Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 15
Drug–Drug–Gene Interactions in Cardiovascular Medicine
Authors Asiimwe IG ![]() , Pirmohamed M
, Pirmohamed M ![]()
Received 23 August 2022
Accepted for publication 21 October 2022
Published 2 November 2022 Volume 2022:15 Pages 879—911
DOI https://doi.org/10.2147/PGPM.S338601
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Innocent G Asiimwe, Munir Pirmohamed
The Wolfson Centre for Personalized Medicine, MRC Centre for Drug Safety Science, Department of Pharmacology and Therapeutics, Institute of Systems, Molecular and Integrative Biology, University of Liverpool, Liverpool, UK
Correspondence: Innocent G Asiimwe; Munir Pirmohamed, Tel +44 151 7955387 ; +44 151 794 5549, Fax +44 151 794 5059, Email [email protected]; [email protected]
Abstract: Cardiovascular disease remains a leading cause of both morbidity and mortality worldwide. It is widely accepted that both concomitant medications (drug–drug interactions, DDIs) and genomic factors (drug–gene interactions, DGIs) can influence cardiovascular drug-related efficacy and safety outcomes. Although thousands of DDI and DGI (aka pharmacogenomic) studies have been published to date, the literature on drug–drug–gene interactions (DDGIs, cumulative effects of DDIs and DGIs) remains scarce. Moreover, multimorbidity is common in cardiovascular disease patients and is often associated with polypharmacy, which increases the likelihood of clinically relevant drug-related interactions. These, in turn, can lead to reduced drug efficacy, medication-related harm (adverse drug reactions, longer hospitalizations, mortality) and increased healthcare costs. To examine the extent to which DDGIs and other interactions influence efficacy and safety outcomes in the field of cardiovascular medicine, we review current evidence in the field. We describe the different categories of DDIs and DGIs before illustrating how these two interact to produce DDGIs and other complex interactions. We provide examples of studies that have reported the prevalence of clinically relevant interactions and the most implicated cardiovascular medicines before outlining the challenges associated with dealing with these interactions in clinical practice. Finally, we provide recommendations on how to manage the challenges including but not limited to expanding the scope of drug information compendia, interaction databases and clinical implementation guidelines (to include clinically relevant DDGIs and other complex interactions) and work towards their harmonization; better use of electronic decision support tools; using big data and novel computational techniques; using clinically relevant endpoints, preemptive genotyping; ensuring ethnic diversity; and upskilling of clinicians in pharmacogenomics and personalized medicine.
Keywords: drug–drug interactions, drug–gene interactions, drug–drug–gene interactions, drug–gene–gene interactions, pharmacogenomics
Introduction
Cardiovascular disease remains a leading cause of mortality, with heart diseases having caused the highest number of deaths in the United States (21% of ~3.4 million deaths) according to provisional leading cause-of-death rankings for 2020.1 Globally, cardiovascular diseases (led by ischaemic heart disease, stroke, and hypertensive heart disease) accounted for approximately 17.8 million (95% confidence intervals/CI 17.1 to 19.7 million) deaths and 393 million (95% CI 368 to 417 million) disability-adjusted life years (DALYs) in 2019, making cardiovascular disease the leading cause of both deaths and DALYs.2 Moreover, the prevalence of cardiovascular disease is increasing due to disparate trends in mortality versus incidence.3 For instance, the number of new cardiovascular cases (55.5 million, 95% CI 52.3 to 58.9 million) was thrice the number of cardiovascular deaths (17.8 million), leading to an estimated total prevalence of 523 million (95% CI 497 to 550 million) by the end of 2019.2
Drug–drug–gene interactions (DDGIs) arise when both another drug (drug–drug interaction, DDI) and an individual’s genetic profile (drug–gene interaction, DGI) alter the efficacy and/or safety profile of a specified drug.4 Advancements in medical knowledge (earlier disease diagnosis, more effective treatments, a realization that lifestyle factors such as smoking can impact disease etc.) have resulted in a remarkable gain in life expectancy.3 An increasingly ageing population is the main driver of multimorbidity, defined by the Academy of Medical Sciences as the coexistence of two or more chronic health conditions.5 Multimorbidity is common in patients with cardiovascular disease (being estimated to be 91% and 93% in 229205 UK,6 and 3478 Chinese,7 participants, respectively). Furthermore, this is not restricted to high-income countries.8 Multimorbidity usually necessitates the administration of multiple medication regimens (to manage the multiple health conditions), often resulting in polypharmacy (most commonly defined as five or more daily medications9), higher DDI/DGI/DDGI frequencies, medication-related harm/adverse outcomes (such as adverse drug reactions, longer hospitalizations, and mortality), and increased healthcare costs.10–15
A systematic review reported that the median percentage of preventable drug-related hospital admissions was 3.7% (range 1.4 to 15.4, 13 studies) and that most preventable drug-related admissions (n = 1406, 9 studies) involved antiplatelets including aspirin (n = 225, 16.0%), diuretics (n = 223, 15.9%), nonsteroidal anti-inflammatory drugs (n = 155, 11.0%) or anticoagulants (n = 117, 8.3%).16 In a later systematic review that also reported hospitalization resulting from adverse drug reactions (ADRs) or adverse drug events (ADEs), all included studies reported involvement of cardiovascular medicines, with these medicines being responsible for a median of 33.9% (interquartile range/IQR 19.9 to 58.6%) of ADRs (n = 21 studies) and 42.3% (IQR 30 to 72.2%) of ADEs (n = 6 studies).17 More recently, in a prospective observational study conducted in the Liverpool University Hospital Foundation National Health Service (NHS) Trust, England, 218 (18.4%) of 1187 patients were admitted with an adverse drug reaction, with these admissions being estimated to cost the UK NHS approximately £2.21 billion each year.12 In this study, cardiovascular drugs (angiotensin-converting enzyme inhibitors/angiotensin receptor blockers, anticoagulants, antiplatelets, and diuretics) and DDIs were implicated in 82 (37.6%) and 64 (29.4%) of the 218 patient episodes with adverse drug reactions, respectively.12 It should be pointed out that the number of potential DDIs far outweighs the number of clinically relevant adverse reactions.18,19 However, DDIs are more likely to be clinically relevant if they involve drugs with low therapeutic indices (eg, warfarin for anticoagulation or digoxin for atrial fibrillation), are given to vulnerable patients (eg, those who are elderly or have multiple morbidities including renal/hepatic impairment), or involve novel therapeutic agents (whose mechanisms of actions are less likely to be fully understood).18
Pharmacogenomics is the study of the genomic basis of variability in drug response, and it is often used interchangeably with pharmacogenetics, which focuses on specific genes.20 More advanced and cheaper genotyping technology have enabled the conduct of several pharmacogenomic (DGI) studies, including genome-wide association studies, that have increased pharmacogenomic awareness and the realization that most patients would benefit from the use of pharmacogenomic information in their clinical management. For instance, McInnes et al have previously reported that 99.5% of 487,409 participants in the UK Biobank had at least one clinically actionable genotype, defined as a genotype associated with a clinically relevant DGI.21 Other studies have reported similar estimates including 99.8% of 42,092 Estonians,22 91.4% of 9589 Vanderbilt pharmacogenotyping program participants,23 99.5% of 6045 Qataris,24 95.9% 5408 Australians,25 99.0% of 1013 Mayo Clinic Biobank participants,26 98.7% of 713 UK patients,27 99.4% of 498 Dutch participants,28 and, 96.9% of 98 Canadian paediatric patients29 having at least one clinically actionable genotype/diplotype. To enable the translation of pharmacogenomic knowledge into clinical practice, clinical implementation guidelines such as the Clinical Pharmacogenetics Implementation Consortium (CPIC), the Canadian Pharmacogenomics Network for Drug Safety (CPNDS), the Dutch Pharmacogenetics Working Group (DPWG), and the French National Network (Réseau) of Pharmacogenetics (RNPGx) have been developed.30 However, these guidelines, like most pharmacogenetic studies, focus on DGIs, which means evidence pertaining to DDGIs remains limited.15,31 As stated above, multimorbidity and polypharmacy, which are common in cardiovascular disease and ageing populations, increase the frequencies of both DDIs and DGIs, which makes it necessary to increase efforts to avoid these interactions and to understand how DDIs interact with DGIs (DDGIs). Using pharmacogenomic and other evidence, we therefore examine the extent to which DDGIs and other interactions influence efficacy and safety outcomes in the field of cardiovascular medicine. We describe the different categories of DDIs and DGIs and illustrate how they interact to produce DDGIs and other complex interactions. We provide examples of studies that have reported the prevalence of clinically relevant interactions and the most implicated cardiovascular medicines, outline the challenges associated with dealing with these interactions in clinical practice and recommendations on how to manage these challenges.
Cardiovascular Medicine
Cardiovascular medicine deals with the diagnosis and treatment of cardiovascular disease (CVD), which is a general term that describes a group of disorders affecting the heart and/or blood vessels.32 The major disorders vary in terms of underlying pathologies, other organ systems involved (eg endocrine, hematologic, immune, neurologic and/or pulmonary), and the population segments affected.33 Although each CVD disorder has a distinct pathology, they all have a common set of risk factors including atherosclerosis (build-up of fatty deposits within arteries), hypertension and related organ damage, infection (including streptococcal-related heart valve damage or rheumatic heart disease), and anatomic deformities (both congenital and acquired).32–35 Examples of CVD disorders include those involving the heart muscles (eg, atrial fibrillation and myocardial infarction), heart valves (eg, rheumatic heart disease) and blood vessels. Blood vessel disorders include ischaemic heart disease (occlusion of the coronary arteries), cerebrovascular disease (blockage of brain-supplying blood vessels), peripheral arterial disease (restriction of blood supply to the arm and leg muscles) and deep vein thrombosis (clots/thrombi forming in the deep veins found in the legs, calf or elsewhere).32,34 When thrombi in deep veins become dislodged, they can travel to the lungs and block the pulmonary vessels resulting in a condition termed pulmonary embolism.32,34 Strokes, on the other hand, result from the blockage (eg, by atherosclerotic plaques or blood clots or emboli) of the arteries supplying the brain (ischaemic strokes), although they can also be caused by other events (eg bleeding in haemorrhagic strokes).32,34
CVD management generally involves three main stages.34,36 In the first, an assessment is conducted to evaluate the causes, if any, of the CVD disorder, evaluate the severity (for instance damage to other organs) and determine concomitant or underlying conditions (such as diabetes) that may add to the cardiovascular burden. Depending on the initial assessment (severity of the disease, risk factors, etc.), both non-pharmacological (second stage) and pharmacological (third stage) interventions may be offered at the same or different time(s), acutely or chronically and therapeutically or prophylactically (primary or secondary prophylaxis). Non-pharmacological treatment includes advice on lifestyle interventions (ie, weight reduction, diet changes, alcohol consumption, smoking and exercise) and mechanical interventions (such as elastic compression stockings or percutaneous coronary intervention/coronary angioplasty). If non-pharmacological treatment is insufficient, pharmacotherapy (cardiovascular medicines) to aid symptom relief, control the disease, retard disease progression, prevent complications, reduce risk factors and improve the length and quality of life is (are) required.34,36,37 Due to the high prevalence of cardiovascular disease, it is unsurprising that cardiovascular medicines are among the most commonly used drugs.38–40 For example, based on the US National Health and Nutrition Examination Survey (2015–2016) and Canadian Health Measures Survey (2016–2017), lipid-lowering drugs were used by 45.0% and 34.3% of US and Canadian adults aged 60–79, respectively.39 Example categories of cardiovascular medicines/drugs are shown in Table 1.
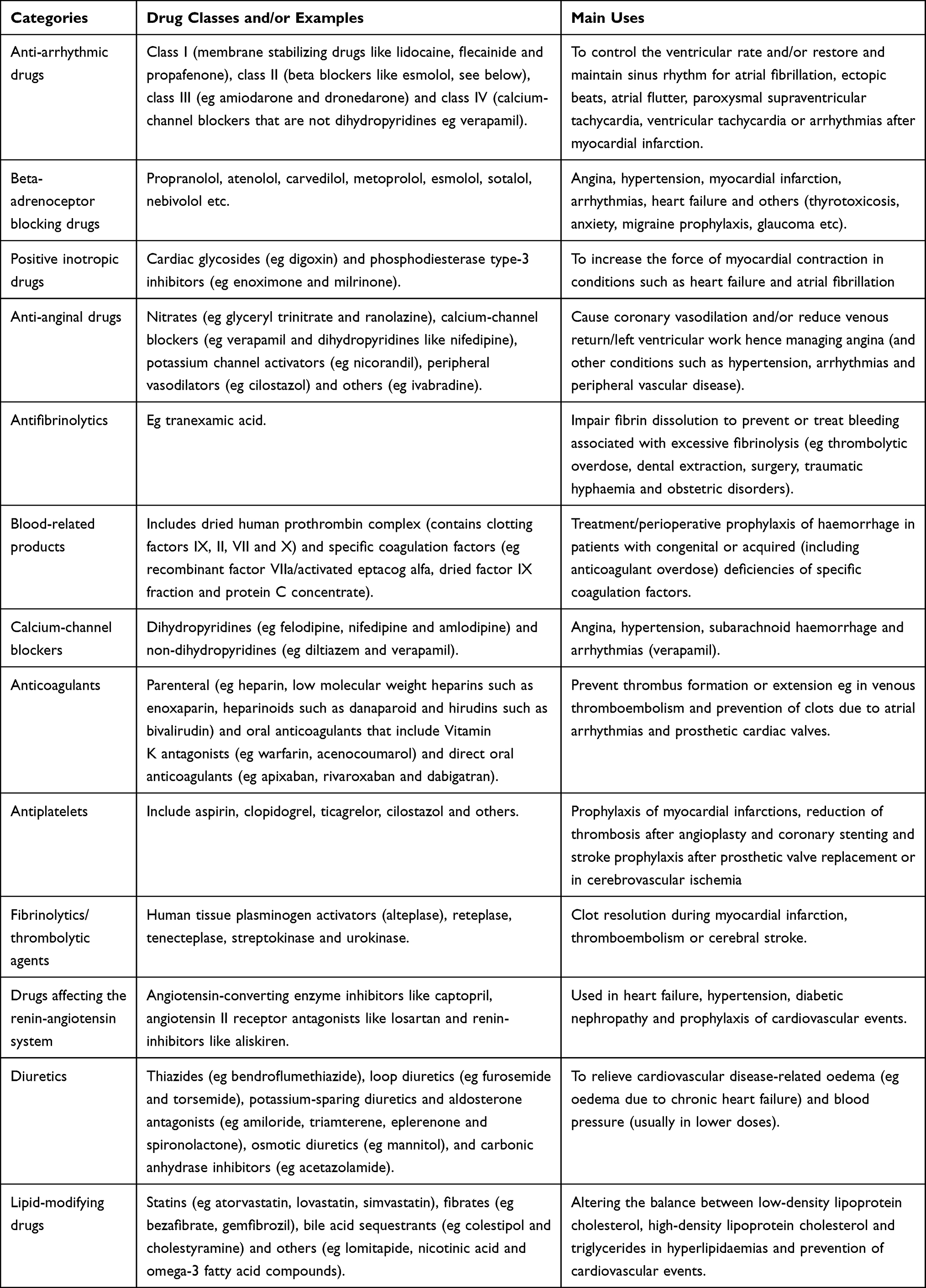 |
Table 1 Example Categories of Cardiovascular Medicines/Drugs |
Drug–Drug Interactions
Drug–drug interactions (DDIs) occur when one drug (the perpetrator drug) affects how the body acts on a victim drug (pharmacokinetic effects) and/or how the victim drug acts on the body (pharmacodynamic effects).4,18 There is a third mechanism by which drugs can interact (pharmaceutical DDIs, caused by inappropriate mixing of drugs before administration eg, precipitation of phenytoin solution for injection when mixed with a glucose solution)18 but these are not common and are therefore not discussed further.
Pharmacokinetic DDIs
Many DDIs are pharmacokinetic in nature and occur when a perpetrator drug alters the absorption, distribution, metabolism (Figure 1A and B), elimination/excretion and/or transcellular transport of the victim drug, resulting in increased or decreased exposure of the victim drug.18
Figure 1 Continued. Figure 1 Drug-drug, drug-gene, and drug-drug-gene interactions.13,52 (A) Normal metabolism: expected drug exposure (black dotted line in plasma drug concentration–time curve, single exposure) for a drug/substrate that is metabolized by two cytochrome P450 (CYP) enzymes. (B) Drug–drug interaction (DDI): inhibition (eg null activity) or induction (eg increased enzyme copies) of CYP A by co-medications resulting in increased (red line in plasma drug concentration–time curve) or decreased (green line in plasma drug concentration–time curve) drug exposures, respectively. (C) Drug–gene interaction (DGI): genetic variation inactivates/reduces (loss-of-function/LoF variant) or increases (gain-of-function/GoF variant) CYP B activity resulting in increased or decreased drug exposures, respectively. (D) Drug-drug-gene interaction (DDGI): cumulative effects of comedications (DDIs) and genetic variations (DGIs). In a category 1 DDGI, a DDI and DGI on the same pathway (eg CYP A) and direction (eg inhibitor with a LOF variant) interact to significantly increase (or decrease) drug exposure while in a category 2 DDGI, the DDI and DGI act on different pathways but still in the same direction to also increase (or decrease) drug exposure. Lastly, category 3 comprises DDIs and DGIs with opposing effects (eg inhibitor with a GOF variant) that leads to increased (inhibitor effects greater than GOF variant effects), decreased (inhibitor effects lower than GOF variant effects) or unchanged (inhibitor effects similar to GOF variant effects) drug exposure. The above interactions also apply to bioactivation of prodrugs (in which decreased metabolism results in decreased systemic exposure) and other pathways (eg drug- and/or gene-mediated changes to drug transporters or drug targets). If a drug has a comparable clinical effect with its metabolites, the effects of metabolism-based DDIs, DGIs and DDGIs may not be apparent. Any compensation by CYP A for the loss (or increase) in CYP B’s activity, and vice-versa, is not depicted/accounted for in this over-simplified schematic.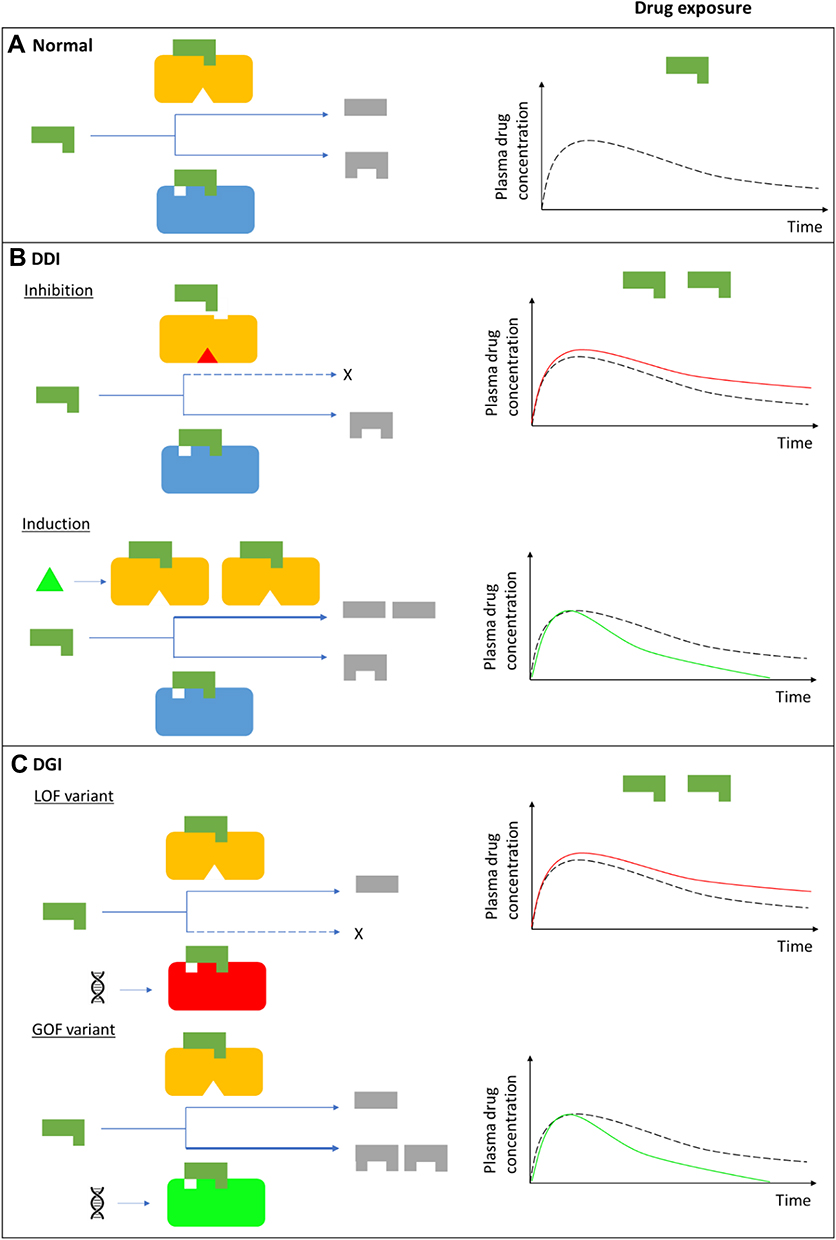
Absorption
During absorption in the gastro-intestinal tract, DDIs can occur when one drug alters: a) intestinal blood flow/motility (eg, metoclopramide increases gastric emptying and can increase the rate but not extent of absorption of some drugs); b) the stomach pH/acidity (eg, proton pump inhibitors, histamine type 2 receptor antagonists or antacids that increase gastric pH facilitate the absorption of weakly acidic drugs like aspirin); c) the formulation of the victim drug (eg, some antacids can damage enteric coatings that are designed to prevent dissolution in the stomach); d) the proportion of drug available for absorption (eg, some antacids can bind drugs such as tetracycline); e) bacterial flora resident in the intestine (eg, broad-spectrum antibiotics can decrease the populations of gut microbes that are important in modulating the bioavailability of oral drugs such as oral contraceptives), among other mechanisms.18,37,41
Distribution
Distribution-related DDIs mostly occur through two mechanisms.18 The first involves the distribution of a victim drug to its site of action, with perpetrator drugs that can alter cardiac output (eg, inotropic drugs) or tissue perfusion (eg, vasodilators or vasoconstrictors) resulting in either increased exposure/drug effect (increased cardiac output and/or tissue vasodilation) or reduced exposure/drug effect (decreased cardiac output and/or tissue vasodilation). The second mechanism involves displacement of highly protein-bound drugs such as warfarin (~99% protein-bound), with the addition of other highly protein-bound drugs such as tizoxanide resulting in the displacement of warfarin, an increase in the unbound (free) fraction, and an increase in pharmacodynamic response.18,42,43 However, these effects are likely to be short-lived and of limited clinical significance since the metabolism of the displaced drug usually increases, which offsets the increase in the unbound drug fraction.18,42
Metabolism
The most studied pharmacokinetic DDIs are those involving the family of cytochrome P450 (P450 or CYP) hepatic metabolizing isoenzymes, with CYP3A4 being the most commonly implicated enzyme.18 Examples of substrates, inducers and inhibitors for the CYP metabolizing enzymes are shown in Table 2 (cardiovascular medicines are highlighted in bold). Although some DDIs can involve only cardiovascular drugs (eg, the antiarrhythmic amiodarone increasing plasma concentrations of the anticoagulant warfarin, through the inhibition of CYP2C9), other pairs of interacting drugs involve drugs from other drug classes (eg, the triazole antifungal fluconazole also potentiating warfarin’s anticoagulation effect through CYP2C9 inhibition or the macrolide antibiotic clarithromycin inhibiting the CYP3A4-mediated metabolism of simvastatin, increasing its exposure and the risk of myopathy/rhabdomyolysis). Therefore, Table 2 is not restricted to cardiovascular drugs. Additionally, as stated earlier, patients with CVD usually take multiple medications including non-cardiovascular drugs, and some non-cardiovascular drug pairs can produce clinically significant cardiovascular side effects (eg, increased risk of QT interval prolongation and Torsades de Pointes when the antihistamine terfenadine is co-administered with the antibiotic erythromycin),49 which makes it important to also study these drugs.
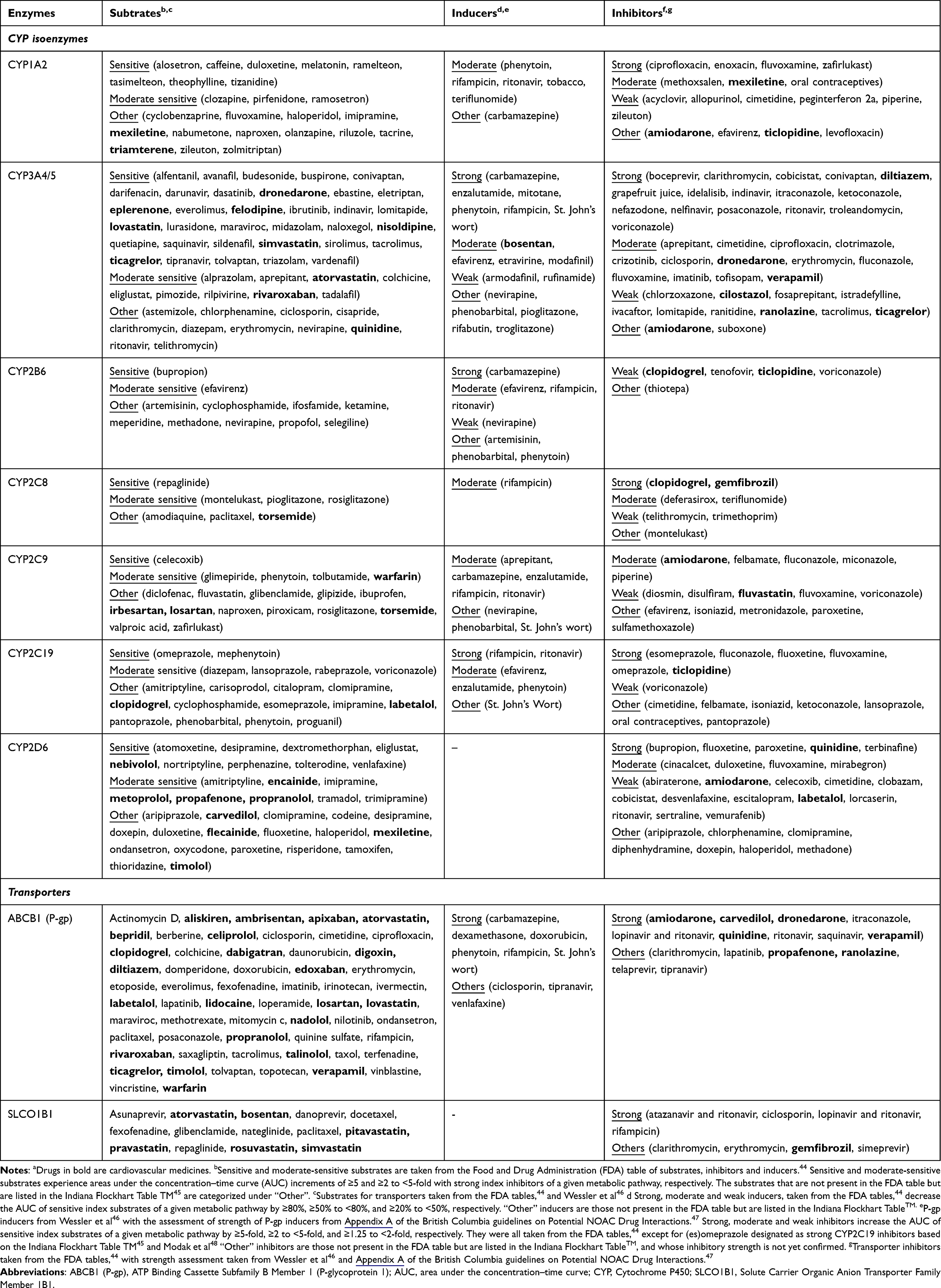 |
Table 2 Examples of Substrates, Inducers and Inhibitors for Cytochrome P450 Isoenzymes and Transportersa (Data from Turner et al27) |
It is important to note that for drugs that are administered as prodrugs such as clopidogrel (metabolism by a CYP enzyme is required to generate the active drug), DDIs will have the opposite effect eg CYP2C19 induction by rifampicin, will increase the rate of production of clopidogrel’s active metabolite, increasing the likelihood of toxicity (haemorrhagic events). Another thing worth pointing out is that enzyme inhibition can take many forms including non-competitive, competitive, uncompetitive, and mixed-type inhibition, with the two most common types being non-competitive (the inhibitor binds at an allosteric site and is not affected by substrate concentration) and competitive (inhibitor binds at the active site and “competes” with the substrate).50 Inhibition can also be reversible (inhibitor noncovalently binds to the enzyme) or irreversible (inhibitor covalently binds to the enzyme) and in terms of clinical relevance means the effects of reversible inhibitors are usually short-lived, once the inhibitor is withdrawn. Lastly, whereas the effects of enzyme inhibition usually occur relatively early after administration of the inhibitor, those of drug induction usually take some time (1–2 weeks) as these require the formation of new enzyme.18
Excretion
The most common excretion/elimination DDIs involve drugs that are excreted by the kidney, with drugs that reduce renal elimination eg aminoglycoside antibiotics (alter glomerular filtration rate) or some nonsteroidal anti-inflammatory drugs (NSAIDs, compete for renal tubular excretion) increasing the risk of toxicity for drugs that are predominantly excreted by the kidneys, such as the cardiac glycoside digoxin.18
Transcellular Transport
P-glycoprotein and organic anion transporter polypeptides (such as Solute Carrier Organic Anion Transporter Family Member 1B1 [SLCO1B1]) can also mediate DDIs;27 examples of substrates, inducers and inhibitors for these drug transporters are shown in Table 2. As P-glycoprotein is an efflux transporter, its inhibition and induction usually increase and decrease bioavailability/exposure of the victim drug, respectively.51 P-glycoprotein is expressed in many tissues and affects several pharmacokinetic attributes, including absorption (expressed in the intestines), distribution (blood–brain barrier and placenta), metabolism (liver) and elimination (kidney).4,18,51 When two substrates are co-prescribed, DDIs can also occur through competition for the transport protein (similar to competitive CYP inhibition, except that the concentration of the inhibitor or second substrate is also clinically relevant).
Pharmacodynamic DDIs
Pharmacodynamic DDIs occur when a perpetrator drug modulates the pharmacological effects of a victim drug in the body.52 Modulation of effects can be synergistic or additive (similar pharmacological actions) or antagonistic (opposing pharmacological actions) and is usually the case when drugs share the same target (eg, enzyme) or physiological system (eg, blood coagulation).18,51 If one drug has greater efficacy than another interacting drug (eg, the full opioid agonist morphine vs the partial agonist buprenorphine), its actions will be antagonized even when the two drugs have similar pharmacological actions.18 Pharmacodynamic DDIs can be beneficial (eg, when different antihypertensives are used for their synergistic effects) or harmful. Examples of synergistic pharmacodynamic DDIs that are harmful and involve different targets but the same physiological system are many in cardiovascular disease and include anticoagulants with NSAIDs (increase the risk of gastrointestinal haemorrhage), anticoagulants with antiplatelets (bleeding), calcium channel blockers with benign prostatic hyperplasia alpha-adrenergic antagonists (orthostatic hypotension and falls), angiotensin-converting enzyme (ACE) inhibitors with potassium-sparing diuretics (hyperkalaemia), diuretics with digoxin (digoxin toxicity), and a pair of QT-interval prolonging drugs (torsade de pointes).18 On the other hand, examples of antagonistic DDIs include ACE inhibitors and NSAIDs (decreased blood pressure lowering effect) and vitamin K oral anticoagulants and vitamin K supplements (decreased anticoagulation effect).51
Clinical Relevance, DDI Prevalence and Implicated Cardiovascular Medicines
For an interaction to be clinically relevant, plasma concentrations of the victim drug should either be increased above (toxicity) or decreased below (lack of efficacy) the therapeutic window, with the likelihood of clinical relevance ie out-of-therapeutic range doses, depending on the steepness of the substrate’s dose–response curve, the perpetrator’s dosage, and the perpetrator’s induction/inhibition strength.18 The strength of inducers/inhibitors is obtained from pharmacokinetic/mechanistic studies, with the Food and Drug Administration considering strong, moderate and weak inducers to be those that, respectively, decrease the area under the plasma concentration–time curve of sensitive index substrates of a given metabolic pathway by ≥80%, ≥50% to <80%, and ≥20% to <50%, as stated in Table 2.44
It is difficult to recruit and monitor many patients to evaluate the prevalence and clinical impact of DDIs in large populations, which means that most mechanistic DDI studies are small-sized.15 Additionally, the large number of drug combinations and the potentially multiple pharmacokinetic and pharmacodynamic pathways for each drug make it an insurmountable task to cover all DDIs in clinical trials.15 Consequently, most DDI literature reports potential DDIs, which are predicted based on an individual’s medication list and the known interacting drug pairs within that list. Prediction may also be undertaken through simulation using methods such as pharmacokinetic-pharmacodynamic (PK-PD) and physiologically based pharmacokinetic (PB-PK) modelling. As Hahn and Roll discuss, inducer/inhibitor strength can be used to distinguish between clinically relevant DDIs (ie, those involving strong/moderate inducers/inhibitors) and those that are not (ie, those involving weak inducers/inhibitors), although this might be misleading due to a phenomenon called “phenoconversion” that is discussed in the next sections.4 DDIs can also be classified by severity into major (“life-threatening or involving permanent damage”), moderate (“requiring additional treatment”), and minor (“unnoticeable or not sufficient to affect therapeutic outcome”).18
Several DDI prevalence studies have been previously reported. For example, of 20,534 patients who were referred for pharmacogenetic testing and drug interaction screening in a US pharmacogenetic testing laboratory, 69.1% patients had at least one reported interaction (DDI/DGI/DDGI), with metoprolol (1484 patients), clopidogrel (1415 patients), and warfarin (1234 patients) being the first, second and fourth most implicated medications, respectively.53 Of the total number of interactions (n = 33,665), 16,924 were rated as severe (ie, rated at a level of “change medication” or “consider changing medication or adjusting dose” by study clinicians) and of these 53.0% were DDIs.53 In another US study of 1143 individuals with known CYP2D6, CYP2C19 and CYP2C9 genotypes, 357 (31.2%) of the individuals had a DDI with the top four interacting medications being metoprolol, clopidogrel, simvastatin, and aspirin.52 A total of 1053 potential major or substantial interactions (including DGIs and DDGIs) were identified in 501 (43.8%) individuals, with potential DDIs accounting for 696 (66.1%) of these interactions.52 As a third example, data mining of a spontaneous reporting database in Italy to identify adverse drug reactions associated with DDIs revealed that of 17,700 reports with at least two drugs, there were 5345 (30.2%) potential DDIs, and 1159 (21.7%) of these reports had a related adverse drug reaction.54 Additionally, digoxin and diuretics (95 reports) was the most frequently reported DDI, while the combination of anticoagulants and antiplatelets (50 reports) had the greatest number of serious reactions (100% of the 50 reports) and deaths (14% of the 50 reports).54 A French study that included more than 6.9 million outpatient dispensed medicines estimated the prevalence of dispensing drugs contraindicated or cautioned because of DDIs further highlights the importance of cardiovascular drugs in DDI literature.55 Specifically, the most frequently contraindicated drug pair was bisoprolol and flecainide (n = 5036, 37.9%), with eight of the ten most represented pairs involving cardiovascular drugs. For the cautioned category, ramipril and spironolactone (n = 4741, 5.0%) was the most frequent pair, with nine of the ten most represented pairs involving cardiovascular drugs.55 The prevalence and/or impacts of DDIs in other clinical settings have been reported in multiple other studies.56–63
The above studies were not restricted to CVD cohorts. Some, however, like Turner et al27 who investigate DDIs in hospital-discharged patients following a non-ST elevation acute coronary syndrome (NSTE-ACS) have. DDIs were based on drug inhibition and/or induction of the metabolizing enzymes (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4/5) and drug transporters (P-gp and SLCO1B1), with a DDI being present if a patient was given both a victim drug and a perpetrator drug that influenced the latter through at least one of the above enzymes/transporters, without consideration of autoinhibition and autoinduction. Of 652 patients with drug use and actionable genotype information available (the “interaction” cohort), 342 (52.5%) patients had at least one DDI of which 186 (28.5%) patients had at least one substantial interaction (defined as DDIs due to strong inhibitors/inducers, with the assessment of the strength of transporter inhibitors/inducers being based on relevant literature).44–48,64 In a Moroccan study conducted in 138 hospitalized cardiac patients, DDI prevalence was 68.1% (94 patients) with the most common DDIs including aspirin and clopidogrel (12.2% prevalence), aspirin and heparin (8.3%) and furosemide and spironolactone (5.8%).65 In this study, there were 726 prescribed drugs with drugs of the cardiovascular system (n = 372, 51.2%) and the blood and hematopoietic organs (n = 288, 39.7%) being the most common. Lastly, out of a total of 360 interactions, there were 40 (11.1%) and 134 (37.2%) DDIs classified as major and moderate severity, respectively.65 Another study investigated potential DDIs in 2343 hospitalized cardiac Pakistani patients and found that 91.6% patients had at least one potential DDI, with 86.3% and 84.5% having at least one major and moderate potential DDI, respectively.66 Of 5109 identified potential DDIs, 45% and 55% were or major and moderate severity, respectively, and all the top 10 most common potential DDIs included at least one cardiovascular drug (aspirin and clopidogrel combination [n = 489] being the most common).66 The prevalence and/or impacts of DDIs in CVD patients have been reported in other studies.67–69
Due to high prevalence, cardiovascular medicines are the most included drug category in DDI-related guidance. For example, the Society for Post-Acute and Long-Term Care Medicine, based on a survey of physicians and pharmacists and three criteria (clinical significance and potential to cause harm, frequency of occurrence, and frequency of being prescribed in nursing homes), compiled a list of the top 10 particularly dangerous drug interactions in post-acute and long-term care medicine.70 Out of the 10 pairs (ACE inhibitors + potassium supplements, ACE inhibitors + spironolactone, digoxin + amiodarone, digoxin + verapamil, theophylline + quinolones, warfarin + macrolides, warfarin + NSAIDs, warfarin + phenytoin, warfarin + quinolones, and warfarin + sulfa drugs), nine involved at least one cardiovascular drug, with warfarin appearing in five pairs.70 Another study that aimed to establish an international consensus list of potentially clinically significant DDIs in people aged ≥65 years and that included 29 experts (geriatricians and clinical pharmacists among these) from 8 European countries came up with 66 potentially clinically significant DDIs, of which about two thirds of the DDIs included at least one cardiovascular drug (ACE inhibitors/ARBs, anti-arrhythmics, anticoagulants, antiplatelets, calcium channel blockers, digoxin, diuretics, and lipid-modifying agents).71
Drug–Gene Interactions
Drug–gene interactions (DGIs) occur when an individual’s genotype affects the pharmacokinetics (pharmacokinetic DGIs, Figure 1C) and/or pharmacodynamics (pharmacodynamic DGIs) of a victim drug.4 For example, polymorphisms in the cytochrome P450 metabolizing enzymes (or drug transporters) can lead to five different phenotypes: poor, intermediate, extensive/normal, rapid, and ultra-rapid metabolizers (or transporters).52,72 Normal metabolizers/transporters respond as expected to standard drug doses as they do not have genetic variants that alter drug metabolism/transport. Poor metabolizers or poor function transporters usually have two copies of loss-of-function (LoF) genetic variants, while intermediate metabolizers usually have one or two copies of reduction-of-function (RoF) genetic variants or one copy of a LoF genetic variant. If metabolism is reduced, drug concentrations increase, which might increase efficacy (patients respond to lower doses) and/or lead to adverse effects. By contrast, decreased metabolism for prodrugs means decreased concentration of the active metabolites, which might decrease efficacy and/or adverse effects. On the other hand, ultra-rapid (usually two or more copies of a gain-of-function [GoF] genetic variant on the same chromosome) and rapid (usually one or two copies of a GoF genetic variant) metabolizers/transporters have increased drug metabolism/transport, with increased drug metabolism resulting in decreased drug exposure and decreased efficacy/adverse effects.52,72 Of note is that non-genetic factors including age, sex, comedications (DDIs), smoking, kidney and liver function can also alter the capacity to metabolize/transport drugs, which might lead to a mismatch between the individual’s genotype-based prediction of drug metabolism/transport and the actual/observed metabolism/transport, a phenomenon termed as phenoconversion.4,73
Pharmacogenomic Studies and Clinical Implementation Guidelines
Thousands of pharmacogenomic studies (aka DGI studies) have explored how genetic/genomic factors influence drug response variability.74 As of 5 July 2022, the “Variant, Gene and Drug Relationship Data” that contains relationships summarized from the Pharmacogenomics Knowledge Base (PharmGKB) annotations contained 9695 unique gene-chemical/gene-drug response relationships (“associated”, “not associated”, and “ambiguous”), reporting the influence of 1933 genes on 1307 drugs.75 The US FDA Table of Pharmacogenetic Associations (last updated 24 May 2022) describes 121 DGIs (influence of 21 genes on 111 drugs) in three sections: Section 1 (“Pharmacogenetic Associations for which the Data Support Therapeutic Management Recommendations”, n = 60 DGIs), Section 2 (“Pharmacogenetic Associations for which the Data Indicate a Potential Impact on Safety or Response”, n = 21 DGIs), and Section 3 (‘Pharmacogenetic Associations for which the Data Demonstrate a Potential Impact on Pharmacokinetic Properties Only’, n = 40 DGIs).72 Of the 121 DGIs, 12 (9.9%) involve cardiovascular drugs including five in Section 1 (clopidogrel:CYP2C19, propafenone:CYP2D6, warfarin:CYP2C9, warfarin: CYP4F2, and warfarin:VKORC1), two in Section 2 (carvedilol:CYP2D6 and simvastatin:SLCO1B1), and five in Section 3 (atorvastatin:SLCO1B1, metoprolol:CYP2D6, nebivolol:CYP2D6, propranolol: CYP2D6, and rosuvastatin:SLCO1B1). It should be noted that although cardiovascular drugs represent less than 10% of the FDA Pharmacogenetic Associations, they are among the most highly ranked drugs in terms of prescription volume, which increases their impact. For example, of the 111 unique drugs mentioned in the US FDA Table of Pharmacogenetic Associations, 41 (36.9%) featured in the 2019 list of the top 300 most prescribed drugs in the US (Figure 2),40 representing a total of 658 million prescriptions and 155 million patients. Only 9 (22.0%) of the 41 drugs were cardiovascular medicines; however, they represented 48.2% (318 million) and 44.5% (69 million) of the total prescriptions and number of patients, respectively.
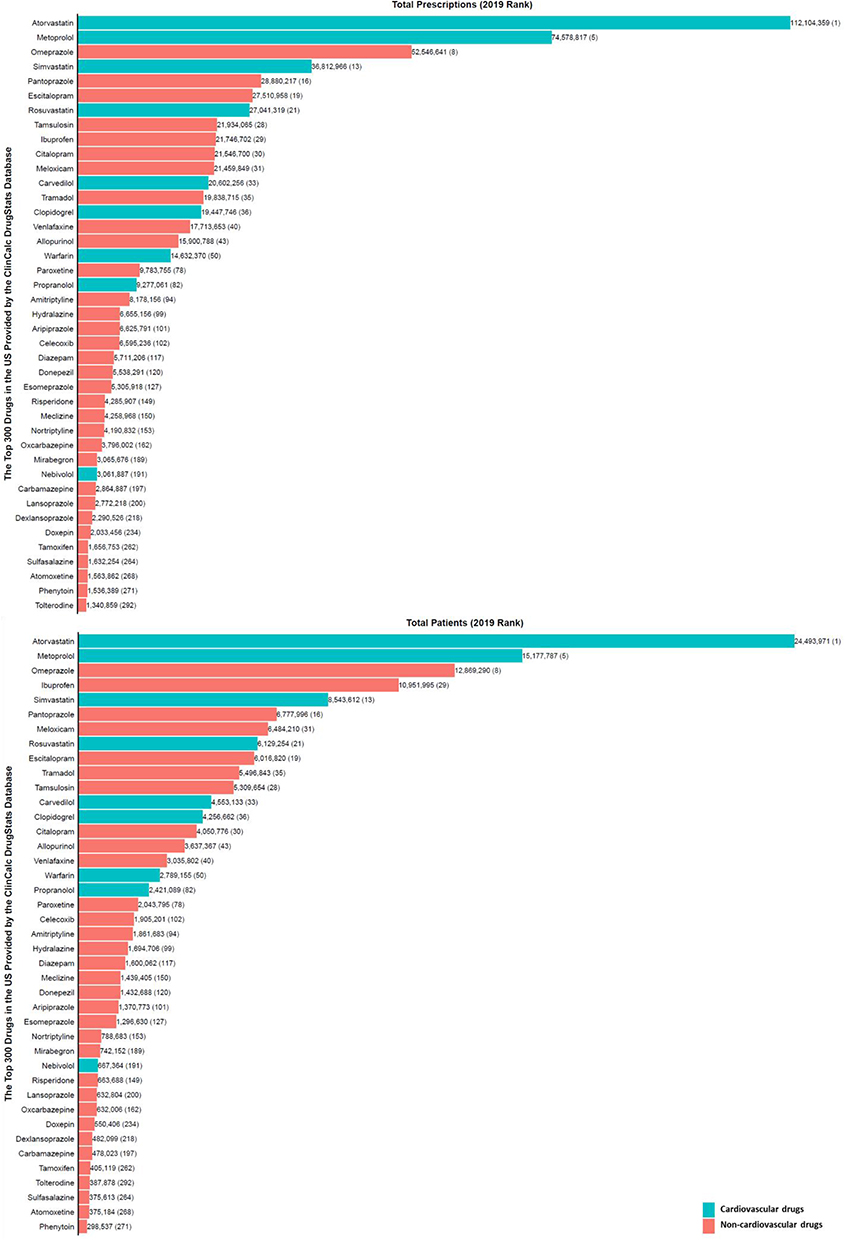 |
Figure 2 Commonly used medications influenced by pharmacogenes. The bar chart shows the total number of prescriptions (top panel) or patients receiving the prescriptions (bottom panel) in 2019 in the United States. Corresponding ranks are shown in parentheses. Data from the ClinCalc DrugStats Database40 that used the Medical Expenditure Panel Survey 2013–2019 (Agency for Healthcare Research and Quality) as a prescription data source. |
Based on a summation of available evidence that ranges from case reports, retrospective cohorts, mechanistic and pharmacokinetic studies to prospective cohorts and randomized control trials,76 several pharmacogenomic clinical implementation guidelines including the Clinical Pharmacogenetics Implementation Consortium (CPIC), the Canadian Pharmacogenomics Network for Drug Safety (CPNDS), the Dutch Pharmacogenetics Working Group (DPWG), and the French National Network (Réseau) of Pharmacogenetics (RNPGx) have been developed.30 For example, as of 28 Jul 2022, CPIC (https://cpicpgx.org/genes-drugs/) reported 442 DGIs between 119 genes and 271 drugs. Only the clinically actionable DGIs have corresponding guidelines and as of 26 Mar 2021, there were 26 CPIC guidelines (https://cpicpgx.org/guidelines/)77 that documented the influence of 23 genes on 95 drugs, with three guidelines78–80 documenting the influence of six genes on nine cardiovascular medicines. Abdullah-Koolmees et al have previously summarized DGIs from the above four clinical implementation guidelines and about a sixth of the listed drugs were cardiovascular medicines, which are shown in Table 3.
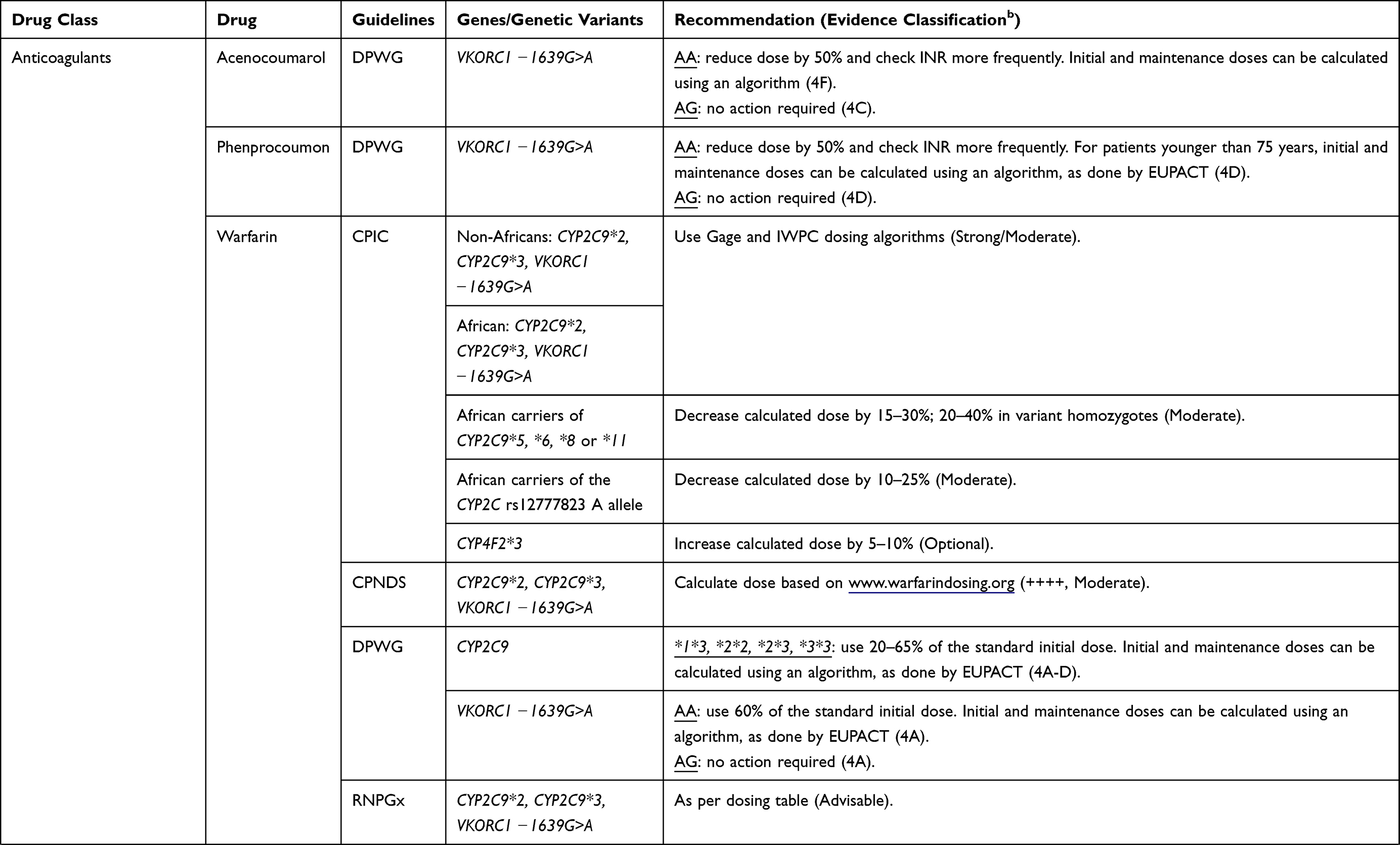 | 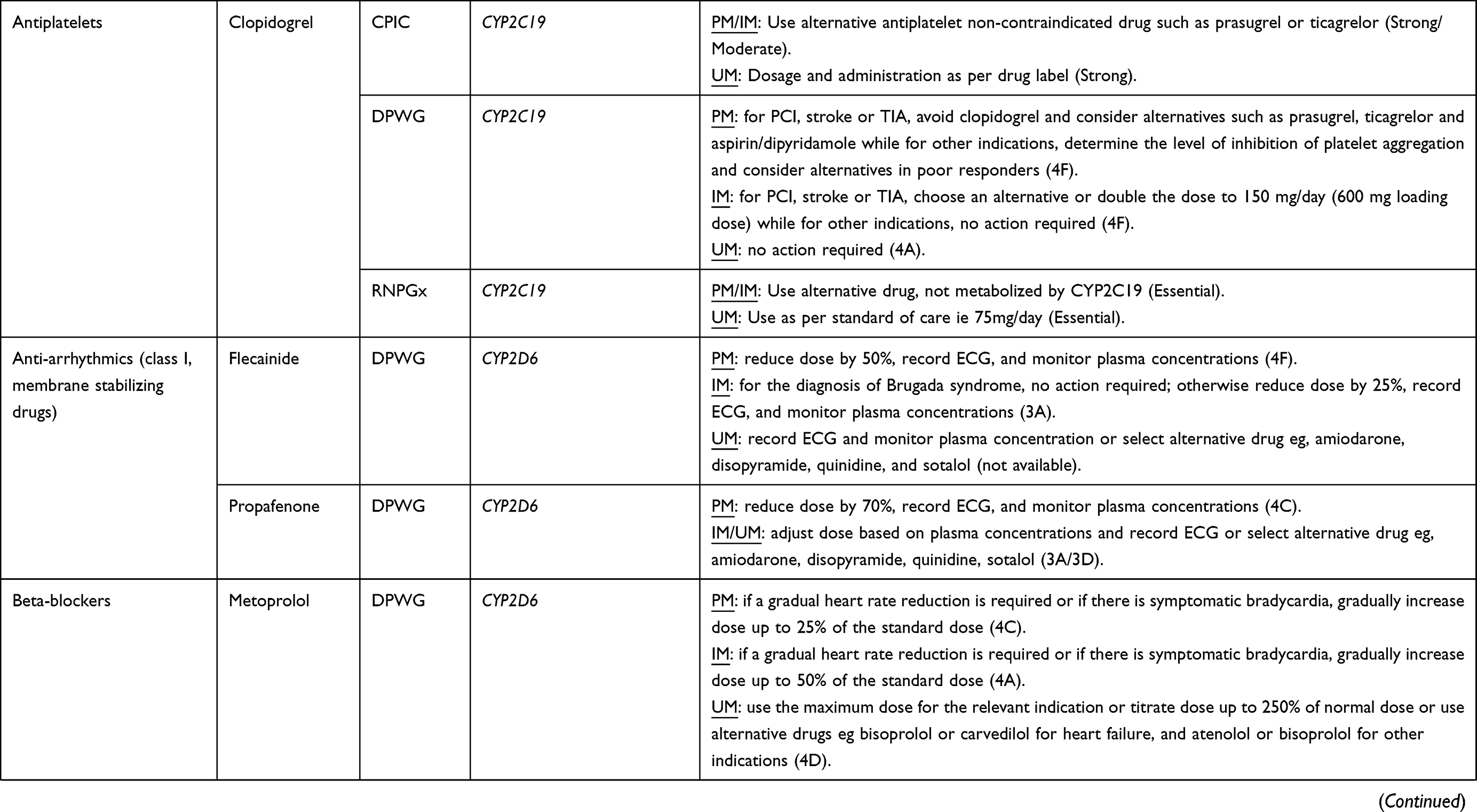 | 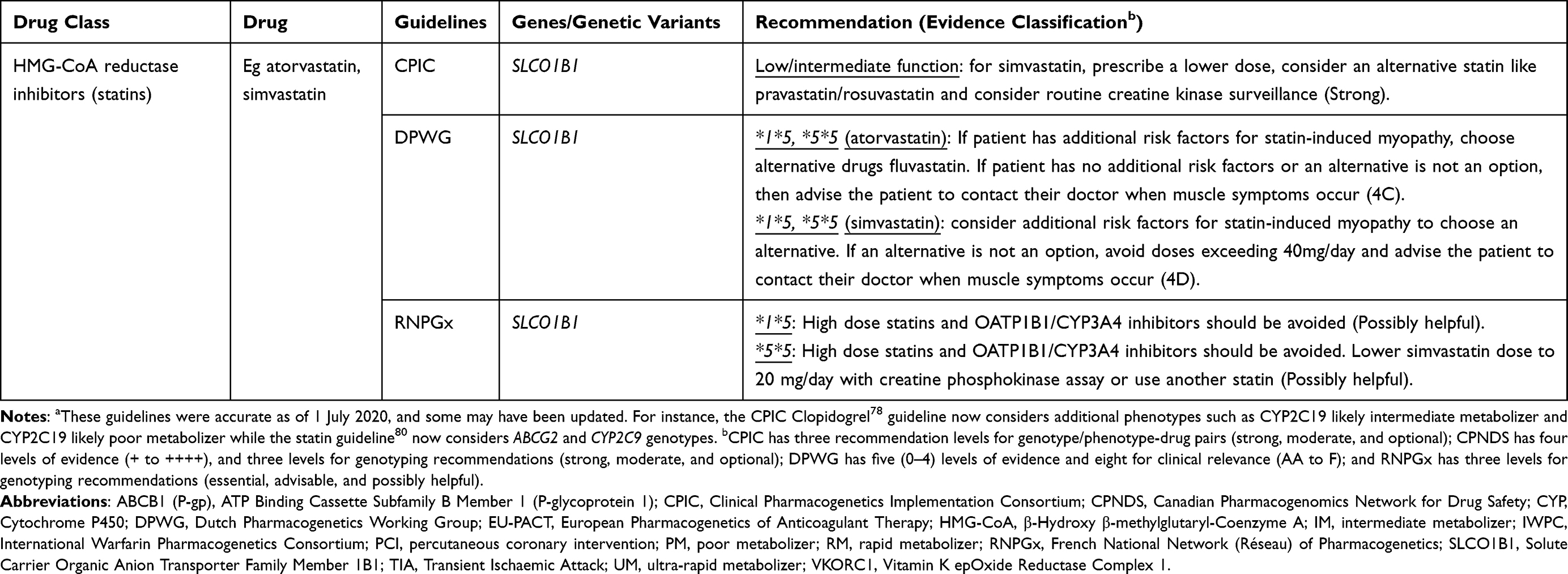 |
Table 3 A Summary of Clinical Implementation Pharmacogenetic Guidelines for Cardiovascular Drugs (Data from Abdullah-Koolmeesa et al)30 |
DGI Prevalence and Implicated Cardiovascular Medicines
In terms of the prevalence of DGIs in the general population, one retrospective US study of 1143 individuals with known CYP2D6, CYP2C19 and CYP2C9 genotypes reported that 138 (12%) of the individuals had a DGI, with DGIs (n = 155) accounting for 14.7% of all potential major or substantial interactions.52 Another US pharmacogenetic testing and drug interaction screening study of 20,534 patients reported that of 16,924 severe interactions (with a guidance of “change medication” or “consider changing medication/adjusting dose”), 24.6% were DGIs,53 while DGI prevalence in 316 (es)citalopram-treated patients from the Northern part of the Netherlands has been estimated to be 47%.81 In another personalized medicine program that recruited 705 US patients, clinically significant (moderate, major or contraindicated) DGIs were identified in 514 (72.9%) of patients and the most common actionable DGIs were for opioid, psychotropic and cardiovascular medications.63 A total of 1295 drugs were prescribed to patients with a detected DGI for that drug, with the majority being psychotropics (34%), cardiovascular medicines (21%) and analgesics (21%). The top cardiovascular medications included clopidogrel, warfarin, beta-blockers (including metoprolol, carvedilol and nebivolol), losartan and statins (simvastatin or atorvastatin) with 69.2% (36/52), 45.3% (24/53), 50.0% (73/146), 38.1% (16/42) and 26.0% (39/150) of those taking these medications having clinically significant DGIs.63 In a Dutch study of 9.7 million patients with 51.3 million drug exposures, a quarter of exposures (12.4 million, 24.1%) were risky DGIs ie had clinical significance that could result in decreased drug efficacy and/or adverse drug reactions.28 About 60%, 22% and 12.4% of the risky exposures were attributable to CYP2D6, SLCO1B1 and CYP2C19 actionable genotypes, while cardiovascular medications (eg, simvastatin and atorvastatin), gastroenterology (eg, omeprazole and pantoprazole), psychiatry/neurology and analgesic/anaesthetic medications were the most prescribed comprising 43%, 29%, 15% and 7% of the prescribed drugs, respectively. Individually, the most issued drugs were metoprolol (16% of the prescriptions), simvastatin (15%), omeprazole (14%) and pantoprazole (10%).28 Lastly, to determine the prevalence of potential DGIs in CVD patients, Turner et al conducted a UK study and of 652 post-NSTE-ACS patients, 384 (58.9%) patients had at least one DGI mediated by the genes CYP2C9 (8 patients), CYP2C19 (275 patients), CYP2D6 (19 patients), CYP3A5 (1 patient), SLCO1B1 (175 patients), and VKORC1 (7 patients).27 Fifty (7.7%) patients experienced at least one substantial interaction, defined as DGIs due to drugs with pharmacogenomic clinical recommendations and variant homozygous/compound heterozygous actionable genotypes.
Drug–Drug–Gene Interactions
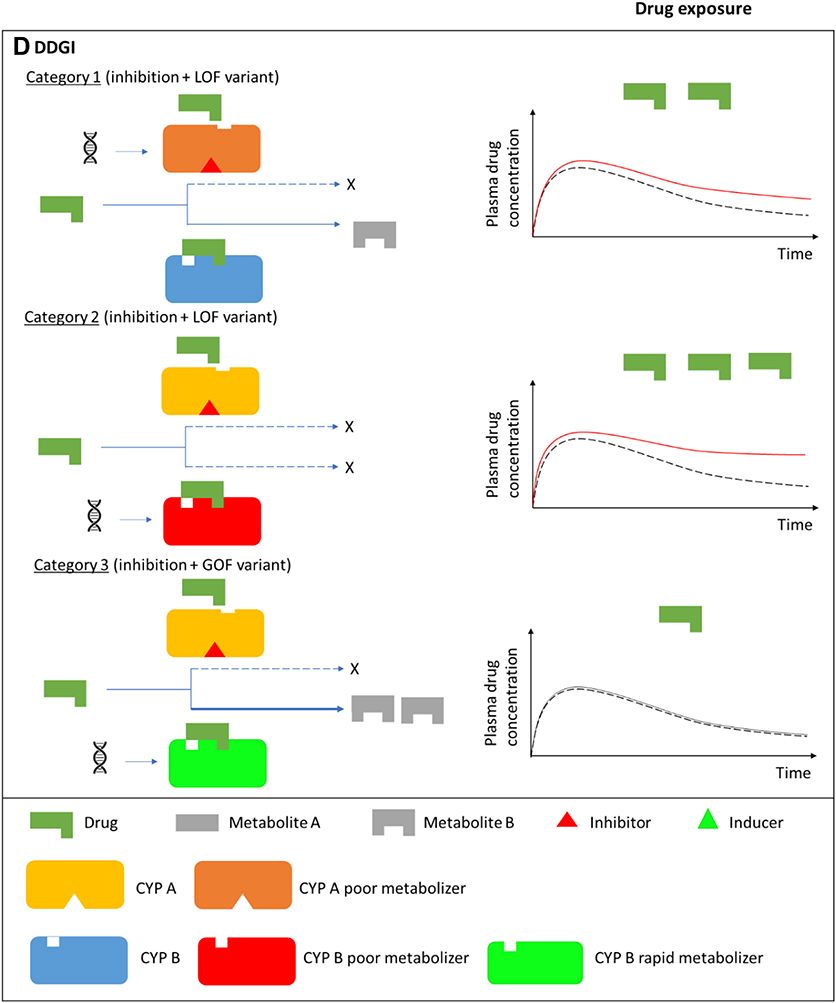
Drug–drug–gene interactions (DDGIs) occur when the individual’s genotype and another drug affect the pharmacokinetics and/or pharmacodynamics of a victim drug ie are a cumulative effect of DDIs and DGIs (Figure 1D).4,52 Like DDIs/DGIs, it is possible to divide DDGIs into pharmacokinetic and pharmacodynamic categories, with a possibility of a third category that involves both pharmacokinetic and pharmacodynamic effects. Malki and Pearson separate the DDGIs into inhibitory, induction and phenoconversion interactions,15 while Bruckmueller and Cascorbi report a slightly different classification in which DDGIs that boost clinically relevant interactions (either inhibition or induction) on the same pathway are classified under category 1, those that boost clinically relevant interactions on different pathways as category 2 and those whose constituent DDIs and DGIs lead to opposing effects as category 3.13 The latter classification is discussed further below.
Category 1 DDGIs
In this category, DDIs and DGIs that share the same pathway and have similar effects produce a DDGI (Figure 1D). For example, in 115 Swiss patients starting acenocoumarol, 35 (30.4%) had DDIs mediated by CYP2C9 inhibitors (amiodarone, clopidogrel, fluconazole, fluoxetine, fluvastatin, irbesartan, losartan, metronidazole, and pantoprazole). This resulted in an age and sex adjusted hazard ratio of 2.50 (95% confidence intervals [CI] 1.38 to 4.53) for the time to first International Normalized Ratio [INR] ≥ 4 (which represents an increased risk of over-anticoagulation). Forty-one (35.7%) patients had CYP2C9 RoF mutations (*2 and *3) with this DGI resulting in an age and sex adjusted hazard ratio of 1.68 (95% CI 1.12 to 2.45) for the time to first INR ≥ 4.82 The cumulative effect of the DDI and DGI in 14 patients who were given a CYP2C9 inhibitor and who also had the CYP2C9*2/*3 alleles tripled the over-anticoagulation risk (age and sex adjusted hazard ratio = 3.04, 95% CI 1.29 to 7.15). Of note is that the cumulative effects are not always additive (ie, DDGI effects do not always equal the sum of the independent effects of the DDI and DGI). For instance, if the maximum reduction of enzyme activity has already been reached as is the case for patients carrying two LoF variants, then a perpetrating inhibitor cannot decrease the activity any further, with the converse being true (the role of genetic variants may be limited in patients given very strong inhibitors).13 Other category 1 DDGIs that include at least one cardiovascular medicine are shown in Table 4. These DDGIs also apply to prodrugs, with the clopidogrel (CYP2C19 substrate) + proton pump inhibitors (CYP2C19 inhibitors) + CYP2C19*2/*3 (CYP2C19 LoF/RoF mutations) DDGI increasing the likelihood of clopidogrel resistance.15,94 Although metabolizing enzymes are the most-frequently studied/reported, transporter-mediated DDGIs also exist. For instance, the SLCO1B1 c.521 T>C RoF mutation significantly increases the magnitude of the interaction between pravastatin (SLCO1B1 substrate) + cyclosporine (SLCO1B1 inhibitor), with the TT genotype increasing susceptibility to the inhibitory effects of cyclosporine compared to C allele carriers.130
 |
Table 4 Examples of Drug–Drug–Gene Interactions Involving Cardiovascular medicines15,31,83 |
Category 2 DDGIs
In this category, the constituent DDIs and DGIs have similar effects but different pathways, which can happen when a substrate drug is metabolized by two or more enzymes (Figure 1D). For example, in a pharmacokinetic study, 32 healthy participants were administered the broad-spectrum triazole antifungal voriconazole that is metabolized by CYP2C19 and to a lesser extent by CYP3A4.135 Co-administration with ritonavir-boosted atazanavir that strongly inhibits CYP3A4 (a DDI) in eight CYP2C19 poor metabolizers increased the voriconazole area under the curve and maximum plasma concentration by 5.6-fold (90% CI 4.5-fold to 7.0-fold) and 4.4-fold (90% CI 3.6-fold to 5.4-fold), respectively. Examples of CVD category 2 DDGIs are shown in Table 4.
Category 3 DDGIs
In this category, constituent DDIs and DGIs lead to opposing effects, which can diminish the clinical relevance of the interaction or lead to phenoconversion in which the individual’s DDI-based or DGI-based prediction of drug metabolism/transport differs from the observed metabolism/transport (Figure 1D).4,13 An example is provided by a pharmacokinetic study of five ultra-rapid metabolizers with a CYP2D6 gene duplication or triplication.136 Due to the CYP2D6 ultra-rapid metabolizer phenotype, the five participants had subtherapeutic concentrations of nortriptyline (dosed at 25 mg twice a day for a week). In the second and third weeks, the CYP2D6 inhibitor paroxetine (10 mg or 20 mg twice a day) was co-administered, which normalized the CYP2D6 metabolic status (increase of nortriptyline plasma concentrations). Three CVD examples are provided in Table 4, with one (a phenoconversion example) being that of a patient taking clopidogrel (CYP2C19 substrate) who has the GoF polymorphism CYP2C19*17, where the administration of a proton pump inhibitor which inhibits CYP2C19 can change the metaboliser status from ultra-rapid to poor, resulting in decreased clopidogrel efficacy.15,95
DDGI Prevalence and Implicated Cardiovascular Medicines
Few studies have explored potential or actual DDGIs. Some have been mentioned above including a US study (n = 1143 individuals, of whom 137 [12.0%] had a potential DDGI, with DDGIs [n = 202] accounting for 19.2% of all potential major or substantial interactions),52 another US study (n = 20,534 patients who had 16,924 severe interactions, of which 22.4% were DDGIs),53 and a study in the Netherlands (n = 316 participants, of whom 8.5% were exposed to DDGIs).81 The Turner study that included only CVD patients reported that 106 (16.3%) patients had at least one DDGI mediated by the genes CYP2C9 (1 patient), CYP2C19 (74 patients), CYP2D6 (3 patients), and SLCO1B1 (34 patients).27 Eighty-eight (13.5%) of these patients experienced at least one substantial interaction, defined as DDGIs in which the constituent interactions acted in the same direction eg the constituent DDIs and DGIs did not lead to opposing effects as is seen with category 3 DDGIs.
The cardiovascular drugs that have been previously implicated in DDGIs are shown in Table 4 and these were mainly obtained from two systematic reviews. The first reviewed DDGI case reports and out of 34 cases, 7 (20.6%) involved at least one cardiovascular medicine.83 The second review included clinical, observational and case studies involving CYP2D6, CYP2C9, and CYP2C19-mediated drug interactions with 66 and 39 studies, respectively, reporting the impact of pharmacogenetics on DDIs and DDGIs, of which 38 (57.6%) and 5 (13.9%) included at least one cardiovascular medicine.31
Other Interactions
There are other kinds of interactions such as drug–gene–gene interactions (DGGIs) that involve the influence of more than one genetic factor on the pharmacokinetics and/or pharmacodynamics of a victim drug.4 For instance, inhibition of one of several drug metabolism pathways by a genetic polymorphism might have minimal effect due to redundancy, but the interaction can become clinically significant when the alternative pathway enzyme is also affected by a polymorphism.4 Some drugs such as warfarin have genes affecting both pharmacokinetics (eg, CYP2C9) and pharmacodynamics (eg, VKORC1) leading to a DGGI.27 These two DGGI scenarios were reported in Turner et al’s study in which 10 (1.5%) patients had at least one substantial DGGI (defined as DGGIs in which the constituent interactions were synergistic) and included six amitriptyline/CYP2D6/CYP2C19 and four warfarin/CYP2C9/VKORC1 DGGIs.27
More complex interactions can involve more than one DDI/DGI. For example, in a randomized three-way crossover study of 27 healthy subjects, the additive antiplatelet effect of cilostazol and clopidogrel was maximized in participants with both the CYP2C19 poor metabolizer and CYP3A5*3/*3 genotypes, which represented a drug–drug–gene–gene interaction (DDGGI, two drugs and two genetic factors).137 A drug–drug–drug–gene interaction (DDDGI) arises when clopidogrel (CYP2C19/CYP3A4 substrate) is prescribed with proton pump inhibitors (CYP2C19 inhibitors) and calcium channel blockers (CYP3A4 inhibitors) in CYP2C19*2 carriers, which increases the risk of adverse cardiovascular events.138
Interactions can also involve disease status (most commonly kidney or hepatic impairment), with a systematic review of case reports reporting 25 cases of drug-drug-disease interactions of which 12 (48%) interactions included at least one cardiovascular medicine.83 In this review, four cases of drug-drug-gene-disease interactions were reported with one (dextromethorphan 30 mg + metoprolol 40 mg/day + CYP2D6*1/*10 + chronic renal failure leading to myoclonus) including a cardiovascular medicine.83,139
As stated earlier, non-genetic factors such as age and sex can alter the capacity to metabolize/transport drugs, and these could lead to more complex interactions. For instance, in a large-scale analysis of Brazilian electronic health records (1,025,754 distinct drug pair co-administrations), women had a 60% increased risk of DDIs as compared to men, and a 90% increased risk when only DDIs known to lead to major adverse drug reactions were considered. DDI risk also increased substantially with age.140
Challenges in Clinical Practice
Drug Information Compendia, Interaction Databases and Clinical Implementation Guidelines
Several organisations have used existing literature to develop interaction databases and other resources that help to predict and/or detect DDIs (eg, the British National Formulary, Micromedex, Stockley’s Interactions Checker, etc.) and DGIs (eg, PharmGKB annotations, the FDA Table of Pharmacogenetic Associations, the Drug-Gene Interaction database,141 the CPIC, CPNDS, DPWG and RNPGx clinical implementation guidelines, etc.).15,30 However, most have considered DDIs or DGIs separately, which might lead to the underestimation of the impact or clinical relevance of a DDI or DGI when it is boosted by another interaction, as in the case of category 1 and 2 DDGIs, or the prediction of a wrong phenotype as can happen with DDGIs that lead to phenoconversion. Missing or underestimating clinically relevant interactions and phenotype misclassification can also lead to incorrect study findings including the failure to replicate previous associations, poor translation of study findings to the clinic, potentially damaging clinical recommendations and/or implementation guidelines that contradict each other, which compromises the potential to advance personalized medicine.4,27
As stated earlier, DDGIs are a subset of DDIs and DGIs, which means that contradictory DDI/DGI information will negatively impact DDGI evidence. Unfortunately, there are several discrepancies in the listing and clinical severity ratings between DDI142–147 and DGI information sources. For example, out of four DDI compendia (Drug Interaction Facts, Drug Interactions: Analysis and Management, Evaluations of Drug Interactions, and the MicroMedex DRUG-REAX program), only 9 (2.2%) of 406 major DDIs were listed in all four compendia.142 Another study compared three major online DDI information resources (the British National Formulary [BNF], Thesaurus, and Micromedex), which, respectively, contained 51,481, 38,037 and 65,446 drug pairs involved in DDIs.147 Only 6970 (13.5% of BNF, 18.3% of Thesaurus and 10.7% of Micromedex) DDIs were common across all three DDI information sources. Of the above DDIs, 12,644 (24.6%), 15,728 (41.3%) and 47,443 (72.5%) had critical severity ratings for the BNF (“severe” rating), Thesaurus (“Contraindicated” and “Not recommended” ratings) and Micromedex (“Contraindicated” and “Major” ratings), respectively. For DGIs, out of a total of 202 drugs for which there is pharmacogenetic guidance from the European Medicines Agency (EMA), the US Food and Drug Administration (FDA), CPIC and DPWG, only one (0.5%) of the drugs (abacavir) is present in all guidance.148 The CPIC and DPWG are consistent in guidance for the majority of reported actionable DGIs; however, their ranking criteria and methodology are different.28,149 For example, DPWG rates the metoprolol:CYP2D6 drug–gene combination as level 4 (the highest rank), whereas CPIC (https://cpicpgx.org/genes-drugs/) rates this drug–gene combination as B/C (“Prescribing actionability based on genetics is not clear without further evidence review”). Some recommendations like for warfarin dosing (Table 3) can result in differences in dosing recommendations of ≥20%.28,149
Electronic Decision Support Tools
Due to the high number of possible DDIs, the main strategy for their detection in a clinical setting has been the use of electronic decision support tools with DDIs presented as interruptive alerts.68,150 However, too many alerts coupled with discrepancies between predicted and observed DDIs can lead to alert fatigue,4,68 which is likely to be exaggerated when DGIs, DDGIs and other interactions are added to these electronic decision support tools. To deal with alert fatigue, clinicians override these alerts or the number of DDI alerts may be reduced by eliminating minor, moderate and/or low probability DDIs, which carries risks that clinically relevant DDIs may be missed, leading to serious adverse events.68,150–152
To incorporate DGIs into electronic decision support tools, several institutions have established pharmacogenomics initiatives. Samwald et al153 list examples of USA institutions/health systems that have incorporated pharmacogenetic testing into practice. They include Vanderbilt University Medical Center,154 St. Jude Children’s Research Hospital,155,156 University of Florida and Shands Hospital,157 University of Illinois at Chicago,158 Mayo Clinic,159 University of Maryland,160 and Mount Sinai Medical Center.159 Despite these examples, there is a slow pace in translating pharmacogenomics into the clinic, which has been due to challenges that the Royal College of Physicians and British Pharmacological Society have described as being related to the design of the pharmacogenomics clinical service, standardizing the consent process, genotyping and laboratory considerations, clinical decision support, funding, prescriber knowledge and education, patient engagement, perspectives and managing expectations, clinical governance, and research.161
Real-World Evidence and “Big Data”
Recruiting and monitoring the extremely large sample sizes needed to evaluate the prevalence and clinical impact of the large number of drug–drug, drug–gene and drug–drug–gene combinations is very difficult.15 Fortunately, increasing advances in “big data” (including the availability of adverse drug reaction databases, online literature repositories and millions of electronic primary healthcare records that are linked to genetic data eg the UK biobank) as well as novel technology (including data mining strategies that can identify DDI/DGI-related adverse drug reactions), have made it possible to evaluate the real-world impact of these interactions.15,18,162 However, the use of big-data raises additional challenges including the need for additional novel computational techniques, validated phenotyping algorithms and technical/clinical expertise. Additionally, routinely collected health data, which is present in most electronic healthcare records, such as the Clinical Practice Research Datalink is obtained for administrative/clinical purposes without pre-specified research goals, and will have several biases (including selection bias, confounding, information bias, missing data and misclassification) that will need to be accounted for.163,164
Clinically Relevant Endpoints
Most current evidence comes from extrapolations from case reports and in vitro studies,165 and uses pharmacokinetic outcomes such the area under the concentration–time curve or clearance (Table 4). These may not necessarily translate into adverse clinical outcomes and is one of the reasons why the number of potential DDIs far outweighs the number of clinically relevant adverse reactions.18,19 Clinically relevant endpoints that can be used to quantify actual DDI, DGI or DDGI-related harm are well known. For instance, McDonough provides a detailed protocol for designing a cardiovascular pharmacogenomics study and gives examples of both dichotomous and continuous efficacy (eg, blood pressure response to thiazide diuretics in hypertensive patients; [prevention of adverse] cardiovascular outcomes after clopidogrel treatment in patients undergoing percutaneous coronary intervention) and safety/toxicity (eg, bleeding events after warfarin treatment in atrial fibrillation patients; myopathy after simvastatin treatment in patients with hypercholesterolemia) outcomes that can be used.166 However, whereas collecting these endpoints is easy in well-designed randomized controlled trials, it might not be possible with lower quality, often incomplete, real-world data such as the electronic health care records discussed above. For DDIs/DGIs/DDGIs, clinical endpoints may require expert adjudication (eg, causality-assessed adverse drug reactions), which further makes it harder to use them on a large scale.
Pre-Emptive Genotyping
It is important to genotype multiple genetic factors in order to be able to capture all clinically relevant gene-based interactions, especially in polypharmacy patients, which makes it necessary to use a panel approach rather than test for individual variants.63 As highlighted in the introduction, there is growing evidence that the majority of patients (some studies reporting more than 99% of the population) have at least one clinically actionable genotype.4,21–29 This makes pre-emptive genotyping (screening for pharmacogenetic variants before a pharmacological intervention, as opposed to reactive methods in which screening occurs when a high-risk medication is prescribed or after unexplained adverse effects occur) using a test panel that includes all actionable variants a way to maximize clinical impact.167 However, as pointed out by the Royal College of Physicians and the British Pharmacological Society, genotyping considerations and determining which variants to include on a panel remain a challenge to clinical implementation.161 Additionally, and although a few small-sized randomized studies have indicated that the clinical utility of pharmacogenetic panel testing is promising, the cost-effectiveness of pre-emptive genotyping needs further study.167,168
Ethnic Diversity
Actionability of genotypes is race/ancestry-dependent with minor allele frequencies of a genetic variant determining its actionability in a given population/race.74 However, most of the existing pharmacogenetic evidence is applicable to Whites and Asians, which means existing databases or guidelines may not be useful for other races.74,169 As of 8 July 2021, 86% of the genome-wide association studies had been conducted in individuals of European ancestry,170,171 which demonstrates a lack of ethnic diversity in genomic evidence. Based on the CPIC guidelines, warfarin, clopidogrel and statins are the three cardiovascular medicine drug classes ready for clinical implementation; however, there is also a lack of diversity in these medicine fields. For example, the CPIC,172 CPNDS,173 and DPWG174 clinical implementation guidelines for warfarin DGIs rely on two algorithms (Gage and International Warfarin Pharmacogenetics Consortium)175,176 that mostly rely on genetic variants discovered in Whites (rs1799853 [CYP2C9*2], rs1057910 [CYP2C9*3] and rs9923231 [VKORC1 −1639G>A]) and miss out important variants such as rs7900194 [CYP2C9*8], rs28371685 [CYP2C9*11], and CYP2C rs12777823 that are likely to be more relevant to Blacks and some Hispanic populations.169 Like for warfarin, where the key genetic variants have varying MAFs (CYP2C9*2 African = 0.01, American = 0.10, East Asian ~ 0.00, European = 0.12, South Asian = 0.04; CYP2C9*3 African ~ 0.00, American = 0.04, East Asian = 0.03, European = 0.07, South Asian = 0.11; CYP2C9*8 African = 0.05, Other ~ 0.00; and CYP2C9*11 African = 0.02, Other ~ 0.00; VKORC1 −1639G>A African = 0.05, American = 0.41, East Asian = 0.89, European = 0.39, South Asian = 0.15), the MAFs for the key genetic variants for clopidogrel (rs4244285 [CYP2C19*2] African = 0.17, American = 0.11, East Asian = 0.31, European = 0.15, South Asian = 0.36; rs4986893 [CYP2C19*3] East Asian = 0.06, South Asian = 0.01, Other ~ 0.00; and rs12248560 [CYP2C19*17] African = 0.24, American = 0.12, East Asian = 0.02, European = 0.22, South Asian = 0.14) and statins (rs4149056 [SLCO1B1*5] African = 0.01, American = 0.13, East Asian = 0.12, European = 0.16, South Asian = 0.04) also differ,177 with clinical implications in terms of translating evidence from one population to another. As DGIs are a component of DDGIs, non-representative DGI evidence directly impacts the relevance of existing/future DDGIs in underserved populations.
Recommendations
Managing drug- and gene-based interactions involves managing both existing and future interactions. For existing DDIs, an international panel recommends treatment modification (reduce dose, discontinue and/or substitute drug, add protective drug) for about three-quarters of DDIs with drug therapy monitoring recommended for the rest.71 For minimizing [future] DDIs, Kennedy, Brewer, and Williams suggest various steps that are outlined in Box 1.18 These steps/recommendations can be extended to DGIs, DDGIs and other complex interactions and should supplement the proposed solutions (Table 5) to the clinical practice challenges mentioned above.
 |
Box 1 Minimizing Drug–Drug Interactions (DDIs)18 |
 |
Table 5 Proposed Solutions to Clinical Practice Challenges |
Polypills, fixed-dose combinations of cardiovascular medications, are increasingly being recommended for preventing cardiovascular disease as they can improve patient adherence, reduce prescription barriers leading to greater use of guideline-concordant medications, can be availed in numerous formulations to aid dosing flexibility. They have been demonstrated to be safe and effective in reducing the incidence of cardiovascular events in several clinical settings.188 In the context of this review, the recommendations for polypills are no different to the individual drugs being given separately in that drugs with clinically relevant DDIs should not be included in the same (future) polypills.
Conclusion
Using evidence from pharmacogenomics, this review has illustrated how DDIs and DGIs interact to produce DDGIs, including in the cardiovascular medicine field. Current DDGI evidence is scarce and sometimes contradictory, and electronic decision support tools do not incorporate DDGIs and their management. In addition to direct patient harm when drugs that should be contraindicated due to DDGIs are administered, harm can also result when insufficient evidence on interactions means that a prescriber is less inclined to use a safe and efficacious medicine leading to sub-optimal patient outcomes.165 To improve translation to the clinic, we have provided several recommendations including, expanding the scope of drug information compendia, interaction databases and clinical implementation guidelines (to include clinically relevant DDGIs and other complex interactions) and working towards their harmonization; better use of electronic decision support tools; using big data and novel computational techniques; preemptive genotyping; ensuring ethnic diversity; and upskilling of clinicians in pharmacogenomics and personalized medicine. The cost-effectiveness of incorporating DDGIs should be evaluated, similar to the work done by the Ubiquitous Pharmacogenomics Consortium in the Pharmacogenomic testing for prevention of Adverse drug Reactions (PREPARE) study.189–191 Better evidence (clinical trials, real-world evaluation of drug- and gene-based interactions) and better electronic decision support tools should help reduce DDIs, DGIs, DDGIs and associated adverse drug events, which should improve drug-related outcomes in cardiovascular disease patients, who often experience multimorbidity and polypharmacy. All populations should be well represented in the evidence base to ensure health equity.
Funding
This work was supported by the Medical Research Council [MR/V033867/1; Multimorbidity Mechanism and Therapeutics Research Collaborative].
Disclosure
MP has received partnership funding for the following: MRC Clinical Pharmacology Training Scheme (co-funded by MRC and Roche, UCB, Eli Lilly and Novartis); and a PhD studentship jointly funded by EPSRC and AstraZeneca. He also has unrestricted educational grant support for the UK Pharmacogenetics and Stratified Medicine Network from Bristol-Myers Squibb. He has developed an HLA genotyping panel with MC Diagnostics, but does not benefit financially from this. He is part of the IMI Consortium ARDAT (www.ardat.org). None of the funding MP received is related to the current paper. IGA reports no conflicts of interest in this work.
References
1. Ahmad FB, Anderson RN. The leading causes of death in the US for 2020. JAMA. 2021;325(18):1829–1830.
2. GBD 2019 Diseases and Injuries Collaborators. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet. 2020;396(10258):1204–1222.
3. Christensen K, Doblhammer G, Rau R, Vaupel JW. Ageing populations: the challenges ahead. Lancet. 2009;374(9696):1196–1208.
4. Hahn M, Roll SC. The influence of pharmacogenetics on the clinical relevance of pharmacokinetic drug-drug interactions: drug-gene, drug-gene-gene and drug-drug-gene interactions. Pharmaceuticals. 2021;14(5):32.
5. The Academy of Medical Sciences. Multimorbidity: a priority for global health research; 2018.
6. Tran J, Norton R, Conrad N, et al. Patterns and temporal trends of comorbidity among adult patients with incident cardiovascular disease in the UK between 2000 and 2014: a population-based cohort study. PLoS Med. 2018;15(3):e1002513.
7. Liu G, Xue Y, Liu Y, Wang S, Geng Q. Multimorbidity in cardiovascular disease and association with life satisfaction: a Chinese national cross-sectional study. BMJ Open. 2020;10(12):e042950.
8. Thienemann F, Ntusi NAB, Battegay E, Mueller BU, Cheetham M. Multimorbidity and cardiovascular disease: a perspective on low- and middle-income countries. Cardiovasc Diagn Ther. 2020;10(2):376–385.
9. Masnoon N, Shakib S, Kalisch-Ellett L, Caughey GE. What is polypharmacy? A systematic review of definitions. BMC Geriatr. 2017;17(1):230.
10. Parekh N, Ali K, Stevenson JM, et al. Incidence and cost of medication harm in older adults following hospital discharge: a multicentre prospective study in the UK. Br J Clin Pharmacol. 2018;84(8):1789–1797.
11. Cheung JTK, Yu R, Woo J. Is polypharmacy beneficial or detrimental for older adults with cardiometabolic multimorbidity? Pooled analysis of studies from Hong Kong and Europe. Fam Pract. 2020;37(6):793–800.
12. Osanlou R, Walker L, Hughes DA, Burnside G, Pirmohamed M. Adverse drug reactions, multimorbidity and polypharmacy: a prospective analysis of 1 month of medical admissions. BMJ Open. 2022;12(7):e055551.
13. Bruckmueller H, Cascorbi I. Drug-drug-gene interactions: a call for clinical consideration. Clin Pharmacol Ther. 2021;110(3):549–551.
14. Abolhassani N, Vollenweider P, Waeber G, Marques-Vidal P. Ten-year trend in polypharmacy in the Lausanne population. J Patient Saf. 2021;17(4):e269–e273.
15. Malki MA, Pearson ER. Drug-drug-gene interactions and adverse drug reactions. Pharmacogenomics J. 2020;20(3):355–366.
16. Howard RL, Avery AJ, Slavenburg S, et al. Which drugs cause preventable admissions to hospital? A systematic review. Br J Clin Pharmacol. 2007;63(2):136–147.
17. Al Hamid A, Ghaleb M, Aljadhey H, Aslanpour Z. A systematic review of hospitalization resulting from medicine-related problems in adult patients. Br J Clin Pharmacol. 2014;78(2):202–217.
18. Kennedy C, Brewer L, Williams D. Drug interactions. Medicine. 2020;48(7):450–455.
19. Magro L, Moretti U, Leone R. Epidemiology and characteristics of adverse drug reactions caused by drug-drug interactions. Expert Opin Drug Saf. 2012;11(1):83–94.
20. Nebert DW. Pharmacogenetics and pharmacogenomics: why is this relevant to the clinical geneticist? Clin Genet. 1999;56(4):247–258.
21. McInnes G, Lavertu A, Sangkuhl K, Klein TE, Whirl-Carrillo M, Altman RB. Pharmacogenetics at scale: an analysis of the UK Biobank. Clin Pharmacol Ther. 2021;109(6):1528–1537.
22. Reisberg S, Krebs K, Lepamets M, et al. Translating genotype data of 44,000 biobank participants into clinical pharmacogenetic recommendations: challenges and solutions. Genet Med. 2019;21(6):1345–1354.
23. Van Driest SL, Shi Y, Bowton EA, et al. Clinically actionable genotypes among 10,000 patients with preemptive pharmacogenomic testing. Clin Pharmacol Ther. 2014;95(4):423–431.
24. Jithesh PV, Abuhaliqa M, Syed N, et al. A population study of clinically actionable genetic variation affecting drug response from the Middle East. NPJ Genom Med. 2022;7(1):10.
25. Mostafa S, Kirkpatrick CMJ, Byron K, Sheffield L. An analysis of allele, genotype and phenotype frequencies, actionable pharmacogenomic (PGx) variants and phenoconversion in 5408 Australian patients genotyped for CYP2D6, CYP2C19, CYP2C9 and VKORC1 genes. J Neural Transm. 2019;126(1):5–18.
26. Ji Y, Skierka JM, Blommel JH, et al. Preemptive pharmacogenomic testing for precision medicine: a comprehensive analysis of five actionable pharmacogenomic genes using next-generation DNA sequencing and a customized CYP2D6 genotyping cascade. J Mol Diagn. 2016;18(3):438–445.
27. Turner RM, de Koning EM, Fontana V, Thompson A, Pirmohamed M. Multimorbidity, polypharmacy, and drug-drug-gene interactions following a non-ST elevation acute coronary syndrome: analysis of a multicentre observational study. BMC Med. 2020;18(1):367.
28. Alshabeeb MA, Deneer VHM, Khan A, Asselbergs FW. Use of pharmacogenetic drugs by the Dutch population. Front Genet. 2019;10:567.
29. Cohn I, Paton TA, Marshall CR, et al. Genome sequencing as a platform for pharmacogenetic genotyping: a pediatric cohort study. NPJ Genom Med. 2017;2:19.
30. Abdullah-Koolmees H, van Keulen AM, Nijenhuis M, Deneer VHM. Pharmacogenetics guidelines: overview and comparison of the DPWG, CPIC, CPNDS, and RNPGx guidelines. Front Pharmacol. 2020;11:595219.
31. Bahar MA, Setiawan D, Hak E, Wilffert B. Pharmacogenetics of drug-drug interaction and drug-drug-gene interaction: a systematic review on CYP2C9, CYP2C19 and CYP2D6. Pharmacogenomics. 2017;18(7):701–739.
32. World Health Organisation. Cardiovascular disease. World Health Organisation; 2021. Available from: https://www.who.int/cardiovascular_diseases/about_cvd/en/.
33. Moran A, Forouzanfar M, Sampson U, Chugh S, Feigin V, Mensah G. The epidemiology of cardiovascular diseases in sub-Saharan Africa: the global burden of diseases, injuries and risk factors 2010 study. Prog Cardiovasc Dis. 2013;56(3):234–239.
34. Koda-Kimble MA, Young LY, Alldredge BK, et al. Applied Therapeutics: The Clinical Use of Drugs.
35. Cappuccio FP, Miller MA. Cardiovascular disease and hypertension in sub-Saharan Africa: burden, risk and interventions. Intern Emerg Med. 2016;11(3):299–305.
36. Kumar P, Clark M. Clinical Medicine. Elsevier; 2005.
37. Joint Formulary Committee. British National Formulary 80 September 2020 – March 2021; London: BMJ Group and Pharmaceutical Press; 2020: 80
38. Audi S, Burrage DR, Lonsdale DO, et al. The ‘top 100’ drugs and classes in England: an updated ‘starter formulary’ for trainee prescribers. Br J Clin Pharmacol. 2018;84(11):2562–2571.
39. Hales C, Servais J, Martin C, Kohen D. Prescription drug use among adults aged 40–79 in the United States and Canada. NCHS data brief, no 347; 2019. Available from: https://www.cdc.gov/nchs/data/databriefs/db347-h.pdf.
40. ClinCalc DrugStats Database. The Top 300 for 2019; 2021. Available from: https://clincalc.com/DrugStats/Top300Drugs.aspx.
41. Zhang X, Han Y, Huang W, Jin M, Gao Z. The influence of the gut microbiota on the bioavailability of oral drugs. Acta Pharm Sin B. 2021;11(7):1789–1812.
42. Sands CD, Chan ES, Welty TE. Revisiting the significance of warfarin protein-binding displacement interactions. Ann Pharmacother. 2002;36(10):1642–1644.
43. Mullokandov E, Ahn J, Szalkiewicz A, Babayeva M. Protein binding drug-drug interaction between warfarin and tizoxanide in human plasma. Austin J Pharmacol Ther. 2014;2(7):1–3.
44. Food and Drug Administration. Drug development and drug interactions: table of substrates, inhibitors and inducers; 2017. Available from: https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table-substrates-inhibitors-and-inducers.
45. Flockhart DA. Drug interactions: cytochrome P450 drug interaction table. Indiana University School of Medicine; 2007. Available from: https://drug-interactions.medicine.iu.edu/MainTable.aspx.
46. Wessler JD, Grip LT, Mendell J, Giugliano RP. The P-glycoprotein transport system and cardiovascular drugs. J Am Coll Cardiol. 2013;61(25):2495–2502.
47. BCGuidelines.ca. Use of non-vitamin K antagonist oral anticoagulants (NOAC) in non-valvular atrial fibrillation. Appendix A: potential NOAC drug interaction; 2015. Available from: https://www2.gov.bc.ca/assets/gov/health/practitioner-pro/bc-guidelines/anticoag_2015november_full.pdf.
48. Modak AS, Klyarytska I, Kriviy V, Tsapyak T, Rabotyagova Y. The effect of proton pump inhibitors on the CYP2C19 enzyme activity evaluated by the pantoprazole-(13)C breath test in GERD patients: clinical relevance for personalized medicine. J Breath Res. 2016;10(4):046017.
49. Cheng JW, Frishman WH, Aronow WS. Updates on cytochrome p450-mediated cardiovascular drug interactions. Dis Mon. 2010;56(3):163–179.
50. Delaune KP, Alsayouri K. Physiology, Noncompetitive Inhibitor. In: StatPearls. Treasure Island (FL); 2022.
51. Cascorbi I. Drug interactions--principles, examples and clinical consequences. Dtsch Arztebl Int. 2012;109(33–34):546–555; quiz 556.
52. Verbeurgt P, Mamiya T, Oesterheld J. How common are drug and gene interactions? Prevalence in a sample of 1143 patients with CYP2C9, CYP2C19 and CYP2D6 genotyping. Pharmacogenomics. 2014;15(5):655–665.
53. Hocum BT, White JR
54. Leone R, Magro L, Moretti U, et al. Identifying adverse drug reactions associated with drug-drug interactions: data mining of a spontaneous reporting database in Italy. Drug Saf. 2010;33(8):667–675.
55. Letinier L, Cossin S, Mansiaux Y, et al. Risk of drug-drug interactions in out-hospital drug dispensings in France: results from the DRUG-drug interaction prevalence study. Front Pharmacol. 2019;10:265.
56. Tulner LR, Frankfort SV, Gijsen GJ, van Campen JP, Koks CH, Beijnen JH. Drug-drug interactions in a geriatric outpatient cohort: prevalence and relevance. Drugs Aging. 2008;25(4):343–355.
57. Marzolini C, Elzi L, Gibbons S, et al. Prevalence of comedications and effect of potential drug-drug interactions in the Swiss HIV Cohort Study. Antivir Ther. 2010;15(3):413–423.
58. Mino-Leon D, Galvan-Plata ME, Doubova SV, Flores-Hernandez S, Reyes-Morales H. Estudio farmacoepidemiológico de potenciales interacciones farmacológicas y sus determinantes, en pacientes hospitalizados [A pharmacoepidemiological study of potential drug interactions and their determinant factors in hospitalized patients]. Rev Invest Clin. 2011;63(2):170–178. Spanish.
59. Zerah L, Henrard S, Wilting I, et al. Prevalence of drug-drug interactions in older people before and after hospital admission: analysis from the OPERAM trial. BMC Geriatr. 2021;21(1):571.
60. Glintborg B, Andersen SE, Dalhoff K. Drug-drug interactions among recently hospitalised patients--frequent but mostly clinically insignificant. Eur J Clin Pharmacol. 2005;61(9):675–681.
61. Obreli-Neto PR, Nobili A, de Oliveira Baldoni A, et al. Adverse drug reactions caused by drug-drug interactions in elderly outpatients: a prospective cohort study. Eur J Clin Pharmacol. 2012;68(12):1667–1676.
62. Kohler GI, Bode-Boger SM, Busse R, Hoopmann M, Welte T, Boger RH. Drug-drug interactions in medical patients: effects of in-hospital treatment and relation to multiple drug use. Int J Clin Pharmacol Ther. 2000;38(11):504–513.
63. Reynolds KK, Pierce DL, Weitendorf F, Linder MW. Avoidable drug-gene conflicts and polypharmacy interactions in patients participating in a personalized medicine program. Per Med. 2017;14(3):221–233.
64. Karlgren M, Vildhede A, Norinder U, et al. Classification of inhibitors of hepatic organic anion transporting polypeptides (OATPs): influence of protein expression on drug-drug interactions. J Med Chem. 2012;55(10):4740–4763.
65. Fettah H, Moutaouakkil Y, Sefrioui MR, et al. Detection and analysis of drug-drug interactions among hospitalized cardiac patients in the Mohammed V Military Teaching Hospital in Morocco. Pan Afr Med J. 2018;29:225.
66. Murtaza G, Khan MY, Azhar S, Khan SA, Khan TM. Assessment of potential drug-drug interactions and its associated factors in the hospitalized cardiac patients. Saudi Pharm J. 2016;24(2):220–225.
67. Kovacevic M, Vezmar Kovacevic S, Miljkovic B, Radovanovic S, Stevanovic P. The prevalence and preventability of potentially relevant drug-drug interactions in patients admitted for cardiovascular diseases: a cross-sectional study. Int J Clin Pract. 2017;71(10):34.
68. Kovacevic M, Vezmar Kovacevic S, Radovanovic S, Stevanovic P, Miljkovic B. Adverse drug reactions caused by drug-drug interactions in cardiovascular disease patients: introduction of a simple prediction tool using electronic screening database items. Curr Med Res Opin. 2019;35(11):1873–1883.
69. Smithburger PL, Kane-Gill SL, Seybert AL. Drug-drug interactions in cardiac and cardiothoracic intensive care units: an analysis of patients in an academic medical centre in the US. Drug Saf. 2010;33(10):879–888.
70. The Society for Post-Acute and Long-Term Care Medicine. Top 10 particularly dangerous drug interactions in PA/LTC; 2022. Available from: https://paltc.org/top-10-particularly-dangerous-drug-interactions-paltc.
71. Anrys P, Petit AE, Thevelin S, et al. An international consensus list of potentially clinically significant drug-drug interactions in older people. J Am Med Dir Assoc. 2021;22(10):2121–2133.
72. US Food and Drug Administration. Table of pharmacogenetic associations; 2022. Available from: https://www.fda.gov/medical-devices/precision-medicine/table-pharmacogenetic-associations#resources.
73. Klomp SD, Manson ML, Guchelaar HJ, Swen JJ. Phenoconversion of cytochrome P450 metabolism: a systematic review. J Clin Med. 2020;9(9):34.
74. Davis BH, Limdi NA. Translational pharmacogenomics: discovery, evidence synthesis and delivery of race-conscious medicine. Clin Pharmacol Ther. 2021;110(4):909–925.
75. Whirl-Carrillo M, McDonagh EM, Hebert JM, et al. Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther. 2012;92(4):414–417.
76. Turner RM, Magavern EF, Pirmohamed M. Pharmacogenomics: relevance and opportunities for clinical pharmacology. Br J Clin Pharmacol. 2022. doi:10.1111/bcp.15329
77. Relling MV, Klein TE. CPIC: clinical pharmacogenetics implementation consortium of the pharmacogenomics research network. Clin Pharmacol Ther. 2011;89(3):464–467.
78. Lee CR, Luzum JA, Sangkuhl K, et al. Clinical pharmacogenetics implementation consortium guideline for CYP2C19 genotype and clopidogrel therapy: 2022 update. Clin Pharmacol Ther. 2022. doi:10.1002/cpt.2526
79. Johnson JA, Caudle KE, Gong L, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for pharmacogenetics-guided warfarin dosing: 2017 update. Clin Pharmacol Ther. 2017;102(3):397–404.
80. Cooper-DeHoff RM, Niemi M, Ramsey LB, et al. The clinical pharmacogenetics implementation consortium guideline for SLCO1B1, ABCG2, and CYP2C9 genotypes and statin-associated musculoskeletal symptoms. Clin Pharmacol Ther. 2022;111(5):1007–1021.
81. Bahar MA, Lanting P, Bos JHJ, Sijmons RH, Hak E, Wilffert B. Impact of drug-gene-interaction, drug-drug-interaction, and drug-drug-gene-interaction on (es)Citalopram Therapy: the PharmLines Initiative. J Pers Med. 2020;10(4):23.
82. Gschwind L, Rollason V, Boehlen F, et al. Impact of CYP2C9 polymorphisms on the vulnerability to pharmacokinetic drug-drug interactions during acenocoumarol treatment. Pharmacogenomics. 2013;14(7):745–753.
83. Storelli F, Samer C, Reny JL, Desmeules J, Daali Y. Complex drug-drug-gene-disease interactions involving cytochromes p450: systematic review of published case reports and clinical perspectives. Clin Pharmacokinet. 2018;57(10):1267–1293.
84. Meyer Zu Schwabedissen HE, Siegmund W, Kroemer HK, Rollnik JD. Creatine kinase elevation caused by a combination of fluvastatin and telmisartan in a patient heterozygous for the CYP2C9*3 and ABCC2-24C > T variants: a case report. BMC Res Notes. 2014;7:688.
85. Gunes A, Bilir E, Zengil H, Babaoglu MO, Bozkurt A, Yasar U. Inhibitory effect of valproic acid on cytochrome P450 2C9 activity in epilepsy patients. Basic Clin Pharmacol Toxicol. 2007;100(6):383–386.
86. Fischer TL, Pieper JA, Graff DW, et al. Evaluation of potential losartan-phenytoin drug interactions in healthy volunteers. Clin Pharmacol Ther. 2002;72(3):238–246.
87. Visser LE, van Schaik RH, van Vliet M, et al. Allelic variants of cytochrome P450 2C9 modify the interaction between nonsteroidal anti-inflammatory drugs and coumarin anticoagulants. Clin Pharmacol Ther. 2005;77(6):479–485.
88. Beinema MJ, de Jong PH, Salden HJ, van Wijnen M, van der Meer J, Brouwers JR. The influence of NSAIDs on coumarin sensitivity in patients with CYP2C9 polymorphism after total Hip replacement surgery. Mol Diagn Ther. 2007;11(2):123–128.
89. Andersson ML, Eliasson E, Lindh JD. A clinically significant interaction between warfarin and simvastatin is unique to carriers of the CYP2C9*3 allele. Pharmacogenomics. 2012;13(7):757–762.
90. Tomisti L, Del Re M, Bartalena L, et al. Effects of amiodarone, thyroid hormones and CYP2C9 and VKORC1 polymorphisms on warfarin metabolism: a review of the literature. Endocr Pract. 2013;19(6):1043–1049.
91. Malhi H, Atac B, Daly AK, Gupta S. Warfarin and celecoxib interaction in the setting of cytochrome P450 (CYP2C9) polymorphism with bleeding complication. Postgrad Med J. 2004;80(940):107–109.
92. Cheng S, Flora DR, Rettie AE, Brundage RC, Tracy TS. Pharmacokinetic modeling of warfarin capital I, Ukrainian - model-based analysis of warfarin enantiomers with a target mediated drug disposition model reveals CYP2C9 genotype-dependent drug-drug interactions of S-warfarin. Drug Metab Dispos. 2022:3:45.
93. Liu Q, Dang DS, Chen YF, Yan M, Shi GB, Zhao QC. The influence of omeprazole on platelet inhibition of clopidogrel in various CYP2C19 mutant alleles. Genet Test Mol Biomarkers. 2012;16(11):1293–1297.
94. Furuta T, Iwaki T, Umemura K. Influences of different proton pump inhibitors on the anti-platelet function of clopidogrel in relation to CYP2C19 genotypes. Br J Clin Pharmacol. 2010;70(3):383–392.
95. Depta JP, Lenzini PA, Lanfear DE, et al. Clinical outcomes associated with proton pump inhibitor use among clopidogrel-treated patients within CYP2C19 genotype groups following acute myocardial infarction. Pharmacogenomics J. 2015;15(1):20–25.
96. Ieiri I, Kimura M, Irie S, Urae A, Otsubo K, Ishizaki T. Interaction magnitude, pharmacokinetics and pharmacodynamics of ticlopidine in relation to CYP2C19 genotypic status. Pharmacogenet Genomics. 2005;15(12):851–859.
97. Chen BL, Chen Y, Tu JH, et al. Clopidogrel inhibits CYP2C19-dependent hydroxylation of omeprazole related to CYP2C19 genetic polymorphisms. J Clin Pharmacol. 2009;49(5):574–581.
98. Hata M, Shiono M, Akiyama K, et al. Incidence of drug interaction when using proton pump inhibitor and warfarin according to cytochrome P450 2C19 (CYP2C19) genotype in Japanese. Thorac Cardiovasc Surg. 2015;63(1):45–50.
99. Uno T, Sugimoto K, Sugawara K, Tateishi T. The role of cytochrome P2C19 in R-warfarin pharmacokinetics and its interaction with omeprazole. Ther Drug Monit. 2008;30(3):276–281.
100. Desmeules JA, Oestreicher MK, Piguet V, Allaz AF, Dayer P. Contribution of cytochrome P-4502D6 phenotype to the neuromodulatory effects of dextromethorphan. J Pharmacol Exp Ther. 1999;288(2):607–612.
101. Pope LE, Khalil MH, Berg JE, Stiles M, Yakatan GJ, Sellers EM. Pharmacokinetics of dextromethorphan after single or multiple dosing in combination with quinidine in extensive and poor metabolizers. J Clin Pharmacol. 2004;44(10):1132–1142.
102. Eap CB, Lessard E, Baumann P, et al. Role of CYP2D6 in the stereoselective disposition of venlafaxine in humans. Pharmacogenetics. 2003;13(1):39–47.
103. Lessard E, Yessine MA, Hamelin BA, O’Hara G, LeBlanc J, Turgeon J. Influence of CYP2D6 activity on the disposition and cardiovascular toxicity of the antidepressant agent venlafaxine in humans. Pharmacogenetics. 1999;9(4):435–443.
104. Abolfathi Z, Fiset C, Gilbert M, Moerike K, Belanger PM, Turgeon J. Role of polymorphic debrisoquin 4-hydroxylase activity in the stereoselective disposition of mexiletine in humans. J Pharmacol Exp Ther. 1993;266(3):1196–1201.
105. Turgeon J, Fiset C, Giguere R, et al. Influence of debrisoquine phenotype and of quinidine on mexiletine disposition in man. J Pharmacol Exp Ther. 1991;259(2):789–798.
106. Feifel N, Kucher K, Fuchs L, et al. Role of cytochrome P4502D6 in the metabolism of brofaromine. A new selective MAO-A inhibitor. Eur J Clin Pharmacol. 1993;45(3):265–269.
107. Muralidharan G, Hawes EM, McKay G, Korchinski ED, Midha KK. Quinidine but not quinine inhibits in man the oxidative metabolic routes of methoxyphenamine which involve debrisoquine 4-hydroxylase. Eur J Clin Pharmacol. 1991;41(5):471–474.
108. Funck-Brentano C, Turgeon J, Woosley RL, Roden DM. Effect of low dose quinidine on encainide pharmacokinetics and pharmacodynamics. Influence of genetic polymorphism. J Pharmacol Exp Ther. 1989;249(1):134–142.
109. Lessard E, Hamelin BA, Labbe L, O’Hara G, Belanger PM, Turgeon J. Involvement of CYP2D6 activity in the N-oxidation of procainamide in man. Pharmacogenetics. 1999;9(6):683–696.
110. Birgersdotter UM, Wong W, Turgeon J, Roden DM. Stereoselective genetically-determined interaction between chronic flecainide and quinidine in patients with arrhythmias. Br J Clin Pharmacol. 1992;33(3):275–280.
111. Morike KE, Roden DM. Quinidine-enhanced beta-blockade during treatment with propafenone in extensive metabolizer human subjects. Clin Pharmacol Ther. 1994;55(1):28–34.
112. Funck-Brentano C, Kroemer HK, Pavlou H, Woosley RL, Roden DM. Genetically-determined interaction between propafenone and low dose quinidine: role of active metabolites in modulating net drug effect. Br J Clin Pharmacol. 1989;27(4):435–444.
113. Lim KS, Cho JY, Jang IJ, et al. Pharmacokinetic interaction of flecainide and paroxetine in relation to the CYP2D6*10 allele in healthy Korean subjects. Br J Clin Pharmacol. 2008;66(5):660–666.
114. Lim KS, Jang IJ, Kim BH, et al. Changes in the QTc interval after administration of flecainide acetate, with and without coadministered paroxetine, in relation to cytochrome P450 2D6 genotype: data from an open-label, two-period, single-sequence crossover study in healthy Korean male subjects. Clin Ther. 2010;32(4):659–666.
115. Funck-Brentano C, Becquemont L, Kroemer HK, et al. Variable disposition kinetics and electrocardiographic effects of flecainide during repeated dosing in humans: contribution of genetic factors, dose-dependent clearance, and interaction with amiodarone. Clin Pharmacol Ther. 1994;55(3):256–269.
116. Labbe L, O’Hara G, Lefebvre M, et al. Pharmacokinetic and pharmacodynamic interaction between mexiletine and propafenone in human beings. Clin Pharmacol Ther. 2000;68(1):44–57.
117. Ujhelyi MR, O’Rangers EA, Fan C, Kluger J, Pharand C, Chow MS. The pharmacokinetic and pharmacodynamic interaction between propafenone and lidocaine. Clin Pharmacol Ther. 1993;53(1):38–48.
118. Sharma A, Pibarot P, Pilote S, et al. Modulation of metoprolol pharmacokinetics and hemodynamics by diphenhydramine coadministration during exercise testing in healthy premenopausal women. J Pharmacol Exp Ther. 2005;313(3):1172–1181.
119. Sharma A, Pibarot P, Pilote S, et al. Toward optimal treatment in women: the effect of sex on metoprolol-diphenhydramine interaction. J Clin Pharmacol. 2010;50(2):214–225.
120. Hamelin BA, Bouayad A, Methot J, et al. Significant interaction between the nonprescription antihistamine diphenhydramine and the CYP2D6 substrate metoprolol in healthy men with high or low CYP2D6 activity. Clin Pharmacol Ther. 2000;67(5):466–477.
121. Damy T, Pousset F, Caplain H, Hulot JS, Lechat P. Pharmacokinetic and pharmacodynamic interactions between metoprolol and dronedarone in extensive and poor CYP2D6 metabolizers healthy subjects. Fundam Clin Pharmacol. 2004;18(1):113–123.
122. Werner D, Wuttke H, Fromm MF, et al. Effect of amiodarone on the plasma levels of metoprolol. Am J Cardiol. 2004;94(10):1319–1321.
123. Werner U, Werner D, Rau T, Fromm MF, Hinz B, Brune K. Celecoxib inhibits metabolism of cytochrome P450 2D6 substrate metoprolol in humans. Clin Pharmacol Ther. 2003;74(2):130–137.
124. Bebawi E, Jouni SS, Tessier AA, Frenette AJ, Brindamour D, Dore M. A metoprolol-terbinafine combination induced bradycardia. Eur J Drug Metab Pharmacokinet. 2015;40(3):295–299.
125. Duricova J, Perinova I, Jurckova N, Kacirova I, Grundmann M. Clinically important interaction between metoprolol and propafenone. Can Fam Physician. 2013;59(4):373–375.
126. Wang Y, Zhou L, Dutreix C, et al. Effects of imatinib (Glivec) on the pharmacokinetics of metoprolol, a CYP2D6 substrate, in Chinese patients with chronic myelogenous leukaemia. Br J Clin Pharmacol. 2008;65(6):885–892.
127. Somer M, Kallio J, Pesonen U, Pyykko K, Huupponen R, Scheinin M. Influence of hydroxychloroquine on the bioavailability of oral metoprolol. Br J Clin Pharmacol. 2000;49(6):549–554.
128. Yang WH, Zeng ZS, Ren XW, et al. Simvastatin-induced myopathy with concomitant use of cyclosporine: case report. Int J Clin Pharmacol Ther. 2011;49(12):772–777.
129. Hu M, Mak VW, Tomlinson B. Simvastatin-induced myopathy, the role of interaction with diltiazem and genetic predisposition. J Clin Pharm Ther. 2011;36(3):419–425.
130. Aquilante C, Page R, Brieke A, et al. SLCO1B1 Genotype Influences the Drug-Drug Interaction between Cyclosporine and Pravastatin. J Heart Lung Transplant. 2013;32(4S):S292–S293.
131. Marusic S, Lisicic A, Horvatic I, Bacic-Vrca V, Bozina N. Atorvastatin-related rhabdomyolysis and acute renal failure in a genetically predisposed patient with potential drug-drug interaction. Int J Clin Pharm. 2012;34(6):825–827.
132. Kosuge K, Jun Y, Watanabe H, et al. Effects of CYP3A4 inhibition by diltiazem on pharmacokinetics and dynamics of diazepam in relation to CYP2C19 genotype status. Drug Metab Dispos. 2001;29(10):1284–1289.
133. Michaud V, Mouksassi MS, Labbe L, et al. Inhibitory effects of propafenone on the pharmacokinetics of caffeine in humans. Ther Drug Monit. 2006;28(6):779–783.
134. Dilger K, Greiner B, Fromm MF, Hofmann U, Kroemer HK, Eichelbaum M. Consequences of rifampicin treatment on propafenone disposition in extensive and poor metabolizers of CYP2D6. Pharmacogenetics. 1999;9(5):551–559.
135. Zhu L, Bruggemann RJ, Uy J, et al. CYP2C19 genotype-dependent pharmacokinetic drug interaction between voriconazole and ritonavir-boosted atazanavir in healthy subjects. J Clin Pharmacol. 2017;57(2):235–246.
136. Laine K, Tybring G, Hartter S, et al. Inhibition of cytochrome P4502D6 activity with paroxetine normalizes the ultrarapid metabolizer phenotype as measured by nortriptyline pharmacokinetics and the debrisoquin test. Clin Pharmacol Ther. 2001;70(4):327–335.
137. Kim HS, Lim Y, Oh M, et al. The pharmacokinetic and pharmacodynamic interaction of clopidogrel and cilostazol in relation to CYP2C19 and CYP3A5 genotypes. Br J Clin Pharmacol. 2016;81(2):301–312.
138. Harmsze AM, van Werkum JW, Souverein PC, et al. Combined influence of proton-pump inhibitors, calcium-channel blockers and CYP2C19*2 on on-treatment platelet reactivity and on the occurrence of atherothrombotic events after percutaneous coronary intervention. J Thromb Haemost. 2011;9(10):1892–1901.
139. Tanaka A, Nagamatsu T, Yamaguchi M, et al. Myoclonus after dextromethorphan administration in peritoneal dialysis. Ann Pharmacother. 2011;45(1):e1.
140. Correia RB, de Araújo Kohler LP, Mattos MM, Rocha LM. City-wide electronic health records reveal gender and age biases in administration of known drug–drug interactions. NPJ Digit Med. 2019;2:74.
141. Freshour SL, Kiwala S, Cotto KC, et al. Integration of the Drug-Gene Interaction Database (DGIdb 4.0) with open crowdsource efforts. Nucleic Acids Res. 2021;49(D1):D1144–D1151.
142. Abarca J, Malone DC, Armstrong EP, et al. Concordance of severity ratings provided in four drug interaction compendia. J Am Pharm Assoc (2003). 2004;44(2):136–141.
143. Olvey EL, Clauschee S, Malone DC. Comparison of critical drug-drug interaction listings: the Department of Veterans Affairs medical system and standard reference compendia. Clin Pharmacol Ther. 2010;87(1):48–51.
144. Vitry AI. Comparative assessment of four drug interaction compendia. Br J Clin Pharmacol. 2007;63(6):709–714.
145. Fung KW, Kapusnik-Uner J, Cunningham J, Higby-Baker S, Bodenreider O. Comparison of three commercial knowledge bases for detection of drug-drug interactions in clinical decision support. J Am Med Inform Assoc. 2017;24(4):806–812.
146. Fulda T, Valuck R, Vander Zanden J, Parker S, Byrns P. Disagreement among drug compendia on inclusion and ratings of drug-drug interactions. Curr Ther Res. 2000;61(8):540–548.
147. Kontsioti E, Maskell S, Bensalem A, Dutta B, Pirmohamed M. Similarity and consistency assessment of three major online drug-drug interaction resources. Br J Clin Pharmacol. 2022. doi:10.1111/bcp.15341
148. Lauschke VM, Zhou Y, Ingelman-Sundberg M. Novel genetic and epigenetic factors of importance for inter-individual differences in drug disposition, response and toxicity. Pharmacol Ther. 2019;197:122–152.
149. Bank PCD, Caudle KE, Swen JJ, et al. Comparison of the guidelines of the clinical pharmacogenetics implementation consortium and the Dutch pharmacogenetics working group. Clin Pharmacol Ther. 2018;103(4):599–618.
150. Schreiber R, Gregoire JA, Shaha JE, Shaha SH. Think time: a novel approach to analysis of clinicians’ behavior after reduction of drug-drug interaction alerts. Int J Med Inform. 2017;97:59–67.
151. Ahn EK, Cho SY, Shin D, Jang C, Park RW. Differences of reasons for alert overrides on contraindicated co-prescriptions by admitting department. Healthc Inform Res. 2014;20(4):280–287.
152. Bryant AD, Fletcher GS, Payne TH. Drug interaction alert override rates in the Meaningful Use era: no evidence of progress. Appl Clin Inform. 2014;5(3):802–813.
153. Samwald M, Xu H, Blagec K, et al. Incidence of exposure of patients in the United States to multiple drugs for which pharmacogenomic guidelines are available. PLoS One. 2016;11(10):e0164972.
154. Pulley JM, Denny JC, Peterson JF, et al. Operational implementation of prospective genotyping for personalized medicine: the design of the Vanderbilt PREDICT project. Clin Pharmacol Ther. 2012;92(1):87–95.
155. Crews KR, Hicks JK, Pui CH, Relling MV, Evans WE. Pharmacogenomics and individualized medicine: translating science into practice. Clin Pharmacol Ther. 2012;92(4):467–475.
156. Hoffman JM, Haidar CE, Wilkinson MR, et al. PG4KDS: a model for the clinical implementation of pre-emptive pharmacogenetics. Am J Med Genet C Semin Med Genet. 2014;166C(1):45–55.
157. Weitzel KW, Elsey AR, Langaee TY, et al. Clinical pharmacogenetics implementation: approaches, successes, and challenges. Am J Med Genet C Semin Med Genet. 2014;166C(1):56–67.
158. Nutescu EA, Drozda K, Bress AP, et al. Feasibility of implementing a comprehensive warfarin pharmacogenetics service. Pharmacotherapy. 2013;33(11):1156–1164.
159. Dunnenberger HM, Crews KR, Hoffman JM, et al. Preemptive clinical pharmacogenetics implementation: current programs in five US medical centers. Annu Rev Pharmacol Toxicol. 2015;55:89–106.
160. Shuldiner AR, Palmer K, Pakyz RE, et al. Implementation of pharmacogenetics: the university of Maryland personalized anti-platelet pharmacogenetics program. Am J Med Genet C Semin Med Genet. 2014;166C(1):76–84.
161. Royal College of Physicians and British Pharmacological Society. Personalised Prescribing: Using Pharmacogenomics to Improve Patient Outcomes. Report of a Working Party. London: Royal College of Physicians and British Pharmacological Society; 2022.
162. Ventola CL. Big data and pharmacovigilance: data mining for adverse drug events and interactions. P T. 2018;43(6):340–351.
163. Bots SH, Groenwold RHH, Dekkers OM. Using electronic health record data for clinical research: a quick guide. Eur J Endocrinol. 2022;186(4):E1–E6.
164. Benchimol EI, Smeeth L, Guttmann A, et al. The REporting of studies Conducted using Observational Routinely-collected health Data (RECORD) statement. PLoS Med. 2015;12(10):e1001885.
165. Bykov K, Gagne JJ. Generating evidence of clinical outcomes of drug-drug interactions. Drug Saf. 2017;40(2):101–103.
166. McDonough CW. Pharmacogenomics in cardiovascular diseases. Curr Protoc. 2021;1(7):e189.
167. van der Wouden CH, van Rhenen MH, Jama WOM, et al. Development of the PGx-Passport: a panel of actionable germline genetic variants for pre-emptive pharmacogenetic testing. Clin Pharmacol Ther. 2019;106(4):866–873.
168. Meaddough EL, Sarasua SM, Fasolino TK, Farrell CL. The impact of pharmacogenetic testing in patients exposed to polypharmacy: a scoping review. Pharmacogenomics J. 2021;21(4):409–422.
169. Asiimwe IG, Pirmohamed M. Ethnic diversity and warfarin pharmacogenomics. Front Pharmacol. 2022;13:866058.
170. Fatumo S, Chikowore T, Choudhury A, Ayub M, Martin AR, Kuchenbaecker K. A roadmap to increase diversity in genomic studies. Nat Med. 2022;28(2):243–250.
171. Buniello A, MacArthur JAL, Cerezo M, et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019;47(D1):D1005–D1012.
172. Johnson JA, Caudle KE, Gong L, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for pharmacogenetics-guided warfarin dosing: 2017 update. Clin Pharmacol Ther. 2017;102(3):397–404.
173. Shaw K, Amstutz U, Kim RB, et al. Clinical practice recommendations on genetic testing of CYP2C9 and VKORC1 variants in warfarin therapy. Lippincott Williams & Wilkins; 2015.
174. The Dutch Pharmacogenetics Working Group. CYP2C9; 2016: 1–16. Available from: https://www.g-standaard.nl/risicoanalyse/B0006234.PDF.
175. Gage BF, Eby C, Johnson JA, et al. Use of pharmacogenetic and clinical factors to predict the therapeutic dose of warfarin. Clin Pharmacol Ther. 2008;84(3):326–331.
176. Klein TE, Altman RB; International Warfarin Pharmacogenetics Consortium. Estimation of the warfarin dose with clinical and pharmacogenetic data. N Engl J Med. 2009;360(8):753–764.
177. Auton A, Brooks LD; Genomes Project C. A global reference for human genetic variation. Nature. 2015;526(7571):68–74.
178. O’Mahony D, O’Sullivan D, Byrne S, O’Connor MN, Ryan C, Gallagher P. STOPP/START criteria for potentially inappropriate prescribing in older people: version 2. Age Ageing. 2015;44(2):213–218.
179. Stouras I, Papaioannou TG, Tsioufis K, Eliopoulos AG, Sanoudou D. The Challenge and Importance of integrating drug-nutrient-genome interactions in personalized cardiovascular healthcare. J Pers Med. 2022;12(4):34.
180. Tod M, Nkoud-Mongo C, Gueyffier F. Impact of genetic polymorphism on drug-drug interactions mediated by cytochromes: a general approach. AAPS J. 2013;15(4):1242–1252.
181. Steelandt J, Jean-Bart E, Goutelle S, Tod M, Prediction A. Model of drug exposure in cirrhotic patients according to Child-Pugh classification. Clin Pharmacokinet. 2015;54(12):1245–1258.
182. Min JS, Bae SK. Prediction of drug-drug interaction potential using physiologically based pharmacokinetic modeling. Arch Pharm Res. 2017;40(12):1356–1379.
183. Johnson D, Wilke MAP, Lyle SM, et al. A systematic review and analysis of the use of polygenic scores in pharmacogenomics. Clin Pharmacol Ther. 2022;111(4):919–930.
184. Pasea L, Chung SC, Pujades-Rodriguez M, et al. Personalising the decision for prolonged dual antiplatelet therapy: development, validation and potential impact of prognostic models for cardiovascular events and bleeding in myocardial infarction survivors. Eur Heart J. 2017;38(14):1048–1055.
185. Malki MA, Dawed AY, Haywood C, Doney A, Pearson ER. Utilizing large electronic medical record data sets to identify novel drug-gene interactions for commonly used drugs. Clin Pharmacol Ther. 2021;110(3):816–825.
186. McInnes G, Altman RB. Drug response pharmacogenetics for 200,000 UK biobank participants. Pac Symp Biocomput. 2021;26:184–195.
187. Shaikh AS, Thomas AB, Chitlange SS. Herb-drug interaction studies of herbs used in treatment of cardiovascular disorders-A narrative review of preclinical and clinical studies. Phytother Res. 2020;34(5):1008–1026.
188. Wang JT. The Polypill at 20 - What have we learned? N Engl J Med. 2022;387(11):1034–1036.
189. van der Wouden CH, Bohringer S, Cecchin E, et al. Generating evidence for precision medicine: considerations made by the Ubiquitous Pharmacogenomics Consortium when designing and operationalizing the PREPARE study. Pharmacogenet Genomics. 2020;30(6):131–144.
190. van der Wouden CH, Cambon-Thomsen A, Cecchin E, et al. Implementing Pharmacogenomics in Europe: design and Implementation Strategy of the Ubiquitous Pharmacogenomics Consortium. Clin Pharmacol Ther. 2017;101(3):341–358.
191. Blagec K, Swen JJ, Koopmann R, et al. Pharmacogenomics decision support in the U-PGx project: results and advice from clinical implementation across seven European countries. PLoS One. 2022;17(6):e0268534.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.