")
Back to Journals » Transplant Research and Risk Management » Volume 7
Drugs in development for prophylaxis of rejection in kidney-transplant recipients
Authors Sanders ML, Langone A
Received 2 May 2015
Accepted for publication 30 June 2015
Published 18 August 2015 Volume 2015:7 Pages 59—69
DOI https://doi.org/10.2147/TRRM.S61446
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Qing Yi
Marion Lee Sanders,1 Anthony James Langone2
1Department of Medicine, Division of Nephrology and Hypertension, University of Iowa, Iowa City, IA, 2Department of Medicine, Division of Nephrology and Hypertension, Vanderbilt University Medical Center, Nashville, TN, USA
Abstract: Transplantation is the preferred treatment option for individuals with end-stage renal disease. Individuals who undergo transplantation must chronically be maintained on an immunosuppression regimen for rejection prophylaxis to help ensure graft survival. Current rejection prophylaxis consists of using a combination of calcineurin inhibitors, mTOR inhibitors, antimetabolite agents, and/or corticosteroids. These agents have collectively improved the short-term outcomes of renal transplantation, but improvements in late/chronic graft loss and recipient survival have lagged significantly behind challenging the field of transplantation to develop novel prophylactic agents. There have been several clinical trials conducted within the last 5 years in an attempt to bring such novel agents to the commercial market. These trials have resulted in the US Food and Drug Administration (FDA) approval of extended-release tacrolimus, as well as belatacept, which has the potential to replace calcineurin inhibitors for rejection prophylaxis. Other trials have focused on the development of novel calcineurin inhibitors (voclosporin), costimulation blockade (ASKP1240 and alefacept), kinase inhibitors (tofacitinib and sotrastaurin), and inhibitors of leukocyte migration (efalizumab). While these later agents have not been FDA-approved for use in transplantation, they remain noteworthy, as these agents explore pathways not previously targeted for allograft-rejection prophylaxis. The purpose of this review was to consolidate available clinical trial data with regard to the recent developments in rejection prophylaxis in kidney transplantation.
Keywords: rejection, prophylaxis, immunosuppression, transplantation
Introduction
Transplantation is the preferred treatment for individuals with end-stage renal disease, and has consistently been associated with superior survival compared to long-term dialysis. The early years of kidney transplantation were marked by early graft failure, due to the lack of safe and effective immunosuppressive drug therapy. Early graft failure due to immunologic rejection is now a rare occurrence in the modern era, where noncompliance is not a factor, due to significant advances in immunosuppression.
Transplant immunosuppression typically includes the use of an induction agent at the time of transplantation followed by a maintenance regimen for the duration of graft survival. Induction agents are typically antibodies (antithymocyte globulins) or an interleukin (IL)2-receptor antagonist (basiliximab). Alemtuzumab is also used as a lymphocyte-depleting induction agent in renal transplantation at some centers, as it targets the CD52 protein on the surface of mature lymphocytes. While alemtuzumab has officially been removed from the US market, it is still available through a special manufacturer program. The drug classes that currently comprise the majority of maintenance regimens are calcineurin inhibitors (CNIs; tacrolimus and cyclosporine), mammalian target of rapamycin (mTOR) inhibitors (sirolimus and everolimus), antimetabolite agents (azathioprine and mycophenolic acid derivatives), and corticosteroids. Kidney Disease: Improving Global Outcomes clinical practice guidelines suggest that first-line agents should include basiliximab induction for low-risk patients and an antithymocyte globulin for high-risk patients in conjunction with maintenance immunosuppression of tacrolimus and mycophenolate.1
The improvement in early graft survival using the aforementioned immunosuppression agents has unfortunately not equally translated into improved long-term graft or recipient survival, as late/chronic graft loss and recipient survival have remained virtually unchanged. Chronic allograft injury as a result of immunosurveillance in addition to unfavorable medication side effects contribute to this unfortunate observation. Side effects, such as new-onset diabetes, dyslipidemia, and hypertension, contribute to cardiovascular disease that is the major cause of recipient death posttransplant. Immunosuppression medication can also contribute to increased infection and malignancy risk, which are also major causes of recipient morbidity and mortality. Therefore, continued research and development of novel immunosuppression agents for the prevention of rejection while simultaneously minimizing long-term morbidity and mortality in transplant recipients is warranted.
There have been several clinical trials conducted within the last 5 years in an attempt to bring such novel agents to the commercial market for use in transplantation (Table 1). Some trials have resulted in new medications approved (extended-release tacrolimus, belatacept), while other efforts have unfortunately fallen short. This review discusses developments and clinical trial outcomes with regard to new agents for the prophylaxis of rejection in kidney-transplant recipients. Novel agents for the treatment of acute rejection are outside the scope of this review.
 | Table 1 Summary of investigational and new agents approved for rejection prophylaxis in renal transplantation |
Calcineurin-inhibitor derivatives
Calcineurin is a calcium-dependent serine/threonine phosphatase that activates nuclear factor of activated T-cells (NFAT), allowing it to translocate to the nucleus, where it upregulates IL2, resulting in an escalation of the immune system. Historically, the introduction of CNIs changed the face of transplantation by dramatically improving graft survival in transplant recipients, and CNIs have become the backbone of immunosuppressive therapy to prevent allograft rejection.1,2 While CNI use has led to excellent short-term graft-survival rates, CNIs have a relatively narrow therapeutic window between rejection prevention and an unfavorable side-effect profile, including neurotoxicity and nephrotoxicity.3 Standard CNIs (tacrolimus and cyclosporine) also require twice-daily dosing, and recipient compliance is a potential issue.4
Extended-release tacrolimus
Prolonged or extended-release tacrolimus has been introduced in an effort to potentially reduce the peak-level neurologic side effects and improve adherence due to single daily dosing.5 In a Phase III, open-label, comparative, noninferiority study, 638 subjects receiving de novo kidney transplants were randomized to one of three treatment arms: daily tacrolimus extended release (Astagraf XL®), tacrolimus (Prograf®) twice daily, or cyclosporine A (CsA) twice daily. All subjects received basiliximab induction, mycophenolate mofetil, and corticosteroids. After 4 years, 129 Astagraf XL, 113 Prograf, and 79 CsA patients had continued follow-up, and Kaplan–Meier estimates of patient survival were 93.2%, 91.2%, and 91.7%, while graft survival was 84.7%, 82.7%, and 93.9%, respectively. Renal function was not significantly different between Astagraf XL and Prograf, but was significantly different comparing Astagraf XL and CsA with higher mean renal function in the Astagraf XL group. While overall efficacy and safety profiles were similar between arms, the notable exception was that the incidence of new-onset diabetes (HbA1c >6.5%) was significantly higher for both extended and regular tacrolimus formulations (41.1% and 33.6%, respectively) compared to the CsA group (21.3%).6
A Phase III trial examining the efficacy and safety of another extended-release formulation (LCP-tacrolimus [LCPT]) vs twice-daily tacrolimus (Prograf) in de novo renal transplants has also been completed. A total of 543 patients were randomized, and the 12-month composite treatment-failure end point (death, graft failure, biopsy-proven acute rejection [BPAR], or loss to follow-up) was 18.3% in the LCPT group vs 19.6% in the twice-daily tacrolimus group. No significant differences in adverse events were noted between groups.7
Another Phase III noninferiority trial examined the efficacy and safety of converting stable renal transplant recipients from twice-daily tacrolimus (Prograf) to LCPT. Controls for the study were maintained on twice-daily tacrolimus. A total of 326 patients were randomized, and at 12 months there were four efficacy failures (death, graft failure, BPAR, or loss to follow-up) in each group. The mean daily dose of LCPT was significantly lower than preconversion tacrolimus dose at each follow-up visit, illustrating that LCPT required lower doses to achieve target trough levels. The incidence of adverse events was also similar between the two groups.8
The collective results of these Phase III clinical trials illustrate that an extended-release formulation of tacrolimus is a safe, efficacious, and effective immunosuppressive option in renal transplantation. These results also led to the approval for use in renal transplantation in the USA in July 2013. There are several ongoing clinical trials further evaluating the extended-release formulation of tacrolimus in organ transplantation (Table 2). Continued observation is warranted to determine if the extended-release formulation with its once-daily dose requirement translates into improved chronic allograft outcomes.
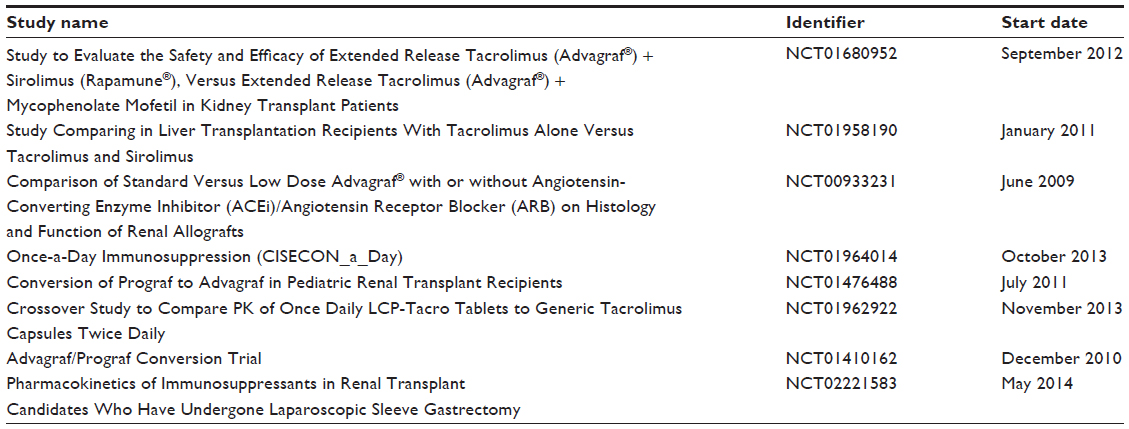 | Table 2 Active ongoing clinical trials further evaluating extended-release tacrolimus in organ transplantation |
Voclosporin
Voclosporin (VCS; ISA247) is an analog of CsA developed in the mid-1990s by the addition of a single-carbon extension to amino acid 1 of CsA. This single-carbon extension as a trans isomer yields a more potent CNI compared to CsA, due to improved interaction between the modified amino acid 1 residue and cyclophilin.9 Amino acid 1 is also the primary site of metabolism for CsA, but modification of this site in VCS changes the primary site for VCS metabolism to the amino acid 9 position, yielding IM9 as the primary metabolite, which has approximately 10% of the parent-compound activity. IM9 is produced in significantly smaller amounts than AM1, the major CsA metabolite, resulting in less competitive antagonism of the metabolite with the parent VCS compound.10 This combination of increased potency and a more favorable metabolite profile for VCS allows for administration of lower doses compared to CsA and an improved safety profile, as demonstrated initially in both in vitro and in vivo animal models, followed by significantly longer renal allograft-survival times in nonhuman primates.11,12
Pharmacokinetic and pharmacodynamic studies in humans have demonstrated that over 99% of the drug is eliminated as metabolite, that the parent molecule is 97% protein-bound, and that VCS inhibits calcineurin activity in a dose-related fashion, with maximal inhibition peaking at 3.0 mg/kg. VCS was safe and well tolerated in healthy human volunteers after oral administration at doses ranging from 0.25 to 4.5 mg/kg. Decreased drug exposure occurs with simultaneous food administration, with a more pronounced effect following a high-fat meal.10 Administration of VCS with strong inhibitors and inducers of CYP3A results in increased and decreased VCS exposure, respectively, and administration of VCS with inhibitors and substrates of P-glycoprotein results in increased VCS and substrate exposure, respectively.13 Appropriate safety monitoring would be needed with coadministration of VCS with CYP3A inhibitors/inducers and P-glycoprotein inhibitors/substrates.
VCS has also been evaluated in human subjects with renal and hepatic impairment. A total of 33 subjects were enrolled into one of four groups based on renal function, as defined by creatinine clearance (>80 mL/min, 50–80 mL/min, 30–50 mL/min, and <30 mL/min), and 18 subjects were enrolled into one of three groups based on hepatic function defined by Child–Pugh class (normal, class A, and class B). Results demonstrated that VCS can be administered safely to patients with mild-to-moderate renal impairment without dose modification, but dose adjustments are recommended for patients with severe renal impairment, as well as for patients with hepatic impairment.14
PROMISE was a 6-month Phase IIB multicenter, randomized, open-label clinical trial comparing three doses of VCS (0.4 mg/kg, 0.6 mg/kg, and 0.8 mg/kg) vs tacrolimus (0.05 mg/kg) in combination with a standard immunosuppression regimen (anti-CD25 antibody induction, mycophenolate mofetil, and corticosteroids) in 334 low-risk renal transplant recipients. Rejection rates, the primary end point for the study, for all three VCS doses were noninferior to tacrolimus (10.7%, 9.1%, and 2.3% vs 5.8%, respectively). Secondary objectives included renal function, new-onset diabetes after transplant, hyperlipidemia, hypertension, and pharmacokinetic–pharmacodynamic evaluation. Renal function was clinically similar (68–72 mL/min) at 6 months after transplantation among all groups. Compared to the tacrolimus group (16.4%), the incidence of new-onset diabetes after transplantation was significantly lower in the 0.4 mg/kg VCS group (1.6%), but not in the 0.6 mg/kg and 0.8 mg/kg VCS arms (5.7% and 17.7%, respectively). There were no clinically significant patterns of hyperlipidemia or hypertension development among the groups. There was exceptional correlation between trough and area under the curve for the VCS groups (r2=0.97), and there was no difference in mycophenolic acid exposure for the VCS groups compared to the tacrolimus group. The incidence of adverse events was similar among all four treatment groups, with no specific safety concerns raised. The major limitation of this trial was that only low-risk patients with immediate graft function were studied, but the results showed great promise for use of VCS for prevention of graft loss.15
INSPIRE was a Phase III randomized, multicenter, open-label, concentration-controlled safety and efficacy study to compare VCS and tacrolimus in renal transplant recipients. The trial was scheduled to begin in March 2013, with a plan for completion by December 2015 (NCT01586845). The primary end point for this noninferiority study was to assess efficacy failure at 12 months postrandomization; however, the study was withdrawn prior to enrollment for unclear reasons.
The Special Access for the Use of Voclosporin for Kidney Transplant study (NCT01236287) is listed as an active renal transplantation trial where subjects previously participating in the PROMISE study may be eligible to continue to receive VCS. Under this compassionate-release program, subjects previously taking VCS may continue to receive the study medication until the drug is US Food and Drug Administration (FDA)-approved and commercially available in the US; however, the status of this study has not been verified in more than 2 years. Outside the scope of renal transplantation, the use of VCS has been evaluated in the treatment of keratoconjunctivitis sicca, noninfectious uveitis, and plaque psoriasis, and is currently under evaluation to achieve complete remission of active lupus nephritis (NCT02141672).
Costimulation blockade
T-cells require two signals to become fully activated. The first signal is antigen-specific, and is provided through interaction between the peptide contained within the major histocompatibility-complex structure on the antigen-presenting cell and the T-cell receptor on the T-cell. The second signal is an antigen-independent costimulatory signal provided by interaction between other molecules on the antigen-presenting and T-cells. Of importance, without costimulation, T-cell activation does not occur, and anergy of the T-cell ensues.16,17 The T-cell requirement for costimulation provides a novel target for development of effective non-CNI immunosuppressant agents.
Belatacept
The interaction between B7 molecules (CD80 and CD86) on the antigen-presenting cell and the CD28 molecule on the T-cell is the most characterized costimulation pathway. Cytotoxic T-lymphocyte-associated protein 4 (CTLA4) is also expressed on T-cells, and can competitively bind with the CD80 and CD86 molecules, but this interaction prevents T-cell activation.18 Abatacept (Orencia®) was the initial CTLA4 analog, combining the extracellular binding domain of CTLA4 with a modified fragment-crystallizable domain of human IgG1, which resulted in a soluble fusion protein.19 Abatacept is FDA-approved for treatment of moderate-to-severe rheumatoid arthritis and was even effective in murine transplant models, but unfortunately it was not effective in nonhuman primate transplant models.20,21
It was determined that abatacept rapidly dissociated from the B7 molecules, and this likely explained its ineffectiveness in the nonhuman primate transplant models. Amino acid changes were made to the abatacept molecule: leucine was substituted for glutamate in position 104, and alanine was substituted for tyrosine in position 29 (LEA29Y, belatacept, Nulojix®). These amino acid changes doubled the avidity for CD80 and increased the avidity for CD86 fourfold compared to abatacept, in addition to increasing the capacity to inhibit T-cell activation tenfold. This modified LEA29Y molecule, belatacept, was then demonstrated to be effective for rejection prevention in a nonhuman primate renal transplant model and transitioned to human studies.22
BENEFIT was a 3-year, randomized, active-controlled, parallel-group, multicenter Phase III study that assessed a more intensive (MI) or less intensive (LI) regimen of belatacept vs CsA in adults receiving a kidney transplant from living or standard-criteria deceased donors. First-time recipients with a panel-reactive antibody of ≥50%, retransplants with a panel-reactive antibody of ≥30%, recipients of prior or concurrent nonrenal solid organ transplants, and recipients of extended-criteria donor kidneys were excluded. All randomized patients received induction therapy with basiliximab and maintenance therapy with daily mycophenolate mofetil and corticosteroids. The coprimary end points were patient/graft survival, a composite renal impairment end point (percentage with measured glomerular filtration rate [GFR] <60 mL/min at month 12 or a decrease in measured GFR ≥10 mL/min in months 3–12), and the incidence of acute rejection. A total of 527 patients completed month 12 of the study (173 MI, 181 LI, and 173 CsA). At month 12, both belatacept regimens had similar patient/graft survival vs CsA (95% and 97% vs 93%, respectively) and were associated with superior renal function as measured by the composite renal end point (55% and 54% vs 78%, respectively). Measured GFR was 65 mL/min and 63 mL/min vs 50 mL/min, respectively. Belatacept patients experienced a higher incidence (22% and 17% vs 7%, respectively) and grade of acute rejection episodes compared to CsA. Almost 100% of rejections occurred within the first 6 months posttransplantation.23
At 3 years, a total of 471 patients had completed ≥3 years of therapy (158 MI, 170 LI, 143 CsA). A total of 92% MI, 92% LI, and 89% CsA patients survived with a functioning graft. Belatacept-treated patients continued to have a higher GFR compared with CsA-treated recipients, with mean calculated GFR of 65.2 mL/min, 65.8 mL/min, and 44.4 mL/min, respectively. Between years 1 and 2, an additional eight patients experienced an acute rejection episode (four MI, zero LI, and four CsA, all nonrecurrent). There were no new cases of acute rejection in the belatacept groups from years 2 to 3, whereas one patient in the CsA group experienced an acute rejection episode. Mean blood pressure was significantly lower and the rise in non-high-density lipid cholesterol was significantly less in belatacept-treated patients, while the incident of new-onset diabetes was not significantly different between groups. Overall safety was generally similar, but posttransplant lymphoproliferative disorder (PTLD) was more common in the belatacept groups (three MI, two LI, and one CsA). Four of these six patients had known risk factors (Epstein–Barr virus-negative serology and/or received lymphocyte-depleting therapy as treatment for an acute rejection).24
BENEFIT-EXT was a Phase III trial with similar study design and end points as BENEFIT, but was conducted in recipients of extended-criteria donors, defined as ≥60 years old, ≥50 years old and at least two other risk factors (cerebrovascular accident, hypertension, serum creatinine >1.5 mg/dL), anticipated cold ischemia time of at least 24 hours, or donation after cardiac death. Lymphocyte-depleting therapy was permitted for anticipated delayed graft function only in the CsA-treated patients and for the treatment of rejection in all patient groups. A total of 387 patients were analyzed at 12 months (133 MI, 129 LI, and 125 CsA), and results demonstrated that both belatacept regimens had similar patient/graft survival vs cyclosporine (86% and 89% vs 85%, respectively), and were associated with superior renal function as measured by the composite renal end point (71% and 77% vs 85%, respectively). Measured GFR was 52 mL/min, 50 mL/min, and 45 mL/min, respectively. The incidence of acute rejection was also similar across groups (18%, 18%, and 14%, respectively).25
At 3 years, a total of 323 patients had completed ≥3 years of therapy (109 MI, 114 LI, and 100 CsA). A total of 80% MI, 82% LI, and 80% CsA patients survived with a functioning graft. Measured GFR was 42.7 mL/min, 42.2 mL/min, and 31.5 mL/min, respectively. More CsA-treated patients (44%) progressed to GFR <30 mL/min than belatacept-treated patients (27%–30%). Acute rejection rates were also similar between groups (18%, 19%, and 16%, respectively). No significant difference in mean blood pressure, rise in non-high-density lipid cholesterol, or incidence of new-onset diabetes was noted. PTLD was again more common in the belatacept groups (two MI, six LI, and one CsA).26
In June 2011, the US FDA approved belatacept for the prophylaxis of organ rejection in adult kidney-transplant recipients. The recommended dosing is 10 mg/kg administered prior to transplantation, on day 5, and at the end of weeks 2, 4, 8, and 12, followed by 5 mg/kg every 4 weeks, which corresponds to the LI regimen tested in the aforementioned clinical trials. There is a listed contraindication for patients that are Epstein–Barr virus-seronegative, due to the observed increased occurrence of PTLD in the clinical trials. There are several ongoing clinical trials further evaluating belatacept in organ transplantation (Table 3).
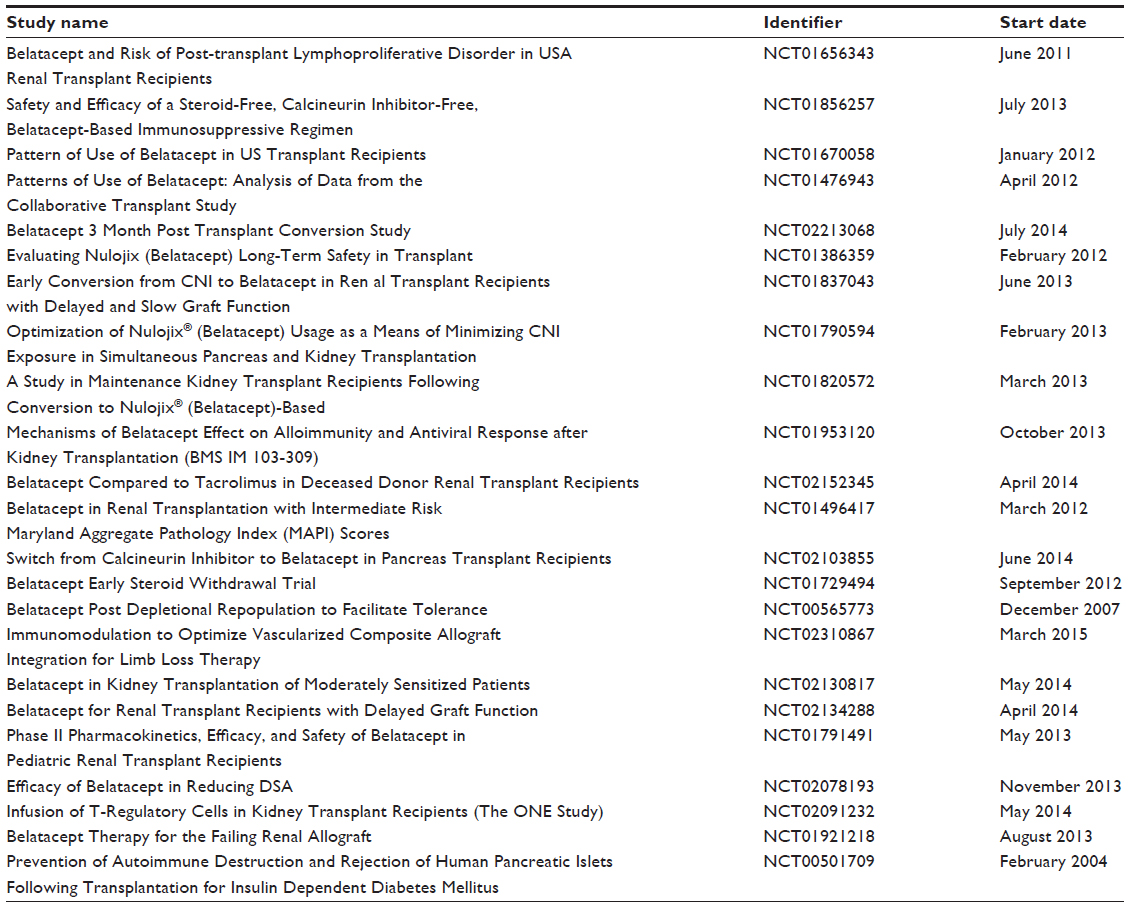 | Table 3 Active ongoing clinical trials further evaluating belatacept in organ transplantation |
ASKP1240
Another attempt at developing a novel immunosuppressant agent through costimulation blockade focused on the CD40–CD154 interaction. Efforts first focused on developing anti-CD154 monoclonal antibodies, which resulted in potent immunosuppression in nonhuman primates, but research was discontinued because these monoclonal antibodies stimulated platelet activation and increased thrombi.27–31 Focus then shifted to the development of anti-CD40 monoclonal antibodies.
ASKP1240 is a fully human anti-CD40 monoclonal antibody, and has demonstrated initial promise by significantly prolonging kidney, liver, and pancreatic islet survival in nonhuman primate transplant models with no serious side effects, including no thromboembolic complications.32–35 The first human Phase I study in healthy subjects (72 active drug vs 36 placebo) examined single ascending doses of intravenous ASK1240 (0.00003–10 mg/kg), and demonstrated that the antibody was both safe and well tolerated. The most common treatment-emergent adverse events experienced were headache, upper respiratory tract infection, and cough, but no thromboembolic events were reported.36 A Phase IIA randomized, open-label, active-control, multicenter study to assess the efficacy and safety of ASKP1240 in de novo kidney transplantation is ongoing (NCT01780844), with estimated completion scheduled for December 2016.
Alefacept
Alefacept (Amevive®) is a combination of the CD2-binding portion of lymphocyte function-associated antigen (LFA) 3 with the fragment-crystallizable domain of human IgG1, resulting in a soluble fusion protein. It acts by binding to the CD2 receptor, which is upregulated on effector memory T-cells, thereby inhibiting effector memory T-cell function and evoking apoptosis. Alefacept has demonstrated a dose-dependent effect on memory T-cells, and is currently FDA-indicated for treatment of moderate-to-severe chronic plaque psoriasis.37
In a Phase II study, patients were randomized in a double-blind fashion to receive alefacept (n=105) or placebo (n=107) for 3 months, and were then followed for an additional 3 months. Alefacept was administered at the time of transplantation, postoperatively on day 3, and then weekly to complete 3 months. All patients also received tacrolimus, mycophenolic acid, and corticosteroids. The primary efficacy end point – the incidence of BPAR through month 6 – was not significantly different (11% drug vs 7% placebo). Patient and graft survival as well as renal function were also similar between groups. Safety and tolerability were generally similar between the two arms, except that the occurrence of malignancy was higher in the alefacept arm (5.7%) compared to the placebo group (0.9%).38 Alefacept was voluntarily withdrawn from the market by Astellas Pharmaceuticals in November 2011, due to “business needs” and not based on any specific safety concern or FDA mandate.39 There are currently no active clinical trials using alefacept in organ transplantation.
Novel mechanisms
In addition to targeting the interactions between antigen-presenting and T-cells necessary for T-cell activation and dampening IL2 amplification via calcineurin inhibition, other novel targets have been explored. Agents that inhibit biologic pathways involved in T-cell growth and differentiation, leukocyte trafficking and adhesion, and cytokine production have also been developed. While each agent represented an exciting novel therapeutic target, mixed efficacy results have been obtained.
Tofacitinib
Tofacitinib (CP-690550, Xeljanz®) is a selective inhibitor of Janus kinase 3 (JAK3). Interleukin signaling during T-cell activation occurs through the IL2Rγ chain, which is modulated in part by JAK3. Inhibition of this interleukin signaling pathway decreases T-cell growth and differentiation, leading to immunosuppression.40
A pilot study in de novo kidney-allograft recipients compared two separate doses of tofacitinib (15 mg twice daily vs 30 mg twice daily) in 20 patients each arm vs 21 patients receiving tacrolimus. All patients received an IL2-receptor antagonist with concomitant mycophenolic acid and corticosteroids. There were one, four, and one BPARs at 6 months in each group, respectively. Mycophenolic acid was discontinued in the 30 mg twice-daily tofacitinib group due to a high incidence of BK nephropathy (four total cases). Cytomegalovirus occurred in two patients receiving tofacitinib 15 mg twice daily and in four patients receiving 30 mg twice daily, while no cases of cytomegalovirus occurred in the tacrolimus group. There were also modest lipid elevations and a trend toward more frequent anemia and neutropenia during the first 6 months in the tofacitinib arms compared to tacrolimus.41
A Phase IIB study examined 331 low- to moderate-risk de novo kidney-transplant recipients randomized to an MI (15 mg twice daily in months 1–6, then 10 mg twice daily in months 7–12) or LI (15 mg twice daily in months 1–3, then 10 mg twice daily in months 4–12) regimen of tofacitinib vs CsA (target trough levels of 125–400 ng/mL in months 1–3 and 100–300 ng/mL in months 4–12). All patients received basiliximab induction, mycophenolic acid, and corticosteroids. Primary end points were incidence of BPAR at month 6 and measured GFR at month 12. Similar 6-month incidences of BPAR (11%, 7%, and 9%, respectively) were observed and measured GFRs were higher at month 12 for the tofacitinib groups vs CsA (65, 65, and 54 mL/min, respectively). Serious infections developed in 45%, 37%, and 25% of the patients, respectively, while anemia, neutropenia, and PTLD occurred more frequently in the tofacitinib groups compared with CsA.42 Currently, an extension of this trial is ongoing to assess the long-term (72-month) safety and efficacy of tofacitinib in renal allograft recipients (NCT00658359) by following patients who completed the Phase IIB study, and is estimated to be completed in 2015.
Efalizumab
Efalizumab (Raptiva®) is a humanized anti-CD11a monoclonal antibody directed against LFA1. The binding of efalizumab to LFA1 inhibits adhesion and trafficking of leukocytes.43 The ability of efalizumab to effectively decrease adhesion and trafficking of leukocytes without lymphocyte depletion led to its FDA approval for use in the treatment of moderate-to-severe plaque psoriasis.44
A Phase I/II trial using efalizumab in organ transplantation produced initially promising results. A total of 38 patients undergoing their first renal transplant (living or deceased donor) were randomized to receive weekly subcutaneous efalizumab at two different doses (0.5 mg/kg vs 2 mg/kg). Patients were maintained on full-dose CsA, mycophenolate mofetil and corticosteroids, or half-dose CsA, sirolimus, and prednisone. At 6 months following transplant, overall patient survival was 97% and overall graft survival 95%. There were four BPARs (11%) in the first 6 months after transplantation. Three patients (8%) developed PTLD, all treated with the higher dose of efalizumab.45 Additional studies were initiated for efalizumab use in transplantation, but use of the drug was discontinued in these studies after the medication was withdrawn from the market in April 2009 due to an observed increased risk for progressive multifocal leukoencephalopathy.46–49 There are currently no active clinical trials using efalizumab in organ transplantation.
Sotrastaurin
Sotrastaurin (STN; AEB071) is a selective inhibitor of protein kinase C (PKC) isoforms θ and α. PKCθ is involved with IL2 production, while PKCα is involved with interferon (IFN)γ production. Inhibition of these cytokine-production pathways dampens the effect of T-cell activation.50,51 Three separate Phase II trials have looked for a potential role for STN in renal transplantation.
Friman et al randomized de novo renal transplant recipients with immediate graft function in a 1:2 fashion to receive either tacrolimus or STN. All 125 patients received basiliximab, mycophenolic acid, and corticosteroids. The study was terminated early, due to higher BPAR rates in the STN group compared to the tacrolimus group (23.6% vs 4.5%, respectively).52
The efficacy and safety of STN in combination with tacrolimus was assessed in a dose-ranging noninferiority study using 298 de novo renal transplant recipients. Patients were randomized in a 1:1:1:1 fashion to receive either 100 mg twice-daily STN plus standard-dose tacrolimus (5–12 ng/mL predose trough levels) or 200 mg twice daily STN plus standard-dose tacrolimus or 300 mg twice daily STN plus reduced tacrolimus (2–5 ng/mL predose trough levels) or mycophenolic acid plus standard-dose tacrolimus. All patients received basiliximab and corticosteroids. Composite efficacy failure (treated BPAR ≥ grade 1A, graft loss, death, or loss to follow-up) rates at month 12 were 18.8%, 12.4%, 10.9%, and 14%, respectively, for the four groups. Episodes of leukopenia were higher in the mycophenolic acid group compared to the STN groups (16.5% vs 1.3%–5.5%, respectively), but more dose-dependent gastrointestinal side effects were experienced in the STN groups.53
The efficacy and safety of STN in a CNI-free regimen in de novo kidney-transplant recipients has also been assessed. Stage 1 of the study randomized 2:1 131 patients to STN 300 mg twice daily or CsA, respectively. Stage 2 randomized 180 patients in a 1:1:1 fashion to STN 300 mg twice daily, STN 200 mg twice daily, or CsA. All patients received basiliximab, everolimus, and prednisone. Composite efficacy failure (treated BPAR, graft loss, death, or loss to follow-up) rates were higher in the STN arms compared to CsA (stage 1 16.5% vs 10.9%; stage 2 34.5%, 27.2%, and 19.4%, respectively). Gastrointestinal and cardiac events were also more frequent with STN.54
The efficacy and safety of STN in combination with tacrolimus has also been assessed in a multicenter randomized Phase II study in de novo liver-transplant recipients. Patients were randomized in a 1:1:1:1 fashion to receive either 200 mg twice daily STN plus standard-dose tacrolimus (5–10 ng/mL predose trough levels), 200 mg twice-daily STN plus reduced-dose tacrolimus (2–5 ng/mL predose trough levels), or 300 mg twice-daily STN plus reduced-dose tacrolimus or mycophenolic acid plus standard tacrolimus. All patients received corticosteroids. Composite efficacy failure (treated BPAR ≥ grade 1A, graft loss, or death) rates at month 6 were 25.0%, 16.5%, 20.9%, and 15.9%, respectively. The study was terminated early, given the higher composite efficacy failure rates in the STN group.55 Currently, there are no active clinical trials using STN in organ transplantation.
Conclusion
Transplantation provides improved survival and quality-of-life benefits compared with long-term dialysis in end-stage renal disease patients. The ability to adequately suppress the immune response of the transplant recipient toward the allograft has significantly reduced short-term rejection rates, but chronic rejection and unfavorable side-effect profiles remain significant obstacles. The search for effective immunosuppressant agents with limited adverse events remains an important target in the field of solid organ transplantation.
Maintenance regimens have predominantly consisted of a combination of CNIs, mTOR inhibitors, antimetabolite agents, and/or corticosteroids, but newer agents continue to be explored. Extended-release tacrolimus has been approved for use in renal transplantation, while VCS, a novel CNI, has been evaluated but has not demonstrated superior efficacy outcomes. Targeting T-cell costimulation blockade has resulted in approval of a novel non-CNI immunosuppressant, belatacept, for use in renal transplantation, while other costimulation blockers, ASKP1240 and alefacept, have been evaluated. A Phase II study with ASKP1240 is ongoing, but alefacept has been voluntarily withdrawn from the market. A JAK3 inhibitor (tofacitinib), an LFA1 inhibitor (efalizumab), and a PKC inhibitor sotrastaurin have also been evaluated as novel immunosuppressant agents in renal transplant recipients. Evaluation of the long-term safety and efficacy of tofacitinib in renal allograft recipients is ongoing, while there are no active ongoing trials with sotrastaurin or efalizumab, suggesting that these agents will likely not be studied further in transplantation.
Currently, there are few ongoing trials evaluating novel immunosuppressant agents for the prophylaxis of rejection in kidney-transplant recipients. While the development of new agents should remain a focus for continued transplantation research, optimization of existing agents, including recently approved extended-release tacrolimus and belatacept, should also be a continued emphasis of study in solid organ transplantation. Cost will unfortunately remain a significant barrier to the development of novel agents, as the combination of standard tacrolimus and a mycophenolic acid derivative is effective and relatively inexpensive. The quest for an immunosuppression regimen that improves long-term graft and patient survival while simultaneously reducing chronic graft injury and unfavorable side effects remains a high-priority area of continued drug development.
Disclosure
The authors report no conflicts of interest in this work.
References
Kidney Disease: Improving Global Outcomes (KDIGO) Transplant Work Group. KDIGO clinical practice guideline for the care of kidney transplant recipients. Am J Transplant. 2009;9 Suppl 3:S1–S155. | |
Andreoni KA, Brayman KL, Guidinger MK, Sommers CM, Sung RS. Kidney and pancreas transplantation in the United States, 1996–2005. Am J Transplant. 2007;7(5 Pt 2):1359–1375. | |
Tolou-Ghamari Z. Nephro and neurotoxicity of calcineurin inhibitors and mechanisms of rejections: a review on tacrolimus and cyclosporine in organ transplantation. J Nephropathol. 2012;1(1):23–30. | |
Butler JA, Roderick P, Mullee M, Mason JC, Peveler RC. Frequency and impact of nonadherence to immunosuppression after renal transplantation: a systematic review. Transplantation. 2004;77(5):769–776. | |
Prendergast MB, Gaston RS. Optimizing medication adherence: an ongoing opportunity to improve outcomes after kidney transplantation. Clin J Am Soc Nephrol. 2010;5(7):1305–1311. | |
Silva HT Jr, Yang HC, Meier-Kriesche HU, et al. Long-term follow-up of a phase III clinical trial comparing tacrolimus extended-release/MMF, tacrolimus/MMF, and cyclosporine/MMF in de novo kidney transplant recipients. Transplantation. 2014;97(6):636–641. | |
Budde K, Bunnapradist S, Grinyo JM, et al. Novel once-daily extended-release tacrolimus (LCPT) versus twice-daily tacrolimus in de novo kidney transplants: one-year results of phase III, double-blind, randomized trial. Am J Transplant. 2014;14(12):2796–2806. | |
Bunnapradist S, Ciechanowski K, West-Thielke P; et al. Conversion from twice-daily tacrolimus to once-daily extended release tacrolimus (LCPT): the phase III randomized MELT trial. Am J Transplant. 2013;13(3):760–769. | |
Kuglstatter A, Mueller F, Kusznir E, et al. Structural basis for the cyclophilin A binding affinity and immunosuppressive potency of E-ISA247 (voclosporin). Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 2):119–123. | |
Mayo PR, Huizinga RB, Ling SY, Freitag DG, Aspeslet LJ, Foster RT. Voclosporin food effect and single oral ascending dose pharmacokinetic and pharmacodynamics studies in healthy human subjects. J Clin Pharmacol. 2013;53(8):819–826. | |
Aspeslet L, Freitag D, Trepanier D, et al. ISA(TX)247: a novel calcineurin inhibitor. Transplant Proc. 2001;33(1–2):1048–1051. | |
Gregory CR, Kyles AE, Bernsteen L, et al. Compared with cyclosporine, ISATX247 significantly prolongs renal-allograft survival in a nonhuman primate model. Transplantation. 2004;78(5):681–685. | |
Ling SY, Huizinga RB, Mayo PR, et al. Cytochrome P450 3A and P-glycoprotein drug-drug interactions with voclosporin. Br J Clin Pharmacol. 2014;77(6):1039–1050. | |
Ling SY, Huizinga RB, Mayo PR, Freitag DG, Aspeslet LJ, Foster RT. Pharmacokinetics of voclosporin in renal impairment and hepatic impairment. J Clin Pharmacol. 2013;53(12):1303–1312. | |
Busque S, Cantarovich M, Mulgaonkar S, et al. The PROMISE study: a phase 2B multicenter study of voclosporin (ISA247) versus tacrolimus in de novo kidney transplantation. Am J Transplant. 2011;11(12):2675–2684. | |
Bretscher PA. A two-step, two-signal model for the primary activation of precursor helper T cells. Proc Natl Acad Sci U S A. 1999;96(1):185–190. | |
Yamada A, Salama AD, Sayegh MH. The role of novel T cell costimulatory pathways in autoimmunity and transplantation. J Am Soc Nephrol. 2002;13(2):559–575. | |
Alegre ML, Frauwirth KA, Thompson CB. T-cell regulation by CD28 and CTLA-4. Nat Rev Immunol. 2001;1(3):220–228. | |
Kremer JM, Westhovens R, Leon M, et al. Treatment of rheumatoid arthritis by selective inhibition of T-cell activation with fusion protein CTLA4Ig. N Engl J Med. 2003;349(20):1907–1915. | |
Pearson TC, Alexander DZ, Winn KJ, Linsley PS, Lowry RP, Larsen CP. Transplantation tolerance induced by CTLA4-Ig. Transplantation. 1994;57(12):1701–1706. | |
Levisetti MG, Padrid PA, Szot GL, et al. Immunosuppressive effects of human CTLA4Ig in a non-human primate model of allogeneic pancreatic islet transplantation. J Immunol. 1997;159(11):5187–5191. | |
Larsen CP, Pearson TC, Adams AB, et al. Rational development of LEA29Y (belatacept), a high-infinity variant of CTLA4-Ig with potent immunosuppressive properties. Am J Transplant. 2005;5(3):443–453. | |
Vincenti F, Charpentier B, Vanrenterghem Y, et al. A phase III study of belatacept-based immunosuppression regimens versus cyclosporine in renal transplant recipients (BENEFIT study). Am J Transplant. 2010; 10(3):535–546. | |
Vincenti F, Larsen CP, Alberu J, et al. Three-year outcomes from BENEFIT, a randomized, active-controlled, parallel-group study in adult kidney transplant recipients. Am J Transplant. 2012;12(1):210–217. | |
Durrbach A, Pestana JM, Pearson T, et al. A phase III study of belatacept versus cyclosporine in kidney transplants from extended criteria donors (BENEFIT-EXT study). Am J Transplant. 2010;10(3):547–557. | |
Pestana JO, Grinyo JM, Vanrenterghem Y, et al. Three-year outcomes from BENEFIT-EXT: a phase III study of belatacept versus cyclosporine in recipients of extended criteria donor kidneys. Am J Transplant. 2012;12(3):630–639. | |
Kirk AD, Burkly LC, Batty DS, et al. Treatment with humanized monoclonal antibody against CD154 prevents acute renal allograft rejection in nonhuman primates. Nat Med. 1999;5(6):686–693. | |
Preston EH, Xu H, Dhanireddy KK, et al. IDEC-131 (anti-CD154), sirolimus and donor-specific transfusion facilitate operational tolerance in non-human primates. Am J Transplant. 2005;5(5):1032–1041. | |
Kanmaz T, Fechner JJ Jr, Torrealba J, et al. Monotherapy with the novel human anti-CD154 monoclonal antibody ABI793 in rhesus monkey renal transplantation model. Transplantation. 2004;77(6):914–920. | |
Kawai T, Andrews D, Colvin RB, Sachs DH, Cosimi AB. Thromboembolic complications after treatment with monoclonal antibody against CD40 ligand. Nat Med. 2000;6(2):114. | |
Andre P, Prasad KS, Denis CV, et al. CD40L stabilizes arterial thrombi by a beta3 integrin – dependent mechanism. Nat Med. 2002;8(3):247–252. | |
Imai A, Suzuki T, Sugitani A, et al. A novel fully human anti-CD40 monoclonal antibody, 4D11, for kidney transplantation in cynomolgus monkeys. Transplantation. 2007;84(8):1020–1028. | |
Aoyagi T, Yamashita K, Suzuki T, et al. A human anti-CD40 monoclonal antibody, 4D11, for kidney transplantation in cynomolgus monkeys: induction and maintenance therapy. Am J Transplant. 2009;9(8):1732–1741. | |
Oura T, Yamashita K, Suzuki T, et al. Long-term hepatic allograft acceptance based on CD40 blockade by ASKP1240 in nonhuman primates. Am J Transplant. 2012;12(7):1740–1754. | |
Watanabe M, Yamashita K, Suzuki T, et al. ASKP1240, a fully human anti-CD40 monoclonal antibody, prolongs pancreatic islet allograft survival in nonhuman primates. Am J Transplant. 2013;13(8):1976–1988. | |
Goldwater R, Keirns J, Blahunka P, et al. A phase I, randomized ascending single-dose study of antagonist anti-human CD40 ASKP1240 in healthy subjects. Am J Transplant. 2013;13(4):1040–1046. | |
Krueger GG. Clinical response to alefacept: results of a phase 3 study of intravenous administration of alefacept in patients with chronic plaque psoriasis. J Eur Acad Dermatol Venereol. 2003;17 Suppl 2:17–24. | |
Rostaing L, Charpentier B, Gylda M, et al. Alefacept combined with tacrolimus, mycophenolate mofetil and steroids in de novo kidney transplantation: a randomized controlled trial. Am J Transplant. 2013;13(7):1724–1733. | |
National Psoriasis Foundation. Amevive (alefacept) voluntarily discontinued in the US Jan 2011. Available from: http://www.psoriasis.org/page.aspx?pid=2311. Accessed April 18, 2015. | |
Zand MS. Tofacitinab in renal transplantation. Transplant Rev. 2013; 27(3):85–89. | |
Busque S, Leventhal J, Brennan DC, et al. Calcineurin-inhibitor-free immunosuppression based on the JAK inhibitor CP-690,550: a pilot study in de novo kidney allograft recipients. Am J Transplant. 2009;9(8):1936–1945. | |
Vincenti F, Tedesco Silva H, Busque S, et al. Randomized phase 2B trial of tofacitinib (CP-690,550) in de novo kidney transplant patients: efficacy, renal function and safety at 1 year. Am J Transplant. 2012; 12(9):2446–2456. | |
Nicolls MR, Gill RG. LFA-1 (CD11a) as a therapeutic target. Am J Transplant. 2006;6(1):27–36. | |
Frampton JE, Plosker GL. Efalizumab: a review of its use in the management of chronic moderate-to-severe plaque psoriasis. Am J Clin Dermatol. 2009;10(1)51–72. | |
Vincenti F, Mendez R, Pescovitz M, et al. A phase I/II randomized open-label multicenter trial of efalizumab, a humanized anti-CD11a, anti-LFA-1 in renal transplantation. Am J Transplant. 2007;7(7):1770–1777. | |
Posselt AM, Bellin MD, Tavakol M, et al. Islet transplantation in type 1 diabetics using an immunosuppressive protocol based on the anti-LFA-1 antibody efalizumab. Am J Transplant. 2010;10(8):1870–1880. | |
Turgeon NA, Avila JG, Cano JA, et al. Experience with a novel efalizumab-based immunosuppressive regimen to facilitate single donor islet cell transplantation. Am J Transplant. 2010;10(9):2082–2091. | |
Posselt AM, Szot GL, Frassetto LA, et al. Islet transplantation in type 1 diabetic patients using calcineurin inhibitor-free immunosuppressive protocols based on T-cell adhesion or costimulation blockade. Transplantation. 2010;90(12):1595–1601. | |
US Food and Drug Administration. FDA statement on the voluntary withdrawal of Raptiva from the US market [press release]. Silver Spring (MD): FDA; 2009 [April 8]. Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm149561.htm. Accessed April 18, 2015. | |
Evenou JP, Wagner J, Zenke G, et al. The potent protein kinase C-selective inhibitor AEB071 (sotrastaurin) represents a new class of immunosuppressive agents affecting early T-cell activation. J Pharmacol Exp Ther. 2009;330(3):792–801. | |
Matz M, Naik M, Mashreqhi MF, Glander P, Neumayer HH, Budde K. Evaluation of the novel protein kinase C inhibitor sotrastaurin as immunosuppressive therapy after renal transplantation. Expert Opin Drug Metab Toxicol. 2011;7(1):103–113. | |
Friman S, Arns W, Nashan B, et al. Sotrastaurin, a novel small molecule inhibiting protein-kinase C: randomized phase II study in renal transplant recipients. Am J Transplant. 2011;11(7):1444–1455. | |
Russ GR, Tedesco-Silva H, Kuypers DR, et al. Efficacy of sotrastaurin plus tacrolimus after de novo kidney transplantation: randomized, phase II trial results. Am J Transplant. 2013;13(7):1746–1756. | |
Tedesco-Silva H, Kho MM, Hartmann A, et al. Sotrastaurin in calcineurin inhibitor-free regimen using everolimus in de novo kidney transplant recipients. Am J Transplant. 2013;13(7):1757–1768. | |
Pascher A, De Simone P, Pratschke J, et al. Protein kinase C inhibitor sotrastaurin in de novo liver transplant recipients: a randomized phase II trial. Am J Transplant. 2015;15(5):1283–1292. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.