Back to Archived Journals » Orphan Drugs: Research and Reviews » Volume 5
Drugs in development and dietary approach for Duchenne muscular dystrophy
Authors Angelini C ![]() , Tasca E
, Tasca E
Received 11 April 2015
Accepted for publication 11 May 2015
Published 11 August 2015 Volume 2015:5 Pages 51—60
DOI https://doi.org/10.2147/ODRR.S55677
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Lise Aagaard
Corrado Angelini, Elisabetta Tasca
Neuromuscular Laboratory, Fondazione San Camillo Hospital IRCCS, Venice, Italy
Abstract: Therapeutic trials studying Duchenne muscular dystrophy (DMD) in Europe and the USA have been done using a protocol that includes manual muscle testing and functional testing, and have shown the efficacy of steroid drugs in various doses and regimens. Further, drisapersen and eteplirsen (exon skipping drugs) and ataluren (a drug to overcome stop codon mutations) have achieved some clinical improvement. Cardioprotective drugs are efficacious in DMD, and eplerenone, an aldosterone inhibitor and diuretic, is now being used to treat the disease. The dietary approach should be used in wheelchair-bound DMD children in combination with respiratory assistance. The importance of some of the treatments proposed is that they might also be useful in other genetic disorders where stop codon mutations are present; moreover, it is possible that these new treatments will improve quality of life for many patients.
Keywords: Duchenne muscular dystrophy, steroids, ataluren, drisapersen, eplerenone, eteplirsen
Introduction
Duchenne muscular dystrophy (DMD) is a relatively common muscle disorder caused by dystrophin deficiency. Over the past 25 years, several clinical trials have been carried out with agents directed to reducing the increase in connective tissue and with agents designed to induce production of dystrophin by frameshifting deletions of one or more exons abutting exon 51 or by stop codon read-through.1,2 Secondary approaches have been tried to compensate for the lack of dystrophin, such as reduction of the fibrosis or inflammation, and consequences of nitric oxide synthase deficiency. Cardiac insufficiency is common in the advanced stages of DMD, and seems to benefit from beta-blockers, angiotensin-converting enzyme (ACE) inhibitors, and diuretics.3,4
It is important in DMD drug studies that patients are carefully selected, using clinical and molecular criteria so that no atypical cases are included. A positive X-linked family history or dystrophin gene and protein analysis (when available) is required. Although a medication might theoretically be most helpful in the preclinical stage of the disease (detected by elevated creatine kinase in at-risk individuals), it is difficult to assess the efficacy of treatment in DMD boys younger than 3–5 years of age. Most studies have involved DMD patients 5 years of age or older who have at least some detectable weakness but are not so advanced that a drug may no longer be able to stop or reverse the dystrophic process.5–8
For inclusion in most trials, DMD patients must be able to walk and present at least two of the following criteria: a genetic diagnosis of sporadic DMD with mutation type (stop codon, deletion, missense); an X-linked family history with a proven genetic diagnosis; and muscle biopsy changes consistent with DMD (ie, absence of dystrophin).
Patients should be excluded if they have facial muscle weakness, skin rash, or sensory abnormalities, and when affected male relatives are still ambulating at the age of 16 years. Such patients could be included in a separate treatment group corresponding to Becker’s dystrophy. To include sporadic cases of DMD when X-linked inheritance cannot be demonstrated, a molecular diagnosis (either on a protein or genetic basis) is required.
Patients who have received systemic medications other than antibiotics or drugs to treat a specific illness should also be excluded. Elective surgery (ie, release of contractures or Achilles tenotomy) should not be performed during the drug trial. Physical therapy must be similar for all patients at a single center, but no specific therapy program is recommended, and it is expected that range of motion of the extremities will be recorded periodically. However, the rate of exercise of extremities should not be formally assessed since this is quite variable and difficult to analyze.
Natural history
In the natural history of DMD, a constant improvement of strength and functional test performance is recorded up to 7 years of age, when muscle strength and motor ability reaches a plateau. After this age, there is an increase in connective tissue in skeletal muscles and loss of muscle fibers, which causes progressive worsening of muscle function (Figure 1). The inability to walk at least 10 meters without aid is defined as “loss of ambulation”.
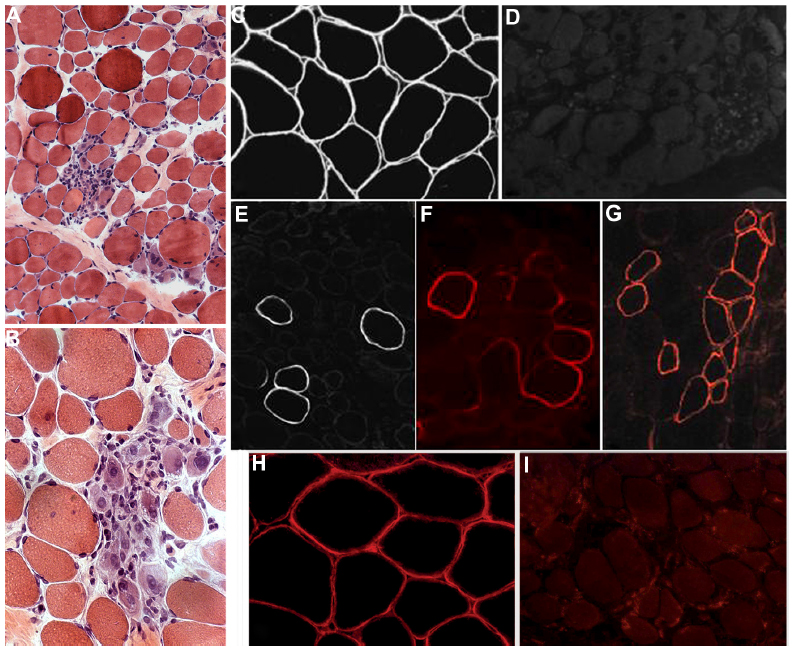 | Figure 1 Muscle histopathology and dystrophin immunohistochemistry in DMD. |
An important issue is that walking ability is lost around 10.5 years of age, with some variation depending mainly on the amount of residual dystrophin.9 Prolongation of independent walking ability is therefore an important primary target in long-term clinical trials, although this might be influenced by external factors, such as surgical interventions, steroid therapy, excessive weight gain, and traumatic bone fractures. Once the child is wheelchair-bound, progressive loss of strength in the respiratory and trunk muscles determines the degree of ventilatory insufficiency and the onset of kyphoscoliosis.
Manual muscle testing
The strength of most of the major muscles of the limbs and trunk can be readily recorded and remeasured using manual testing. Several studies have demonstrated that the degree and rate of muscle weakness and rate of deterioration of individual muscles varies in a predictable fashion in DMD, depending on the muscle tested.5,7,8 Therefore, muscle testing can be simplified by omitting muscles that are either too weak to be accurately tested or are not expected to deteriorate rapidly enough to detect any drug effect. Accordingly, muscles in the upper extremities (eg, deltoid and triceps brachii) and lower extremities (eg, quadriceps and iliopsoas) should be scored for muscle strength using the Medical Research Council (MRC) recommendations. All muscles are tested with the limb held at 90°. Recently, a more simple revised MRC scale with only four grades has been proposed;10 its usefulness should be evaluated in future trials.
Functional muscle testing
A fast and efficient functional test that has already been validated, known as the GSGC (Gait, Stairs, Gowers, Chair) score, consists of several functional maneuvers that can be graded according to disease severity.11,12 These tests are adapted from a standardized series of protocols and are given considerable emphasis because they do not require the same level of cooperation as manual muscle strength testing. Although functional tests are easy to perform, even in younger children who cannot cooperate with manual muscle testing, a major disadvantage of these tests is that their results may abruptly worsen at 9–10 years of age. In ambulatory patients, the function of the upper extremities usually remains quite good, while a progressive deterioration can be expected in the function of the lower extremities. For this reason, these functional tests place greater emphasis on the lower extremities.
Standard 9 inch stairs with four steps seems to be a reliable functional test for DMD. A series of chairs is used to evaluate the function of “arising from a chair” to ensure that the child’s legs are at 90° with the feet on the floor. It is emphasized that all tests should be performed in a uniform manner, with each child using the same equipment for each examination.
Six-minute walking test
Many studies conducted in the last 10 years, especially those involving the new molecular drugs, have used this functional test as an outcome,12 but in DMD children there is extreme variability in its performance because of their variable motivation or early fatigability, and these factors can affect the reliability of the test. Therefore, the results of drug trials using only this parameter as a functional outcome should be interpreted with caution.
Drugs in development
Several categories of drugs are in use, and act on different pathogenetic mechanisms in DMD (Table 1).
 | Table 1 Properties of drugs in development for DMD |
Corticosteroids
Corticosteroids continue to be the gold standard for the treatment of DMD. However, the choice of steroid and mode of administration is still a matter of debate. Use of corticosteroids should be discussed with parents and not started before 3 years of age; however, the steroid is usually stopped when boys become wheelchair-bound.
In two trials, we established that deflazacort (Lepetit Company, Milan, Italy; Teofarma, Pavia, Italy; EG Labnet, Edison, NJ, USA) at 2 mg/kg on alternate days or 0.9 mg/kg stabilized muscle strength in DMD when compared with a placebo group.5,13 Further, deflazacort appeared to cause less weight gain than prednisone, and bone sparing was more evident with deflazacort than with prednisone.10
Deflazacort has demonstrated good efficacy in slowing progression of DMD and delaying loss of independent ambulation.5,14 Many corticosteroid trials have been conducted in DMD, and hundreds of patients have been treated in many double-blind, randomized, controlled trials.5–8,13,15 These trials have shown the benefit of steroids, with a marked initial improvement in the first 3–6 months and a subsequent slowing of disease progression for over 3 years. The mechanism of action of steroids is unknown, but they may act in a number of ways, probably by inhibiting fibrotic and connective tissue replacement of the muscle or by increasing the synthesis of dystrophin and of other synergistic sarcolemmal proteins, such as utrophin.
Different steroids with different regimens (daily, alternate day, pulsed) have been used with the aim of obtaining maximal efficacy with fewer side effects. Many uncertainties remain about when to begin therapy, but earlier (after 3–5 years of age) is thought to be better. An open-label trial on DMD and bone mineral density16 found that prednisone 0.35 mg has the same positive effects on muscle strength and function as higher doses. Another trial8 compared two dosages of prednisone, ie, 0.3 mg/kg day and 0.75 mg/kg day, and patients on 0.75 mg/kg day improved more than patients on 0.3 mg/kg day.
Two trials reported that deflazacort had fewer side effects than prednisone.11 This oxazolidine derivative has been shown to prevent bone loss in children with other diseases.17 We conducted a controlled, randomized, double-blind trial to compare deflazacort 0.9 mg/kg/day with prednisone 0.75 mg/kg/day in 18 boys with DMD.13 The effects of the steroids were examined after 12 months by comparing the status of the treated patients with that of historical controls. The two steroids were equally effective in improving motor function and functional performance. The average increase in weight at 9 months with respect to baseline was 5% (2 kg) in the deflazacort group and 18% in the prednisone group (P<0.005), and the change remained significant after 12 months (P<0.05). After 2 years, the increase in body weight was 19% in the deflazacort group and 41% in the prednisone group. Mild cataracts were observed in three patients in the deflazacort group and in one patient in the prednisone group.
At a European Neuromuscular Center workshop,18 Reitter reported a large multicenter trial comparing deflazacort and prednisone in 100 boys with DMD in Germany, in which 14 patients dropped out prematurely due to intolerable weight gain; the majority of these patients were treated with prednisone. Among the 80 patients who completed the study, the weight gain was significantly higher in those receiving prednisone. However, more boys in the deflazacort group developed cataract. Both studies used manual muscle testing and timed motor functions (eg, running, climbing stairs), and the outcomes were similar. Growth was impaired in all boys regardless of the type of steroid administered. The conclusion was that deflazacort appeared to cause less severe side effects than prednisone, particularly with regard to weight gain, which could be important to maximize motor performance. Both drugs were equally effective in preserving muscle performance, and in order to slow down the disorder it is important to taper them according to the side effects.
The worldwide experience with steroids in DMD has been summarized in a chapter.19 The ability of long-term corticosteroids to significantly improve muscle strength and to prolong the ability to walking independently beyond the age of 10 years was confirmed by a Cochrane systematic review of corticosteroids in DMD.20
Deflazacort is available in Europe and Canada, but is not available in the USA, although Marathon Pharmaceuticals (Northbrook, IL, USA) has submitted a proposal to the National Institutes of Health for a study in children with DMD.
Oxandrolone
Oxandrolone (Pfizer Inc., Walton Oaks, UK) is an anabolic (androgenic) steroid widely used to promote growth in children with Turner syndrome and in those with constitutional delay of growth and puberty. Oxandrolone has been demonstrated to increase protein content in muscle and fat-free mass. A 3-month open-label trial of oxandrolone 0.1 mg/kg day was performed in ten DMD boys,21 and showed an average muscle score improvement of 0.315 against the estimated loss of 0.1 in the known natural history of DMD. In this trial, muscle strength was significantly improved after 1 month. A subsequent study by Fenichel et al22 did not confirm the long-term effect of oxandrolone, which is currently not recommended for long-term treatment in DMD.
Givinostat
Givinostat (ITF2357; Italfarmaco, Rome, Italy) is a histone deacetylase inhibitor with a potential anti-inflammatory effect. This drug had a positive effect in dystrophin-deficient Mdx mice, with increased muscle fiber size and decreased amounts of connective tissue noted.23 Givinostat has received an orphan drug designation and is undergoing a multicenter trial in seven centers in Italy.
Gentamicin
Gentamicin was the first drug identified to be able to stop codon dystrophin gene mutations.24 Its administration had a positive effect, but the possibility of side effects including renal toxicity or deafness have limited its use. In cell cultures, gentamicin interacts with the ribosomal subunit in the transcription of RNA, suppressing the termination codon and inserting in its place another amino acid. In a study of 12 DMD patients performed over a 6-month period, expression of dystrophin was observed in six patients, but without any clinical effect.25
Ataluren
A subset of dystrophin gene mutations that cause DMD are single base variations that change a codon to a premature stop codon (nonsense mutations that are present in about 10% of DMD cases). Screening procedures were carried out to identify drugs that would modulate the translational machinery of ribosomes so that these drugs could read through the stop codons. Ataluren (PTC124; PTC Therapeutics Inc., South Plainfield, NJ, USA) was then developed to increase dystrophin in DMD patients with stop codon mutations.
Ataluren is an orally administered drug which, in theory, would have the same effect as gentamicin. Multiple clinical studies of ataluren in DMD have been carried out, but so far there has been no report on the effects of the drug on production of dystrophin in muscle. Baseline and placebo data have been published for a Phase IIb/III trial in 174 patients,26,27 but no clear drug efficacy has been reported. This trial used two different doses of ataluren. The low-dose arm (three doses per day; 10, 10, and 20 mg/kg in morning, noon, and evening, respectively) achieved a 31.3 m improvement in the 6-minute walking test compared with placebo, which was slightly statistically significant, but the high-dose arm (80 mg/day) did not show any improvement in this test. This paradoxical effect was explained by the fact that even in vitro a low dose seems to restore dystrophin in certain conditions, and is more effective in doing so than the high dose. A bell-shaped concentration curve was found using PTC124, but not with gentamicin, when studying suppression of stop codons.28
Clinical data on this Phase IIb/III trial has been reported, including in a press release regarding European Medicine Agency validation of a marketing authorization application seeking conditional approval for ataluren and including a prospective Phase III trial. This Phase III trial is now underway using a lower dose (40 mg/day), with 220 patients to be recruited.
Muscle biopsies were taken as part of the Phase IIa and IIb trials, but the dystrophin measurements for these biopsies have not been reported. Dystrophin assays using either immunostaining or immunoblotting typically involve antibody studies. Interpretation of immunodetection for dystrophin is complicated by the requirement for well-preserved biopsies, and muscle specimens can be difficult to obtain in a multicenter clinical trial. Immunostaining signals are also challenging to quantify because of the presence of dystrophin-positive revertant fibers in about 40% of DMD boys, the numbers of which increase with age.9 Dystrophin immunoblotting is technically challenging for the large (427 kDa) and relatively low abundance of dystrophin protein. Difficulties in measuring dystrophin have been suggested as a motivation to embark on multicenter Phase III trials rather than seeking accelerated approval based on dystrophin expression as a surrogate biomarker. An open question is whether ataluren has sufficient activity to read through stop codons and rescue biochemical defects.29–31
Drisapersen
An alternative approach to increasing production of dystrophin in muscle is exon skipping.32 This approach involves anti-sense oligonucleotide (AON) drugs that are able to bind specifically to dystrophin RNA while it is being transcribed and processed/spliced (see Figure 2). Exon skipping has emerged as a promising therapeutic approach for DMD. The general strategy is to introduce an AON molecule that pairs with a target sequence in the pre-mRNA for dystrophin. This pairing causes the nuclear splicing machinery to skip over the target sequence, thereby excluding one or more mutation-bearing exons that would prevent production of the dystrophin protein. The end result is a shorter but still functional dystrophin molecule.
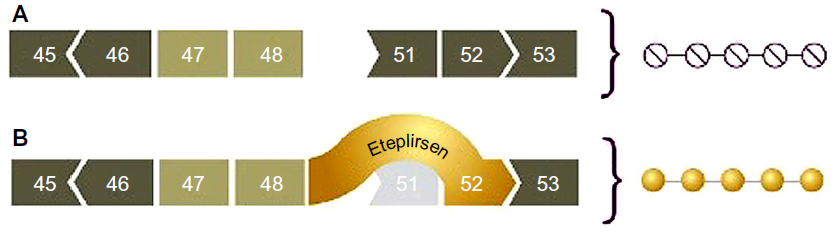 | Figure 2 Mechanism of exon 51 skipping in the dystrophin gene. |
AON drugs are designed to bind to an exon neighboring a patient’s deletion mutation, modulating splicing of the RNA to exclude an additional exon. In this manner, an out-of-frame mRNA transcript, which is unable to make dystrophin protein, is converted to an in-frame transcript capable of producing dystrophin. A shortened dystrophin will be produced, similar to what happens in dystrophin-positive revertant fibers or in Becker-like dystrophin.
The initial target for development for exon skipping was exon 51, since about 13% of boys with DMD have a frameshift mutation that can be treated by inducing a skip of exon 51 (although there is some disagreement about the actual percentage, with some estimates being as high as 17%). The skipping of exons other than 51 (eg, 44 and 53) is under consideration.
Two different AON molecules have been developed by the pharmaceutical industry and taken to clinical trials. Drisapersen (PRO051, GSK2402968) was developed by a partnership between Prosensa Therapeutics (Leiden, the Netherlands) and GSK (Brentford, UK), and was constructed using 2′-O-methyl phosphorothioate chemistry. This compound has the advantage of extensive clinical experience in drug programs for other genetic disorders (eg, cystic fibrosis). Prosensa Therapeutics was the early developer of the exon skipping approach in DMD. Phase II trials were carried out, and these included functional tests such as the 6-minute walking test, as well as investigation of muscle biopsies to monitor production of dystrophin as the primary endpoint.33,34 The investigators observed that 64%–97% of muscle fibers expressed the dystrophin protein in amounts ranging from 17% to 35%. While there was evidence of de novo dystrophin, its levels appeared low and variable, keeping in mind the presence of dystrophin produced by revertant fibers. However, the biochemical assay (Western blotting or semiquantitative immunohistochemistry) used to quantify dystrophin remains difficult to evaluate.
A subset of patients in Phase II extension trials showed stabilization of disease progression on drisapersen,34 but only marginal improvement in the 6-minute walking test, which was used as a secondary functional outcome measure. A Phase III trial in 186 patients that used the 6-minute walking test as the primary outcome measure found no statistically significant improvement on drisapersen. In fact, in a trial using 6 mg/kg/week administered by subcutaneous injection, there was an improvement in the 6-minute walking test of 31 meters,35 and GSK terminated the trial in September 2013. An extension study was allowed only recently. The implications of these results for exon 51 and other exon-skipping drugs are under discussion.
Eteplirsen
Another molecule for exon 51 skipping presently in clinical trials is a phosphorodiamidate morpholino oligomer (PMO) compound36,37 developed by Sarepta Therapeutics Inc. (Cambridge, MA, USA). This AON drug appears to have the advantage of lower toxicity and a considerably broader therapeutic window.32,38 However, there is still little clinical experience with this drug. Preclinical studies in mouse and dog models have shown dystrophin expression in muscle, and the extent of dystrophin expression appears to be dose-dependent.39–42 A single-blind, placebo-controlled study in humans gave proof-of-concept to the drug.43
Two Phase II studies have been published for an exon 51 morpholino drug known as eteplirsen (AVI-4658). Both Phase II trials of eteplirsen used de novo production of dystrophin in muscle as the primary outcome measure. In the studies by Cirak et al,44,45 increasing doses of PMO were injected into cohorts of DMD boys aged 5–15 years; from the third cohort onwards, while receiving doses of 2 mg/kg/week and above, dystrophin-positive fibers increased and 8%–18% dystrophin expression was seen on Western blotting. Biopsy samples showed a reduction in cytotoxic T-cells, which appears consistent with the fact that most patients received concomitant steroids.2
Mendell et al46 tried two different doses (30 mg/kg/week and 50 mg/kg/week by intravenous administration) in 12 DMD boys, and was able to meet the primary outcome and to obtain dystrophin-positive fibers. Moreover, in the 6-minute walking test, while the control group declined, there was stabilization of walking ability with a difference of about 67.3 meters. It is clear that 12 children are few, while Sarepta is pursuing accelerated approval for eteplirsen and conducted additional studies.
The amount of de novo production of dystrophin in DMD muscle was variable in the AON studies, but the amount of expression appeared greater than with the 2′-O-methyl phosphorothioate compound in the study by Cirak et al.44 Sarepta issued a press release in 2013 concerning a new drug application for eteplirsen in the treatment of DMD, providing information on its approach and interactions with the US Food and Drug Administration. In this press release, Sarepta stated that it intended to seek accelerated approval based on results for Phase IIb de novo dystrophin expression in the muscles of DMD patients, but the US Food and Drug Administration requested additional information related to the methodology and verification of dystrophin quantification. This is the first attempt of a company to seek accelerated drug development approval from regulatory authorities based on biomarker data in DMD muscle, such as measurement of dystrophin.
The results of AON trials are hindered by several main factors:
- Poor penetrance of AON in tissues. Delivery of AON to cardiac muscle remains problematic. A recent new AON drug (tricyclo-DNA oligomers), on systemic delivery in the Mdx mouse, seems to be able to promote a higher degree of rescued dystrophin in skeletal muscle, the heart, and to a less extent, the brain.47
- Most AON drugs have to be delivered either by intramuscular or intravenous administration.
- Dystrophin in tissues such as skeletal muscle in the limbs and diaphragm is a structural protein of the sarcolemma and ameliorates the disease only when the dystrophin complex is restored.
Eplerenone
DMD boys develop progressive weakness and lose the ability to walk by 10 years of age, and then develop dilated cardiomyopathy, a leading cause of death. In this context, Raman et al48 presented a positive placebo-controlled randomized trial in 42 DMD boys from three US centers treated with eplerenone (Pfizer Inc.) or placebo in association with steroids and on a background of ACE inhibitors or angiotensin receptor blockers. Eplerenone works by blocking the hormone aldosterone (and the aldosterone receptor), and, because it lowers the amount of sodium and water retained in the body, it is used as a diuretic in many cardiac conditions.
Forty-two DMD boys (aged 11–19 years) were administered eplerenone 25 mg/daily or placebo in addition to other cardioprotective drugs and were followed for 12 months. Significantly decreased left ventricular circumferential strain as a primary outcome was found on cardiac magnetic resonance imaging.48 A longer follow-up would be necessary to determine if the combination of eplerenone and other cardioprotective treatments results in prolonged survival. Another potential limitation of this trial is the relative small cohort of patients, but in a disorder like DMD it is difficult to recruit adequate numbers of patients and perform blinded cardiac magnetic resonance imaging studies.
Prolonged survival due to improvement in respiratory clinical care, such as non-invasive positive pressure ventilation, has led to an increased incidence of cardiomyopathy being seen in DMD patients.49 Two patterns of cardiac involvement are recognized: left ventricular wall motion abnormality, which is frequent in the early stage of the disease; and dilated cardiomyopathy,4 which is more frequent in the second decade of life, such as in the patients recruited for the study reported by Raman et al.48
In a retrospective analysis, Markham et al50 demonstrated that steroid-naïve patients (aged 10 years or younger) were four times more likely to have decreased cardiac function than patients in the same age group who had received steroids, while patients older than 10 years who had not received steroids were 15 times more likely to have decreased cardiac function than patients in the same age group who had taken steroids. Prophylactic use of cardioprotective drugs is also worth pursuing. In a small randomized trial in France, early ACE inhibitor (ie, perindopril) monotherapy in DMD patients aged 9.5–13.0 years delayed the onset and progression of left ventricular dysfunction, monitored by decline in ejection fraction.51
The beneficial effect of a combination of a beta-blocker and ACE inhibitor was established in 56 wheelchair-bound patients (mean age 19.5 years), with 5-year and 7-year survival rates of 93% and 84%, respectively.52
One possible limitation of the study by Raman et al48 is that their DMD patients were taking several different drugs in addition to eplerenone or placebo. In fact, duration of ACE inhibitors, angiotensin receptor blockers, and beta-blocker assumption was about 1.6 years. Further, most of the patients included (35/42, 83%) were on steroids (either prednisone 30 mg or deflazacort 25 mg) in addition to eplerenone or placebo for an average of 5.5 years. It is conceivable that in steroid-treated cases, an aldosterone inhibitor could be more beneficial due to the secondary mineralocorticoid effect of steroids. On the other hand, treatment with corticosteroids might directly influence the development of cardiomyopathy,14,49,50 and various types of steroids, eg, deflazacort or prednisone, might have different effects on the heart. Of note, a body weight increase might negatively impact cardiac function, and a differential weight increase has been found with deflazacort versus prednisone.11
Eplerenone appears to be a promising drug for use in DMD, and a future trial could assess the effects of eplerenone on left ventricular function in younger patients and evaluate their motor outcomes. It is important to realize that medications used for adult heart failure do not necessarily benefit the heart in children. The dystrophic process in heart and muscle is different in time frame and distribution; in the heart, necrosis starts from the posterobasal region of the left ventricle, after which remodeling occurs, with ventricular dilatation and reduction of ejection fraction.49
The lack of important side effects suggests that eplerenone could be used to improve and prolong the life of patients with DMD. The prognosis for this disease, once considered to be fatal by 20 years of age, has now shifted, with many patients surviving up to their fourth decade with various clinical care combinations. Future trials should investigate whether treating cardiomyopathy with eplerenone may benefit not only patients with DMD, but also those with other muscular dystrophies and severe cardiomyopathy, such as Becker muscular dystrophy, sarcoglycanopathies, and laminopathies.
Eplerenone has been used in a pilot study in a 22-year-old female DMD carrier, in whom muscle edema was assessed by magnetic resonance imaging. Treatment with eplerenone in this carrier resulted in a reduction of leg circumferences attributed to decreased edema, with the leg muscles becoming stronger with a lower water content and no progression of dystrophy.53 Although eplerenone has not yet been tested for its antifibrotic effect in skeletal muscle, the positive results on cardiac and smooth muscle support a putative beneficial effect.
Sildenafil
Nitric oxide stimulates soluble guanylate cyclase in such a way to produce cyclic guanosine monophosphate (cGMP), and in the absence of dystrophin, the nitric oxide-soluble guanylate cyclase-cGMP pathway is disrupted, impairing the microcirculation in the heart and muscle. Nucleotide phosphodiesterases (PDEs) hydrolyze cGMP and regulate the downstream signaling. PDE5 expression in cardiomyocytes is low at baseline and increases in response to ischemia or pressure overload from heart failure. Impaired blood flow in muscle and heart tissue in dystrophin-deficient Mdx and nitric oxide synthase-deficient Mdx mice was rescued by inhibition of PDE5.54 Nitric oxide inhibitors to treat cardiac and skeletal muscle are undergoing trials in dystrophinopathies. Unfortunately, in patients with DMD or Becker muscular dystrophy, a clinical trial with sildenafil (Pfizer Inc.), a PDE5 inhibitor, did not improve cardiomyopathy, with 30% of patients progressing to ventricular dilatation.55
Dietary approach
As far as the impact of nutritional status and energy requirements in children with DMD is concerned, micronutrients are needed and vitamin D and calcium should be supplemented, especially in patients receiving steroids.
Whole grains, vegetables, fruits, legumes, sea vegetables, saturated fats and essential fats, vitamin E, and coenzyme Q10 (400 mg/day) may possibly promote bone and general health. L-carnitine and N-acetyl-cysteine may help DMD children to maintain their energy.56
A small group of DMD patients were evaluated over a 6-month period while receiving a standardized diet rich in fructose, since this sugar seems to be better utilized by dystrophic muscle.57,58 According to Ellis,57 there is more incorporation of triglycerides in muscle using glucose-labeled 14C then fructose, therefore this sugar should not increase deposition of triglyceride droplets. Carrol et al59 demonstrated that palmitic acid, a component of triglycerides, is poorly oxidized in DMD muscle. Fourteen DMD patients with a fructose diet in their natural history were compared with 132 DMD patients with a normocaloric diet in their natural history. Patients on the fructose diet had better functional test results, which were significant for Gowers maneuver (P<0.001) after 2 months of the diet, and the percent MRC index of muscle strength also improved.
Another diet trial was conducted using branched-chain amino acids (a mixture of leucine, valine, and isoleucine; 0.4 g/kg/day) for one year in 18 DMD patients. These branched-chain amino acids are indeed directed to reducing degradation of muscle protein. No significant difference in ability to perform the Gowers maneuver was found between the branched-chain amino acid group and the placebo group. The only noticeable adverse event was the difficulty in getting children to ingest this mixture of amino acids, which can be unpalatable for children.58
Long-term dietary supplementation of L-arginine (a nitric oxide synthase substrate) was also not a viable therapy for dystrophinopathy,60 but administering antioxidants that attenuate attack by superoxides and restore the bioactive nitric oxide level might be a useful approach for the treatment of these disorders.
Perspectives
Research on muscular dystrophy is characterized by unsuccessful therapeutic efforts, but several drugs are now clinically available for various stages of the disease, ie, corticosteroids, cardioprotective drugs, and possibly also eplerenone. Much attention has been given to molecular therapies, ie, AONs to skip specific deletion mutations and ataluren to bypass stop codon mutations.1 Their benefit remains unclear, given that molecular drugs bring about only a modest increase in the 6-minute walking test; however, it is also still unknown if they increase independent walking ability and survival. These drugs await open validation in a larger cohort of patients. An appropriate number of patients is required to evaluate new drugs using the various methodologies proposed. The number of patients required to study the effect of different steroid regimens in the ongoing FOR-DMD trial is about 300, that will be recruited according to planned functional outcomes. Further, molecular therapies are restricted to selected patients, ie, ataluren would be of benefit to only 13% of DMD patients1 and AONs are tailored for individual mutations.
Although molecular drugs like AONs and ataluren are now in development, the available data suggest that the most useful treatment for DMD is steroids, which are recommended until patients lose the ability to walk. The side effect profile of deflazacort appears to be better than that of other steroids, and should be used in ambulant DMD patients. For prevention of cardiac insufficiency, a beta-blocker and the combination of ACE inhibitors and eplerenone has to be considered. A controlled diet, low in calories and glucose and rich in fructose and calcium could be useful to control body weight. The beneficial effects of cardioprotective drugs in DMD patients need further study using clinical endpoints such as death or hospitalization for heart failure.
Conclusion
In sum, we have overviewed the possibility of using the above-mentioned drugs to prolong independent ambulation in DMD patients aged 3–4 years. According to the published standard of care,61 this approach improves respiratory function, and when associated with ventilatory night support and cardioprotection, extends the life expectancy in DMD patients beyond two decades. Molecular medicine could be applied to obtain exon skipping and bypass stop codon mutations in other genetic and neuromuscular disorders, leading to benefit in many patients. While no single approach provides a cure for patients with DMD, all approaches have the potential to limit the disease.
Disclosure
The authors report no conflicts of interest in this work.
References
Finkel RS, Flanigan KM, Wong B, et al. Phase 2a study of ataluren-mediated dystrophin production in patients with nonsense mutation Duchenne muscular dystrophy. PLoS One. 2013;8:e81302. | |
Angelini C. Neuromuscular diseases: advances in therapy and diagnosis. Lancet Neurol. 2012;11:15–17. | |
Bushby K, Finkel R, Birnkrant DJ, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9:77–93. | |
Melacini P. Cardiac problems in DMD. In: Angelini C, editor. Muscular Dystrophy: Causes and Management. New York, NY, USA: Nova Science Publishing; 2013:367–380. | |
Angelini C, Pegoraro E, Turella E, Intino MT, Pini A, Costa C. Deflazacort in Duchenne dystrophy: study of long-term effect. Muscle Nerve. 1994;17:386–391. | |
Mesa L, Dubrovsky AL, Corderi J, Marco P, Flores D. Steroids in Duchenne muscular dystrophy: deflazacort trial. Neuromuscul Disord. 1991;1:261–266. | |
Mendell JR, Moxley RT, Griggs RG, et al. Randomized, double-blind six-month trial of prednisone in Duchenne’s muscular dystrophy. N Engl J Med. 1989;320:1592–1597. | |
Griggs RC, Moxley RT, Mendell JR, et al. Prednisone in Duchenne dystrophy: a randomized, controlled trial defining the time course and dose response. Arch Neurol. 1991;48:383–388. | |
Fanin M, Danieli GA, Cadaldini M, Miorin M, Vitiello L, Angelini C. Dystrophin-positive fibers in Duchenne dystrophy: origin and correlation to clinical course. Muscle Nerve. 1995;18:1115–1120. | |
Vanhoutte EK, Faber CG, van Nes SI, et al. Modifying the Medical Research Council grading system through Rasch analyses. Brain. 2012;135:1639–1649. | |
Angelini C. The role of corticosteroids in muscular dystrophy: a critical appraisal. Muscle Nerve. 2007;36:424–435. | |
Semplicini C, Angelini C. Clinical scales for the evaluation of neuromuscular patients. In: Angelini C, editor. Muscular Dystrophy: Causes and Management. New York, NY, USA: Nova Science Publishing; 2013:55–65. | |
Bonifati DM, Ruzza G, Bonometto P, et al. A multicenter, double-blind, randomized trial of deflazacort versus prednisone in Duchenne muscular dystrophy. Muscle Nerve. 2000;23:1344–1347. | |
Biggar WD, Harris VA, Eliasoph L, Alman B. Long-term benefits of deflazacort treatment for boys with Duchenne muscular dystrophy in their second decade. Neuromuscul Disord. 2006;16:249–255. | |
Fenichel GM, Mendell JR, Moxley RT, et al. A comparison of daily and alternate-day prednisone therapy in the treatment of Duchenne muscular dystrophy. Arch Neurol. 1991;48:575–579. | |
Backman E, Henriksson KG. Low-dose prednisolone treatment in Duchenne and Becker muscular dystrophy. Neuromuscul Disord. 1995;5:233–241. | |
Loftus J, Allen R, Hesp R, et al. Randomized double-blind trial of deflazacort versus prednisone in juvenile chronic (or rheumatoid) arthritis: a relatively bone sparing effect of deflazacort. Pediatrics. 1991;88:428–436. | |
Dubowitz V. 75th European Neuromuscular Center International Workshop: second workshop on the treatment of muscular dystrophy, December 10–12, 1999, Naarden, The Netherlands. Neuromuscul Disord. 2000;10:313–320. | |
Dubrovsky AL, Angelini C. Treatment with steroids of DMD. In: Angelini C, editor. Muscular Dystrophy: Causes and Management. New York, NY, USA: Nova Science Publishing; 2013:439–453. | |
Manzur AY, Kuntzer T, Pike M, Swan A. Glucocorticoid corticosteroids for Duchenne muscular dystrophy. Cochrane Database Syst Rev. 2008;1:CD003725. | |
Fenichel G, Pestronk A, Florence J, Robison V, Hemelt V. A beneficial effect of oxandrolone in the treatment of Duchenne muscular dystrophy: a pilot study. Neurology. 1997;48:1225–1226. | |
Fenichel GM, Griggs RC, Kissel J, et al. A randomized efficacy and safety trial of oxandrolone in the treatment of Duchenne dystrophy. Neurology. 2001;56:1075–1079. | |
Consalvi S, Mozzetta C, Bettica P, et al. Preclinical studies in the Mdx mouse model of Duchenne muscular dystrophy with the histone deacetylase inhibitor givinostat. Mol Med. 2013;19:79–87. | |
Politano L, Nigro G, Nigro V, et al. Gentamicin administration in Duchenne patients with premature stop codon. Preliminary results. Acta Myol. 2003;22:15–21. | |
Wagner KR, Hamed S, Hadley DW, et al. Gentamicin treatment of Duchenne and Becker muscular dystrophy due to nonsense mutations. Ann Neurol. 2001;49:706–711. | |
McDonald CM, Henricson EK, Abresch RT, et al. The 6-minute walk test and other endpoints in Duchenne muscular dystrophy: longitudinal natural history observations over 48 weeks from a multicenter study. Muscle Nerve. 2013;48:343–356. | |
McDonald CM, Henricson EK, Abresch RT, et al. The 6-minute walk test and other clinical endpoints in Duchenne muscular dystrophy: reliability, concurrent validity, and minimal clinically important differences from a multicenter study. Muscle Nerve. 2013;48:357–368. | |
Welch EM, Barton ER, Zhuo J, et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007;447:87–91. | |
Auld DS, Thorne N, Maguire WF, Inglese J. Mechanism of PTC124 activity in cell-based luciferase assays of nonsense codon suppression. Proc Natl Acad Sci U S A. 2009;106:3585–3590. | |
Auld DS, Lovell S, Thorne N, et al. Molecular basis for the high-affinity binding and stabilization of firefly luciferase by PTC124. Proc Natl Acad Sci U S A. 2010;107:4878–4883. | |
McElroy SP, Nomura T, Torrie LS, et al. A lack of premature termination codon read-through efficacy of PTC124 (Ataluren) in a diverse array of reporter assays. PLoS Biol. 2013;11:e1001593. | |
Hoffman EP, Bronson A, Levin AA, et al. Restoring dystrophin expression in Duchenne muscular dystrophy muscle progress in exon skipping and stop codon read through. Am J Pathol. 2011;179:12–22. | |
Van Deutekom JC, Janson AA, Ginjaar IB, et al. Local dystrophin restoration with antisense oligonucleotide PRO051. N Engl J Med. 2007;357:2677–2686. | |
Goemans NM, Tulinius M, van den Akker JT, et al. Systemic administration of PRO051 in Duchenne’s muscular dystrophy. N Engl J Med. 2011;364:1513–1522. | |
Voit T, Topaloglu H, Straub V, et al. Safety and efficacy of drisapersen for the treatment of Duchenne muscular dystrophy (DEMAND II): an exploratory, randomised, placebo-controlled phase 2 study. Lancet Neurol. 2014;13:987–996. | |
Fletcher S, Honeyman K, Fall AM, Harding PL, Johnsen RD, Wilton SD. Dystrophin expression in the Mdx mouse after localised and systemic administration of a morpholino antisense oligonucleotide. J Gene Med. 2006;8:207–216. | |
Alter J, Lou F, Rabinowitz A, et al. Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nat Med. 2006;12:175–177. | |
Sazani P, Ness KP, Weller DL, Poage D, Nelson K, Shrewsbury AS. Chemical and mechanistic toxicology evaluation of exon skipping phosphorodiamidate morpholino oligomers in Mdx mice. Int J Toxicol. 2011;30:322–333. | |
Malerba A, Thorogood FC, Dickson G, Graham IR. Dosing regimen has a significant impact on the efficiency of morpholino oligomer-induced exon skipping in mdx mice. Hum Gene Ther. 2009;20:955–965. | |
Yokota T, Lu QL, Partridge T, et al. Efficacy of systemic morpholino exon-skipping in Duchenne dystrophy dogs. Ann Neurol. 2009;65:667–676. | |
Yokota T, Nakamura A, Nagata T, et al. Extensive and prolonged restoration of dystrophin expression with vivo-morpholino-mediated multiple exon skipping in dystrophic dogs. Nucleic Acid Ther. 2012;22:306–315. | |
Wu B, Lu P, Benrashid E, et al. Dose-dependent restoration of dystrophin expression in cardiac muscle of dystrophic mice by systemically delivered morpholino. Gene Ther. 2010;17:132–140. | |
Kinali M, Arechavala-Gomeza V, Feng L, et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009;8:918–928. | |
Cirak S, Arechavala-Gomeza V, Guglieri M, et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet. 2011;378:595–605. | |
Cirak S, Feng L, Anthony K, et al. Restoration of the dystrophin-associated glycoprotein complex after exon skipping therapy in Duchenne muscular dystrophy. Mol Ther. 2012;20:462–467. | |
Mendell JR, Rodino-Klapac LR, Sahenk Z, et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol. 2013;74:637–647. | |
Goyenvalle A, Griffith G, Babbs A, et al. Functional correction in mouse models of muscular dystrophy using exon-skipping tricycle-DNA oligomers. Nat Med. 2015;21:270–275. | |
Raman SV, Hor KN, Mazur W, et al. Eplerenone for early cardiomyopathy in Duchenne muscular dystrophy: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2015;14:153–161. | |
Melacini P, Vianello A, Villanova C, et al. Cardiac and respiratory involvement in advanced stage Duchenne muscular dystrophy. Neuromuscul Disord. 1996;6:367–376. | |
Markham LW, Spicer RL, Khoury PR, Wong BL, Mathews KD, Cripe LH. Steroid therapy and cardiac function in Duchenne muscular dystrophy. Pediatr Cardiol. 2005;26:768–771. | |
Duboc D, Meune C, Lerebours G, Devaux JY, Vaksmann G, Becane HM. Effect of perindopril on the onset and progression of left ventricular dysfunction in Duchenne muscular dystrophy. J Am Coll Cardiol. 2005;45:855–857. | |
Ogata H, Ishikawa Y, Ishikawa Y, Minami R. Beneficial effects of beta-blockers and angiotensin-converting enzyme inhibitors in Duchenne muscular dystrophy. J Cardiol. 2009;53:72–78. | |
Lehman-Horn F, Weber MA, Nagel AM, et al. Rationale for treating oedema in Duchenne muscular dystrophy with eplerenone. Acta Myol. 2012;31:31–39. | |
Percival JM, Whitehead NP, Adams ME, et al. Sildenafil reduces respiratory muscle weakness and fibrosis in the Mdx mouse model of Duchenne muscular dystrophy. J Pathol. 2012;228:77–87. | |
Leung DG, Herzka DA, Thompson WR, et al. Sildenafil does not improve cardiomyopathy in Duchenne/Becker muscular dystrophy. Ann Neurol. 2014;76:541–549. | |
Chen JY, Yen MH, Chen HS, et al. Health support: health promotion for families with a DMD child. In: Angelini C, editor. Muscular Dystrophy: Causes and Management. New York, NY, USA: Nova Science Publishing; 2013:319–337. | |
Ellis DA. Intermediary metabolism of muscle in Duchenne muscular dystrophy. Br Med Bull. 1980;36:165–171. | |
Angelini C, Tasca E. Management and clinical trials in Duchenne dystrophy. In: Honorio S, editor. Duchenne Muscular Dystrophy: Symptoms, Management and Prognosis. New York, NY, USA: Nova Science Publishing; 2015. In press. | |
Carrol JE, Norris BJ, Brooke MJ. Defective (U-C14) palmitic oxidation in Duchenne muscular dystrophy. Neurology. 1985;35:96–98. | |
Wehling-Henricks M, Jordan MC, Gotoh T, Grody WW, Roos KP, Tidball JG. Arginine metabolism by macrophages promotes cardiac and muscle fibrosis in mdx muscular dystrophy. PLoS One. 2010;5:e10763. | |
Sejerson T, Bushby K, TREAT-NMD EU Network of Excellence. Standards of care for Duchenne muscular dystrophy: brief TREAT- NMD recommendations. Adv Exp Med Biol. 2009;652:13–21. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.