Back to Journals » International Journal of Nanomedicine » Volume 15
Dual Receptor-Targeted and Redox-Sensitive Polymeric Micelles Self-Assembled from a Folic Acid-Hyaluronic Acid-SS-Vitamin E Succinate Polymer for Precise Cancer Therapy
Authors Yang Y, Li Y, Chen K, Zhang L, Qiao S, Tan G, Chen F, Pan W ![]()
Received 11 February 2020
Accepted for publication 8 April 2020
Published 24 April 2020 Volume 2020:15 Pages 2885—2902
DOI https://doi.org/10.2147/IJN.S249205
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Linlin Sun
Yue Yang,1 Yunjian Li,1 Kai Chen,1 Ling Zhang,2 Sen Qiao,1 Guoxin Tan,1 Fen Chen,1,3,4 Weisan Pan1
1School of Pharmacy, Shenyang Pharmaceutical University, Shenyang 110016, People’s Republic of China; 2Department of Biotherapy, Cancer Research Institute, The First Affiliated Hospital of China Medical University, Shenyang 110001, People’s Republic of China; 3Key Laboratory of Ministry of Education for TCM Viscera-State Theory and Applications, Liaoning University of Traditional Chinese Medicine, Shenyang 110847, People’s Republic of China; 4Zhejiang Jingxin Pharmaceutical Co., Ltd, Zhejiang 312500, People’s Republic of China
Correspondence: Weisan Pan
School of Pharmacy, Shenyang Pharmaceutical University, Shenyang 110016, People’s Republic of China
Tel/ Fax +86 24 4352 0533
Email [email protected]
Fen Chen
Key Laboratory of Ministry of Education for TCM Viscera-State Theory and Applications, Liaoning University of Traditional Chinese Medicine, Shenyang 110847, People’s Republic of China
Tel/ Fax +86 24 3120 7125
Email [email protected]
Purpose: Poor site-specific delivery and insufficient intracellular drug release in tumors are inherent disadvantages to successful chemotherapy. In this study, an extraordinary polymeric micelle nanoplatform was designed for the efficient delivery of paclitaxel (PTX) by combining dual receptor-mediated active targeting and stimuli response to intracellular reduction potential.
Methods: The dual-targeted redox-sensitive polymer, folic acid-hyaluronic acid-SS-vitamin E succinate (FHSV), was synthesized via an amidation reaction and characterized by 1H-NMR. Then, PTX-loaded FHSV micelles (PTX/FHSV) were prepared by a dialysis method. The physiochemical properties of the micelles were explored. Moreover, in vitro cytological experiments and in vivo animal studies were carried out to evaluate the antitumor efficacy of polymeric micelles.
Results: The PTX/FHSV micelles exhibited a uniform, near-spherical morphology (148.8 ± 1.4 nm) and a high drug loading capacity (11.28% ± 0.25). Triggered by the high concentration of glutathione, PTX/FHSV micelles could quickly release their loaded drug into the release medium. The in vitro cytological evaluations showed that, compared with Taxol or single receptor-targeted micelles, FHSV micelles yielded higher cellular uptake by the dual receptor-mediated endocytosis pathway, thus leading to significantly superior cytotoxicity and apoptosis in tumor cells but less cytotoxicity in normal cells. More importantly, in the in vivo antitumor experiments, PTX/FHSV micelles exhibited enhanced tumor accumulation and produced remarkable tumor growth inhibition with minimal systemic toxicity.
Conclusion: Our results suggest that this well-designed FHSV polymer has promising potential for use as a vehicle of chemotherapeutic drugs for precise cancer therapy.
Keywords: micelles, paclitaxel, dual-targeted, redox-sensitive, cytotoxicity, antitumor
Introduction
Malignant tumors seriously threaten human life and health and are a great challenge for treatment worldwide. The development of numerous approaches to treat cancers is urgent. At present, chemotherapy is the most common strategy in cancer treatment.1 Among the most commonly used chemotherapeutic drugs, Paclitaxel (PTX), a microtubule-stabilizing agent, has a considerably wide chemotherapeutic range against various malignant tumors.2–4 However, the therapeutic capacity of PTX is extremely inhibited by its low therapeutic index and poor water solubility.5,6 To address these problems, Taxol®, a commercial formulation of PTX, was formulated by dissolving PTX in a 50:50 (v/v) mixture of Cremophor EL and dehydrated ethanol. Nevertheless, a series of Cremophor EL-dependent side effects, including hypersensitivity, myelosuppression and neurotoxicity, heavily restrict the clinical effects of Taxol®.7,8 As a result, in 2005, an albumin-bound PTX nanoparticle (Abraxane®) was approved by the United States Food and Drug Administration (FDA). Although avoiding the side effects of Cremophor EL, Phase III clinical trials of Abraxane® still reveal the appearance of neutropenia and sensory neuropathy in patients.9,10 In view of this, a low-toxicity and efficient delivery system for PTX is urgently needed.
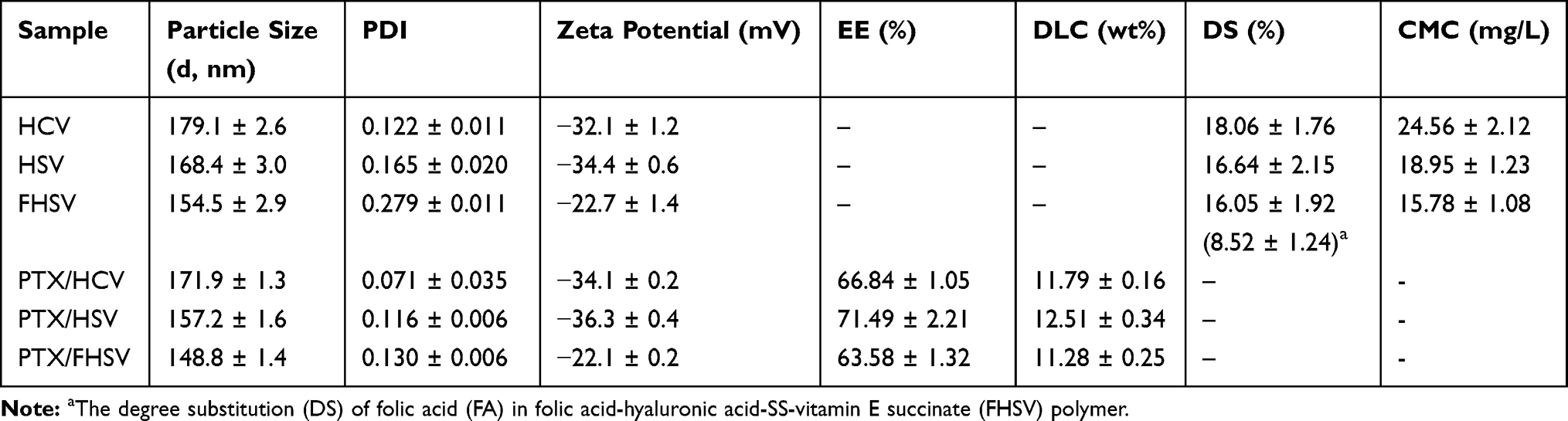 |
Table 1 Physicochemical Characteristics of PTX-Loaded Micelles (n=3) |
Recently, many new drug delivery platforms for PTX, including prodrugs,11 liposomes,12 solid lipid nanoparticles13 and polymeric micelles,14 have emerged to circumvent the drawbacks of PTX. Polymeric micelles have been regarded as an effective nanoplatform because of their ability to encapsulate hydrophobic agents into their “core-shell” structure and deliver these agents to tumor regions by the enhanced permeability and retention (EPR) effect. Nevertheless, this EPR effect only facilitates the accumulation of micelles in tumors, and insufficient intracellular uptake remains a great challenge for effective chemotherapy.15,16 To overcome the limitation of the EPR effect, researchers have proposed that tumor cell-specific ligands be utilized as targeting moieties to improve the internalization of micelles within tumor sites. Accordingly, modified micelles bearing multiple tumor-targeted ligands, such as peptides,17 transferrin,18 folic acid (FA)19 and hyaluronic acid (HA),20 have been developed, which can readily enter cancer cells via receptor-mediated endocytosis rather than by relying on the EPR effect alone, thus obviously enhancing the antitumor efficacy and reducing systematic toxicity. Owning to its nontoxicity, low immunogenicity, high biodegradability and biocompatibility, HA has been widely applied in pharmaceutical fields.21 Moreover, HA possesses a selective binding affinity to receptors, such as cluster determinant 44 (CD44) receptor, that are overexpressed within numerous tumors and thus can act as a ligand for active tumor targeting.22,23 Based on the strengths mentioned above, a variety of hydrophobic agents, such as deoxycholic acid,24 poly(L-histidine)25 and stearic acid,26 have been chemically conjugated to the backbones of HA to assemble into micelles for targeted delivery of anticancer drugs. However, the aggregation and saturation of receptors might be nonnegligible blocking factors for CD44-mediated internalization, resulting in compromised targeting efficiency.27 To address these complicated problems, dual receptor-targeted strategies have drawn great attention from researchers.27–29 Dual ligand-modified micelles can be developed to recognize two different receptors of tumor cells, thus producing a synergistic targeting capability. As reported, FA not only has low immunogenicity, storage stability and ready availability but also possesses a high binding affinity to FA receptor (FR) which is overexpressed in several carcinomas but only at low levels in normal cells.30,31 When FA-modified nanoparticles encounter with FR presented on the surface of tumor cells, they are easily captured into cells by the ligand-receptor binding effect. More importantly, the simultaneous overexpression of the CD44 receptor and FR on several tumor cells has been reported.32–34 For example, Lee et al synthesized amphiphilic HA derivative along with 6.5% FA substitution.35 Similarly, Liu et al also adopted FA- and HA-modified micelles to enhance cellular uptake.34 However, we noticed that these micelles exhibit incomplete and delayed drug release after internalization by tumor cells, which has become a crucial explanation for the compromised treatment outcomes. Hence, intelligent dual-targeted micelles based on the binding of HA to CD44 receptor and FA to FR should be rationally designed, which can not only improve the targeting capacity of micelles but also guarantee on-demand intracellular drug release.
Recently, stimuli-responsive micelles have been developed to trigger rapid and complete drug release in tumor cells according to the unique physiological conditions of the tumor microenvironment.36,37 It is generally known that the concentration of glutathione (GSH) in the tumor intracellular milieu is much higher than that of the extracellular milieu.38 This redox-heterogeneous environment provides the basis of on-demand drug release at tumor sites. Among them, disulfide bonds have been commonly used as redox-responsive linkages. They are relatively stable in blood circulation but cleave by means of thiol-disulfide exchanges in the presence of GSH stimuli once entering tumor cells.39 As a result, shell-sheddable micelles with disulfide linkages between hydrophilic shells and hydrophobic cores can be exploited to respond to overproduced GSH in tumor cells, achieving precise and rapid drug release.
In this study, we, for the first time, synthesized an amphiphilic folic acid-hyaluronic acid-SS-vitamin E succinate (FHSV) polymer by utilizing both HA and FA as tumor-targeted ligands and disulfide bonds as redox-responsive linkages for on-demand intracellular delivery of PTX in tumor therapy. (Figure 1) In addition, vitamin E succinate (VES) was selected as the hydrophobic core due to its highly selective toxicity towards tumor cells but low toxicity towards healthy cells.40 The redox sensitivity and tunable drug release behavior were studied under high concentration of GSH. Furthermore, in vitro cytotoxicity, apoptosis and cellular uptake studies were performed in human breast cancer cell (MCF-7) and mice embryonic fibroblast cells (NIH3T3 cells). Last, a series of in vivo studies were comprehensively conducted to investigate the anticancer activity and biological safety of this innovative and targeted drug delivery system.
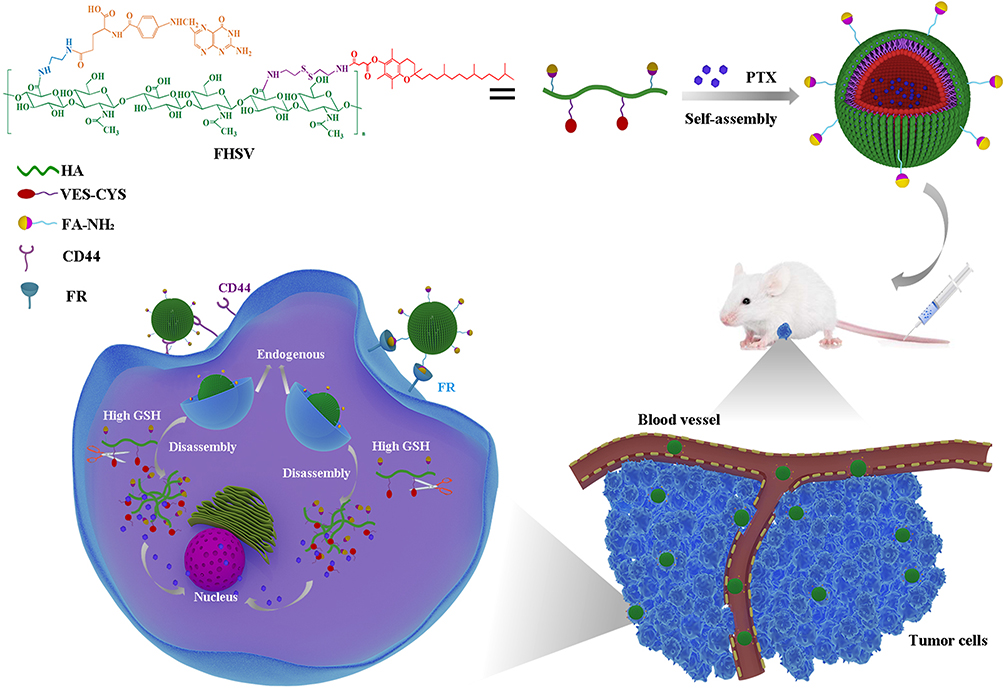 |
Figure 1 Illustration of self-assembly, tumor accumulation and intracellular release of PTX/FHSV micelles. |
Materials and Methods
Materials
Hyaluronic acid (HA, MW=8000 kDa) was obtained from Freda Biochem Co., Ltd. (Shandong, China). Vitamin E succinate (VES), cystamine dihydrochloride (CYS•2HCl), coumarin 6 (C6), glutathione (GSH) and folic acid (FA) were all bought from Aladdin Bio-Chem Technology Co., Ltd (Shanghai, China). Ethylene diamine (EDA) was purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Paclitaxel (PTX) of purity 99% and DiR were bought from Dalian Meilun Biotechnology Co., Ltd. (Dalian, China). 1-Ethyl-3(3-dimethylaminopropyl) carbodiimide (EDC), N-hydroxysuccinimide (NHS) and N, N’-dicyclohexylcarbodiimide (DCC) were obtained from Shanghai Medpep Co., Ltd (Shanghai, China). 4ʹ,6-diamidino-2-phenylindole (DAPI), 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT), 4% paraformaldehyde and Annexin V-FITC/propodium iodide (Annexin V-FITC/PI) Apoptosis Detection Kit were provided by Beyotime Institute of Biotechnology (Shanghai, China).
Cell Culture
MCF-7 cells and NIH3T3 cells were provided by the Chinese Academy of Sciences Cell bank (Shanghai, China). MCF-7 cells were cultured in Roswell Park Memorial Institute 1640 (RPMI 1640) medium, and NIH3T3 cells were cultured in Dulbecco’s modified eagle’s medium (DMEM). Both cell culture media were supplemented with 10% (v/v) FBS and 1% penicillin-streptomycin solution in a humidified atmosphere with 5% CO2 at 37 °C.
Synthesis of VES-CYS and VES-EDA Linkers
VES was modified with cystamine (CYS) via an amidation reaction. In detail, VES (1 mmol), EDC (2 mmol) and NHS (2 mmol) were dispersed in 20 mL of DMF to activate the carboxylic groups of VES for 2 h. Later, 5 mL of DMF solution of CYS•2HCl (3 mmol) and triethylamine (TEA, 400 μL) stirred for 1 h in advance were added. After 24 h of stirring at ambient temperature, the solution was extracted several times with distilled water and dichloromethane to remove excess activators and CYS and then dried under vacuum to obtain VES-CYS. The structure of VES-CYS was characterized by 1H-nuclear magnetic resonance (1H-NMR) in DMSO-d6 (Figure S1A): δ (ppm) 0.78 (12H, 4CH3), 0.98–1.54 (21H, the aliphatic methylene protons), 2.78 (4H, 2CH2S), 3.12 (2H, CH2NH), 3.32 (2H, CH2NH2). As a control, the nonreductive VES-EDA linker was synthesized as described above using EDA instead of CYS and TEA. The structure of VES-EDA was confirmed by 1H-NMR in DMSO-d6 (Figure S1A): δ (ppm): 0.78 (12H, 4CH3), 0.98–1.54 (21H, the aliphatic methylene protons), 2.59 (2H, CH2NH), 3.01 (2H, CH2NH2).
Synthesis of HA-CYS-VES (HSV) and HA-EDA-VES (HCV) Polymers
The amphiphilic HSV and HCV polymers were prepared by conjugating VES-CYS and VES-EDA to the backbone of HA, respectively, as reported previously with some modifications.41 Briefly, HA (1 mmol) was activated for 2 h in 20 mL of formamide with the help of EDC (1.5 mmol) and NHS (1.5 mmol) in an ice bath. Next, 0.5 mol of VES-CYS or VES-EDA in 80 mL of DMF was added dropwise and then stirred overnight. Excess precooled acetone was added to precipitate the reaction solution. After rinsing thrice with cold acetone, the precipitation was redissolved in water and then dialyzed against excess distilled water for 3 days. Subsequently, HSV and HCV polymers were collected by lyophilization.
Synthesis of Amphiphilic FHSV Polymer
To synthesize the FHSV polymer, the amination of FA was first performed according to the published literature with some modifications.42 Briefly, DCC, NHS and FA were first dispersed in 20 mL of DMSO at a molar ratio of 1.5: 1.5: 1. After stirring at 50 °C for 6 h, excess EDA and pyridine were added, and that mixture continued to react overnight at ambient temperature. Then, the reaction solution was precipitated in diethyl ether thrice. The product was dried under vacuum for 12 h to obtain the yellow powder of FA-NH2. The FA-NH2 was characterized by 1H-NMR in DMSO-d6 (Figure S1B): δ (ppm): 2.60 (2H, CH2NH), 3.07 (2H, CH2NH2), 6.65–7.64 (4H, the protons of benzene ring), 8.65 (1H, the proton of pteridine ring).
HSV, EDC and NHS with a molar ratio of 1: 1.5: 1.5 were mixed in 20 mL of formamide with stirring for 2 h in an ice bath, followed by the addition of 0.15 mmol FA-NH2 dissolved in 80 mL of DMSO under vigorous stirring. After 24 h at ambient temperature, the solution was purified by dialysis against DMSO/water (3:1–1:1, v/v) for 3 days to remove the unreacted FA-NH2, followed by dialysis against water for 2 days to remove DMSO. Eventually, the resultant FHSV polymer was collected by lyophilization.
Characteristics of HSV, HCV and FHSV Polymers
1H-NMR Analysis
The chemical structures of HA, HSV, HCV and FHSV were determined by 1H-NMR (BRUKER AVANCE Ⅲ, Switzerland). The degree of substitution (DS) of VES, which was defined as the number of VES per 100 sugar units of HA, was calculated according to a previous report.43 In addition, the DS of FA in the FHSV polymer was analyzed via an Ultraviolet-Visible spectrophotometer at 361 nm.44
Critical Micelle Concentration (CMC)
The determination of CMC of the graft polymers were undertaken using pyrene as a fluorescent probe.24 Briefly, after evaporation of 1 mL of 6×10−6 M pyrene acetone solution under a nitrogen flow, a series of block polymer solutions with concentration varying from 1×10−3 to 0.5 mg/mL were added into each volumetric flask resulting in a final pyrene concentration of 6×10−7 M, followed by sonication in a water bath for 30 min. Thereafter, all samples were incubated at 50 °C for 1 h and then left at room temperature overnight. Next, the fluorescence intensity of pyrene at excitation wavelength of 333 nm and 338 nm was monitored with a fixed emission wavelength of 390 nm. The CMC values were obtained by plotting the intensity ratio of I338/I333 versus the Log-transformed polymer concentration.
Redox-Triggered Disassembly of Blank Micelles
The polymer (HCV, HSV or FHSV) was dispersed in phosphate buffered saline (PBS, pH 7.4, 10 mM) and the solution was sonicated for 15 min at 100 W in an ice bath. Then, the resulting solution was filtered through a 0.45 μm microfiltration membrane to obtain the blank HCV micelles, blank HSV micelles or blank FHSV micelles. The size distribution of blank micelles under stimulation with various concentration of GSH was monitored via the dynamic light scattering (DLS) method. Briefly, blank HSV and FHSV micellar solution were incubated with various concentration of GSH in a thermostatic vibrating incubator at 37 °C. After 24 h, their size changes were investigated by DLS. For comparison, the change in particle size of blank HCV micellar solution in the presence of 20 mM of GSH was also analyzed.
Preparation and Characterizations of PTX-Loaded Micelles
A dialysis method was applied to formulate drug-loaded micelles according to a reference with some modifications.24 First, 12 mg of amphiphilic polymer (HCV, HSV or FHSV) was dissolved in 4 mL of water. Then, a PTX/ethanol stock solution was injected into the polymer solution (PTX/polymer = 20%, w/w). After stirring for 2 h, the organic solvent was removed by dialysis in water for 10 h. Then, the mixture was sonicated for 15 min at 100 W in an ice bath. The final micellar dispersion (PTX/FHSV, PTX/HSV or PTX/HCV) was obtained by filtration through a 0.45 μm microfiltration membrane.
The particle size and zeta potential were determined by DLS at 25 °C with a scattering angle of 90° in triplicate. In addition, the morphology of PTX-loaded micelles was observed by transmission electron microscopy (TEM) through a JEOL JEM 1200 TEM microscope (Tokyo, Japan) at 120 kV. To calculate the drug loading capacity (DLC) and encapsulation efficiency (EE), 0.2 mL PTX-loaded micelles were mixed with 10 mL ethanol, and the solution was homogenized by ultrasonication for 20 min. The PTX amount was analyzed using high-performance liquid chromatography (HPLC).
Power X-ray diffraction (PXRD) analysis was conducted to investigate the change in the crystalline state of PTX in the micelles. All samples were scanned from 3° to 40° (2θ) at a scanning speed of 3°/min with a step size of 0.03°. The potential and current of X-ray tube were set to 40 mV and 30 mA, respectively.
In vitro Drug Release
The cumulative release of PTX from micelles was determined using a dialysis method. 1 mL of self-prepared Taxol or PTX-loaded micellar solution containing about 80 μg PTX was immersed in 50 mL of 0.01 M PBS solution (pH 7.4) containing 0.5% (w/v) Tween 80. Various concentration of GSH (0 mM or 10 mM) were added to the buffer solution and all samples were shaken at 100 rpm for 60 h in a 37 °C thermostatic oscillator. At specific time intervals, 1 mL samples were collected and replaced by an equivalent volume of fresh buffer. The cumulative release of PTX was calculated by HPLC.
Hemolysis Study
The biosafety of micelles was preliminarily investigated by a hemolysis assay. Briefly, rabbit erythrocytes were collected by centrifugation and suspended in saline at 2.0% (v/v) hematocrit. Different PTX-loaded formulations were mixed with an equivalent volume of red blood cells (RBC) suspensions, and the mixed solution was shaken at 37 °C for 3 h. The RBC suspensions incubated with distilled water and physiological saline were set as the positive control and negative control, respectively. After 15 min of centrifugation at 5000 rpm, the hemolysis ratio was calculated using a UV–visible spectrophotometer at 540 nm.
In vitro Cytotoxicity Study
The toxicity of blank carriers and PTX-loaded formulations was determined by the MTT assay. MCF-7 or NIH3T3 cell suspensions were seeded in 96-well plates and incubated for 24 h. Then, the cells were cultured with 100 μL of fresh medium containing a series of concentrations of blank carriers and PTX-loaded formulations for 48 h. Untreated cells were used as controls. After that, the culture medium was replaced by 90 μL of fresh FBS-free medium and 10 μL of MTT solution (5 mg/mL), and the cells were further incubated for 4 h. Next, the supernatant was aspirated, and the generated formazan was dissolved in 150 μL of DMSO. The absorbance values were recorded using a microplate reader (Varioskan Flash, Thermo Scientific, USA) at 490 nm. Additionally, the IC50 values were determined with SPSS Statistics 22.0 (IBM, New York, USA).
To investigate the receptor-mediated effect, competitive inhibitory experiments were carried out according to the published reports with some modifications.34,45,46 MCF-7 cells were precultured either with free HA (10 mg/mL) or with both free HA (10 mg/mL) and free FA (1 mM) for 1 h prior to treatment with PTX/FHSV micelles. The cytotoxicity was measured by MTT assay as described above.
In vitro Cell Apoptosis Study
The apoptosis rate of MCF-7 cells induced by various drug-loaded formulations was quantitatively evaluated by Annexin V-FITC/PI staining method. The cells were cultured with Taxol, PTX/HCV, PTX/HSV and PTX/FHSV micelles at an equivalent PTX concentration. Untreated cells were used as controls. After 12 h, the cells were harvested for Annexin V-FITC/PI staining. Finally, the stained cells were analyzed by flow cytometer (BD FACSAriaTM Ⅲ, USA).
Cellular Uptake
With respect to the evaluation of cellular uptake of micelles, fluorescence probe C6, as a substitute for PTX, was entrapped in polymeric micelles in line with the protocol of PTX-loaded micelles.47 C6 solution prepared with reference to Taxol was used as a control. When the MCF-7 cells reached approximately 80% confluence, various C6-loaded formulations at equivalent C6 concentration of 0.5 μg/mL were added to incubate with cells for additional 1 h and 4 h. In the competitive inhibitory experiment, the cells were precultured either with free HA (10 mg/mL) or with both free HA (10 mg/mL) and free FA (1 mM) for 1 h to saturate the receptors expressed on cell surface. Finally, the cells were harvested by trypsinization for flow cytometry evaluation.
Furthermore, the ability of MCF-7 cells to capture micelles was qualitatively analyzed by confocal laser scanning microscopy (LSM710, CarlZeiss, Oberkochen, Germany). First, MCF-7 cells were seeded on coverslips in 12-well plates and incubated to reach 70–90% confluence. Then, the cells were treated with fresh medium containing different C6-loaded formulations (0.5 μg/mL C6). In the competitive experiments, the cellular pretreatment was similar to that for flow cytometry. After cultured for 1 h or 4 h, the cells were rinsed thrice with cold PBS, fixed with 4% paraformaldehyde for 30 min. The cell nucleus was stained with DAPI solution for 3 min. The intracellular distribution of C6 was visualized through CLSM.
Animals and Tumor Xenograft Model
Kunming mice (female, 18 ± 4 g) were Liaoning Changsheng biotechnology Co. Ltd. All animal experimental procedures were approved by the Animal Ethics Committee of Shenyang Pharmaceutical University (Shenyang, China) and were conducted according to the Principles of Laboratory Animals Care. S180 cells suspension was inoculated into the left axillae of mice to develop a subcutaneous tumor model.
In vivo Biodistribution and Tumor-Targeting Ability
The near-infrared fluorescence (NIRF) imaging method was adapted to observe the tumor-targeted potency and in vivo biodistribution of micelles. DiR was chosen to replace PTX for in vivo imaging experiments.47 When the tumor volume was increased to approximately 300–400 mm3, DiR solution prepared with reference to Taxol and various DiR-loaded micelles were injected intravenously at a dosage of 2 mg DiR/kg (n=3). After the mice were sacrificed at predetermined time points, NIRF images of tumor-bearing mice were obtained by Bruker MI SE imaging system (Bruker Corporation, Germany). In addition, the major tissues were dissected after 24 h of administration to observe the biodistribution of DiR-loaded formulations in each organ.
In vivo Chemotherapy of Tumors
The S180 tumor-bearing mice were randomly assigned to five groups (n=5) when the tumor volume reached approximately 100 mm3, and each mouse was injected intravenously every other day with Taxol and PTX-loaded micelles at a dose of 10 mg PTX/kg on days 1, 3, 5, 7, 9 and 11, using saline injection as a control. Moreover, the tumor size and body weight of mice were monitored throughout the treatment. On day 13, tumor tissues and major organs were excised and stained with hematoxylin and eosin (H&E) for histological analysis. In addition, terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) and anti-CD31 staining were carried out to analyze the apoptosis and angiogenesis of tumor cells, respectively. The tumor inhibitory rate (TIR) was measured according to Equation 1:48
(1)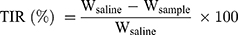
where Wsaline and Wsample are the average tumor weights of mice treated with saline and PTX-loaded formulations, respectively.
Statistical Analysis
All quantitative data are shown as the mean ± standard deviation (SD). Statistical significance was determined with one-way analysis of variance (ANOVA) and Student′s t-test at 95% confidence levels.
Results and Discussion
Synthesis and Characterizations of HCV, HSV and FHSV Polymers
Synthesis of HCV, HSV and FHSV Polymers
Herein, the synthetic scheme of HCV, HSV and FHSV polymers was described in Figure S2. Initially, VES was covalently coupled to the backbone of HA via an amide reaction, utilizing disulfide bonds and alkyl bonds as the linkages, to synthesize CD44-targeted redox-sensitive HSV and redox-insensitive HCV, respectively. Second, amine-functionalized FA was conjugated to HSV using EDC/NHS as catalysts, thus obtaining both CD44- and FR-targeted redox-sensitive FHSV. The chemical structures of HCV, HSV and FHSV were verified by 1H-NMR in DMSO-d6/D2O (Figure 2A and B). The signal peak at 1.94 ppm was assigned to the N-acetyl groups of HA, and the broad multiplet between 3.16 ppm and 4.44 ppm belonged to the methylene protons in the sugar rings of HA. In the spectrum of HSV, the characteristic peaks at 2.83 ppm and 0.78 ppm were attributed to the methylene protons of CYS (4H, 2CH2S) and the methyl protons of VES (12H, 4CH3), respectively, suggesting the successful preparation of HSV polymer. Similarly, the successful conjugation of VES-EDA onto the backbone of HA was confirmed by the appearance of peaks at 0.83 ppm and 2.89 ppm, which belonged to the methyl protons of VES (12H, 4CH3) and the methylene protons of EDA (4H, 2CH2NH). Compared to the spectra of HSV and HCV, the newly emerged signal peaks at 6.76 ppm, 7.65 ppm and 8.74 ppm in the 1H-NMR spectrum of FHSV were ascribed to the protons of benzene rings and pteridine rings of FA, suggesting the successful conjugation of FA to HSV.
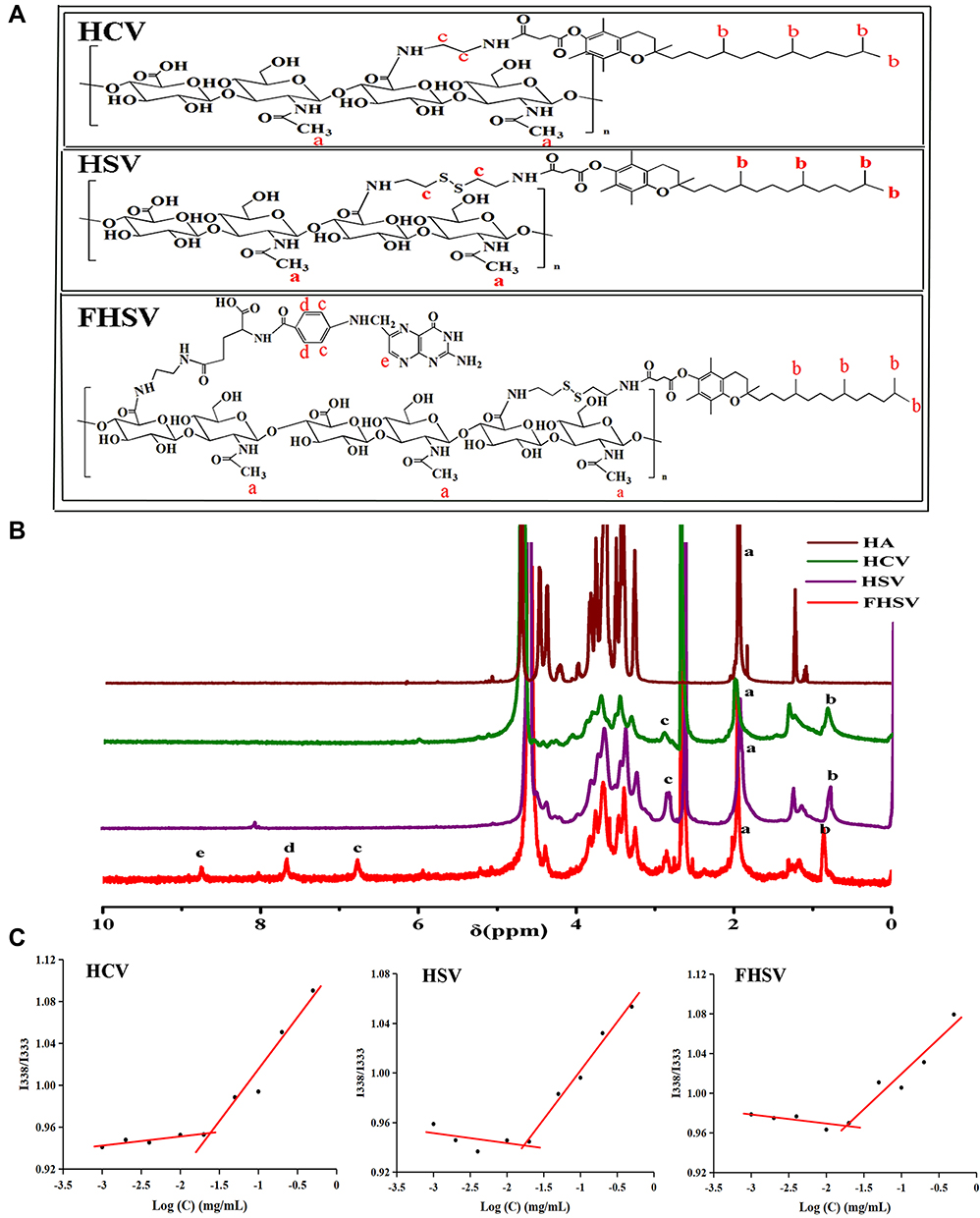 |
Figure 2 (A) Chemical structures of HCV, HSV and FHSV polymers. (B) 1H-NMR spectra of HA in D2O, HCV, HSV and FHSV polymers in DMSO-d6/D2O (v/v, 1:1). (C) The CMC of HCV, HSV and FHSV polymers. |
The DS of VES for HCV, HSV and FHSV polymers were quantitatively determined by the ratio of integration area of the methyl protons of VES (~0.78 ppm) to the N-acetyl groups of HA (1.94 ppm), which were 18.06 ± 1.76%, 16.64 ± 2.15% and 16.05 ± 1.92%, respectively. In addition, the DS of FA for FHSV measured by UV-vis spectrophotometry was 8.52 ± 1.24%.
Critical Micelle Concentration (CMC)
The CMC values of amphiphilic polymers were determined by fluorescence spectroscopy. As shown in Figure 2C, the CMC values of HCV, HSV and FHSV were 24.56 ± 2.12 mg/L, 18.95 ± 1.23 mg/L and 15.78 ± 1.08 mg/L, respectively. The CMC of the well-designed polymers were significantly lower than those of low molecular weight surfactants, such as Pluronic F127 (0.24 mg/mL).49 Moreover, the relatively low CMC was beneficial for maintaining the stability of polymeric micelles in blood circulation before reaching the target sites.26
Redox-Triggered Disassembly of Blank Micelles
To illustrate the redox-sensitive breakage of disulfide bonds in the micelles, the size distribution of blank HSV and FHSV micelles upon exposure to various concentration of GSH over time was measured with blank HCV micelles as the control. As apparent from Figure 3, the particle size of all blank micelles exhibited no obvious change after incubation in PBS for 24 h. In contrast, the presence of 10 mM GSH led to an increase in the particle size of FHSV micelles and HSV micelles exhibiting the multipeak phenomenon in distribution. Furthermore, significant size growth and a broader size distribution were observed under the condition of 20 mM GSH, suggesting the reduction-triggered breakage of disulfide bonds would rely on the amount of the surrounding GSH.24 However, simulation of the redox potential of extracellular compartment by 10 μM GSH caused negligible size variation in FHSV and HSV micelles under otherwise unchanged conditions. As a control, the particle size of redox-insensitive HCV micelles was almost constant even after exposure to 20 mM GSH. Therefore, these results confirmed that redox-sensitive FHSV micelles and HSV micelles could be stable under normal physiological conditions but rapidly disassemble under GSH-rich environment, such as in the cytoplasm of tumor cells, which excellently resolved the contradiction between extracellular stability and intracellular release.
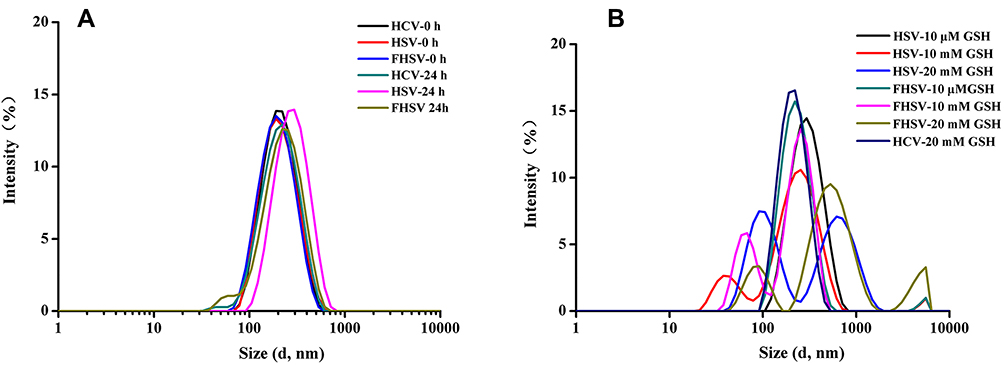 |
Figure 3 Size distribution of blank micelles after incubation (A) in PBS (10 mM, pH 7.4) for 0 h and 24 h (B) in PBS (10 mM, pH 7.4) with 10 μM, 10 mM and 20 mM GSH for 24 h. |
Preparation and Characterizations of PTX-Loaded Micelles
A simple dialysis method was applied to encapsulate hydrophobic PTX into the hydrophobic cores of polymeric micelles. Table 1 summarizes a variety of properties of blank micelles and drug-loaded micelles. The hydrodynamic diameters of PTX/HCV micelles, PTX/HSV micelles and PTX/FHSV micelles were 171.9 ± 1.3 nm, 157.2 ± 1.6 nm, and 148.8 ± 1.4 nm, respectively, which were larger than those of corresponding blank micelles. This phenomenon might be because the drug-loading process resulted in a more compact nanostructure through hydrophobic interactions between PTX and the hydrophobic inner cores of polymeric micelles.50 Furthermore, the polydispersity index (PDI) of all drug-loaded micelles was in a narrow range of 0.071 to 0.130, indicating a uniform particle size distribution. The morphology was further detected by TEM (Figure 4A). The TEM images all displayed uniform, near-spherical structures and narrow size distributions. However, the average size of TEM measurement was smaller than that of DLS measurement, which might be attributed to the removal of water from micelles during TEM sample preparation leading to the shrinkage of hydrophilic shell of micelles.51 The zeta potential of all drug-loaded micelles was negative because of the existence of free carboxyl groups on the backbone of HA, which helped to reduce their binding to serum proteins and prolong the in vivo circulation time of the drug. More notably, PTX/FHSV micelles had a higher potential (−22.1 ± 0.2 mV) than PTX/HSV micelles (−36.3 ± 0.4 mV) and PTX/HCV micelles (−34.1 ± 0.2 mV). This might be caused by the reduction in the number of free carboxyl groups in FHSV polymer after connecting the carboxyl groups of HA with the primary amines of FA-NH2. As shown in Table 1, due to the similar DS of VES, the DLC and EE of the three drug-loaded micelles were comparable, and were in the range of 11.28–12.51% and 63.58–71.49%, respectively. Moreover, modification of FA has a small influence on the DLL and EE.
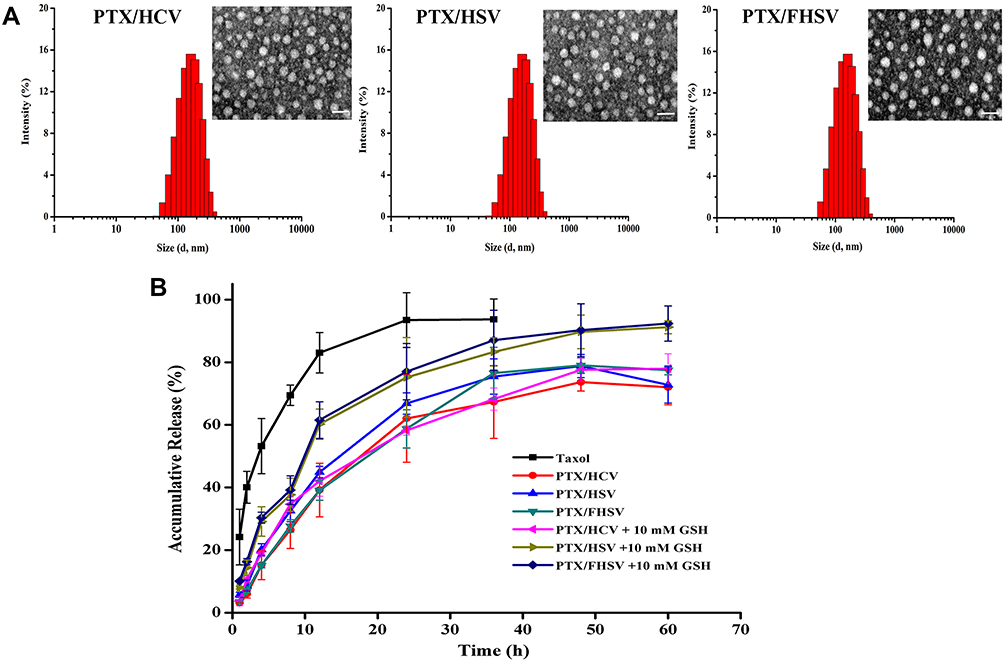 |
Figure 4 (A) Size distribution and TEM images of PTX-loaded micelles. Scale bar represents 50 μm. (B) In vitro drug release of Taxol and PTX-loaded micelles in PBS (10 mM, pH 7.4) with or without 10 mM GSH. Data are presented as mean ± SD (n = 3). |
PXRD analysis was carried out to verify the existence state of PTX in the PTX-loaded micelles. As shown in Figure S3, PTX exhibited several intense peaks at 2θ values of 5.64°, 9.12°, 10.11°, 11.16° and 12.66° and numerous small peaks in the range of 15.67° to 29.97°.52 These characteristic peaks appeared in the XRD pattern of the physical mixture of PTX and FHSV polymer but disappeared in the XRD spectra of PTX-loaded micelles. These observations indicated that PTX was distributed in the micelles in amorphous or molecular state.
In vitro Drug Release
Evidence suggests that tumor cells have a higher level of GSH than normal cells, which is beneficial to cancer therapy.38 Therefore, the cumulative release of micelles was measured in the presence or absence of GSH. As shown in Figure 4B, Taxol showed a rapid release of PTX with 83.26% at pH 7.4 within 12 h. In comparison, all PTX-loaded micelles showed slow and inefficient PTX release at pH 7.4, with only 37.93%, 46.47% and 40.96% of PTX released from PTX/HCV micelles, PTX/HSV micelles and PTX/FHSV micelles within 12 h, respectively. In addition, the cumulative release of all PTX-loaded micelles only reached 72.01–76.80% within 60 h. However, in the same buffer solution with 10 mM GSH, the release rate of PTX from PTX/HSV micelles and PTX/FHSV micelles was drastically accelerated, and the cumulative drug release reached 65.21% and 69.58% within 12 h, and 91.02% and 92.35% within 60 h, respectively. As expected, no obvious release acceleration was observed in PTX/HCV micelles in the presence of 10 mM GSH. These signified that the redox-sensitive micelles could be rapidly disassembled in the high concentration of GSH to achieve fast drug release and enhance their antitumor efficacy.
Hemolysis Study
Blood biocompatibility is a crucial issue in clinical administration, particularly in intravenous administration, so a hemolysis study of PTX-loaded micelles was conducted. Figure S4 demonstrates that none of the micellar solution caused significant hemolysis of rabbit erythrocytes, and the hemolysis ratio was far below the acceptable range (< 5%). Additionally, when the PTX concentration was 100 μg/mL and 200 μg/mL, the hemolytic toxicity of PTX-loaded micelles was significantly lower than that of Taxol (p < 0.05), indicating their good biocompatibility for intravenous injection.
In vitro Cytotoxicity Study
To preliminarily evaluate the pharmaceutical application of micelles, the toxicity of blank micelles and drug-loaded micelles against NIH3T3 cells and MCF-7 cells were estimated by the MTT assay. Cremophor EL and Taxol were considered controls. As shown in Figure S5, compared to Cremophor EL, the cell viability of NIH3T3 cells treated with blank micelles was above 95%, even at high concentrations, indicating their biosafety to normal cells. For MCF-7 cells, the blank micelles showed no toxicity when the concentration of blank micelles was in the range of 5–200 μg/mL. However, at high concentration, the survival rates of MCF-7 cells were appreciably reduced and were 86.02% ± 2.69%, 81.07% ± 4.58% and 77.72% ± 4.75% for blank HCV micelles, blank HSV micelles and blank FHSV micelles, respectively. Moreover, the redox-sensitive blank HSV micelles and blank FHSV micelles showed higher cytotoxicity than redox-insensitive blank HCV micelles, indicating that the dissociation of polymers played an indirect effect on cell viability. According to the literature, VES could decrease the mitochondrial membrane of tumor cells and accelerate cell apoptosis, but had less toxicity or non-toxicity to normal cells.40 Therefore, compared to blank HCV micelles, the rapidly disaggregation of blank HSV micelles and FHSV micelles in high concentration GSH produced more VES to killing tumor cells.
The cytotoxicity of PTX-loaded formulations in NIH3T3 cells and MCF-7 cells was dose-dependent (Figure 5A–C). In MCF-7 cells, the half maximum inhibition concentration (IC50) values of Taxol, PTX/HCV micelles, PTX/HSV micelles and PTX/FHSV micelles were 0.885 ± 0.071 μg/mL, 0.818 ± 0.036 μg/mL, 0.357 ± 0.079 μg/mL and 0.200 ± 0.027 μg/mL, respectively. Compared to Taxol and PTX/HCV micelles, PTX/HSV micelles and PTX/FHSV micelles exhibited a statistically significant (p < 0.001) increase of cytostatic activity. Importantly, PTX/FHSV micelles demonstrated the best toxicity against MCF-7 cells. It was previously reported that MCF-7 cells exhibited high CD44 receptor and FR expression.53 So, the enhanced cytotoxicity of PTX/FHSV micelles might be attributed to the enhanced cellular uptake mediated by the binding of HA to CD44 receptor and FA to FR. Moreover, according to the results of in vitro drug release, PTX/FHSV micelles could rapidly release PTX in the GSH-rich tumor environment, thus leading to fast transport of PTX into the nucleus to restrain the cell proliferation. NIH3T3 cells which expressed a low level of CD44 receptor54 and FR55 were chosen to evaluate the safety of PTX-loaded micelles. It was obvious that all the PTX-loaded formulations revealed lower cytotoxicity than their counterparts in MCF-7 cells, indicating their high selectivity to carcinomatous cell lines. In contrast, Taxol could be readily internalized into NIH3T3 cells by passive diffusion, thus killing cells.
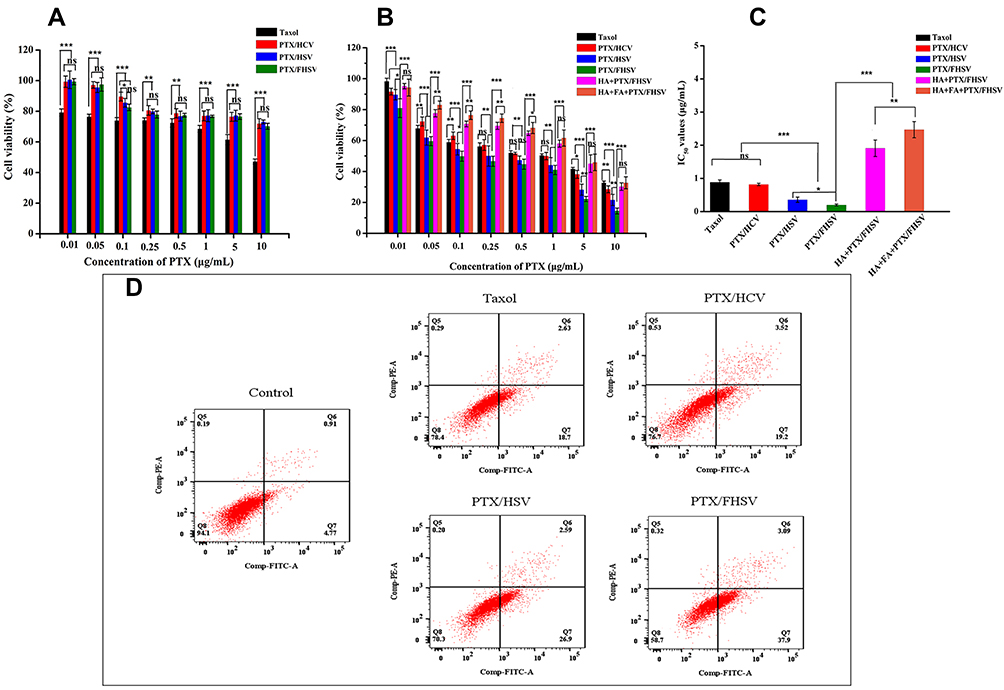 |
Figure 5 Cell viability of (A) NIH3T3 cells and (B) MCF-7 cells treated for 48 h with Taxol and PTX-loaded micelles. In the competitive inhibition experiment, the MCF-7 cells were precultured either with free HA (10 mg/mL) or with both free HA (10 mg/mL) and free FA (1 mM) for 1 h prior to treatment with PTX/FHSV micelles. (C) The IC50 values of PTX-loaded formulations against MCF-7 cells. Data are presented as mean ± SD (n=6). *p < 0.05; **p < 0.01; ***p < 0.001; ns, p > 0.05. (D) Apoptotisis of MCF-7 cells treated for 12 h with Taxol and PTX-loaded micelles. |
To ascertain the effect of receptor-mediated endocytosis on the enhanced cytotoxicity of PTX/FHSV micelles, competitive inhibitory experiments were conducted. Free HA and free FA were used as receptor inhibitors to competitively saturate the CD44 receptor and FR on the surface of MCF-7 cells, respectively. As shown in Figure 5B and C, shows that the cell viability of MCF-7 cells in the receptor inhibitor groups (HA+PTX/FHSV group or HA+FA+PTX/FHSV group) was drastically increased. Additionally, the IC50 value of HA+FA+PTX/FHSV group was about 2.470 ± 0.244 μg/mL, which was extremely higher than 1.910 ± 0.249 μg/mL of HA+PTX/FHSV group (p < 0.01). Therefore, these results underlined that the interaction of HA-CD44 binding and FA-FR binding played an active role in the therapy efficacy of PTX/FHSV micelles.
In vitro Cell Apoptosis Study
Annexin V-FITC/PI staining method was used to detect the apoptosis of MCF-7 cells induced by different PTX-loaded formulations. As shown in Figure 5D, all the PTX formulations produced significantly stronger apoptosis-inducing ability than the untreated control group (p < 0.01). Moreover, a total apoptotic rate (early apoptosis cells plus late apoptotic cells) of MCF-7 cells incubated with PTX/FHSV micelles was 40.99%, which was much higher than the 29.49% of PTX/HSV micelles, 22.72% of PTX/HCV micelles and 21.33% of Taxol. These results were consistent with the findings of MTT assay, indicating that our designed PTX/FHSV micelles would effectively promote the apoptosis of tumor cells and improve the antitumor efficacy of PTX.
Cellular Uptake
The cellular uptake of PTX-load micelles was studied in MCF-7 cells via flow cytometry and CLSM. To better evaluate the uptake behavior of PTX-loaded micelles, the fluorescent probe C6 was selected as a substitute for PTX. Figure 6 suggests that the cellular uptake of different C6 formulations occurred in a time-dependent manner. As expected, C6-loaded micelles exhibited significantly stronger mean fluorescent intensity (MFI) than C6 solution in MCF-7 cells (p < 0.01), which might be due to the enhanced endocytosis mediated by specific ligand-receptor interaction. In addition, after 4 h of incubation, the MFI of cells incubated with C6/FHSV micelles was 1.22-fold and 1.30-fold of those incubated with C6/HSV micelles and C6/HCV micelles, suggesting the high intracellular internalization capacity of C6/FHSV. The findings from the CLSM analysis were consistent with the results of flow cytometry, and the strongest green fluorescence signal was appeared in MCF-7 cells treated with C6/FHSV micelles.
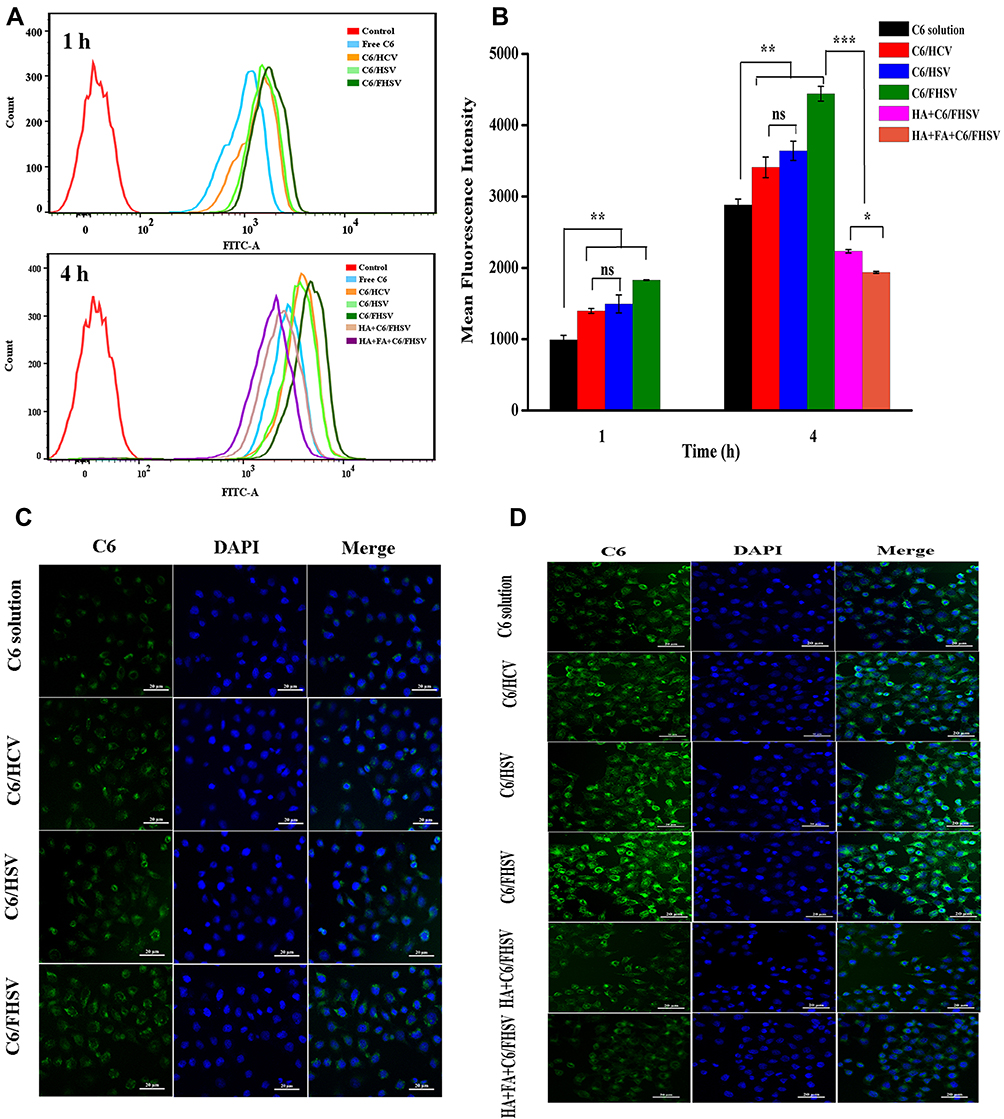 |
Figure 6 Time-dependent cellular uptake of C6-loaded formulations in MCF-7 cells. (A) Flow cytometry profiles and (B) quantification of MFI of MCF-7 cells following 1 h and 4 h incubation with C6-loaded formulations. Data are presented as mean ± SD. (n=3). *p < 0.05; **p < 0.01; ***p < 0.001; ns, p > 0.05. CLSM images of MCF-7 cells incubated for (C) 1 h and (D) 4 h with C6-loaded formulations. The competitive inhibition experiments were performed by treating MCF-7 cells either with free HA (10 mg/mL) or with free HA (10 mg/mL) and FA (1 mM) before adding C6/FHSV micelles. |
To verify that the FHSV micelles were selectively internalized by receptor-mediated endocytosis pathway into tumor cells, competitive experiments were conducted. Figure 6 shows that the C6 fluorescence intensity in MCF-7 cells was dramatically reduced by pretreating the cells with receptor inhibitors prior to incubating with C6/FHSV micelles. In addition, the pretreatment of MCF-7 cells with free HA and FA exhibited the weakest fluorescence intensity, indicating that C6/FHSV micelles were predominantly trafficked into the cells via dual receptor-mediated endocytosis pathway. Moreover, these observations of cellular uptake gave a good explanation to the results of MTT studies, suggesting that our well-designed FHSV micelles could selectively deliver antitumor drugs to tumor cells via specific receptor-mediated endocytosis and effectively kill tumor cells.
In vivo Biodistribution and Tumor-Targeting Assays
The tumor targetability of micelles was estimated via a noninvasive NIR imaging system. The time-dependent biodistribution of DiR-loaded formulations is displayed in Figure 7A. The fluorescence signal of the DiR solution was rapidly distributed throughout the whole body of mice 1 h after injection. The fluorescence signal weakened sharply after 12 h of administration and remained faintly visible at 24 h, indicating its rapid elimination from the blood. In contrast, the fluorescence signal of DiR-loaded micelles exhibited stronger tumor accumulation and remained strong even at 24 h postinjection. Therefore, it demonstrated that the hydrophilic HA shell of DiR-loaded micelles could not only prolong the blood circulation time of micelles but also increase the tumor aggregation of micelles by virtue of the EPR effect and receptor-mediated targeting effect. In addition, during the entire imaging test, the DiR/FHSV micelles displayed the strongest fluorescence signal accumulation at the tumor sites, followed by DiR/HSV micelles, DiR/HCV micelles and the DiR solution. This suggested that the FA functionalization of DiR/FHSV micelles also had a crucial influence on the tumor-specific recognition.
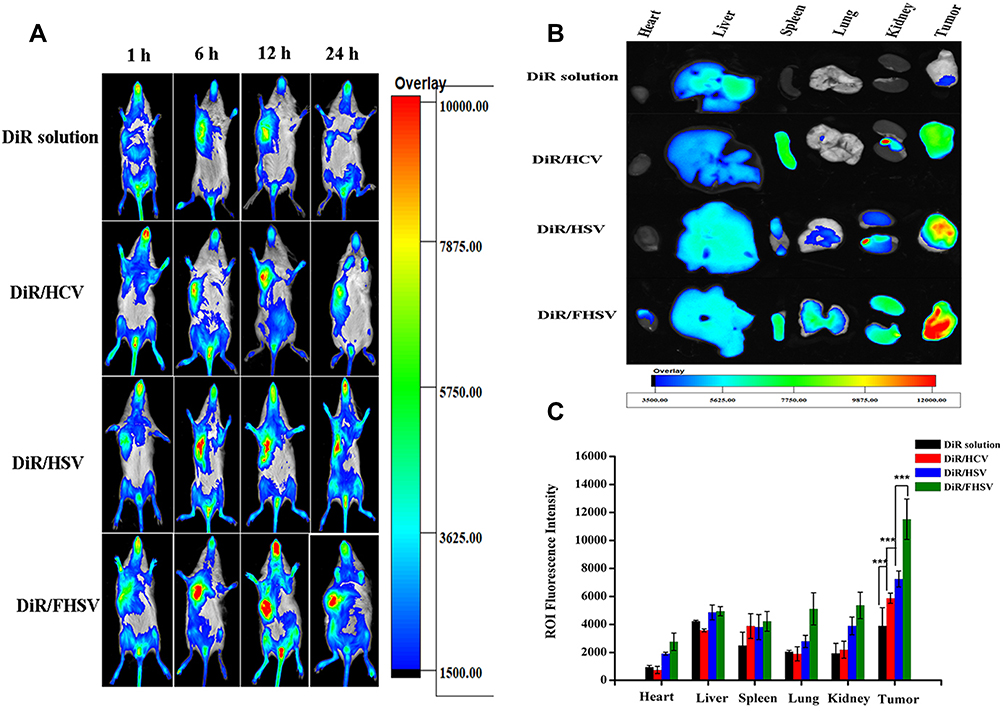 |
Figure 7 Bio-distribution of DiR solution and DiR-loaded micelles. (A) In vivo imaging of DiR solution and DiR-loaded micelles in S180 tumor-bearing mice at 1 h, 6 h, 12 h and 24 h postinjection, respectively. (B) Ex vivo fluorescence imaging of excised organs and tumors at 24 h postinjection. (C) Quantitative results of major organs and tumors accumulation at 24 h post injection (n=3). ***p < 0.001. |
The fluorescence imaging of the excised major organs after 24 h of administration was further studied. As shown in Figure 7B, the DiR solution group showed negligible fluorescence signals in the tumor, while strong accumulation was observed in the liver. In comparison, DiR-loaded micelles exhibited considerable fluorescence signal in the tumor, liver and spleen. Importantly, the fluorescence signal in tumor tissues was dramatically stronger than that in normal organs. Such an increase in the liver and spleen was likely ascribed to the interaction of HA with HARE receptors on liver sinusoidal endothelial cells and the nonspecific uptake by phagocytic cells of reticuloendothelial system (RES).56 Moreover, FHSV micelles showed a significantly higher distribution in the kidney and lung than HSV micelles and HCV micelles, which was likely due to the prolonged blood circulation time of FHSV micelles after modification with FA, resulting in a slower excretion from the body. It was undeniable that the binding of FA to the FR expressed on the proximal kidney tubules also provided an explanation for the increased kidney accumulation.57 Among the three micelles tested, DiR/FHSV micelles showed the greatest amount of tumor accumulation. Quantitative region-of-intensity (ROI) analysis revealed that the fluorescence intensity of DiR/FHSV micelles in tumor tissue was 1.59-fold, 1.96-fold and 2.96-fold stronger than that of DiR/HSV micelles, DiR/HCV micelles and DiR solution, respectively (Figure 7C). These results demonstrated that dual receptor-targeted FHSV micelles exhibited high tumor accumulation and retention. The excellent antitumor efficacy of FHSV micelles could be due to the following reasons: (1) EPR effect, (2) prolonged blood circulation time, (3) enhanced cellular internalization by dual receptor-mediated endocytosis.
In vivo Chemotherapy of Tumors
To assess the therapeutic effect of drug-loaded formulations, Taxol and different PTX-loaded micelles (equivalent to 10 mg PTX/kg) were injected intravenously into the S180 subcutaneous tumor model every other day for six times, while saline acted as a control. In addition, tumor volume and body weight were simultaneously monitored. Figure 8A depicts that the tumor growth was time-related, and the tumors in all drug-treated groups exhibited substantial growth inhibition in comparison to the saline group. For tumor suppression ability, Taxol and PTX/HCV only presented moderate antitumor activity, leading to tumor volumes of 1032.47 ± 160.42 mm3 and 937.29 ± 162.36 mm3 on day 13, respectively. Notably, although the surface properties and particle size of redox-sensitive PTX/HSV micelles and redox-insensitive PTX/HCV micelles, PTX/HSV micelles exhibited more promising antitumor ability than PTX/HCV micelles (p < 0.01), suggesting the advantage of reduction-triggered intracellular drug release strategy. Furthermore, PTX/FHSV micelles exhibited the most outstanding reduction in tumor size compared to other groups, which was in line with the results from MTT and apoptosis assays. The tumor inhibition rates (TIR) were also calculated (Figure 8C). It demonstrated that the highest TIR was observed in the PTX/FHSV group, with a value of 70.33% ± 1.52%, which was remarkably higher than that of PTX/HSV group (58.78% ± 8.73%, p < 0.05), PTX/HCV group (36.36 ± 8.66%, p < 0.001) and Taxol group (30.32% ± 17.12%, p < 0.001). Additionally, the body weight curve of mice during the administration course is presented in Figure 8B. No significant differences in body weight suggested the safety and negligible systemic toxicity of polymeric micelles.
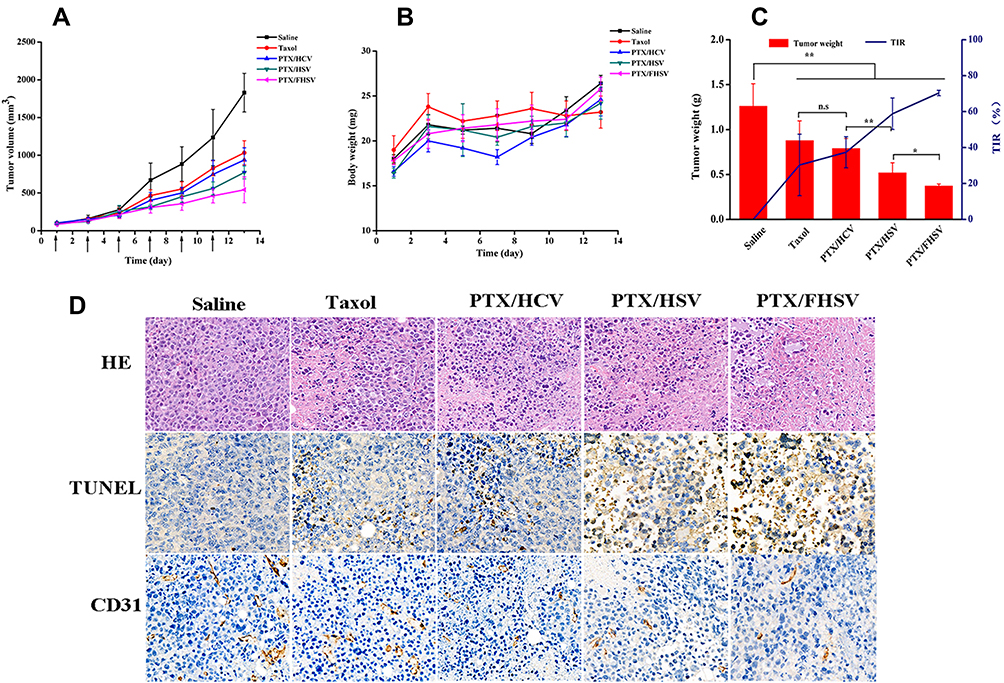 |
Figure 8 In vivo antitumor efficacy against S180 tumor xenograft model. (A) Tumor growth curves of mice treated with saline, Taxol and PTX-loaded micelles. (B) Body weight changes of mice. (C) Tumor weight and TIR of different groups after the last treatments (n=5).*p < 0.05, **p < 0.01, ns, p > 0.05. (D) H&E, TUNEL and CD31 analyses of excised tumors after the last treatments. |
For the purpose of further evaluating the tumor suppression mechanisms of PTX-loaded micelles, necrosis, apoptosis and angiogenesis in the tumor specimens of different treated groups were analyzed by histology and immunohistochemical studies (Figure 8D). H&E staining revealed that different degrees of cell necrosis were observed on day 13 after administration with different PTX-loaded formulations; in particular, almost completely necrotic tumor cells were found in the PTX/FHSV group. In TUNEL staining, the results were in coincidence with those of H&E staining, and the tumor slide of PTX/FHSV group yielded more dark brown apoptotic areas than the other treated groups, which further confirmed the superior antitumor potency of PTX/FHSV micelles. Rapid tumor proliferation is always accompanied by angiogenesis to meet the needs of tumor cells for oxygen and nutrition.47 Therefore, CD31 staining of tumor tissues was executed to evaluate the anti-angiogenic capacity of different PTX-loaded formulations. According to Figure 8D, the tumor microvessel density of the PTX/FHSV group was less than that of the other treated groups, suggesting the strongest anti-angiogenic ability of PTX/FHSV micelles at the tumor sites. These results showed that PTX/FHSV micelles could effectively prevent angiogenesis and promote the apoptosis and necrosis of tumor cells, followed by the growth inhibition of tumors.
Minimal damage of normal organs was essential during the chemotherapy period. It was obvious that pathological changes were not significant in these major tissues of mice administered with PTX-loaded micelles (Figure 9). In contrast, the cardiomyocytes of the mice treated with Taxol exhibited myocardial fiber breakage and vacuolization. Taken together, the well-designed micelles were endowed with superior security owing to their excellent selectivity between tumors and normal organs.
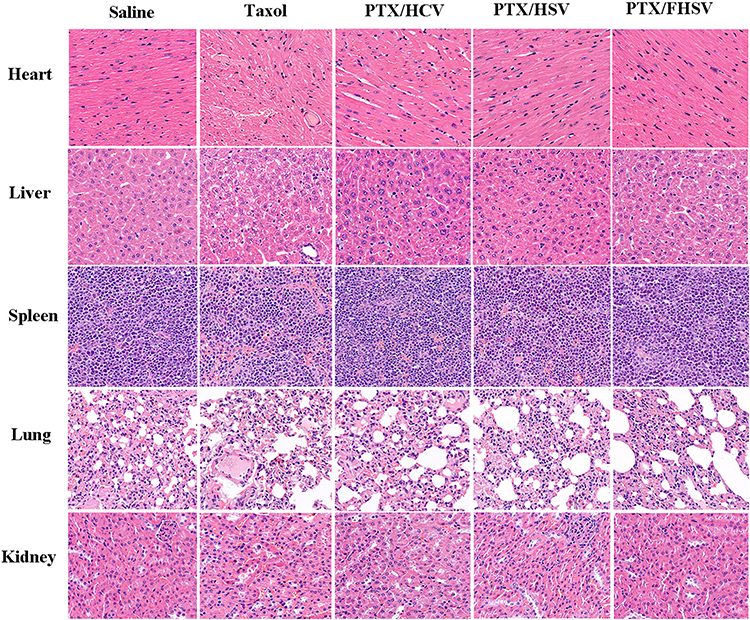 |
Figure 9 H&E staining of major organs at the end of treatments. |
Conclusions
In summary, we successfully designed an innovative amphiphilic block polymer-FHSV polymer with dual receptor-targeted ability and reduction sensitivity to selectively deliver anticancer drugs for tumor therapy. The first-line chemotherapeutic drug PTX was successfully encapsulated into the hydrophobic cores of FHSV to self-assemble into uniformly near-spherical micelles. As expected, the loaded PTX could be released completely from PTX/FHSV micelles under stimulation with 10 mM GSH. In in vitro cytological studies, the PTX/FHSV micelles revealed the most superior cytotoxicity to MCF-7 cells by means of dual receptor-mediated cellular uptake and precise release in the cytoplasm. More importantly, the in vivo study results demonstrated that FHSV micelles exhibited enhanced active targeting ability to promote drug accumulation in tumor regions, which resulted in the best therapeutic effect on solid tumors and reduced toxicity to normal organs. Thus, selective redox-sensitive FHSV micelles have greatly potential applications for tumor therapy.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 81703709, 81703425), China Postdoctoral Science Foundation (No. 2018M641716) and Natural Science Foundation of Liaoning Province (NO. 2019-ZD-0433).
Disclosure
Fen Chen is employed by Zhejiang Jingxin Pharmaceutical Co., Ltd. The authors report no other conflicts of interest in this work.
References
1. Zhang Y, Zhang C, Chen J, et al. Trackable mitochondria-targeting nanomicellar loaded with doxorubicin for overcoming drug resistance. ACS Appl Mater Interfaces. 2017;9(30):25152–25163. doi:10.1021/acsami.7b07219
2. Wang K, Yang B, Ye H, et al. Self-strengthened oxidation-responsive bioactivating prodrug-nanosystem with sequential and synergistically facilitated drug release for treatment of breast cancer. ACS Appl Mater Interfaces. 2019;11:18914–18922. doi:10.1021/acsami.9b03056
3. Herrera DA, Ashai N, Perez-Soler R, Cheng H. Nanoparticle albumin bound-paclitaxel for treatment of advanced non-small cell lung cancer: an evaluation of the clinical evidence. Expert Opin Pharmacother. 2018;20(1):95–102. doi:10.1080/14656566.2018.1546290
4. Mekhail TM, Markman M. Paclitaxel in cancer therapy. Expert Opin Pharmacother. 2002;3(6):755–766. doi:10.1517/14656566.3.6.755
5. Rezazadeh M, Emami J, Hassanzadeh F, et al. Targeted nanostructured lipid carriers for delivery of paclitaxel to cancer cells: preparation, characterization, and cell toxicity. Curr Drug Deliv. 2017;14(8):1189–1200. doi:10.2174/1567201814666170503143646
6. Gorain B, Choudhury H, Pandey M, Kesharwani P. Paclitaxel loaded vitamin E-TPGS nanoparticles for cancer therapy. Mater Sci Eng C. 2018;91:868–880. doi:10.1016/j.msec.2018.05.054
7. Sun B, Luo C, Yu H, et al. Disulfide bond-driven oxidation- and reduction-responsive prodrug nanoassemblies for cancer therapy. Nano Lett. 2018;18:3643–3650. doi:10.1021/acs.nanolett.8b00737
8. Luo C, Wang Y, Chen Q, et al. Advances of paclitaxel formulations based on nanosystem delivery technology. Mini-Rev Med Chem. 2012;12(5):434–444. doi:10.2174/138955712800493924
9. Chakravarthi SS, De S, Miller DW, Robinson DH. Comparison of anti-tumor efficacy of paclitaxel delivered in nano- and microparticles. Int J Pharm. 2010;38(1–2):37–44. doi:10.1016/j.ijpharm.2009.09.004
10. Ravar F, Saadat E, Gholami M, et al. Hyaluronic acid-coated liposomes for targeted delivery of paclitaxel, in-vitro characterization and in-vivo evaluation. J Control Release. 2016;229:10–22. doi:10.1016/j.jconrel.2016.03.012
11. Meng Z, Lv Q, Lu J, et al. Prodrug strategies for paclitaxel. Int J Mol Sci. 2016;17(5):796–818. doi:10.3390/ijms17050796
12. Monteiro LOF, Malachias Â, Pound-Lana G, et al. paclitaxel-loaded ph-sensitive liposome: new insights on structural and physicochemical characterization. Langmuir. 2018;34(20):5728–5737. doi:10.1021/acs.langmuir.8b00411
13. Banerjee I, De K, Mukherjee D, et al. Paclitaxel-loaded solid lipid nanoparticles modified with Tyr-3-octreotide for enhanced anti-angiogenic and anti-glioma therapy. Acta Biomater. 2016;38:69–81. doi:10.1016/j.actbio.2016.04.026
14. Emami J, Rezazadeh M, Hasanzadeh F, et al. Development and in vitro/in vivo evaluation of a novel targeted polymeric micelle for delivery of paclitaxel. Int J Biol Macromol. 2015;80:29–40. doi:10.1016/j.ijbiomac.2015.05.062
15. Fang J, Nakamura H, Maeda H. The EPR effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2010;63:136–151. doi:10.1016/j.addr.2010.04.009
16. Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv Drug Deliv Rev. 2014;66:2–25. doi:10.1016/j.addr.2013.11.009
17. Mao J, Ran D, Xie C, Shen Q, Wang S, Lu W. EGFR/EGFRvIII dual-targeting peptide-mediated drug delivery for enhanced glioma therapy. ACS Appl Mater Interfaces. 2017;9(29):24462–24475. doi:10.1021/acsami.7b05617
18. Yue J, Liu S, Wang R, et al. Transferrin-conjugated micelles: enhanced accumulation and antitumor effect for transferrin-receptor-overexpressing cancer models. Mol Pharm. 2012;9(7):1919–1931. doi:10.1021/mp300213g
19. Cheng L, Jiang Y, Xie Y, Qiu L, Yang Q, Lu H. Novel amphiphilic folic acid-cholesterol-chitosan micelles for paclitaxel delivery. Oncotarget. 2017;8(2):3315–3326. doi:10.18632/oncotarget.13757
20. Yin T, Wang J, Yin L, et al. Redox-sensitive hyaluronic acid–paclitaxel conjugate micelles with high physical drug loading for efficient tumor therapy. Polym Chem. 2015;6:8047–8059. doi:10.1039/C5PY01355K
21. Huang G, Huang H. Application of hyaluronic acid as carriers in drug delivery. Drug Deliv. 2018;25(1):766–772. doi:10.1080/10717544.2018.1450910
22. Arpicco S, Milla P, Stella B, Dosio F. Hyaluronic acid conjugates as vectors for the active targeting of drugs, genes and nanocomposites in cancer treatment. Molecules. 2014;19(3):3193–3230. doi:10.3390/molecules19033193
23. Dosio F, Arpicco S, Stella B, Fattal E. Hyaluronic acid for anticancer drug and nucleic acid delivery. Adv Drug Deliv Rev. 2016;97:204–236. doi:10.1016/j.addr.2015.11.011
24. Li J, Huo M, Wang J, et al. Redox-sensitive micelles self-assembled from amphiphilic hyaluronic acid-deoxycholic acid conjugates for targeted intracellular delivery of paclitaxel. Biomaterials. 2012;33(7):2310–2320. doi:10.1016/j.biomaterials.2011.11.022
25. Lee SJ, Jeong YI. Hybrid nanoparticles based on chlorin e6-conjugated hyaluronic acid-poly(L-histidine) copolymer for theranostic application to tumors. J Mater Chem B. 2018;6(18):2851–2859. doi:10.1039/C7TB03068A
26. Jeong GW, Jeong YI, Nah JW. Triggered doxorubicin release using redox-sensitive hyaluronic acid-g-stearic acid micelles for targeted cancer therapy. Carbohydr Polym. 2019;209:161–171. doi:10.1016/j.carbpol.2019.01.018
27. Hu D, Mezghrani O, Zhang L, Chen Y, Ke X, Ci T. GE11 peptide modified and reduction-responsive hyaluronic acid-based nanoparticles induced higher efficacy of doxorubicin for breast carcinoma therapy. Int J Nanomed. 2016;11:5125–5147. doi:10.2147/IJN.S113469
28. Chen J, Ouyang J, Chen Q, et al. EGFR and CD44 dual-targeted multifunctional hyaluronic acid nanogels boost protein delivery to ovarian and breast cancers in vitro and in vivo. ACS Appl Mater Interfaces. 2017;9(28):24140–24147. doi:10.1021/acsami.7b06879
29. Sawant RR, Jhaveri AM, Koshkaryev A, Qureshi F, Torchilin VP. The effect of dual ligand-targeted micelles on the delivery and efficacy of poorly soluble drug for cancer therapy. J Drug Target. 2013;21(7):630–638. doi:10.3109/1061186X.2013.789032
30. Xu L, Bai Q, Zhang X, Yang H. Folate-mediated chemotherapy and diagnostics: an updated review and outlook. J Control Release. 2017;252:73–82. doi:10.1016/j.jconrel.2017.02.023
31. Cheung A, Bax HJ, Josephs DH, et al. Targeting folate receptor alpha for cancer treatment. Oncotarget. 2016;7(32):52553–52574. doi:10.18632/oncotarget.9651
32. Yan Y, Dong Y, Yue S, Qiu X, Sun H, Zhong Z. Dually active targeting nanomedicines based on a direct conjugate of two purely natural ligands for potent chemotherapy of ovarian tumors. ACS Appl Mater Interfaces. 2019;11(50):46548–46557. doi:10.1021/acsami.9b17223
33. Elamin KM, Motoyama K, Higashi T, Yamashita Y, Tokuda A, Arima H. Dual targeting system by supramolecular complex of folate-conjugated methyl-β-cyclodextrin with adamantane-grafted hyaluronic acid for the treatment of colorectal cancer. Int J Biol Macromol. 2018;113:386–394. doi:10.1016/j.ijbiomac.2018.02.149
34. Liu Y, Sun J, Cao W, et al. Dual targeting folate-conjugated hyaluronic acid polymeric micelles for paclitaxel delivery. Int J Pharm. 2011;421(1):160–169. doi:10.1016/j.ijpharm.2011.09.006
35. Lee JY, Termsarasab U, Park JH, et al. Dual CD44 and folate receptor-targeted nanoparticles for cancer diagnosis and anticancer drug delivery. J Control Release. 2016;236:38–46. doi:10.1016/j.jconrel.2016.06.021
36. Li F, Chen W, You B, et al. Enhanced cellular internalization and on-demand intracellular release of doxorubicin by stepwise pH-/reduction-responsive nanoparticles. ACS Appl Mater Interfaces. 2016;8(47):32146–32158. doi:10.1021/acsami.6b09604
37. Hong SH, Larocque K, Jaunky DB, Piekny A, Oh JK. Dual disassembly and biological evaluation of enzyme/oxidation-responsive polyester-based nanoparticulates for tumor-targeting delivery. Colloids Surf B Biointerfaces. 2018;172:608–617. doi:10.1016/j.colsurfb.2018.09.013
38. Yu C, Gao C, Lü S, Chen C, Huang Y, Liu M. Redox-responsive shell-sheddable micelles self-assembled from amphiphilic chondroitin sulfate-cholesterol conjugates for triggered intracellular drug release. Chem Eng J. 2013;228:290–299. doi:10.1016/j.cej.2013.04.083
39. Raza A, Hayat U, Rasheed T, Bilal M, Iqbal HMN. Redox-responsive nano-carriers as tumor-targeted drug delivery systems. Eur J Med Chem. 2018;157:705–715. doi:10.1016/j.ejmech.2018.08.034
40. Duhem N, Danhier F, Preat V. Vitamin E-based nanomedicines for anti-cancer drug delivery. J Control Release. 2014;182:33–44. doi:10.1016/j.jconrel.2014.03.009
41. Liang D, Wang A, Yang Z, Liu Y, Qi X. Enhance Cancer Cell Recognition and Overcome Drug Resistance Using Hyaluronic Acid and alpha-Tocopheryl Succinate Based Multifunctional Nanoparticles. Mol Pharm. 2015;12(6):2189–2202. doi:10.1021/acs.molpharmaceut.5b00129
42. Liu Y, Zong Y, Yang Z, et al. dual-targeted controlled delivery based on folic acid modified pectin-based nanoparticles for combination therapy of liver cancer. ACS Sustain Chem Eng. 2019;7(3):3614–3623. doi:10.1021/acssuschemeng.8b06586
43. Gao Y, Sarfraz MK, Clas SD, Roa W, Löbenberg R. Hyaluronic acid-tocopherol succinate-based self-assembling micelles for targeted delivery of rifampicin to alveolar macrophages. J Biomed Nanotechnol. 2015;11(8):1312–1329. doi:10.1166/jbn.2015.2091
44. Xu J, Han Y, Pei M, et al. Multi-functionalized hyaluronic acid nanogels crosslinked with carbon dots as dual receptor-mediated targeting tumor theranostics. Carbohydr Polym. 2016;152:391–397. doi:10.1016/j.carbpol.2016.06.109
45. Ganesh S, Iyer AK, Morrissey DV, et al. Hyaluronic acid based self-assembling nanosystems for CD44 target mediated siRNA delivery to solid tumors. Biomaterials. 2013;34(13):3489–3502. doi:10.1016/j.biomaterials.2013.01.077
46. Zhong Y, Zhang J, Cheng R, et al. Reversibly crosslinked hyaluronic acid nanoparticles for active targeting and intelligent delivery of doxorubicin to drug resistant CD44+ human breast tumor xenografts. J Control Release. 2015;205:144–154. doi:10.1016/j.jconrel.2015.01.012
47. Zhang J, Zhao X, Chen Q, et al. Systematic evaluation of multifunctional paclitaxel-loaded polymeric mixed micelles as a potential anticancer remedy to overcome multidrug resistance. Acta Biomater. 2017;50:381–395. doi:10.1016/j.actbio.2016.12.021
48. Song S, Qi H, Xu J, et al. Hyaluronan-based nanocarriers with CD44-overexpressed cancer cell targeting. Pharm Res. 2014;31(11):2988–3005. doi:10.1007/s11095-014-1393-4
49. Lin J, Zhao C, Liu C, et al. Redox-responsive F127-folate/F127-disulfide bond-d-α-tocopheryl polyethylene glycol 1000 succinate/P123 mixed micelles loaded with paclitaxel for the reversal of multidrug resistance in tumors. Int J Nanomed. 2018;13:805–830. doi:10.2147/IJN.S152395
50. Choi KY, Jeon EJ, Yoon HY, et al. Theranostic nanoparticles based on PEGylated hyaluronic acid for the diagnosis, therapy and monitoring of colon cancer. Biomaterials. 2012;33(26):6186–6193. doi:10.1016/j.biomaterials.2012.05.029
51. Mezghrani O, Tang Y, Ke X, et al. Hepatocellular carcinoma dually-targeted nanoparticles for reduction triggered intracellular delivery of doxorubicin. Int J Pharm. 2015;478(2):553–568. doi:10.1016/j.ijpharm.2014.10.041
52. Li J, Yin T, Wang L, Yin L, Zhou J, Huo M. Biological evaluation of redox-sensitive micelles based on hyaluronic acid-deoxycholic acid conjugates for tumor-specific delivery of paclitaxel. Int J Pharm. 2015;483(1–2):38–48. doi:10.1016/j.ijpharm.2015.02.002
53. Zhao H, Yang C, Yan X. Fabrication and bioconjugation of BIII and CrIII co-doped ZnGa2O4 persistent luminescent nanoparticles for dual-targeted cancer bioimaging. Nanoscale. 2016;8(45):18987–18994. doi:10.1039/C6NR06259H
54. Park HK, Lee SJ, Oh JS, Lee SG, Jeong YI, Lee HC. Smart nanoparticles based on hyaluronic acid for redox-responsive and CD44 receptor-mediated targeting of tumor. Nanoscale Res Lett. 2015;10(1):288. doi:10.1186/s11671-015-0981-5
55. Galbiati A, Tabolacci C, Blasco MDR, et al. Targeting tumor cells through chitosan-folate modified microcapsules loaded with camptothecin. Bioconjug Chem. 2011;22(6):1066–1072. doi:10.1021/bc100546s
56. Liang D, Su H, Liu Y, Wang A, Qi X. Tumor-specific penetrating peptides-functionalized hyaluronic acid-D-α-tocopheryl succinate based nanoparticles for multi-task delivery to invasive cancers. Biomaterials. 2015;71:11–23. doi:10.1016/j.biomaterials.2015.08.035
57. Parker N, Turk MJ, Westrick E, et al. Folate receptor expression in carcinomas and normal tissues determined by a quantitative radioligand binding assay. Anal Biochem. 2005;338(2):284–293. doi:10.1016/j.ab.2004.12.026
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.