Nakazawa T, Matsuda R, et al. Antitumor effects of intravenous natural killer cell infusion in an orthotopic glioblastoma xenograft murine model and gene expression profile analysis. Int J Mol Sci. 2024;25(4):2435. PMID: 38397112; PMCID: PMC10889440. doi:10.3390/ijms25042435
16. Li Y, Zhang R, Hei H. Advances in post-translational modifications of proteins and cancer immunotherapy. Front Immunol. 2023;14:1229397. PMID: 37675097; PMCID: PMC10477431. doi:10.3389/fimmu.2023.1229397
17. Pan S, Chen R. Pathological implication of protein post-translational modifications in cancer. Mol Aspects Med. 2022;86:101097. PMID: 35400524; PMCID: PMC9378605. doi:10.1016/j.mam.2022.101097
18. Wang S, Osgood AO, Chatterjee A. Uncovering post-translational modification-associated protein-protein interactions. Curr Opin Struct Biol. 2022;74:102352. PMID: 35334254; PMCID: PMC9464464. doi:10.1016/j.sbi.2022.102352
19. Olsen JV, Blagoev B, Gnad F, et al. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell. 2006;127(3):635–648. PMID: 17081983. doi:10.1016/j.cell.2006.09.026
20. Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol. 2006;7(11):833–846. [PMID: 17057753]. doi:10.1038/nrm2039
21. Tang Q, Chen Y, Li X, et al. The role of PD-1/PD-L1 and application of immune-checkpoint inhibitors in human cancers. Front Immunol. 2022;13:964442. PMID: 36177034; PMCID: PMC9513184. doi:10.3389/fimmu.2022.964442
22. Johnson LN. The regulation of protein phosphorylation. Biochem Soc Trans. 2009;37(Pt 4):627–641. PMID: 19614568. doi:10.1042/BST0370627
23. Arrington JV, Hsu CC, Elder SG, Andy Tao W. Recent advances in phosphoproteomics and application to neurological diseases. Analyst. 2017;142(23):4373–4387. PMID: 29094114; PMCID: PMC5708141. doi:10.1039/c7an00985b
24. Liu X, Zhang Y, Wang Y, Yang M, Hong F, Yang S. Protein phosphorylation in cancer: role of nitric oxide signaling pathway. Biomolecules. 2021;11(7):1009. PMID: 34356634; PMCID: PMC8301900. doi:10.3390/biom11071009
25. Farria A, Li W, Dent SY. KATs in cancer: functions and therapies. Oncogene. 2015;34(38):4901–4913. PMID: 25659580; PMCID: PMC4530097. doi:10.1038/onc.2014.453
26. Sauer M, Schuldner M, Hoffmann N, et al. CBP/p300 acetyltransferases regulate the expression of NKG2D ligands on tumor cells. Oncogene. 2017;36(7):933–941. PMID: 27477692; PMCID: PMC5318661. doi:10.1038/onc.2016.259
27. Cho H, Son WC, Lee YS, et al. Differential effects of histone deacetylases on the expression of NKG2D ligands and NK cell-mediated anticancer immunity in lung cancer cells. Molecules. 2021;26(13):3952. PMID: 34203519; PMCID: PMC8271929. doi:10.3390/molecules26133952
28. Hajji N, Wallenborg K, Vlachos P, Füllgrabe J, Hermanson O, Joseph B. Opposing effects of hMOF and SIRT1 on H4K16 acetylation and the sensitivity to the topoisomerase II inhibitor etoposide. Oncogene. 2010;29(15):2192–2204. PMID: 20118981. doi:10.1038/onc.2009.505
29. Elsheikh SE, Green AR, Rakha EA, et al. Global histone modifications in breast cancer correlate with tumor phenotypes, prognostic factors, and patient outcome. Cancer Res. 2009;69(9):3802–3809. PMID: 19366799. doi:10.1158/0008-5472.CAN-08-3907
30. Harachi M, Masui K, Cavenee WK, Mischel PS, Shibata N. Protein acetylation at the interface of genetics, epigenetics and environment in cancer. Metabolites. 2021;11(4):216. PMID: 33916219; PMCID: PMC8066013. doi:10.3390/metabo11040216
31. Kaypee S, Sudarshan D, Shanmugam MK, Mukherjee D, Sethi G, Kundu TK. Aberrant lysine acetylation in tumorigenesis: implications in the development of therapeutics. Pharmacol Ther. 2016;162:98–119. PMID: 26808162. doi:10.1016/j.pharmthera.2016.01.011
32. Eichler J. Protein glycosylation. Curr Biol. 2019;29(7):R229–R231. PMID: 30939300. doi:10.1016/j.cub.2019.01.003
33. Stowell SR, Ju T, Cummings RD. Protein glycosylation in cancer. Annu Rev Pathol. 2015;10(1):473–510. PMID: 25621663; PMCID: PMC4396820. doi:10.1146/annurev-pathol-012414-040438
34. Bangarh R, Khatana C, Kaur S, et al. Aberrant protein glycosylation: implications on diagnosis and immunotherapy. Biotechnol Adv. 2023;66:108149. PMID: 37030554. doi:10.1016/j.biotechadv.2023.108149
35. Grzesik K, Janik M, Hoja-łukowicz D. The hidden potential of glycomarkers: glycosylation studies in the service of cancer diagnosis and treatment. Biochim Biophys Acta Rev Cancer. 2023;1878(3):188889. PMID: 37001617. doi:10.1016/j.bbcan.2023.188889
36. Pinho SS, Reis CA. Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer. 2015;15(9):540–555. PMID: 26289314. doi:10.1038/nrc3982
37. Kachroo P, Morrow JD, Vyhlidal CA, et al. DNA methylation perturbations may link altered development and aging in the lung. Aging. 2021;13(2):1742–1764. PMID: 33468710; PMCID: PMC7880367. doi:10.18632/aging.202544
38. He PC, He C. m6 A RNA methylation: from mechanisms to therapeutic potential. EMBO J. 2021;40(3):e105977. PMID: 33470439; PMCID: PMC7849164. doi:10.15252/embj.2020105977
39. Zhao S, Chuh KN, Zhang B, et al. Histone H3Q5 serotonylation stabilizes H3K4 methylation and potentiates its readout. Proc Natl Acad Sci U S A. 2021;118(6):e2016742118. PMID: 33526675; PMCID: PMC8017887. doi:10.1073/pnas.2016742118
40. Di Blasi R, Blyuss O, Timms JF, Conole D, Ceroni F, Whitwell HJ. Non-histone protein methylation: biological significance and bioengineering potential. ACS Chem Biol. 2021;16(2):238–250. PMID: 33411495. doi:10.1021/acschembio.0c00771
41. Ambler RP, Rees MW. Epsilon-N-methyl-lysine in bacterial flagellar protein. Nature. 1959;184(4679):56–57. PMID: 13793118. doi:10.1038/184056b0
42. Kim S, Paik WK. Studies on the origin of epsilon-N-methyl-L-lysine in protein. J Biol Chem. 1965;240(12):4629–4634. [PMID: 5846984]. doi:10.1016/S0021-9258(18)97001-8
43. Luo M. Chemical and biochemical perspectives of protein lysine methylation. Chem Rev. 2018;118(14):6656–6705. PMID: 29927582; PMCID: PMC6668730. doi:10.1021/acs.chemrev.8b00008
44. Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet. 2011;12(1):7–18. PMID: 21116306. doi:10.1038/nrg2905
45. Barski A, Cuddapah S, Cui K, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129(4):823–837. PMID: 17512414. doi:10.1016/j.cell.2007.05.009
46. Cao R, Wang L, Wang H, et al. Role of histone H3 lysine 27 methylation in polycomb-group silencing. Science. 2002;298(5595):1039–1043. PMID: 12351676. doi:10.1126/science.1076997
47. Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet. 2007;8(4):286–298. PMID: 17339880. doi:10.1038/nrg2005
48. Zhao Y, Lu Q, Li C, et al. PRMT1 regulates the tumour-initiating properties of esophageal squamous cell carcinoma through histone H4 arginine methylation coupled with transcriptional activation. Cell Death Dis. 2019;10(5):359. PMID: 31043582; PMCID: PMC6494844. doi:10.1038/s41419-019-1595-0
49. Antao AM, Tyagi A, Kim KS, Ramakrishna S. Advances in deubiquitinating enzyme inhibition and applications in cancer therapeutics. Cancers. 2020;12(6):1579. PMID: 32549302; PMCID: PMC7352412. doi:10.3390/cancers12061579
50. Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. 2012;81(1):203–229. PMID: 22524316.6. doi:10.1146/annurev-biochem-060310-170328
51. Wang Y, Dasso M. SUMOylation and deSUMOylation at a glance. J Cell Sci. 2009;122(Pt 23):4249–4252. PMID: 19923268; PMCID: PMC2779127. doi:10.1242/jcs.050542
52. Han ZJ, Feng YH, Gu BH, Li YM, Chen H. The post-translational modification, SUMOylation, and cancer (Review). Int J Oncol. 2018;52(4):1081–1094. PMID: 29484374; PMCID: PMC5843405. doi:10.3892/ijo.2018.4280
53. Müller S, Ledl A, Schmidt D. SUMO: a regulator of gene expression and genome integrity. Oncogene. 2004;23(11):1998–2008. PMID: 15021887. doi:10.1038/sj.onc.1207415
54. Nie M, Boddy MN. Cooperativity of the SUMO and ubiquitin pathways in genome stability. Biomolecules. 2016;6(1):14. PMID: 26927199; PMCID: PMC4808808. doi:10.3390/biom6010014
55. Melchior F, Schergaut M, Pichler A. SUMO: ligases, isopeptidases and nuclear pores. Trends Biochem Sci. 2003;28(11):612–618. PMID: 14607092. doi:10.1016/j.tibs.2003.09.002
56. Eifler K, Vertegaal AC. Mapping the SUMOylated landscape. FEBS J. 2015;282(19):3669–3680. PMID: 26185901; PMCID: PMC4869838. doi:10.1111/febs.13378
57. Choi SG, Kim H, Jeong EI, et al. SUMO-modified FADD recruits cytosolic Drp1 and caspase-10 to mitochondria for regulated necrosis. Mol Cell Biol. 2017;37(2):e00254–16. PMID: 27799292; PMCID: PMC5214857. doi:10.1128/MCB.00254-16
58. Saitoh H, Hinchey J. Functional heterogeneity of small ubiquitin-related protein modifiers SUMO-1 versus SUMO-2/3. J Biol Chem. 2000;275(9):6252–6258. PMID: 10692421. doi:10.1074/jbc.275.9.6252
59. Bergink S, Jentsch S. Principles of ubiquitin and SUMO modifications in DNA repair. Nature. 2009;458(7237):4617. PMID: 19325626. doi:10.1038/nature07963
60. Thomson TM, Guerra-Rebollo M. Ubiquitin and SUMO signalling in DNA repair. Biochem Soc Trans. 2010;38(Pt 1):116–131. PMID: 20074046. doi:10.1042/BST0380116
61. Ulrich HD. Ubiquitin and SUMO in DNA repair at a glance. J Cell Sci. 2012;125(Pt 2):249–254. PMID: 22357966. doi:10.1242/jcs.091801
62. Jackson SP, Durocher D. Regulation of DNA damage responses by ubiquitin and SUMO. Mol Cell. 2013;49(5):795–807. PMID: 23416108. doi:10.1016/j.molcel.2013.01.017
63. Sarangi P, Zhao X. SUMO-mediated regulation of DNA damage repair and responses. Trends Biochem Sci. 2015;40(4):233–242. Erratum in: Trends Biochem Sci. 2015 Jun;40(6):338. PMID: 25778614; PMCID: PMC4380773. doi:10.1016/j.tibs.2015.02.006
64. Bettermann K, Benesch M, Weis S, Haybaeck J. SUMOylation in carcinogenesis. Cancer Lett. 2012;316(2):113–125. PMID: 22138131. doi:10.1016/j.canlet.2011.10.036
65. Eifler K, Vertegaal ACO. SUMOylation-mediated regulation of cell cycle progression and cancer. Trends Biochem Sci. 2015;40(12):779–793. PMID: 26601932; PMCID: PMC4874464. doi:10.1016/j.tibs.2015.09.006
66. Flotho A, Melchior F. SUMOylation: a regulatory protein modification in health and disease. Annu Rev Biochem. 2013;82(1):357–385. PMID: 23746258. doi:10.1146/annurev-biochem-061909-093311
67. Rabellino A, Andreani C, Scaglioni PP. The role of PIAS SUMORE3-ligases in cancer. Cancer Res. 2017;77(7):1542–1547. PMID: 28330929; PMCID: PMC5380518. doi:10.1158/0008-5472.CAN-16-2958
68. Seeler JS, Dejean A. SUMO and the robustness of cancer. Nat Rev Cancer. 2017;17(3):184–197. PMID: 28134258. doi:10.1038/nrc.2016.143
69. Wang T, Ye Z, Li Z, et al. Lactate-induced protein lactylation: a bridge between epigenetics and metabolic reprogramming in cancer. Cell Prolif. 2023;56(10):e13478. PMID: 37060186; PMCID: PMC10542650. doi:10.1111/cpr.13478
70. Zhang D, Tang Z, Huang H, et al. Metabolic regulation of gene expression by histone lactylation. Nature. 2019;574(7779):575–580. PMID: 31645732; PMCID: PMC6818755. doi:10.1038/s41586-019-1678-1
71. Liu R, Ren X, Park YE, et al. Nuclear GTPSCS functions as a lactyl-CoA synthetase to promote histone lactylation and gliomagenesis. Cell Metab. 2025;37(2):377–394.e9. PMID: 39642882; PMCID: PMC11798710. doi:10.1016/j.cmet.2024.11.005
72. Abaza M, Luqmani YA. The influence of pH and hypoxia on tumor metastasis. Expert Rev Anticancer Ther. 2013;13(10):1229–1242. PMID: 24099530. doi:10.1586/14737140.2013.843455
73. Jin J, Yan P, Wang D, et al. Targeting lactylation reinforces NK cell cytotoxicity within the tumor microenvironment. Nat Immunol. 2025;26(7):1099–1112. PMID: 40494934. doi:10.1038/s41590-025-02178-8
74. Du Y, Li R, Fu D, et al. Multi-omics technologies and molecular biomarkers in brain tumor-related epilepsy. CNS Neurosci Ther. 2024;30(4):e14717. PMID: 38641945; PMCID: PMC11031674. doi:10.1111/cns.14717
75. Deng Y, Liu J, Pu Z, et al. Targeting the HLA-E-NKG2A axis in combination with MS-275 enhances NK cell-based immunotherapy against DMG. J Exp Clin Cancer Res. 2025;44(1):133. PMID: 40296045; PMCID: PMC12039099. doi:10.1186/s13046-025-03390-y
76. Fernández-Sánchez A, Baragaño Raneros A, Carvajal Palao R, et al. DNA demethylation and histone H3K9 acetylation determine the active transcription of the NKG2D gene in human CD8+ T and NK cells. Epigenetics. 2013;8(1):66–78. PMID: 23235109; PMCID: PMC3549882. doi:10.4161/epi.23115
77. Li Y, Wang J, Zhou L, et al. DNMT1 inhibition reprograms T cells to NK-like cells with potent antitumor activity. Sci Immunol. 2025;10(105):eadm8251. PMID: 40117344. doi:10.1126/sciimmunol.adm8251
78. Was H, Krol SK, Rotili D, et al. Histone deacetylase inhibitors exert anti-tumor effects on human adherent and stem-like glioma cells. Clin Epigenetics. 2019;11(1):11. PMID: 30654849; PMCID: PMC6337817. doi:10.1186/s13148-018-0598-5
79. Wang LB, Karpova A, Gritsenko MA, et al. Clinical PROTEOMIC TUMOR ANALYSIS CONSORTIUM. Proteogenomic and metabolomic characterization of human glioblastoma. Cancer Cell. 2021;39(4):509–528.e20. PMID: 33577785; PMCID: PMC8044053. doi:10.1016/j.ccell.2021.01.006
80. Suico MA, Nakamura H, Lu Z, et al. SUMO down-regulates the activity of Elf4/myeloid Elf-1-like factor. Biochem Biophys Res Commun. 2006;348(3):880–888. PMID: 16904644. doi:10.1016/j.bbrc.2006.07.151
81. Shanley M, Daher M, Dou J, et al. Interleukin-21 engineering enhances NK cell activity against glioblastoma via CEBPD. Cancer Cell. 2024;42(8):1450–1466.e11. PMID: 39137729; PMCID: PMC11370652. doi:10.1016/j.ccell.2024.07.007
82. Batlle E, Massagué J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity. 2019;50(4):924–940. PMID: 30995507; PMCID: PMC7507121. doi:10.1016/j.immuni.2019.03.024
83. Sloan KE, Eustace BK, Stewart JK, et al. CD155/PVR plays a key role in cell motility during tumor cell invasion and migration. BMC Cancer. 2004;4:73. PMID: 15471548; PMCID: PMC524493. doi:10.1186/1471-2407-4-73
84. Zhu Q, Liang P, Meng H, et al. Stabilization of Pin1 by USP34 promotes Ubc9 isomerization and protein SUMOylation in glioma stem cells. Nat Commun. 2024;15(1):40. PMID: 38167292; PMCID: PMC10762127. doi:10.1038/s41467-023-44349-x
85. Wang L, Ji S. Inhibition of Ubc9-Induced CRMP2 SUMOylation disrupts glioblastoma cell proliferation. J Mol Neurosci. 2019;69(3):391–398. PMID: 31267313. doi:10.1007/s12031-019-01368-y
86. Terrén I, Orrantia A, Vitallé J, Zenarruzabeitia O, Borrego F. NK cell metabolism and tumor microenvironment. Front Immunol. 2019;10:2278. PMID: 31616440; PMCID: PMC6769035. doi:10.3389/fimmu.2019.02278
87. Fuertes MB, Domaica CI, Zwirner NW. Leveraging NKG2D ligands in immuno-oncology. Front Immunol. 2021;12:713158. PMID: 34394116; PMCID: PMC8358801. doi:10.3389/fimmu.2021.713158
88. Huang L, Zhu P, Xia P, Fan Z. WASH has a critical role in NK cell cytotoxicity through Lck-mediated phosphorylation. Cell Death Dis. 2016;7(7):e2301. PMID: 27441653; PMCID: PMC4973352. doi:10.1038/cddis.2016.212
89. Mentlik AN, Sanborn KB, Holzbaur EL, Orange JS. Rapid lytic granule convergence to the MTOC in natural killer cells is dependent on dynein but not cytolytic commitment. Mol Biol Cell. 2010;21(13):2241–2256. PMID: 20444980; PMCID: PMC2893988. doi:10.1091/mbc.e09-11-0930
90. Luo J, Guo M, Huang M, et al. Neoleukin-2/15-armored CAR-NK cells sustain superior therapeutic efficacy in solid tumors via c-Myc/NRF1 activation. Signal Transduct Target Ther. 2025;10(1):78. PMID: 40025022; PMCID: PMC11873268. doi:10.1038/s41392-025-02158-2
91. Wei C, Liao K, Chen HJ, et al. Nuclear mitochondrial acetyl-CoA acetyltransferase 1 orchestrates natural killer cell-dependent antitumor immunity in colorectal cancer. Signal Transduct Target Ther. 2025;10(1):138. PMID: 40289129; PMCID: PMC12034769. doi:10.1038/s41392-025-02221-y
92. Sohn H, Kolicheski A, Poursine-Laurent J, et al. ACLY promotes NK cell effector function by regulating glycolysis and histone acetylation. J Immunol. 2025:vkaf209. PMID: 40853248; PMCID: PMC12380160. doi:10.1093/jimmun/vkaf209
93. Jo DH, Kaczmarek S, Khan AUH, et al. Entinostat, a histone deacetylase inhibitor, enhances CAR-NK cell anti-tumor activity by sustaining CAR expression. Front Immunol. 2025;16:1533044. PMID: 40124378; PMCID: PMC11925867. doi:10.3389/fimmu.2025.1533044
94. Zheng J, Lu Y, Xiao J, et al. Pan-HDAC inhibitors augment IL2-induced proliferation of NK cells via the JAK2-STAT5B signaling pathway. Int Immunopharmacol. 2023;116:109753. PMID: 36738675. doi:10.1016/j.intimp.2023.109753
95. Skořepa O, Pazicky S, Kalousková B, et al. Natural killer cell activation receptor NKp30 oligomerization depends on its N-glycosylation. Cancers. 2020;12(7):1998. PMID: 32708305; PMCID: PMC7409301. doi:10.3390/cancers12071998
96. Rodriguez Benavente MC, Hughes HB, Kremer PG, Subedi GP, Barb AW. Inhibiting N-glycan processing increases the antibody binding affinity and effector function of human natural killer cells. Immunology. 2023;170(2):202–213. PMID: 37218360; PMCID: PMC10524233. doi:10.1111/imm.13662
97. Antillon K, Ross PA, Farrell MP. Directing CAR NK cells via the metabolic incorporation of CAR ligands into malignant cell glycans. ACS Chem Biol. 2022;17(6):1505–1512. PMID: 35648806; PMCID: PMC10061155. doi:10.1021/acschembio.2c00173
98. Benson V, Grobarova V, Richter J, Fiserova A. Glycosylation regulates NK cell-mediated effector function through PI3K pathway. Int Immunol. 2010;22(3):167–177. PMID: 20089585. doi:10.1093/intimm/dxp123
99. Song H, Song J, Cheng M, et al. METTL3-mediated m6A RNA methylation promotes the anti-tumour immunity of natural killer cells. Nat Commun. 2021;12(1):5522. PMID: 34535671; PMCID: PMC8448775. doi:10.1038/s41467-021-25803-0
100. Bugide S, Green MR, Wajapeyee N. Inhibition of Enhancer of zeste homolog 2 (EZH2) induces natural killer cell-mediated eradication of hepatocellular carcinoma cells. Proc Natl Acad Sci U S A. 2018;115(15):E3509–E3518. PMID: 29581297; PMCID: PMC5899497. doi:10.1073/pnas.1802691115
101. Kumar R, Gupta R. Epigenetic regulation of NKG2D ligand and the rise of NK cell-based immunotherapy for cancer treatment. Front Oncol. 2024;14:1456631. PMID: 39161385; PMCID: PMC11330816. doi:10.3389/fonc.2024.1456631
102. Raulet DH, Marcus A, Coscoy L. Dysregulated cellular functions and cell stress pathways provide critical cues for activating and targeting natural killer cells to transformed and infected cells. Immunol Rev. 2017;280(1):93–101. PMID: 29027233; PMCID: PMC5687887. doi:10.1111/imr.12600
103. Rea A, Santana-Hernández S, Villanueva J, et al. Enhancing human NK cell antitumor function by knocking out SMAD4 to counteract TGFβ and activin A suppression. Nat Immunol. 2025;26(4):582–594. PMID: 40119192; PMCID: PMC11957989. doi:10.1038/s41590-025-02103-z
104. Lei W, Liu H, Deng W, et al. Safety and feasibility of 4-1BB co-stimulated CD19-specific CAR-NK cell therapy in refractory/relapsed large B cell lymphoma: a Phase 1 trial. Nat Cancer. 2025;6(5):786–800. Erratum in: Nat Cancer. 2025 Jul;6(7):1295. doi: 10.1038/s43018-025-01026-w. PMID: 40251398; PMCID: PMC12122374. doi:10.1038/s43018-025-00940-3
105. Petillo S, Sproviero E, Loconte L, et al. NEDD8-activating enzyme inhibition potentiates the anti-myeloma activity of natural killer cells. Cell Death Dis. 2023;14(7):438. PMID: 37460534; PMCID: PMC10352239. doi:10.1038/s41419-023-05949-z
106. Liu Y, Su C, Wei X, Wei N, Qian Q, Xu Z. Prospects and applications of NK therapy in the treatment of gliomas (Review). Oncol Rep. 2025;54(2):88. PMID: 40476557; PMCID: PMC12159591. doi:10.3892/or.2025.8921
107. Yuan H, Wu X, Wu Q, et al. Lysine catabolism reprograms tumour immunity through histone crotonylation. Nature. 2023;617(7962):818–826. PMID: 37198486; PMCID: PMC11089809. doi:10.1038/s41586-023-06061-0
108. Jiao D, Sun R, Ren X, et al. Lipid accumulation-mediated histone hypoacetylation drives persistent NK cell dysfunction in anti-tumor immunity. Cell Rep. 2023;42(10):113211. Erratum in: Cell Rep. 2024 Jan 23;43(1):113632. doi: 10.1016/j.celrep.2023.113632. PMID: 37792534. doi:10.1016/j.celrep.2023.113211
109. Rosenstock P, Bezold V, Bork K, Scheffler J, Horstkorte R. Glycation interferes with natural killer cell function. Mech Ageing Dev. 2019;178:64–71. PMID: 30659859. doi:10.1016/j.mad.2019.01.006
110. Peipp M, Lammerts van bueren JJ, Schneider-Merck T, et al. Antibody fucosylation differentially impacts cytotoxicity mediated by NK and PMN effector cells. Blood. 2008;112(6):2390–2399. PMID: 18566325. doi:10.1182/blood-2008-03-144600
111. Zitti B, Molfetta R, Fionda C, et al. Innate immune activating ligand SUMOylation affects tumor cell recognition by NK cells. Sci Rep. 2017;7(1):10445. PMID: 28874810; PMCID: PMC5585267. doi:10.1038/s41598-017-10403-0
112. Molfetta R, Zingoni A, Santoni A, Paolini R. Post-translational mechanisms regulating NK cell activating receptors and their ligands in cancer: potential targets for therapeutic intervention. Front Immunol. 2019;10:2557. PMID: 31736972; PMCID: PMC6836727. doi:10.3389/fimmu.2019.02557
113. Husain Z, Huang Y, Seth P, Sukhatme VP. Tumor-derived lactate modifies antitumor immune response: effect on myeloid-derived suppressor cells and NK cells. J Immunol. 2013;191(3):1486–1495. PMID: 23817426. doi:10.4049/jimmunol.1202702
114. Tong L, Jiménez-Cortegana C, Tay AHM, Wickström S, Galluzzi L, Lundqvist A. NK cells and solid tumors: therapeutic potential and persisting obstacles. Mol Cancer. 2022;21(1):206. PMID: 36319998; PMCID: PMC9623927. doi:10.1186/s12943-022-01672-z
115. Boulpicante M, Darrigrand R, Pierson A, et al. Tumors escape immunosurveillance by overexpressing the proteasome activator PSME3. Oncoimmunology. 2020;9(1):1761205. PMID: 32923122; PMCID: PMC7458623. doi:10.1080/2162402X.2020.1761205
116. Shi Y, Fu Y, Zhang X, et al. Romidepsin (FK228) regulates the expression of the immune checkpoint ligand PD-L1 and suppresses cellular immune functions in colon cancer. Cancer Immunol Immunother. 2021;70(1):61–73. PMID: 32632663; PMCID: PMC7838139. doi:10.1007/s00262-020-02653-1
117. Lightcap ES, Yu P, Grossman S, et al. A small-molecule SUMOylation inhibitor activates antitumor immune responses and potentiates immune therapies in preclinical models. Sci Transl Med. 2021;13(611):eaba7791. PMID: 34524860; PMCID: PMC9719791. doi:10.1126/scitranslmed.aba7791
118. Li G, Wang D, Zhai Y, et al. Glycometabolic reprogramming-induced XRCC1 lactylation confers therapeutic resistance in ALDH1A3-overexpressing glioblastoma. Cell Metab. 2024;36(8):1696–1710.e10. PMID: 39111285. doi:10.1016/j.cmet.2024.07.011
119. Guo Y, Li Z, Parsels LA, et al. H3K27M diffuse midline glioma is homologous recombination defective and sensitized to radiotherapy and NK cell-mediated antitumor immunity by PARP inhibition. Neuro Oncol. 2025:noaf097. PMID: 40207661. doi:10.1093/neuonc/noaf097
120. Sönmez C, Wölfer J, Holling M, et al. Blockade of inhibitory killer cell immunoglobulin-like receptors and IL-2 triggering reverses the functional hypoactivity of tumor-derived NK-cells in glioblastomas. Sci Rep. 2022;12(1):6769. PMID: 35474089; PMCID: PMC9042843. doi:10.1038/s41598-022-10680-4
121. Kirschenbaum D, Xie K, Ingelfinger F, et al. Time-resolved single-cell transcriptomics defines immune trajectories in glioblastoma. Cell. 2024;187(1):149–165.e23. PMID: 38134933. doi:10.1016/j.cell.2023.11.032
122. Ma S, Zhao Y, Lee WC, et al. Hypoxia induces HIF1α-dependent epigenetic vulnerability in triple negative breast cancer to confer immune effector dysfunction and resistance to anti-PD-1 immunotherapy. Nat Commun. 2022;13(1):4118. PMID: 35840558; PMCID: PMC9287350. doi:10.1038/s41467-022-31764-9
123. Dan H, Zhang S, Zhou Y, Guan Q. DNA methyltransferase inhibitors: catalysts for antitumour immune responses. Onco Targets Ther. 2019;12:10903–10916. PMID: 31849494; PMCID: PMC6913319. doi:10.2147/OTT.S217767
124. Huang W, Zhu Q, Shi Z, et al. Dual inhibitors of DNMT and HDAC induce viral mimicry to induce antitumour immunity in breast cancer. Cell Death Discov. 2024;10(1):143. PMID: 38490978; PMCID: PMC10943227. doi:10.1038/s41420-024-01895-7
125. Eisele G, Wischhusen J, Mittelbronn M, et al. TGF-beta and metalloproteinases differentially suppress NKG2D ligand surface expression on malignant glioma cells. Brain. 2006;129(Pt 9):2416–2425. PMID: 16891318. doi:10.1093/brain/awl205
126. Wang S, Jiao B, Geng S, Ma S, Liang Z, Lu S. Combined aberrant expression of microRNA-214 and UBC9 is an independent unfavorable prognostic factor for patients with gliomas. Med Oncol. 2014;31(1):767. PMID: 24277415. doi:10.1007/s12032-013-0767-5
127. Li X, Meng Y. SUMOylation regulator-related molecules can be used as prognostic biomarkers for glioblastoma. Front Cell Dev Biol. 2021;9:658856. PMID: 33898460; PMCID: PMC8063029. doi:10.3389/fcell.2021.658856
128. Fernandez-Gil BI, Schiapparelli P, Navarro-Garcia de Llano JP, et al. Effects of PREOPERATIVE radiotherapy in a preclinical glioblastoma model: a paradigm-shift approach. J Neurooncol. 2024;169(3):633–646. PMID: 39037687. doi:10.1007/s11060-024-04765-5
129. Fares J, Davis ZB, Rechberger JS, et al. Advances in NK cell therapy for brain tumors. NPJ Precis Oncol. 2023;7(1):17. PMID: 36792722; PMCID: PMC9932101. doi:10.1038/s41698-023-00356-1
130. Nakazawa T, Morimoto T, Maeoka R, et al. CIS deletion by CRISPR/Cas9 enhances human primary natural killer cell functions against allogeneic glioblastoma. J Exp Clin Cancer Res. 2023;42(1):205. PMID: 37563692; PMCID: PMC10413513. doi:10.1186/s13046-023-02770-6
131. Huang RS, Lai MC, Shih HA, Lin S. A robust platform for expansion and genome editing of primary human natural killer cells. J Exp Med. 2021;218(3):e20201529. PMID: 33433623; PMCID: PMC7808298. doi:10.1084/jem.20201529
132. Daly J, Sarkar S, Natoni A, et al. Targeting hypersialylation in multiple myeloma represents a novel approach to enhance NK cell-mediated tumor responses. Blood Adv. 2022;6(11):3352–3366. PMID: 35294519; PMCID: PMC9198929. doi:10.1182/bloodadvances.2021006805
133. Zhang Z, Kong X, Ligtenberg MA, et al. RNF31 inhibition sensitizes tumors to bystander killing by innate and adaptive immune cells. Cell Rep Med. 2022;3(6):100655. PMID: 35688159; PMCID: PMC9245005. doi:10.1016/j.xcrm.2022.100655
134. Molfetta R, Milito ND, Zitti B, et al. The Ubiquitin-proteasome pathway regulates Nectin2/CD112 expression and impairs NK cell recognition and killing. Eur J Immunol. 2019;49(6):873–883. PMID: 30888046. doi:10.1002/eji.201847848
135. Pu J, Sharma A, Liu T, Hou J, Schmidt-Wolf IG. Synergistic integration of histone deacetylase inhibitors apparently enhances the cytokine-induced killer cell efficiency in multiple myeloma via the NKG2D pathway. Clin Transl Immunology. 2024;13(3):e1500. PMID: 38529413; PMCID: PMC10961996. doi:10.1002/cti2.1500
136. Bi J, Huang C, Jin X, et al. TIPE2 deletion improves the therapeutic potential of adoptively transferred NK cells. J Immunother Cancer. 2023;11(2):e006002. PMID: 36725083; PMCID: PMC9896240. doi:10.1136/jitc-2022-006002
137. Feinberg D, Ramakrishnan P, Wong DP, Asthana A, Parameswaran R. Inhibition of O-GlcNAcylation decreases the cytotoxic function of natural killer cells. Front Immunol. 2022;13:841299. PMID: 35479087; PMCID: PMC9036377. doi:10.3389/fimmu.2022.841299
138. Long GV, Shklovskaya E, Satgunaseelan L, et al. Neoadjuvant triplet immune checkpoint blockade in newly diagnosed glioblastoma. Nat Med. 2025;31(5):1557–1566. PMID: 40016450; PMCID: PMC12092302. doi:10.1038/s41591-025-03512-1
139. Alonso-Valenteen F, Mikhael S, Wang H, et al. Systemic HER3 ligand-mimicking nanobioparticles enter the brain and reduce intracranial tumour growth. Nat Nanotechnol. 2025;20(5):683–696. PMID: 39984637; PMCID: PMC12095042. doi:10.1038/s41565-025-01867-7
140. Wang W, Li T, Cheng Y, et al. Identification of hypoxic macrophages in glioblastoma with therapeutic potential for vasculature normalization. Cancer Cell. 2024;42(5):815–832.e12. PMID: 38640932. doi:10.1016/j.ccell.2024.03.013
141. Zhang G, Zheng P, Lv Y, Shi Z, Shi F. m6A regulatory gene-mediated methylation modification in glioma survival prediction. Front Genet. 2022;13:873764. Erratum in: Front Genet. 2022 Dec 05;13:1106086. doi: 10.3389/fgene.2022.1106086. PMID: 35559019; PMCID: PMC9086406. doi:10.3389/fgene.2022.873764
142. Wang J, Shen S, You J, et al. PRMT6 facilitates EZH2 protein stability by inhibiting TRAF6-mediated ubiquitination degradation to promote glioblastoma cell invasion and migration. Cell Death Dis. 2024;15(7):524. PMID: 39043634; PMCID: PMC11266590. doi:10.1038/s41419-024-06920-2
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.