Back to Journals » International Journal of Nanomedicine » Volume 13
Dual-functional Brij-S20-modified nanocrystal formulation enhances the intestinal transport and oral bioavailability of berberine
Authors Xiong W, Sang W, Linghu KG, Zhong ZF ![]() , Cheang WS, Li J, Hu YJ, Yu H
, Cheang WS, Li J, Hu YJ, Yu H ![]() , Wang YT
, Wang YT
Received 27 January 2018
Accepted for publication 9 March 2018
Published 28 June 2018 Volume 2018:13 Pages 3781—3793
DOI https://doi.org/10.2147/IJN.S163763
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Wei Xiong,1 Wei Sang,1 Ke Gang Linghu,1 Zhang Feng Zhong,1 Wai San Cheang,1 Juan Li,2 Yuan Jia Hu,1 Hua Yu,1,3,4 Yi Tao Wang1
1Institute of Chinese Medical Sciences, State Key Laboratory of Quality Research in Chinese Medicine, University of Macau, Macao SAR, China; 2Department of Pharmaceutics, China Pharmaceutical University, Nanjing, Jiangsu, China; 3Hong Kong Baptist University Shenzhen Research Center, Shenzhen, Guangdong, China; 4School of Chinese Medicine, Hong Kong Baptist University, Kowloon Tong, Hong Kong SAR, China
Introduction: Berberine (BBR) is a plant-derived benzylisoquinoline alkaloid and has been demonstrated to be a potential treatment for various chronic diseases. The poor water solubility and P-glycoprotein (Pgp)-mediated drug efflux are the main challenges for its further application in a clinical setting.
Materials and methods: In this study, a Brij-S20 (BS20)-modified nanocrystal formulation (BBR-BS20-NCs) has been developed and investigated with the purpose of improving the intestinal absorption of BBR. The physicochemical properties of the developed BBR-BS20-NCs were characterized and the enhancement of the BBR-BS20-NCs on BBR absorption were investigated both in vitro and in vivo.
Results: The results indicated that BS20 could significantly enhance the intracellular uptake of BBR in MDCK-MDR1 cells via a short-term and reversible modulation on the Pgp function, accompanied by a marked increase in Pgp mRNA expression but without significant influence on the Pgp protein expression. Moreover, the morphology of the prepared BBR-BS20-NCs was observed to be prism-like, with a smooth surface and an average diameter of 148.0 ± 3.2 nm. Compared to raw BBR and physical mixture, BBR-BS20-NCs facilitated the dissolution rate and extent of release of BBR in aqueous solution, and further increased the absorption of BBR in MDCK-MDR1 monolayer by overcoming the Pgp-mediated secretory transport (Papp[BL-AP] values of 2.85 ± 0.04 × 10-6 cm/s, 2.21 ± 0.14 × 10-6 cm/s, and 2.00 ± 0.07 × 10-6 cm/s for pure BBR, physical mixture, and BBR-BS20-NCs, respectively). Significant improvements in the maximum concentration observed (Cmax) and area under drug concentration-time curve (AUC0–t) of BBR-BS20-NCs were obtained in pharmacokinetic studies compared to pure BBR, and the relative bioavailability of BBR-BS20-NCs to pure BBR was 404.1%.
Conclusion: The developed BBR-BS20-NCs combine the advantages of nanocrystal formulation and functional excipient. The novel pharmaceutical design provides a new strategy to improve the oral bioavailability of those drugs with both poor water solubility and Pgp-mediated efflux.
Keywords: berberine, poor water solubility, Pgp-mediated drug efflux, Brij, nanocrystals
Introduction
Solubility and permeability are the two most important factors affecting drug oral bioavailability. The US Food and Drug Administration’s biopharmaceutics classification system provides a mechanistic framework for understanding the concept of drug absorption in terms of permeability and solubility.1 In the past decades, numerous methods have been developed to improve the solubility of poorly water-soluble drugs.2,3 The widely reported approaches include solid dispersions,4 soluble cyclodextrin complexes,5 self-emulsifying drug delivery systems,6 various types of nanocarriers,7,8 and nanocrystal formulations.9 On the other hand, to enhance the intestinal absorption of the drugs with P-glycoprotein (Pgp)-mediated drug efflux, co-formulation with Pgp inhibitors or modulators might be one of the most efficient approaches.10–12 However, in order to minimize the unexpected side effects and effects on intestinal functions, the co-formulated inhibitors or modulators should be biologically nonactive and Pgp inhibition reversible. This presents a great challenge for pharmaceutical development of those drugs that are characterized as having poor water solubility and Pgp-mediated efflux.
Berberine (BBR) is a benzylisoquinoline alkaloid that is widely distributed in numerous medicinal plants included in the families of Berberidaceae, Papaveraceae, Mao Langke, Rutaceae, Menispermaceae, and Rhamnaceae, among others (Figure 1).13 BBR is one of the main active ingredients of these medicinal plants and is widely used in clinics due to its potential antidiarrheal14,15 and antibacterial applications.16,17 In recent years, BBR has been demonstrated to be effective against various chronic diseases,18 such as diabetes mellitus,19,20 hyperlipidemia,21 neurodegenerative diseases,22,23 antioxidation,24 inflammation,25,26 and tumors.27–29 All the scientific evidence provides new perspectives for the extended use of BBR in clinical applications. However, the oral bioavailability of BBR has been reported to be extremely poor (<1%), because of its poor aqueous solubility,30 extensive intestinal elimination,31 and hepatobiliary excretion,32 as well as the Pgp-mediated efflux induced low intestinal permeability.33 The medical efficacy of BBR may never be obtained for the patients taking BBR as a therapeutic agent. Therefore, improvement of its solubility and intestinal absorption are the main challenges for further applications of BBR.
 | Figure 1 Plant of Rhizoma Coptidis (A), chemical structure of berberine (B), and Rhizoma Coptidis (C). |
Nanocrystals (NCs) are defined as nanoparticles of pure drug without any matrix material and with an average diameter <1 μm (typically in the range of 100–500 nm).34,35 The NCs could be well dispersed in a liquid medium (also called nanosuspensions) with a minimum amount of surfactant (or polymers). Mechanically, NCs have been demonstrated to enhance the oral bioavailability of poorly soluble drugs by improving the solubility and dissolution rate and extending the bioadhesion of the drugs to the intestinal wall.36,37 Recently, functional NCs modified with Pgp inhibitory surfactants were noticed and reported by some researchers.38,39 The novel formulation design provides a new strategy for those drugs that are characterized as having poor solubility and Pgp-mediated efflux.
Brij surfactants (Brijs) belong to the family of polyoxyethylene ethers and include four chemical types: polyoxyethylene lauryl ether, polyoxyethylene cetyl ether, polyoxyethylene oleyl ether, and polyoxyethylene stearyl ether. Some Brijs have been widely used in pharmaceutical formulations to enhance the solubility of poorly water-soluble drugs.40 Moreover, Brijs have been reported to present inhibitory effects on Pgp functions but without causing effects on the expression of Pgp in cancer cells.41 In our previous study, polyoxyethylene (23) lauryl ether (trade name Brij-35 or Brij-L23) was found to significantly increase the permeation of bis(12)-hupyridone (a substrate of Pgp) through Caco-2 monolayer.42 All the investigations suggested that Brijs might be developed as safe and effective Pgp modulators to enhance the oral bioavailability of those drugs that act as Pgp substrates.
In the present study, the effects of Brij-S20 (BS20) on the expression and function of Pgp in MDCK-MDR1 cells were investigated. Moreover, the physicochemical properties and in vitro characteristics of the developed BS20-modified nanocrystal formulation of BBR (BBR-BS20-NCs) were determined. The enhancement effects of the BBR-BS20-NCs on permeability, overcoming the poor solubility and Pgp-mediated efflux for BBR, were evaluated using MDCK-MDR1 cell monolayer. Finally, the pharmacokinetic study investigated the bioavailability improvement of BBR-BS20-NCs via oral administration in male Sprague Dawley rats.
Materials and methods
Chemicals and reagents
BBR was purchased from Oddfoni Biological Technology Company (Nanjing, China). Verapamil, cyclosporin A (CsA), and Rhodamine123 were purchased from J&K Scientific Ltd. (Beijing, China). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), methanol, dimethyl sulphoxide (DMSO), and Brijs (Brij-L4, decaethylene glycol monododecyl ether [BD], Brij-L23, Brij-52 Brij-C10, Brij-58, Brij-93, Brij-O10, Brij-O20, Brij-S2, Brij-S10, Brij-S20, Brij-S100, D-α-Tocopherol polyethylene glycol 1000 succinate [TPGS]) were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). High-performance liquid chromatography (HPLC) grade acetonitrile was purchased from Merck Company (Darmstadt, Germany).
ATPlite luminescence ATP detection assay system was bought from Perkin Elmer (Waltham, MA, USA). Pgp-Glo assay system with human Pgp membrane was obtained from Promega (Madison, WI, USA). All materials for cell culture were obtained from Thermo Fisher Scientific (Waltham, MA, USA). Antibodies against Pgp and GAPDH, and anti-rabbit IgG were obtained from Cell Signaling Technology (Beverly, MA, USA).
Cell culture
Madin-Darby canine kidney cell line transfected with the multidrug resistance 1 gene (MDCK-MDR1) was a kind gift from Prof P Borst (the Netherlands Cancer Institute, Amsterdam, the Netherlands). The use of MDCK-MDR1 was approved by the Institute of Chinese Medical Sciences, University of Macau, for research purpose. MDCK-MDR1 cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum and 1% penicillin/streptomycin (P/S) at 37°C in humidified 5% CO2 atmosphere.
Cytotoxicity assay
MDCK-MDR1 cells were seeded on 96-well plates (1 × 104 cells/well) and allowed to adhere overnight. The cells were treated with indicated concentrations of BS20, BBR, or BBR-BS20-NCs for 3 h, respectively. Cell viability was examined using the MTT assay.42 Moreover, the cytotoxicity of BS20 to MDCK-MDR1 cells in 24 h was determined with the same protocol. The experimental concentrations for all tested chemicals were less than the maximum nontoxic concentrations (cell survival >90% vs control).
Effects of Brij-S20 on P-glycoprotein
Cellular uptake
The effect of BS20 on Pgp-mediated drug efflux was assessed by determining the cell uptake of BBR in MDCK-MDR1 cells. Briefly, MDCK-MDR1 cells were seeded on six-well plates (5 × 105 cells/well) and allowed to adhere overnight. After the cells reached 70%–80% confluence, the culture medium was replaced with BBR (100 μM) in the absence or presence of verapamil (10 μM), CsA (2 μM), or BS20 (8.1 μM, equaled to that in the nanocrystal formulation) and incubated at 37°C for 90 min. Thereafter, the cells were washed twice with ice-cold PBS and then lysed in 100 μL of methanol-water (80:20, v/v). The content of BBR in the lysate was quantified using a modified ultra-performance liquid chromatography (UPLC) method.43
Cellular P-glycoprotein expression
The effect of BS20 on Pgp expression in MDCK-MDR1 cells was determined using Western blotting assay. After treatment with BS20 (5 or 10 μM) for 3 or 24 h, the cells were harvested, and proteins were extracted with radio-immunoprecipitation assay (RIPA) lysis buffer (Beyotime Biotechnology, Shanghai, China) containing a protease inhibitor cocktail (Thermo Fisher Scientific Inc), phenylmethanesulfonyl fluoride (PMSF) (Beyotime Biotechnology) for 30 min on ice. The sample was centrifuged at 20,800 ×g for 15 min at 4°C. The supernatant was collected, and the protein content was measured using the bicinchoninic acid (BCA) protein assay kit (Thermo Fisher Scientific Inc). Equal amounts of individual protein samples were separated by 8% SDS–PAGE gel and then electro-transferred onto the polyvinylidene difluoride (PVDF) membrane (Bio-Rad Laboratories Inc, Hercules, CA, USA). Membranes were blocked for 30 min with 5% non-fat dry milk (Bio-Rad) in TBST buffer (composed of 50 mM Tris (pH 7.6), 150 mM NaCl, and 0.1% Tween-20) and incubated with the antibodies against Pgp (1:1,000) and GAPDH (1:1,000) overnight at 4°C. After incubation with secondary antibody (1:1,000), the specific protein bands were visualized using a chemiluminescence kit (Thermo Fisher Scientific), and the chemiluminescent signals were quantified using ChemiDoc MP Gel Imaging System (Bio-Rad, Hercules, CA, USA).
Quantitative gene expression analysis
After treatment with BS20 (5 or 10 μM) for 3 or 24 h, the total RNA of MDCK-MDR1 cells was isolated by the TaKaRa MiniBEST universal RNA extraction kit (TaKaRa Bio Inc., Tokyo, Japan) according to the manufacturer’s protocol. The content of total RNA was detected by the Nano Vue spectrophotometer (Biochrom, Cambridge, UK). The cDNA was synthesized using the PrimeScript™ RT reagent kit with gDNA eraser (TaKaRa) in accordance with the manufacturer’s instruction.44 Gene expression levels were assessed by real-time quantitative polymerase chain reaction with the Applied Biosystems ViiA 7 (Thermo Fisher Scientific). The primers were synthesized by Sangon Biotech (Shanghai, China) and sequences were as follows: Pgp, forward 5′TGGCACCCAGCACAATGAA3′ and reverse 5′CTAAGTCATAGTCCGCCTAGAAGCA3′; GAPDH, forward 5′GCACCGCAAGGCTGAGAAC3′ and reverse 5′TGGTGAAGACGCCAGTGGA3′. The amplification procedure was as follows: 1 cycle at 95°C for 30 s, 40 cycles at 95°C for 5 s, and 60°C for 30 s. At the end of PCR reactions, melt curve analyses were performed. Relative gene expression of Pgp was determined after normalizing to GAPDH in each sample using 2−ΔΔCq method.
Intracellular ATP level
The intracellular ATP content was determined using ATPlite Luminescence ATP Detection Assay System with modifications from the manufacturer’s protocol. Briefly, MDCK-MDR1 cells were seeded on 96-well plates (1 × 104 cells/well) and allowed to adhere overnight. After treatment with BS20 (5 or 10 μM) for 3 or 24 h, the cells were lysed by three freeze-thaw cycles in lysis reagent. An equal volume of substrate reagent was added to each well and rocked for 10 min at room temperature. Intracellular ATP content was measured by a luminescent plate reader.
P-glycoprotein ATPase activity
Pgp ATPase activity was measured using the Pgp-Glo assay system with human Pgp membrane according to the manufacturer’s instructions (Promega Co.). The assay relies on the ATP dependence of the light-generating reaction of firefly luciferase. Briefly, 25 μg of Pgp membrane was incubated at 37°C with either Na3VO4 (100 μM), buffer (blank), verapamil (100 μM, positive control), BS20 (5 or 10 μM), or verapamil (100 μM) plus BS20 (5 or 10 μM). The ATPase reaction was initiated by addition of 5 mM MgATP and followed by incubation at 37°C for 120 min. The reaction was stopped, and the remaining unmetabolized ATP was detected as a luciferase-generated luminescence signal by addition of ATP detection reagent. Following a room-temperature signal-stabilization period (30 min), luminescence was read on a microplate luminometer. Pgp ATPase activity was presented as a drop in luminescence of samples compared to that treated with Na3VO4.
Preparation of BBR-BS20-NCs
BBR-BS20-NCs were prepared by a three-phase nanoparticle engineering technology with some modifications.45 In brief, BBR and BS20 were dissolved in methanol with mass ratio of 4:1 (BBR:BS20, w/w) and coprecipitated by evaporating the methanol under reduced pressure. The trace amount of methanol in precipitate was removed under a vacuum with desiccators for 4 h. Subsequently, the precipitate was hydrated in water for 10 min, vortexed, and then sonicated with a bath-type sonicator (output 80 kC, 80 w) for 10 min to form BBR-BS20-NCs. The powdered BBR-BS20-NCs were obtained by lyophilizing the sample with a freeze dryer (Ilshin Lab Co., Ltd., Yangju, Korea). In addition, the nanocrystals of BBR without BS20 (BBR-NCs) were prepared using the same method but without addition of BS20.
Characterization of BBR-BS20-NCs
The size distribution, polydispersity index (PDI), and zeta potential of the prepared BBR-BS20-NCs were determined using a Zeta-sizer Nano instrument (Malvern Instruments, Malvern, UK) at room temperature.
The X-ray diffraction patterns of BBR, BS20, physical mixture, and BBR-BS20-NCs were achieved with a D8 advance X-ray diffractometer (Bruker-AXS, Karlsruhe, Germany) using the Cu Kα radiation generated at a current of 20 mA and a voltage of 30 kV. The data were achieved at the step width of 0.02° with a scan rate of 6°/min from 10° to 40°.
The thermogram of BBR, BS20, physical mixture, and BBR-BS20-NCs were recorded using a differential scanning calorimetry system (DSC-60; Shimadzu, Koyo, Japan). The temperature range was from 50°C to 150°C at a heating rate of 10°C/min with a constant N2 flow of 20 mL/min.
The Fourier transform infrared (FTIR) spectra of BBR, BS20, physical mixture, and BBR-BS20-NCs were recorded with a FTIR spectrometer (Bruker, Ettlingen, Germany) using the KBr method.46
The morphologies of BBR, BS20, physical mixture, and BBR-BS20-NCs were accomplished using scanning electron microscopy (SEM, S-3700; Hitachi, Tokyo, Japan). Briefly, an appropriate amount of each sample was fixed on a SEM stub using double-sided adhesive tape and coated with Au. The coated samples were examined at an acceleration voltage of 15 kV.47
In vitro drug dissolution
The in vitro drug dissolution of BBR, physical mixture, and BBR-BS20-NCs were determined using a dialysis method. Briefly, an equal amount of BBR (1 mg) for each sample was dispersed into 0.5 mL of Milli-Q water and transferred into a dialysis bag (cut off 8,000–12,000 Da). The bag was placed into a conical flask containing 50 mL of water, and then incubated on a water-bath thermostatic oscillator at 37°C ± 0.5°C with a speed of 100 rpm for 2 h. The cumulative drug release at indicated time intervals was determined by the UPLC method.
Bidirectional transport of BBR-BS20-NCs in MDCK-MDR1 monolayer
The harvested MDCK-MDR1 cells were plated onto polycarbonate inserts (22.74 mm ID, 0.4 μm pore size, 4.06 cm2 of growth area; SPL Life Sciences Co., Pochoen, Korea) at a density of 5 × 105 cells/well and cultured for 7 days prior to transport experiments. The integrity of the monolayer was monitored by measuring the transepithelial electrical resistance (TEER) at 37°C with an epithelial voltohmmeter (World Precision Instruments, Inc., Sarasota, FL, USA). Only MDCK-MDR1 monolayers with a TEER value above 200 ohms (after subtracting the background value of the transwell) were employed in the transport study.
The transport study was carried out in Hank’s balanced salt solution (HBSS) buffer (pH 7.4). MDCK-MDR1 cell monolayers were rinsed twice and equilibrated with HBSS buffer at 37°C for 30 min before the transport experiment. In the bidirectional transport study, BBR (10 μM) in the absence of or in the presence of verapamil (10 μM) or CsA (2 μM), or BBR-BS20-NCs (containing 10 μM BBR) in HBSS were loaded onto the apical (AP) (1.5 mL of transport buffer) or basolateral (BL) (2.6 mL of transport buffer) side, the so-called donor side. Aliquots of 1 mL samples were taken from the other (receiver) side at different time points (15, 30, 45, 60, 90, and 120 min) during the experiment. The same volume of blank HBSS buffer was replaced to the receiver chamber after each sampling. All the samples were stored at −20°C before the UPLC analysis. At the end of the experiment, the MDCK-MDR1 cells on the membranes were washed twice with ice-cold PBS and then collected. The BBR inside the cells was extracted with 1 mL methanol-water (80:20, v/v) for the UPLC analysis. The permeability coefficient (Papp), efflux ratio (EfR), absorptive quotient (AQ), and secretory quotient (SQ) were calculated according to our previously reported methods,42 to evaluate the enhancement effect of BBR-BS20-NCs on BBR absorption in MDCK-MDR1 cell model.
In vivo pharmacokinetic study
Male Sprague Dawley rats (8 weeks of age) were fed on a standard laboratory diet with free access to water under the controlled temperature at 20°C–22°C and relative humidity of 50% with 12-hour light/dark cycle. One day prior to drug administration, the rats were surgically cannulated with polyethylene catheters (polyethylene tubing, 0.4 mm internal diameter (ID), 0.8 mm outside diameter (OD); Portex Ltd., Hythe, Kent, England) on the right jugular veins under anesthesia with 10% chloral hydrate. The animals were recovered in individual metabolic cages and fasted but were allowed free access to water overnight. All the experimental protocols (reference No: UMARE-029-2016) were in accordance with the National Institutes of Health guidelines for the Care of Use of Laboratory Animals, and approved by the Animal Research Ethics Committee, University of Macau, Macau SAR, China.
Ten surgically cannulated Sprague Dawley rats were randomly divided into two groups and orally administered 0.5% Carboxymethylcellulose sodium (CMC-Na) suspension liquid of BBR (65 mg/kg) and BBR-BS20-NCs (85 mg/kg, equivalent to 65 mg/kg BBR), respectively. Serial venous blood samples (0.25 mL) were collected from the right jugular vein via the cannulated catheter into heparinized tubes at 0.5, 1, 2, 3, 4, 6, 8, 12, and 24 h after drug administration. After each blood sampling, an equivalent volume of heparinized saline (0.25 mL) was injected into the rats to maintain a constant blood volume. The collected blood samples were centrifuged at 16,000 ×g for 10 min and the plasma was collected and stored at −20°C until the UPLC analysis. The coptisine solution was used as internal standard. The plasma concentrations vs time profiles were analyzed with DAS 2.0 software (Mathematical Pharmacology Professional Committee of China; Shanghai, China). The non-compartmental model was used to estimate the pharmacokinetic parameters.
Statistical analysis
Statistically significant differences between two groups or more than two groups were evaluated by Student’s t-test and one-way analysis of variance, respectively. A value of p < 0.05 was considered significant for all tests.
Results
Effects of Brij-S20 on P-glycoprotein
Cellular uptake
Rhodamine123 (Rho123), a substrate of Pgp, is frequently used in Pgp efflux study.48 Verapamil and CsA were used to inhibit Pgp efflux and increase cellular uptake of Pgp substrate. To select an optimal Brij for the preparation of BBR nanocrystals, the effects of representative Brijs (comprising four chemical types) on the uptake of Rho123 in MDCK-MDR1 cells were determined and compared (Figure 2). The result indicated that polyoxyethylene (n) stearyl ether, especially polyoxyethylene (20) stearyl ether (Brij-S20), presented the most potency on increasing the intracellular accumulation of Rho123 than other types of Brijs. Therefore, Brij-S20 was selected for further studies.
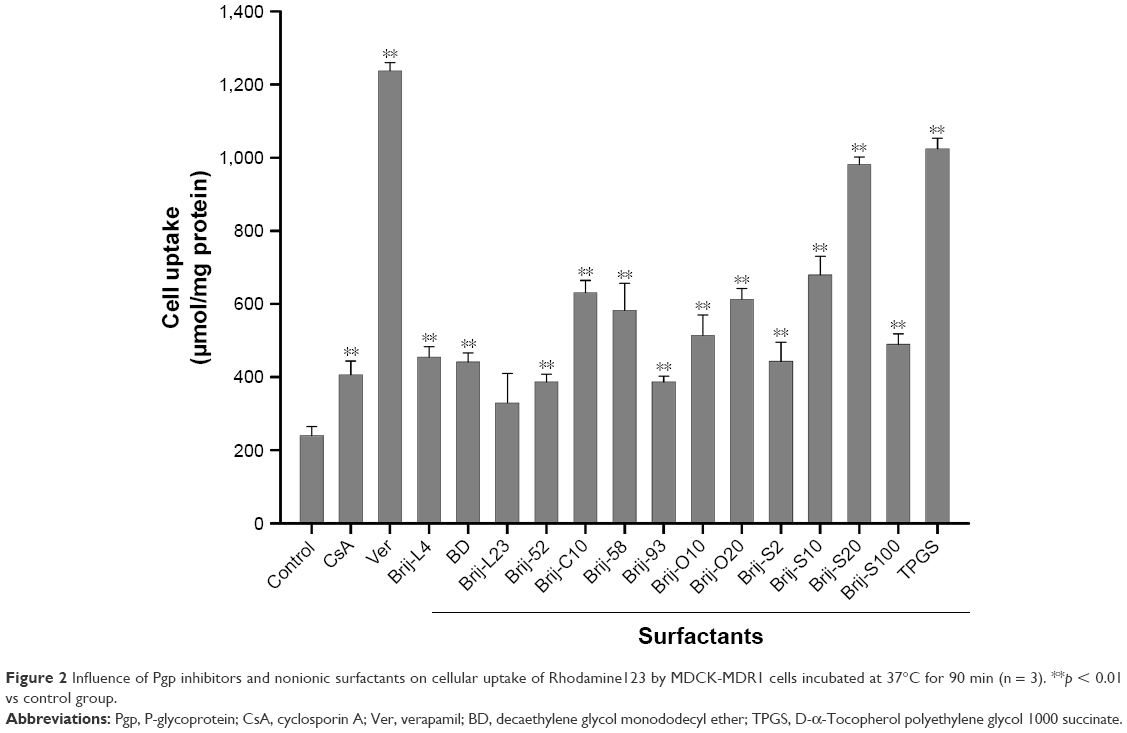 | Figure 2 Influence of Pgp inhibitors and nonionic surfactants on cellular uptake of Rhodamine123 by MDCK-MDR1 cells incubated at 37°C for 90 min (n = 3). **p < 0.01 vs control group. |
The effects of verapamil (10 μM), CsA (2 μM), or BS20 (8.1 μM) on intracellular uptake of BBR in MDCK-MDR1 cells were compared and illustrated (Figure 3). Compared to the BBR group, BS20 and verapamil significantly increased the cellular uptake of BBR by 4.1-fold and 5.4-fold respectively, which was higher than that by CsA (2.9-fold). The result suggested that BS20 might be developed as a potential Pgp modulator to facilitate the intestinal absorption of BBR.
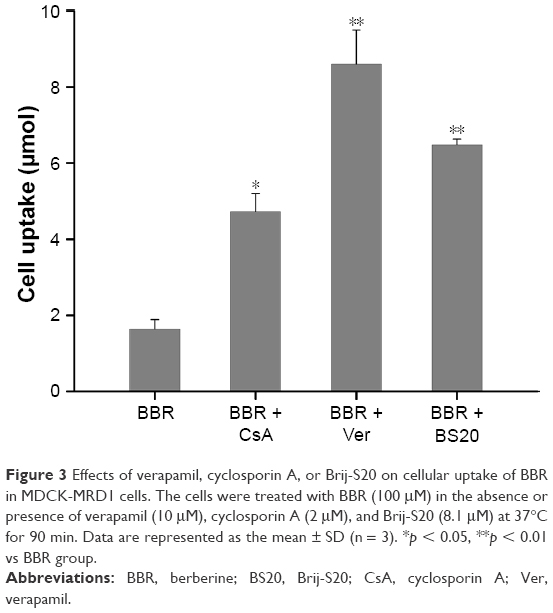 | Figure 3 Effects of verapamil, cyclosporin A, or Brij-S20 on cellular uptake of BBR in MDCK-MRD1 cells. The cells were treated with BBR (100 μM) in the absence or presence of verapamil (10 μM), cyclosporin A (2 μM), and Brij-S20 (8.1 μM) at 37°C for 90 min. Data are represented as the mean ± SD (n = 3). *p < 0.05, **p < 0.01 vs BBR group. |
Potential mechanisms of Brij-S20 on P-glycoprotein modulation
To investigate the potential mechanisms of BS20 on Pgp, the effects of BS20 on cellular Pgp protein expression, mRNA expression, and intracellular ATP level, and Pgp-ATPase activity in MDCK-MDR1 cells were determined.
As shown in Figure 4A, after treatment with BS20 for 3 h and 24 h, no significant changes in Pgp protein expression were observed in MDCK-MDR1 cells. Also, there was no statistically significant change in mRNA expression after 3 h of incubation with BS20 at 5 μM and 10 μM concentrations (0.9- and 1.4-fold, respectively). However, the mRNA expression of Pgp showed a significant change after exposure to BS20 for 24 h. BS20 at 5 μM and 10 μM concentrations showed a concentration-dependent upregulation of mRNA levels to 2.3- and 3.8-fold, respectively (Figure 4B).
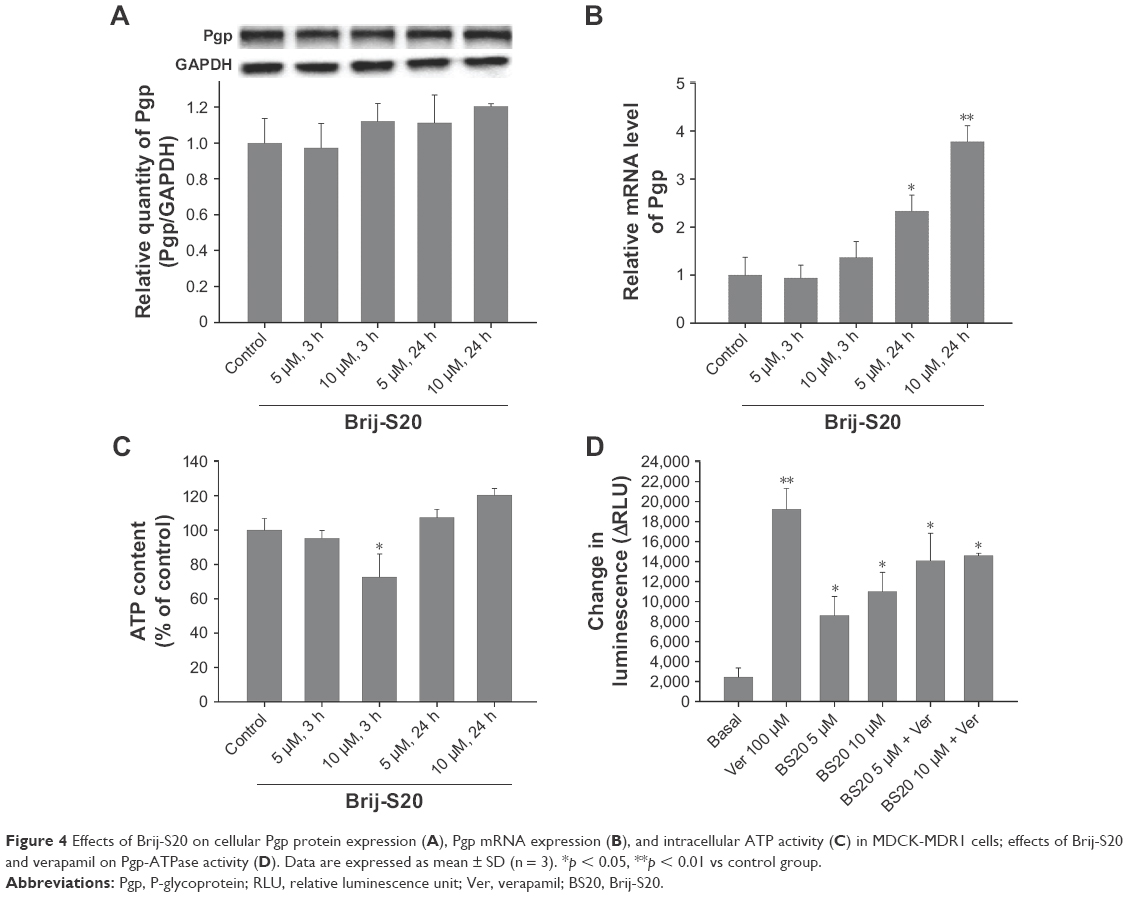 | Figure 4 Effects of Brij-S20 on cellular Pgp protein expression (A), Pgp mRNA expression (B), and intracellular ATP activity (C) in MDCK-MDR1 cells; effects of Brij-S20 and verapamil on Pgp-ATPase activity (D). Data are expressed as mean ± SD (n = 3). *p < 0.05, **p < 0.01 vs control group. |
Interestingly, the activity of intracellular ATP was observed to be dose-dependently inhibited by BS20 in 3 h, and this effect could be reversed to normal level in 24 h (Figure 4C). Moreover, the effects of BS20 on activities of human Pgp-ATPase and verapamil-stimulated human Pgp-ATPase were determined and compared using the Pgp-Glo assay system (Figure 4D). Verapamil (100 μM) strongly stimulated the Pgp-ATPase activity by 7.9-fold compared with that of the normal control (basal level). BS20 at 5 μM and 10 μM concentrations presented a dose-dependent stimulation of the Pgp-ATPase activity by 3.5- and 4.5-fold, respectively. However, combinations of BS20 and verapamil presented the intermediate strength on Pgp-ATPase stimulation (5.8- and 6.0-fold, respectively) between the BS20 and verapamil groups.
Characterization of BBR-BS20-NCs
The size distribution, PDI, and zeta potential of various BBR-NCs and BBR-BS20-NCs were determined and are summarized in Table 1. Although BBR could form nanocrystals alone, the BBR-NCs (size: 230.0 nm, PDI: 0.27, and zeta potential: 0.09 mV) were found to be significantly aggregated and precipitated in 12 h at 4°C in aqueous solution. Moreover, addition of BS20 could improve the stability of BBR nanocrystals; the BBR-BS20-NCs (size: 148.0, PDI: 0.31, and zeta potential: 0.76 mV) were observed to be stable at 24 h at 4°C in aqueous solution.
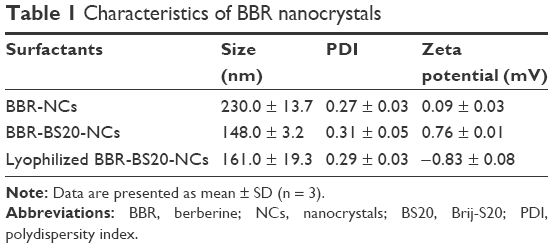 | Table 1 Characteristics of BBR nanocrystals |
The XRD patterns (Figure 5A) showed that BBR and BS20 possessed the crystalline structure with two pronounced diffraction 2θ peaks of 25.52° and 26.33° for BBR as well as of 19.52° and 23.50° for BS20, respectively. Both two 2θ peaks could be observed in the pattern of the physical mixture but were weak in the pattern of BBR-BS20-NCs; these weak peaks suggested the BBR was maintained in the crystalline state in the prepared nanosuspension.
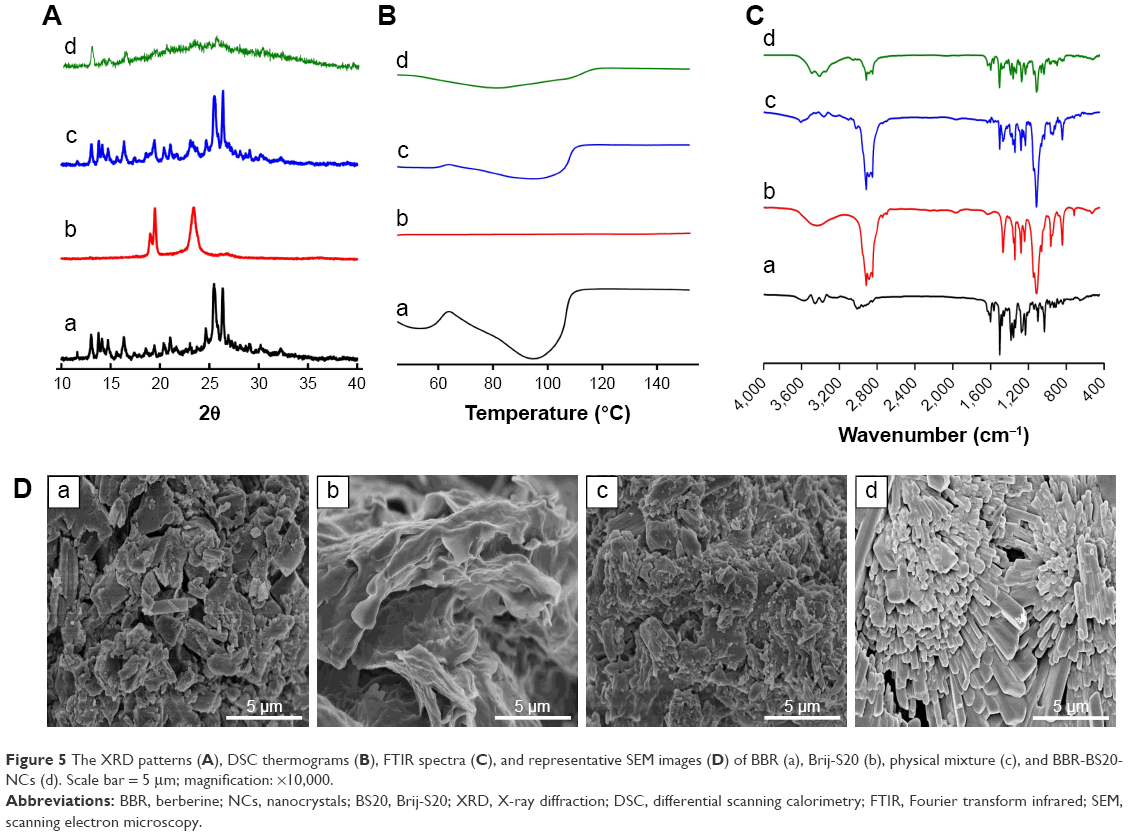 | Figure 5 The XRD patterns (A), DSC thermograms (B), FTIR spectra (C), and representative SEM images (D) of BBR (a), Brij-S20 (b), physical mixture (c), and BBR-BS20-NCs (d). Scale bar = 5 μm; magnification: ×10,000. |
The thermograms of BBR, BS20, physical mixture, and BBR-BS20-NCs are illustrated in Figure 5B. BBR presented a characteristic peak at 95°C predominantly corresponding to drug melting endotherm. The intensity of the characteristic peak was weakened in the thermogram of the physical mixture and almost disappeared in that of BBR-BS20-NCs, indicating much loss of its crystallinity.
The FTIR spectra of BBR, BS20, physical mixture, and BBR-BS20-NCs are illustrated in Figure 5C. The spectra of both the physical mixture and BBR-BS20-NCs presented a simple overlap with those of BBR and BS20. The results indicated that there was no chemical reaction between BBR and BS20 in the formation of BBR-BS20-NCs.
The representative SEM images of BBR, BS20, physical mixture, and BBR-S20-NCs are illustrated in Figure 5D. The pure BBR exhibited irregular crystal structure and rough surface with the size of 1–2 μm, while BS20 exhibited irregular structure and smooth surface with the size >5 μm. Physical mixture (mechanically blended berberine and Brij-S20) was observed with the simple overlay of BBR and BS20 by the surface adherence of BS20 to the surface of BBR. By contrast, BBR-BS20-NCs showed a prism-like morphology with smooth surface, which was quite different to the form of pure BBR.
Dissolution test
The in vitro drug release of BBR, physical mixture, and BBR-BS20-NCs is illustrated in Figure 6. Physical mixture of BBR with BS20 did not influence the dissolution rate and cumulative release of BBR, when compared to those of pure BBR. However, BBR-BS20-NCs significantly improved the dissolution rate and extent of release of BBR, which might be due to the reduced particle size and increased specific area of the nanocrystal formulation.
 | Figure 6 In vitro dissolution profiles for pure BBR, physical mixture (blended berberine and Brij-S20), and BBR-BS20-NCs. Data are presented as mean ± SD (n = 3). |
Transepithelial transport of BBR
The transepithelial transport of BBR-BS20-NCs and BBR (in the absence or presence of Pgp inhibitors) across the MDCK-MDR1 cell monolayer was determined and compared. The results are summarized in Table 2. BBR exhibited a strong efflux in MDCK-MDR1 cell model at the concentration of 10 μM. The secretory permeability coefficient of BBR (Papp[BL-AP]: 2.85 ± 0.04 × 10−6 cm/s) was found to be 7.1-fold its absorptive permeability coefficient (Papp[AB-BL]: 0.40 ± 0.01 × 10−6 cm/s). However, the BBR efflux could be significantly inhibited by the Pgp inhibitors of CsA (2 μM) and verapamil (10 μM) with the decreased EfR values of 4.83 and 2.17, respectively. It is worth noting that BS20 modulated Pgp efflux and increased BBR permeability in both physical mixture group and BBR-BS20-NCs group (EfR values 3.77 and 3.62, respectively).
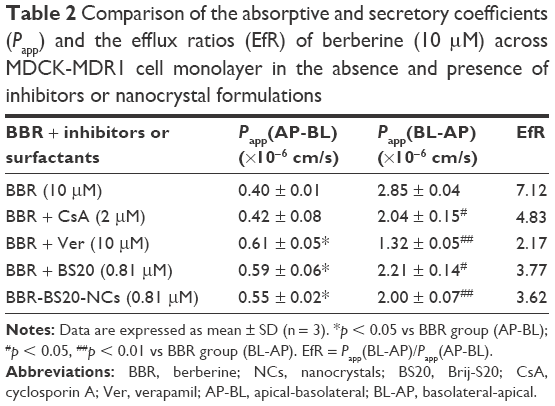 | Table 2 Comparison of the absorptive and secretory coefficients (Papp) and the efflux ratios (EfR) of berberine (10 μM) across MDCK-MDR1 cell monolayer in the absence and presence of inhibitors or nanocrystal formulations |
In vivo pharmacokinetic study
The plasma concentration curve of BBR vs time obtained after oral administration of pure BBR and BBR-BS20-NCs is shown in Figure 7, and the main pharmacokinetic parameters are listed in Table 3. In Figure 7, a double-absorption curve was observed in both pure BBR and BBR-BS20-NCs, and this may be due to enterohepatic circulation as reported by previous studies.49,50 The Cmax and AUC0~t values of BBR-BS20-NCs were 1.9- and 2.4-fold to the values of pure BBR, and the relative bioavailability of BBR-BS20-NCs to pure BBR was 404.1%. The increased absorption and improved bioavailability of BBR-BS20-NCs group proved that the improvement of BBR solubility after preparation of nanocrystal formulation and BS20 is an efficient Pgp modulator to inhibit Pgp-mediated drug efflux.
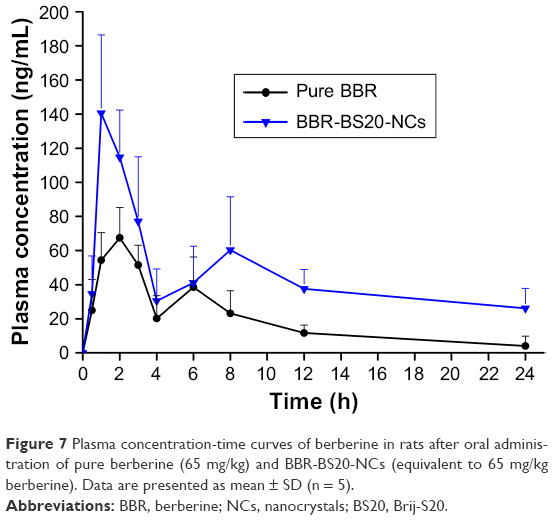 | Figure 7 Plasma concentration-time curves of berberine in rats after oral administration of pure berberine (65 mg/kg) and BBR-BS20-NCs (equivalent to 65 mg/kg berberine). Data are presented as mean ± SD (n = 5). |
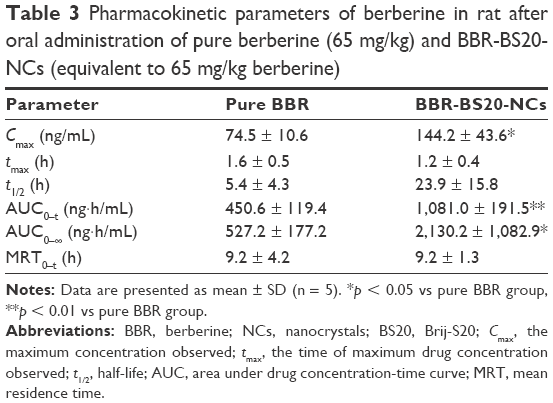 | Table 3 Pharmacokinetic parameters of berberine in rat after oral administration of pure berberine (65 mg/kg) and BBR-BS20-NCs (equivalent to 65 mg/kg berberine) |
Discussion
Pgp, a product of ABCB1 gene (or human MDR1), is composed of 1,280 amino acids with 170 kDa molecular weight and having 2 ATP-binding cassettes and 12 transmembrane helices in its structure.51 In human intestine, Pgp sits on the apical surface of superficial columnar epithelial cells,52 which act as the gatekeepers for cells, controlling cellular uptake and drug efflux.53 Because of the function of Pgp efflux, sufficient oral absorption of BBR would not be expected.
TPGS has been widely studied as a functional excipient for overcoming multidrug resistance (MDR) and as an inhibitor of Pgp for increasing the oral bioavailability of anticancer drugs.54 Recently, the key structures of Brij surfactants that play a role in overcoming MDR in cancer cells were investigated and reported.41 The structure of Brij is similar to that of TPGS, and both of them have an amphiphilic structure with a lipophilic tail of varying lengths of linear hydrocarbon and a hydrophilic polar head of varying lengths of polyethylene glycol. Thus, Brij surfactants might be developed as another functional excipient and can help to improve the oral bioavailability of drugs influenced by Pgp. As shown in Figure 2, although TPGS is the most powerful nonionic surfactant with the function of Pgp inhibition, the critical micelle concentration of BS20 (0.006 mM) is much lower than that of TPGS (0.02 mM). So BS20 was selected to form BBR-BS20-NCs. The reason why we prepared BBR-BS20-NCs is due to their structural simplicity, as BBR-BS20-NCs only require one type of Brij surfactant as an excipient.
Using efflux transporter modulators may be another promising strategy, especially functional excipients that are frequently applied as pharmaceutical additives and can suppress the function of efflux transporters involving Pgp.11 In the present study, BS20 increased the accumulation of BBR in MDCK-MDR1 cells (Figure 3), suggesting that it could inhibit the efflux function of Pgp, thereby increasing the intracellular concentration of Pgp substrate.
Expression of Pgp was evaluated by Western blotting followed by quantitative PCR assay, and the results (Figure 4A and B) indicated that the regulation of BS20 on Pgp expression was observed at the transcription level instead of protein expression level. Actually, Pgp is well equipped for the task of cellular defense, and the rapid upregulation of Pgp aims to protect the cells against a multitude of adverse insults.55 Considerable knowledge about the transcriptional upregulation of the Pgp gene now exists. It has been reported that in vitro treatment of kidney cancer cells with depsipeptide (5 ng/mL, 72 h) resulted in increased expression levels of Pgp (6.3-fold).56 This upregulation of mRNA of Pgp was also observed to known Pgp inhibitors.57 Exposure of human intestinal epithelial T84 cells to Pgp inhibitors such as quinidine, atorvastatin, and amprenavir (10 μM, for 72 h) significantly increased the mRNA levels of Pgp (3.8-, 4.1-, 5.1-fold, respectively). These aforementioned results suggested that the potential protective effect of Pgp is expressed at transcriptional level, which may not be reflected by the increased protein expression.53,58
BS20 inhibited the activity of intracellular ATP in 3 h, and this effect reversed to normal level in 24 h (Figure 4C). The results suggested that BS20 could enhance the oral absorption of BBR by short-term modulation of the function of intestinal Pgp, without causing any influence on the physiological properties of intestinal membrane. BS20 might be considered to be a safe and functional excipient to increase the oral bioavailability of those drugs that exhibit Pgp-mediated efflux. With regard to Pgp ATPase activity, binding of test compounds to the drug-binding cavity in transmembrane domain (TMD) of Pgp will stimulate ATPase activity, and these compounds can be identified as stimulators,59 while inhibitors bind to the site located in nucleotide binding domain of Pgp and inhibit ATP hydrolysis.60 The reduced simulation by the combination of verapamil and BS20 indicated that they competed with one other for interaction with similar drug binding sites inside the TMD of Pgp (Figure 4D).61
Lyophilization has been considered a good technique to improve the long-term stability of nanoparticles.62 A good lyophilizate should be able to be easily reconstituted and maintain the physical and chemical properties of the original product. In this study, the reconstituted size of lyophilized BBR-BS20-NCs (161.0nm, PDI0.29) was not significantly changed compared to that of the freshly prepared nanosuspension, but there was a slight shift in the zeta potential which was found to be −0.83 mV (Table 1). The results indicated the BBR-BS20-NCs exhibited good stability during the freeze-drying process.
The distribution of membrane P-gp plays an important role in the transepithelial transport of drugs across the intestinal membrane. The expression and activity of the intestinal Pgp significantly influence the oral bioavailability of the drugs via the Pgp-mediated drug efflux. The expression of Pgp increases from proximal regions to distal regions in small intestine, which means higher absorption of Pgp substrates could be expected at the proximal intestine instead of at the distal intestine.63 However, the substrates must be released rapidly and become a solution before reaching the proximal intestine, to be absorbed at the proximal intestine. All these results proved the significance of the absorption of Pgp substrates at the proximal intestine in order to escape Pgp-mediated efflux.64 Thus, the speed of dissolution of BBR would be very significant, especially the rapid solubility will play an important role in escaping from Pgp-mediated efflux. BBR-BS20-NCs showed faster and more complete release in comparison with pure BBR and physical mixture (Figure 6). This fast release behavior of BBR- BS20-NCs benefits the escape of Pgp-mediated efflux in the distal intestine and improve the oral absorption of BBR.
In transwell experiment of BBR (Table 2), the results suggested the AQ and SQ values for BBR in the presence of 10 μM verapamil were 0.344 and 1.16 respectively, suggesting Pgp does not much affect the absorptive transport (AQ <0.5) of BBR but highly affects its secretory transport (SQ >0.5) in MDCK-MDR1 cell model. Based on the aforementioned results, it was hypothesized that BS20-modified BBR nanocrystals (BBR-BS20-NCs) might improve the absorptive transport of BBR by combining the advantages of nanocrystal formulation and the modulation of BS20 on Pgp. The results of bidirectional transport of BBR-BS20-NCs indicated that BBR-BS20-NCs presented more potency on enhancing BBR absorption than the physical mixture by decreasing the secretory transport (Papp[BL-AP] values of 2.00 ± 0.07 × 10−6 cm/s for BBR-BS20-NCs and 2.21 ± 0.14 × 10−6 cm/s for physical mixture).
The improvement of the oral bioavailability of BBR is a key determinant for its further clinical application. Poor solubility and Pgp-mediated efflux induced low intestinal permeability which in turn resulted in low bioavailability of BBR after oral administration. The developed BBR-BS20-NCs combine the advantages of nanocrystal formulation (drug solubilization) and functional excipient (Pgp modulation), which improved the speed and extent of release of BBR, as well as increased the permeability of BBR. Thus, in vivo results showed that BBR-BS20-NCs markedly enhanced the oral bioavailability of BBR compared to pure BBR. These results proved that a BS20-modified nanocrystal formulation (BBR-BS20-NCs) is a promising strategy to extend the application of BBR in a clinical setting.
Conclusion
In the present study, the effects and mechanisms of the nonionic surfactant of BS20 on P-gp modulation in MDCK-MDR1 cells have been investigated and reported. BS20 has been demonstrated to inhibit Pgp activity reversibly and in the short term, coupled with a marked increase in Pgp mRNA expression but without significant effects on the protein expression of cellular Pgp. The results suggest that BS20 might be used as a pharmacological nonactive Pgp inhibitor to improve the oral bioavailability of those drugs which are affected by the Pgp-mediated efflux.
Based on the aforementioned results, BS20-modified BBR nanocrystals (BBR-BS20-NCs) have been developed and characterized. BBR-BS20-NCs present smaller particle size and better stability than BBR-NCs (without BS20). Moreover, BBR-BS20-NCs effectively overcome Pgp-mediated BBR efflux (EfR value of 7.12) and significantly enhance BBR absorption (EfR value of 3.62) in MDCK-MDR1 cells. Also, pharmacokinetic study proved that BBR-BS20-NCs statistically enhanced the bioavailability of BBR by combination of the advantages of nanocrystal formulations and Pgp modulation by BS20.
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (No 81470170), the Research Committee of the University of Macau (SRG2015-00060-ICMS-QRCM, MYRG2017-00178-ICMS, MYRG2016-00144-ICMS-QRCM), Macao Science and Technology Development Fund (FDCT 013/2015/A1), and the opening fund of the State Key Laboratory of Quality Research in Chinese Medicine of University of Macau (No SKL-QRCM-2014-2016).
Disclosure
The authors report no conflicts of interest in this work.
References
Chavda HV, Patel CN, Anand IS. Biopharmaceutics classification system. Sys Rev Pharm. 2010;1(1):62–69. | ||
Al-Kassas R, Bansal M, Shaw J. Nanosizing techniques for improving bioavailability of drugs. J Control Release. 2017;260:202–212. | ||
Göke K, Lorenz T, Repanas A, et al. Novel strategies for the formulation and processing of poorly water-soluble drugs. Eur J Pharm Biopharm. 2018;126:40–56. | ||
Jermain SV, Brough C, Williams RO. Amorphous solid dispersions and nanocrystal technologies for poorly water-soluble drug delivery – an update. Int J Pharm. 2018;535(1):379–392. | ||
Loftsson T. Drug solubilization by complexation. Int J Pharm. 2017;531(1):276–280. | ||
Singh B, Beg S, Khurana RK, Sandhu PS, Kaur R, Katare OP. Recent advances in self-emulsifying drug delivery systems (SEDDS). Crit Rev Ther Drug Carrier Syst. 2014;31(2):121–185. | ||
Zhang L, Wang S, Zhang M, Sun J. Nanocarriers for oral drug delivery. J Drug Target. 2013;21(6):515–527. | ||
Pathak K, Raghuvanshi S. Oral bioavailability: issues and solutions via nanoformulations. Clin Pharmacokinet. 2015;54(4):325–357. | ||
Gao L, Liu G, Ma J, et al. Application of drug nanocrystal technologies on oral drug delivery of poorly soluble drugs. Pharm Res. 2013;30(2):307–324. | ||
Zakeri-Milani P, Valizadeh H. Intestinal transporters: enhanced absorption through P-glycoprotein-related drug interactions. Expert Opin Drug Metab Toxicol. 2014;10(6):859–871. | ||
Ankit S, Ravindra Dhar D, Noor A, Vaibhav K, Prem NG. Co-formulation of P-glycoprotein substrate and inhibitor in nanocarriers: an emerging strategy for cancer chemotherapy. Curr Cancer Drug Targets. 2014;14(5):419–433. | ||
Alakhova DY, Kabanov AV. Pluronics and MDR reversal: an update. Mol Pharm. 2014;11(8):2566–2578. | ||
Ayati SH, Fazeli B, Momtazi-Borojeni AA, Cicero AFG, Pirro M, Sahebkar A. Regulatory effects of berberine on microRNome in cancer and other conditions. Crit Rev Oncol Hematol. 2017;116:147–158. | ||
Rabbani GH, Butler T, Knight J, Sanyal SC, Alam K. Randomized controlled trial of berberine sulfate therapy for diarrhea due to enterotoxigenic Escherichia coli and Vibrio cholerae. J Infect Dis. 1987;155(5):979–984. | ||
Chen C, Tao C, Liu Z, et al. A randomized clinical trial of berberine hydrochloride in patients with diarrhea-predominant irritable bowel syndrome. Phytother Res. 2015;29(11):1822–1827. | ||
Čerňáková M, Košťálová D. Antimicrobial activity of berberine – a constituent of Mahonia aquifolium. Folia Microbiol (Praha). 2002;47(4):375–378. | ||
Karaosmanoglu K, Sayar NA, Kurnaz IA, Akbulut BS, Akbulut BS. Assessment of berberine as a multi-target antimicrobial: a multi-omics study for drug discovery and repositioning. Omics. 2014;18(1):42–53. | ||
Cicero AFG, Baggioni A. Berberine and its role in chronic disease. In: Gupta SC, Prasad S, Aggarwal BB, editors. Anti-Inflammatory Nutraceuticals and Chronic Diseases. Cham, Switzerland: Springer International Publishing; 2016:27–45. | ||
Pang B, Zhao L-H, Zhou Q, et al. Application of berberine on treating type 2 diabetes mellitus. Int J Endocrinol. 2015;2015:905749. | ||
Chang W. Non-coding RNAs and berberine: a new mechanism of its anti-diabetic activities. Eur J Pharmacol. 2017;795:8–12. | ||
Pirillo A, Catapano AL. Berberine, a plant alkaloid with lipid- and glucose-lowering properties: from in vitro evidence to clinical studies. Atherosclerosis. 2015;243(2):449–461. | ||
Jiang W, Li S, Li X. Therapeutic potential of berberine against neurodegenerative diseases. Sci China Life Sci. 2015;58(6):564–569. | ||
Ahmed T, Gilani A-U-H, Abdollahi M, Daglia M, Nabavi SF, Nabavi SM. Berberine and neurodegeneration: a review of literature. Pharmacol Rep. 2015;67(5):970–979. | ||
Li Z, Geng Y-N, Jiang J-D, Kong W-J. Antioxidant and anti-inflammatory activities of berberine in the treatment of diabetes mellitus. Evid Based Complement Alternat Med. 2014;2014:289264. | ||
Ni W-J, Ding H-H, Tang L-Q. Berberine as a promising anti-diabetic nephropathy drug: an analysis of its effects and mechanisms. Eur J Pharmacol. 2015;760:103–112. | ||
Habtemariam S. Berberine and inflammatory bowel disease: a concise review. Pharmacol Res. 2016;113(Pt A):592–599. | ||
Guamán Ortiz ML, Lombardi P, Tillhon M, Scovassi IA. Berberine, an epiphany against cancer. Molecules. 2014;19(8):12349–12367. | ||
Wang N, Tan H-Y, Li L, Yuen M-F, Feng Y. Berberine and coptidis rhizoma as potential anticancer agents: recent updates and future perspectives. J Ethnopharmacol. 2015;176:35–48. | ||
Zou K, Li Z, Zhang Y, et al. Advances in the study of berberine and its derivatives: a focus on anti-inflammatory and anti-tumor effects in the digestive system. Acta Pharmacol Sin. 2017;38(2):157–167. | ||
Shen R, Kim JJ, Yao M, Elbayoumi TA. Development and evaluation of vitamin E d-α-tocopheryl polyethylene glycol 1000 succinate-mixed polymeric phospholipid micelles of berberine as an anticancer nanopharmaceutical. Int J Nanomedicine. 2016;11:1687–1700. | ||
Zhaojie M, Ming Z, Shengnan W, et al. Amorphous solid dispersion of berberine with absorption enhancer demonstrates a remarkable hypoglycemic effect via improving its bioavailability. Int J Pharm. 2014;467(1):50–59. | ||
Tsai P-L, Tsai T-H. Hepatobiliary excretion of berberine. Drug Metab Dispos. 2004;32(4):405. | ||
Zhang X, Qiu F, Jiang J, Gao C, Tan Y. Intestinal absorption mechanisms of berberine, palmatine, jateorhizine, and coptisine: involvement of P-glycoprotein. Xenobiotica. 2011;41(4):290–296. | ||
Shi J, Guo F, Zheng A, Zhang X, Sun J. Progress in the study of drug nanocrystals. Die Pharmazie. 2015;70(12):757–764. | ||
Rabinow BE. Nanosuspensions in drug delivery. Nat Rev Drug Discov. 2004;3(9):785–796. | ||
Nagarwal RC, Kumar R, Dhanawat M, Das N, Pandit JK. Nanocrystal technology in the delivery of poorly soluble drugs: an overview. Curr Drug Deliv. 2011;8(4):398–406. | ||
Khajuria DK, Konnur MC, Vasireddi R, Mahapatra DR. Photonic monitoring of chitosan nanostructured alginate microcapsules for drug release. In: Proceedings of SPIE 9303, Photonic Therapeutics and Diagnostics XI, 93033V; San Francisco, CA: February 26, 2015. | ||
Gao L, Liu G, Kang J, et al. Paclitaxel nanosuspensions coated with P-gp inhibitory surfactants: I. Acute toxicity and pharmacokinetics studies. Colloids Surf B Biointerfaces. 2013;111:277–281. | ||
Patel K, Patil A, Mehta M, Gota V, Vavia P. Oral delivery of paclitaxel nanocrystal (PNC) with a dual Pgp-CYP3A4 inhibitor: preparation, characterization and antitumor activity. Int J Pharm. 2014;472(1):214–223. | ||
Ribeiro M, de Moura CL, Vieira MGS, et al. Solubilisation capacity of Brij surfactants. Int J Pharm. 2012;436(1):631–635. | ||
Tang J, Wang Y, Wang D, et al. Key structure of Brij for overcoming multidrug resistance in cancer. Biomacromolecules. 2013;14(2):424–430. | ||
Yu H, Hu YQ, Ip FCF, Zuo Z, Han YF, Ip NY. Intestinal transport of bis(12)-hupyridone in Caco-2 cells and its improved permeability by the surfactant Brij-35. Biopharm Drug Dispos. 2011;32(3):140–150. | ||
Cui HM, Zhang QY, Wang JL, Chen JL, Zhang YL, Tong XL. Poor permeability and absorption affect the activity of four alkaloids from coptis. Mol Med Rep. 2015;12(5):7160–7168. | ||
Jin D, Zhao T, Feng W-W, et al. Schisandra polysaccharide increased glucose consumption by up-regulating the expression of GLUT-4. Int J Biol Macromol. 2016;87:555–562. | ||
Liu F, Park JY, Zhang Y, et al. Targeted cancer therapy with novel high drug-loading nanocrystals. J Pharm Sci. 2010;99(8):3542–3551. | ||
Xiong W, Li L, Wang Y, et al. Design and evaluation of a novel potential carrier for a hydrophilic antitumor drug: auricularia auricular polysaccharide-chitosan nanoparticles as a delivery system for doxorubicin hydrochloride. Int J Pharm. 2016;511(1):267–275. | ||
Xiong W, Zhang Q, Yin F, et al. Auricularia auricular polysaccharide-low molecular weight chitosan polyelectrolyte complex nanoparticles: preparation and characterization. Asian J Pharm Sci. 2016;11(3):439–448. | ||
Guo Y, Chu M, Tan S, et al. Chitosan-g-TPGS nanoparticles for anticancer drug delivery and overcoming multidrug resistance. Mol Pharm. 2014;11(1):59–70. | ||
Shi C, Tong Q, Fang J, Wang C, Wu J, Wang W. Preparation, characterization and in vivo studies of amorphous solid dispersion of berberine with hydrogenated phosphatidylcholine. Eur J Pharm Sci. 2015;74(Suppl C):11–17. | ||
Zhang Z, Chen Y, Deng J, Jia X, Zhou J, Lv H. Solid dispersion of berberine–phospholipid complex/TPGS 1000/SiO2: preparation, characterization and in vivo studies. Int J Pharm. 2014;465(1):306–316. | ||
Chen C-J, Chin JE, Ueda K, et al. Internal duplication and homology with bacterial transport proteins in the mdr1 (P-glycoprotein) gene from multidrug-resistant human cells. Cell. 1986;47(3):381–389. | ||
Didier A, Wenger J, Loor F. Decreased uptake of cyclosporin A by P-glycoprotein (Pgp) expressing CEM leukemic cells and restoration of normal retention by Pgp blockers. Anticancer drugs. 1995;6(5):669–680. | ||
Fojo AT, Ueda K, Slamon DJ, Poplack DG, Gottesman MM, Pastan I. Expression of a multidrug-resistance gene in human tumors and tissues. Proc Natl Acad Sci U S A. 1987;84(1):265–269. | ||
Zhang Z, Tan S, Feng S-S. Vitamin E TPGS as a molecular biomaterial for drug delivery. Biomaterials. 2012;33(19):4889–4906. | ||
Silva R, Vilasboas V, Carmo H, et al. Modulation of P-glycoprotein efflux pump: induction and activation as a therapeutic strategy. Pharmacol Ther. 2015;149(2):1–123. | ||
Robey RW, Zhan Z, Piekarz RL, Kayastha GL, Fojo T, Bates SE. Increased MDR1 expression in normal and malignant peripheral blood mononuclear cells obtained from patients receiving depsipeptide (FR901228, FK228, NSC630176). Clin Cancer Res. 2006;12(5):1547–1555. | ||
Haslam IS, Jones K, Coleman T, Simmons NL. Induction of P-glycoprotein expression and function in human intestinal epithelial cells (T84). Biochem Pharmacol. 2008;76(7):850–861. | ||
Arias A, Rigalli JP, Villanueva SS, et al. Regulation of expression and activity of multidrug resistance proteins MRP2 and MDR1 by estrogenic compounds in Caco-2 cells. Role in prevention of xenobiotic-induced cytotoxicity. Toxicology. 2014; 320:46–55. | ||
Ambudkar SV, Dey S, Hrycyna CA, Ramachandra M, Pastan I, Gottesman MM. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu Rev Pharmacol Toxicol. 1999;39(1):361–398. | ||
Chan KF, Zhao Y, Burkett BA, Wong ILK, Chow LMC, Chan TH. Flavonoid dimers as bivalent modulators for P-glycoprotein-based multidrug resistance: synthetic apigenin homodimers linked with defined-length poly(ethylene glycol) spacers increase drug retention and enhance chemosensitivity in resistant cancer cells. J Med Chem. 2006;49(23):6742–6759. | ||
Kim MK, Choo H, Chong Y. Water-soluble and cleavable quercetin–amino acid conjugates as safe modulators for P-glycoprotein-based multidrug resistance. J Med Chem. 2014;57(17):7216–7233. | ||
Fonte P, Reis S, Sarmento B. Facts and evidences on the lyophilization of polymeric nanoparticles for drug delivery. J Control Release. 2016;225:75–86. | ||
Mouly S, Paine MF. P-Glycoprotein increases from proximal to distal regions of human small intestine. Pharm Res. 2003;20(10):1595–1599. | ||
Chen C, Liu X, Smith BJ. Utility of Mdr1-gene deficient mice in assessing the impact of P-glycoprotein on pharmacokinetics and pharmacodynamics in drug discovery and development. Curr Drug Metab. 2003;4(4):272–291. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.