Back to Journals » International Journal of Nanomedicine » Volume 19
Drug-Loaded Tumor-Derived Microparticles Elicit CD8+ T Cell-Mediated Anti-Tumor Response in Hepatocellular Carcinoma
Authors Chen Y ![]() , Zhang Y, Wang J, Cai X, Chen J, Min X, Xu Y, Qin Q, Wan C
, Zhang Y, Wang J, Cai X, Chen J, Min X, Xu Y, Qin Q, Wan C
Received 14 November 2023
Accepted for publication 14 February 2024
Published 6 March 2024 Volume 2024:19 Pages 2227—2239
DOI https://doi.org/10.2147/IJN.S449694
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lijie Grace Zhang
Yulin Chen,1 Yi Zhang,2 Jianjun Wang,3 Xiong Cai,1 Junzhang Chen,1 Xiaobo Min,1 Yunjie Xu,1 Qi Qin,1 Chidan Wan1
1Department of Hepatobiliary Surgery, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, 430022, People’s Republic of China; 2Hubei Engineering Research Center of Tumor-Targeted Biochemotherapy, Wuhan, 430030, People’s Republic of China; 3Department of Hepatobiliary Surgery, Mianyang Central Hospital, School of Medicine, University of Electronic Science and Technology of China, Mianyang, 621000, People’s Republic of China
Correspondence: Qi Qin; Chidan Wan, Department of Hepatobiliary Surgery, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, No. 1277, Jiefang Avenue, Wuhan, 430022, People’s Republic of China, Email [email protected]; [email protected]
Background: Hepatocellular Carcinoma (HCC) poses significant challenges due to limited effective treatments and high recurrence rates. Immunotherapy, a promising approach, faces obstacles in HCC patients due to T-cell exhaustion and immunosuppression within the tumor microenvironment.
Methods: Using doxorubicin-loaded tumor-derived microparticles (Dox-TMPs), the mice with H22 ascites model and subcutaneous tumors model were treated. Following the treatment, mice were re-challenged with H22 cells to compare the therapeutic effects and recurrence among different groups of mice, alongside examining the changes in the proportions of immune cells within the tumor microenvironment. Furthermore, Dox-TMPs were combined with anti-PD-1 to further validate their anti-tumor efficacy. In vitro studies using various liver cancer cell lines were conducted to verify the tumor-killing effects of Dox-TMPs. Additionally, CD8+ T cells from the abdominal cavity of tumor-free mice were co-cultured with H22 cells to confirm their specific tumor-killing abilities.
Results: Dox-TMPs demonstrate effective anti-tumor effects both in vitro and in vivo. In vivo, their effectiveness primarily involves enhancing CD8+ T cell infiltration, alleviating T cell immunosuppression, and improving the immune microenvironment to combat tumors. When used in combination with anti-PD-1, their anti-tumor effects are further enhanced. Moreover, some mice treated with Dox-TMPs developed anti-tumor immunity, displaying a self-specific T-cell immune response upon re-challenged with tumor cells. This suggests that Dox-TMPs also have the potential to act as a long-term immune response against tumor recurrence, indicating their capability as a tumor vaccine.
Conclusion: Dox-TMPs exhibit a dual role in liver cancer by regulating T cells within the tumor microenvironment, functioning both as an anti-tumor agent and a potential tumor vaccine.
Keywords: liver cancer, drug-loaded microparticles, tumor immune microenvironment, tumor vaccine
Graphical Abstract:

Introduction
Primary liver cancer is the sixth most common cancer worldwide and stands as the third leading cause of cancer-related deaths.1 HCC constitutes the predominant type of primary liver cancer, accounting for 75–86% of cases.2 Despite the development of effective strategies such as radiotherapy, chemotherapy, and liver transplantation in recent years, surgical resection remains the primary treatment method for early-stage patients. Unfortunately, the 5-year survival rate for primary liver cancer remains relatively low, with significant contributing factors being tumor recurrence and metastasis.3,4 The recurrence rate for malignant primary liver tumors reaches as high as 70%, with the majority of these recurrences occurring within the first 1–3 years following clinical resection.5 Currently, there is no widely accepted effective neoadjuvant or adjuvant chemotherapy regimen to reduce the risk of the recurrence.5 Systemic chemotherapy and chemoembolization have shown little effectiveness, and sorafenib has not been successful in preventing recurrence after early HCC resection or ablation.6 Other drugs, such as retinoids, vitamin K2, transarterial ¹³¹I-lipiodol, and interferon, have shown only preliminary positive results.7 While liver transplantation is the most effective option, it is unable to fully meet clinical needs due to donor scarcity and indication issues.5 Therefore, developing effective methods to prevent HCC recurrence is an urgent current need.
The tumor immune microenvironment (TME) plays a pivotal role in HCC progression and recurrence. In particular, the presence and function of CD8+ T cells, a subset of cytotoxic T lymphocytes, are critical in mounting an effective antitumor response. However, the majority of HCC cases are accompanied by T cell exhaustion, and the immunosuppressive environment within the TME leads to a reduction in T cell co-stimulatory factors and an increase in immune checkpoint molecules, resulting in impaired T cell effector function.8 Anti-PD-1/PD-L1 therapy works by blocking the programmed cell death 1(PD-1)/PD-1 ligand (PD-L1) immune checkpoint, thus relieving the inhibition of T cells. This enables T cells to more effectively recognize and attack cancer cells, enhancing the immune system’s response.9 It is a promising clinical approach for cancer treatment. However, in most cases, the durable response rate to anti-PD-1/PD-L1 therapy remains relatively low.10,11 The primary reason for this is the lack of infiltrating T cells in tumor tissue, referred to as “cold tumors”, and the presence of an immune-suppressive tumor microenvironment.12 Therefore, increasing T cell infiltration and reversing the immune-suppressive tumor microenvironment are crucial directions that need to be addressed in the field of cancer immunotherapy.
Microparticles (MPs) are extracellular vesicles released by cells in response to either internal or external stimuli. They typically range in size from 100 to 1000 nm and serve as carriers for the exchange of informational substances between cells.13,14 Under conditions of stimuli, tumor cells can also release MPs. These substructures not only contain tumor antigens but also carry unique information specific to the parent cells,15–17 suggesting that TMPs have the potential to serve as cancer vaccines. It has been demonstrated that tumor-derived microparticles (TMPs) can function as a safe and effective vehicle for delivering chemotherapy drugs to tumor cells.18 Moreover, they can act as tumor vaccines, triggering a T cell-mediated anti-tumor immune response.19,20 This helps in reducing the risk of tumor escape. In this study, TMPs loaded with doxorubicin (Dox-TMPs) were used to treat a mouse model of ascites HCC. We found that Dox-TMPs treatment increased T cell infiltration, ameliorated the tumor immune-suppressive microenvironment, and enhanced T cell-mediated anti-tumor responses. Additionally, it exhibited properties of a tumor vaccine, leading to the development of anti-tumor immunity in treated mice, reducing the risk of tumor recurrence. When used in combination with anti-PD-1, Dox-TMPs demonstrated a more potent anti-tumor effect and greater efficacy in preventing tumor recurrence than when used individually. These findings provide a novel approach for preventing the recurrence and metastasis of liver cancer.
Materials and Methods
Cell Lines and Animals
Murine hepatocarcinoma cell line H22, Hepa1-6 and human hepatocarcinoma cell line Huh7, HepG2 were all purchased from China Center for Type Culture Collection (CCTCC, Wuhan, China). Cells were cultured in RPMI 1640 or DMEM medium (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) containing 10% fetal bovine serum (Gibco, #10099141, Thermo Fisher Scientific, Waltham, MA, USA) and 1% penicillin-streptomycin (PS) (Beyotime, #C0222, Shanghai, China) and maintained at 37°C in a 5% CO2 incubator.
BALB/c mice (Female, 6–8 weeks) were purchased from Hubei BIONT biotechnology Co., Ltd (Wuhan, China) and were housed in the SPF animal facility at Tongji Medical College. H22-ascites model was established by injecting a certain number (5 × 104 or 1×105) of H22 cells into the peritoneal cavity of female BALB/c mice. The subcutaneous tumor model was established by injecting 2×105 H22 cells into the flanks of the BALB/c mice. Tumor volume was calculated according to the formula: volume = (length × width2)/2. In both models, the drug for each mouse was administered at the following doses: 5 μg of Dox, 0.1 U TMPs, 0.1 U Dox-TMPs (containing 5 μg of Dox), and 100 μg of anti-PD-1. The control group received an equivalent volume of saline. In the treatment of ascites model mice with Dox-TMPs, the aforementioned dosage of the drug was administered through intraperitoneal injection on days 3, 4, and 7 after modeling. In the combined treatment of Dox-TMPs and anti-PD-1, mice in different groups received intraperitoneal injections of Dox-TMPs or the corresponding drugs on days 7, 9, 11, 13, and 15 after modeling. On days 8, 10, 12, and 14, intraperitoneal injections of anti-PD-1 or saline were administered according to the respective treatment groups. In the subcutaneous tumor model, when the tumor volume reached approximately 100 mm^3, the drug was administered through intratumoral injection for five consecutive days. All experimental procedures complied with the “Laboratory Animal-Guideline for Ethical Review of Animal Welfare (GB/T 35892–2018)” and were approved by the Institutional Animal Care and Use Committee at Tongji Medical College, Huazhong University of Science and Technology (Wuhan, China).
Reagents and Antibodies
Doxorubicin (Dox) was purchased from Shenzhen WanLe Pharmaceuticals Co., Ltd (Shenzhen, China). PKH26 Red Fluorescent Cell Linker Kit and 5(6)-Carboxyfluorescein diacetate N-succinimidyl ester (CFSE) were purchased from Sigma-Aldrich (#21888, St Louis, MO, USA). 4’,6-Diamidino-2-Phenylindole (DAPI) and antibodies used for flow cytometry analysis were purchased from BioLegend (#422801, San Diego, CA, USA). LDH cytotoxicity Assay kit was purchased from Beyotime (#C0016, Wuhan, China).
Preparation of Dox-TMPs
2 × 108 H22 cells in 20cm culture dish were exposed to ultraviolet irradiation (UVB, 300 J/m2) for 1 h. Twenty-four hours later, the supernatant was collected for centrifugation: first 10 min at 1000× g, and then 2 minutes at 14,000×g to get rid of cell debris. The supernatant was then centrifuged at 14,000× g at 4°C for 1 hour to pellet the TMPs. TMPs were washed three times and resuspended in sterile PBS for further use. Each unit (1 U) is defined as the MPs produced by 1×108 H22 cells using the method described above. MPs was stored in the dark at 4°C. TMPs and Dox were dissolved in sterile PBS at the desired dose, with Dox at a concentration of 50 μg/mL. The mixture was then incubated at 4°C in the dark for 2 hours to obtain Dox-TMPs. The hydrodynamic diameter distribution of TMPs and Dox-TMPs were determined by nanoparticle tracking analysis (NTA) system (Nanosight NS300, Malvern). Their morphology was observed by TEM. The drug content in Dox-TMPs was determined using High-Performance Liquid Chromatography (HPLC).
Cellular Uptake
H22 were cocultured cells with TMPs for 0h, 2h, 4h, 8h and 24h, and following washed with PBS three times. The intracellular fluorescence intensity of PKH26 was measured using NovoCyte 2060R flow cytometry (Agilent). Data were analyzed using Flowjo 10.8.
CFSE-labeled H22 cells and PKH26-labeled TMPs were cocultured for 24 hours, followed by triple washing with PBS. Subsequently, the cells were fixed with 0.4% paraformaldehyde, and cell nuclei were stained with DAPI. After two additional washes with PBS, fluorescent distribution was observed using confocal microscopy (LSM 780, Carl Zeiss). Staining steps were all performed following the manufacturer’s protocol.
Flow Cytometric Analysis
For a phenotype analysis of cells, cells were stained with surface antibodies: CD45 (#103107, #103132, #103114), CD19 (#152403), CD3 (#100236, #103114), CD4 (#100511), CD8 (#100705, #100711, #100733), CD11b (#101212, #101207), F4/80 (#123109), CD86 (#105005), CD206 (#141703) and Ly-6G (#127606). For intracellular markers staining, cells were first treated with Fix/Perm solution (Biolegend, #420801, San Diego, CA, USA) before staining. For intracellular cytokine staining, cells were first surface-stained, and then fixed and permeabilized for subsequent intracellular staining with IL-2 (#503809), Ki-67 (#652409), IFN-γ (#505807), TNF-α (#506303) and Granzyme B (#372207). Analysis was performed using NovoCyte 2060R flow cytometer.
Cell Apoptosis Detection
For H22 cells, 2×10^4 cells were seeded in six-well plates with 1 mL complete RPMI 1640 medium containing 0.5% FBS and treated with Dox (0.5 μg/mL), TMPs (0.05 U), Dox-TMPs (0.05 U, containing 0.5 μg of Dox) or equivalent volume of PBS. After 24 hours of incubation in cell culture incubator, cells were harvested. For Huh7, HepG2 and Hepa1-6 cells, 2×10^4 cells were initially seeded in six-well culture plates with 1mL complete DMEM medium containing 10% FBS. After 6 hours of cell adhesion, the medium was replaced with 0.5% FBS-containing complete DMEM medium `emented with Dox (0.5 μg/mL), TMPs (0.05 U), Dox-TMPs (0.05 U, containing 0.5 μg of Dox) or equivalent volume of PBS. The cells were then cultured for an additional 44 hours before the supernatant and cells were collected after digestion with trypsin without EDTA (Beyotime, #C0205, Shanghai, China). Finally, all four cell types were stained with Annexin V (Biolegend, #640920, San Diego, CA, USA) and Zombie Violet™ Fixable Viability Kit (Biolegend, #423113, San Diego, CA, USA) and analyzed using the Sony ID7000 flow cytometer.
Cell apoptosis was also assessed using LDH Cytotoxicity Assay Kit. The drug treatment conditions were the same as mentioned above, and the detection procedure was performed following the manufacturer’s instructions.
Tumor-Specific CTL Killing Assay
CD8+ T cells were separated from mesenteric lymph nodes of female BALB/c mice using the CD8+ T cell isolation kit (STEMCELL Technologies, #19858, Vancouver, BC, Canada) following the manufacturer’s instructions and were cultured in RPMI 1640 complete medium containing 10% FBS, 1% PS, 50 mmol/L β-mercaptoethanol (ThermoFisher Scientific, #21985023, Waltham, MA, USA), 50 mmol/L EDTA (Beyotime, #C0196, Shanghai, China), 200 mmol/L L-glutamine (Beyotime, #C0212, Shanghai, China) and 20 ng/mL IL-2 (Peprotech, ThermoFisher Scientific, #212-12, Waltham, MA, USA). Next, CD8+ T cells (as effector cells) were co-cultured with H22 cells (as target cells) at various Effector-to-Target (E:T) ratios ranging from 1: 1 to 20: 1. After 24 hours, the cells were harvested, stained with Annexin V and 7-AAD (Biolegend, #420404, San Diego, CA, USA), and analyzed using flow cytometry to determine the apoptosis status of CD45- cells.
Impact of Dox-TMPs on Immune Cells Infiltration in the Subcutaneous Tumor Model
The construction of the subcutaneous tumor model and the drug treatment procedures are as described above. The excised subcutaneous tumor specimens were fixed with 4% paraformaldehyde. Subsequently, specimens were subjected to H&E staining and examined under microscope (CX31, OLYMPUS). Alternatively, fixed tumor sections were stained with DAPI (Beijing Solarbio Science & Technology Co., Ltd., #C0065, Beijing, China), CD3 (1: 4000) (Abcam, #ab237721, Cambridge, UK), CD4 (1: 4000) (Abcam, #ab183685, Cambridge, UK), and CD8 (1: 4000) (Abcam, #ab209775, Cambridge, UK) fluorescent antibodies, and observed under fluorescence microscope (BX53, OLYMPUS).
Statistical Analysis
All experiments were repeated at least three times. Statistical analysis was carried out using GraphPad Prism 10.0 (GraphPad Software, CA) software. For comparison between two groups, unpaired two-tailed Student’s t-test were employed. In the case of multiple groups, one-way ANOVA or two-way ANOVA was used, followed by Tukey’s honest significant difference (HSD) post-hoc test or Bonferroni’s multiple comparisons post-test, unless otherwise specified. Results are presented as means ± SEM. P < 0.05 was considered statistically significant.
Results
Characterization of DOX-TMPs and Effect on Peritoneal Metastasis Model of HCC
Under TEM, TMPs exhibited bilayered, translucent, ellipsoidal vesicular structures with a size range between 100–1000 nm. TMPs loaded with Dox exhibited a size and morphology similar to TMPs (Figure 1a). NTA system analysis revealed that TMPs exhibited a particle size distribution ranging from 100 to 1000 nm, with the majority falling within the range of 100–500 nm (Figure 1b). TMPs were co-incubated with Dox at concentrations of 25 μg/mL, 50 μg/mL, 100 μg/mL, and 200 μg/mL to load Dox into the TMPs. HPLC analysis was performed to determine the drug-loading capacity of the Dox-TMPs. The results showed that as the Dox concentration increased, the drug-loading capacity of Dox-TMPs gradually increased. Subsequently, we assessed the stability of Dox-TMPs and observed that those prepared at Dox concentrations of 100 μg/mL and 200 μg/mL exhibited significant precipitation within 24 hours and the number of TMPs decreased after 24 hours. In contrast, Dox-TMPs obtained at Dox concentrations of 25 μg/mL and 50 μg/mL remained stable in terms of quantity and characteristics for up to 72 hours. Therefore, we used the latter drug concentrations for subsequent experiments (Figure 1c and d).
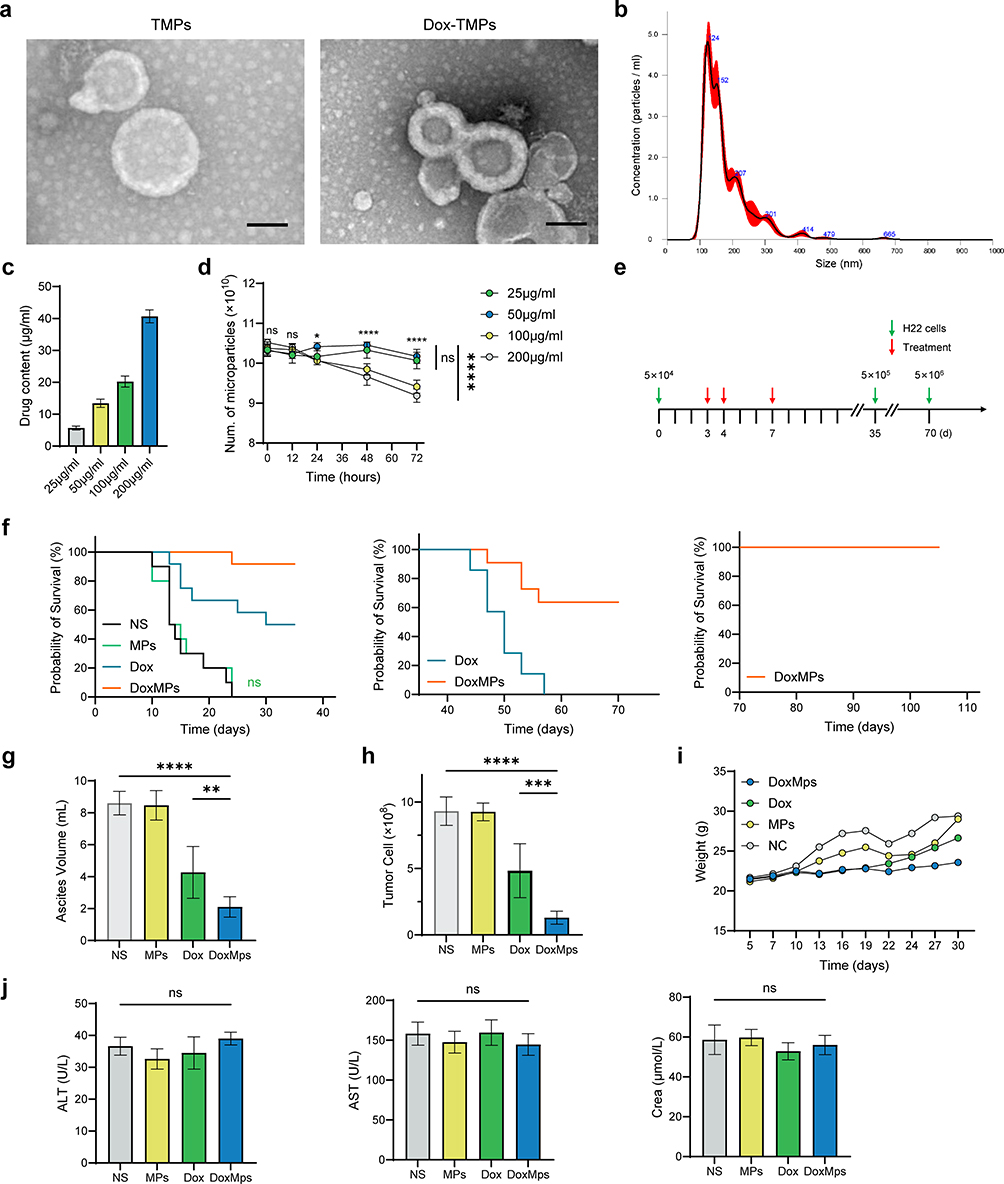 |
Figure 1 Characterization of Dox-TMPs and effect on peritoneal metastasis model of HCC. (a) Morphology of TMPs and Dox-TMPs by TEM. Scale bars, 100 nm. (b) Diameter distribution of TMPs by NTA. (c) Drug loading capacity of TMPs at various drug incubation concentrations (25μg/mL, 50μg/mL, 100μg/mL and 200μg/mL), quantified by HPLC. (d) TMPs were stored at 4°C and concentration was assessed at different time points using NTA (12h, 24h, 48h and 72h). (e) Schematic diagram of re-challenge with H22 cells in H22-ascite mice following Various Treatments. (f) Kaplan–Meier survival plot of H22-ascitic mice re-challenged with H22 cells after intraperitoneal injection of NS, TMPs, Dox or Dox-TMPs at the time points indicated in (e) (n=12). (g and h) Mice were intraperitoneal injected with H22 cells and treated according to (a). On the 15th day, mice were euthanized and the ascites volume (g) and number of CD45- tumor cells (h) were assessed (n=6). (i) Weight changes in mice following intraperitoneal injection of H22 cells and different treatments (n=12). (j) On the 5th day following the final treatment (day 12), venous blood was obtained from mice orbit to assess liver and kidney function (n=6). Data are presented as mean ± SEM. ns, not statistically significant; *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001. |
To investigate the efficacy of Dox-TMPs in the treatment of peritoneal metastasis in HCC, as depicted in Figure 1e, H22-ascites mice were treated with NS, TMPs, Dox and Dox-TMPs. Kaplan-Meier survival curves revealed that treatment with TMPs alone showed no significant difference compared to the control group. In contrast, the Dox-TMPs and Dox treatment groups exhibited survival rates of 91.7% and 50% of mice, respectively, with no development of ascites. Furthermore, the mice in the Dox-TMPs group exhibited a significantly delayed onset of ascites compared to the other three groups. (Figure 1f left). The volume of ascites and the number of tumor cells in the ascites also corroborated the aforementioned results (Figure 1g and h). Changes in mouse body weight among the groups followed a similar pattern. Upon ascites development, there was a temporary, noticeable increase in mouse body weight. In contrast, the Dox-TMPs group of mice displayed a more stable and gradual increase in body weight (Figure 1i). On the 5th day following the final treatment, venous blood was obtained from mice orbit to assess liver and kidney function, and no significant differences in results were observed (Figure 1j). Interestingly, when mice in the Dox and Dox-TMPs groups, which remained free of ascites, were re-challenged intraperitoneally with H22 cells at 10 times the dose used initially without any further treatment, all the mice in the Dox group developed ascites and died, whereas 63.6% of the mice in the Dox-TMPs group remained ascites-free without additional treatment (Figure 1f middle). Subsequently, these ascites-free mice from the Dox-TMPs group were re-challenged with H22 cells at a dose 10 times higher than the previous challenge, and remarkably, none of them developed ascites (Figure 1f right). These results indicate that the mice treated with Dox-TMPs developed a robust immune response against H22 tumor cells, and this aligns with previous reports that TMPs can serve as a tumor vaccine.19 It suggests that Dox-TMPs not only exert a more potent anti-tumor effect compared to Dox alone but also act as a tumor vaccine.
Dox-TMPs Uptake Efficiency and Anti-Tumor Activity in HCC
To further confirm the anti-tumor activity of Dox-TMPs on HCC, we first verified the uptake efficiency of Dox-TMPs by H22 tumor cells. After co-culturing PKH26-labeled Dox-TMPs with H22 cells, uptake of Dox-TMPs by H22 cells was observed as early as 2 hours, and the uptake continued to increase gradually within 24 hours (Figure 2a). Confocal microscopy analysis showed that Dox-TMPs, upon uptake, were predominantly located in the cytoplasm, with a smaller portion present in the cell nucleus (Figure 2b). We selected H22 cells and another murine liver cancer cell line Hepa1-6, as well as two human liver cancer cell lines Huh7 and HepG2, to validate the cytotoxic effect of Dox-TMPs in vitro. Using flow cytometry to assess cell apoptosis, the results showed that Dox-TMPs derived from H22 cells not only exhibited a higher killing efficiency against H22 cells compared to Dox but also demonstrated significant cytotoxicity against another liver cancer cell line, hepa1-6 (Figure 2c and d). TMPs did not exhibit a noticeable tumor-killing effect in vitro (Figure 2e). When tested on human liver cancer cell lines, although the overall cytotoxicity was weaker compared to in murine cell lines, Dox-TMPs still demonstrated a superior anti-tumor effect compared to using Dox alone. The results of LDH release experiments also support the above conclusion. These findings demonstrate that, compared to Dox, Dox-TMPs not only exhibit enhanced cytotoxicity against their source cells but also effectively induce cytotoxicity in similar tumor cell types.
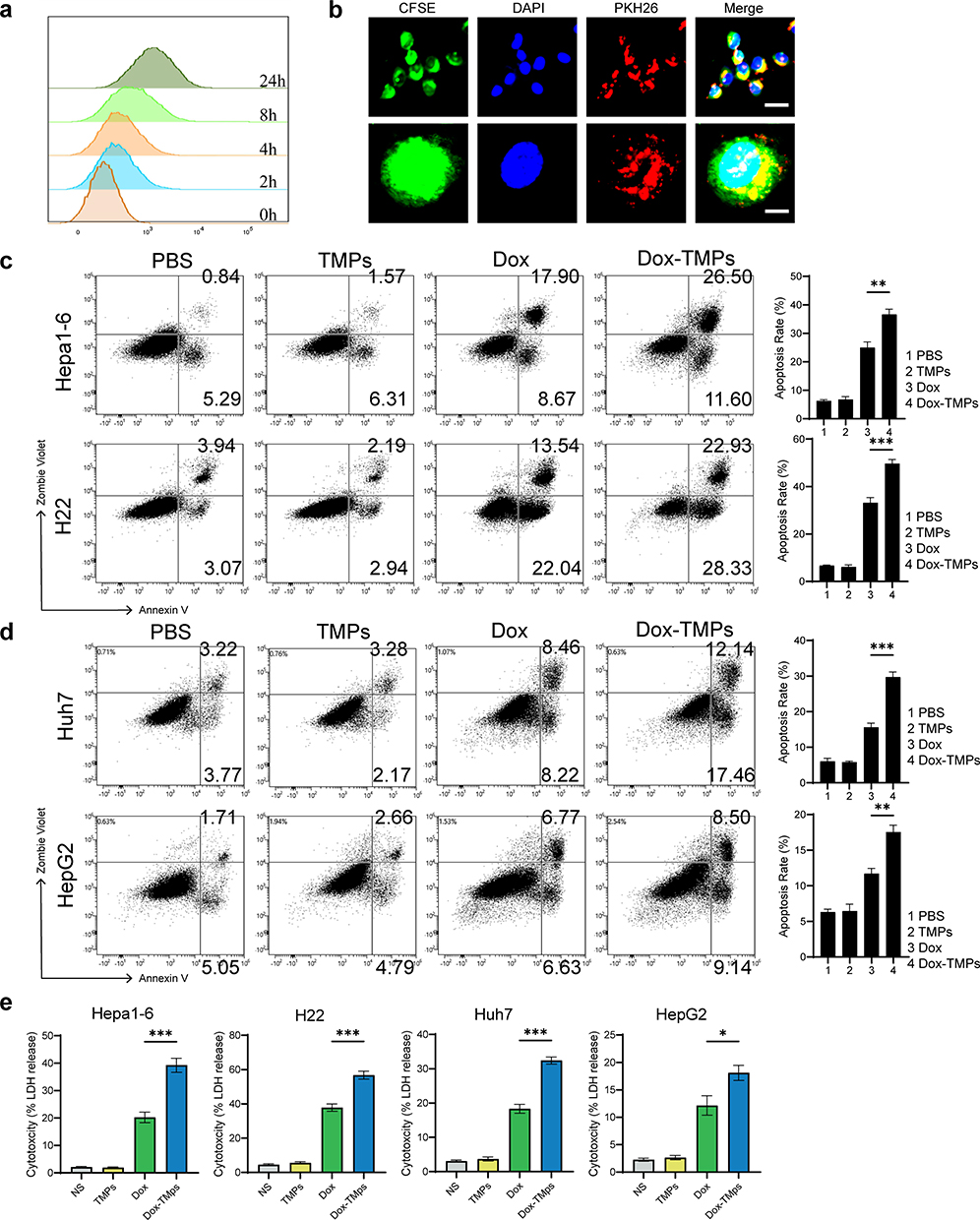 |
Figure 2 Dox-TMPs uptake efficiency and anti-tumor activity in HCC. (a) PKH26-labeled TMPs were co-incubated with H22 cells, and the uptake of H22 cells on TMPs was assessed at different time points (2h, 4h, 8h and 24h) using flow cytometry. (b) CSFE-labeled H22 cells were co-incubated with PKH26-labeled TMPs for 24 hours, and DAPI was used to stain the cell nuclei. The cells were observed under laser confocal microscope. Scale bar: 50μm (above), 10μm (below). (c) Mouse hepatoma cell lines (Hepa1-6 and H22) were treated with PBS, TMPs, Dox, and Dox-TMPs for 48 hours and 24 hours, respectively. Subsequently, tumor cell death was assessed using flow cytometry. (d) Human hepatoma cell lines (Huh7 and HepG2) were treated with PBS, TMPs, Dox, and Dox-TMPs for 48 hours, and subsequently, tumor cell death was assessed using flow cytometry. (e) LDH release of the hepatoma cell lines after different treatments (as in c and d). Data are representative of three independent experiments and presented as mean ± SEM. *P<0.05; **P<0.01; ***P<0.001. |
Modulation of the Tumor Immune Microenvironment by Dox-TMPs
In the treatment of the H22-ascites model (Figure 1e), we observed that some mice treated with Dox-TMPs remained tumor-free upon re-challenge with H22 cells (Figure 3a), indicating that Dox-TMPs exhibited the characteristics of a tumor vaccine. Previous research has indicated that TMPs can promote dendritic cell maturation and activate T cells to generate an anti-tumor immune response through the cGAS/STING-mediated DNA-sensing pathway.19 Hence, Dox-TMPs are likely to modulate the tumor immune microenvironment. To investigate this further, we performed another intraperitoneal injection of 5×10^5 H22 cells into those mice without ascites after being treated with Dox and Dox-TMPs as shown in Figure 1e. At 48 and 120 hours after the injection, we examined the immune cells in the peritoneal cavity of the mice. The results indicated that at 48 hours, the Dox-TMPs group of mice exhibited a significantly lower proportion and number of tumor cells in the peritoneal cavity compared to the Dox group. At 120 hours, the Dox-TMPs group showed a further reduction in tumor cell count, while the Dox group exhibited a significant increase in the number of tumor cells (Figure 3b). At 48 hours, the Dox-TMPs group exhibited higher proportions of both CD4+ and CD8+ T cells compared to the Dox group. And in both groups, there was an increase in the CD4+/CD8+ T cell ratio at 120 hours. However, the Dox-TMPs group displayed a significantly higher increase in the proportion of CD8+ T cells compared to the Dox group. It is worth noting that although the Dox group showed an upward trend in the CD8+ T cell proportion at 120 hours, the absolute count of CD8+ T cells decreased. In contrast, the Dox-TMPs group demonstrated a substantial increase in both the proportion and absolute count of CD8+ T cells (Figure 3c). From 48 to 120 hours, the proportion of B cells in the Dox group exhibited a noticeable decrease, while the Dox-TMPs group showed no significant change (Figure 3d). The results for MDSCs indicated that in the Dox group, the intraperitoneal MDSCs were at relatively high levels at both time points. In the Dox-TMPs group, the proportion of MDSCs was remarkably low at 48 hours and showed a slight increase at 120 hours (Figure 3e). In the Dox group, there was no significant change in the proportion of M1-type macrophages, while the proportion of M2-type macrophages showed a clear increase. In the Dox-TMPs group, both M1 and M2-type macrophages exhibited an increase in proportion (Figure 3f and g). The results above indicate that compared to Dox, Dox-TMPs treatment can modulate the murine peritoneal immune microenvironment, allowing it to activate primarily T-cell-mediated immune responses upon re-challenge with H22 cells, leading to the clearance of tumor cells.
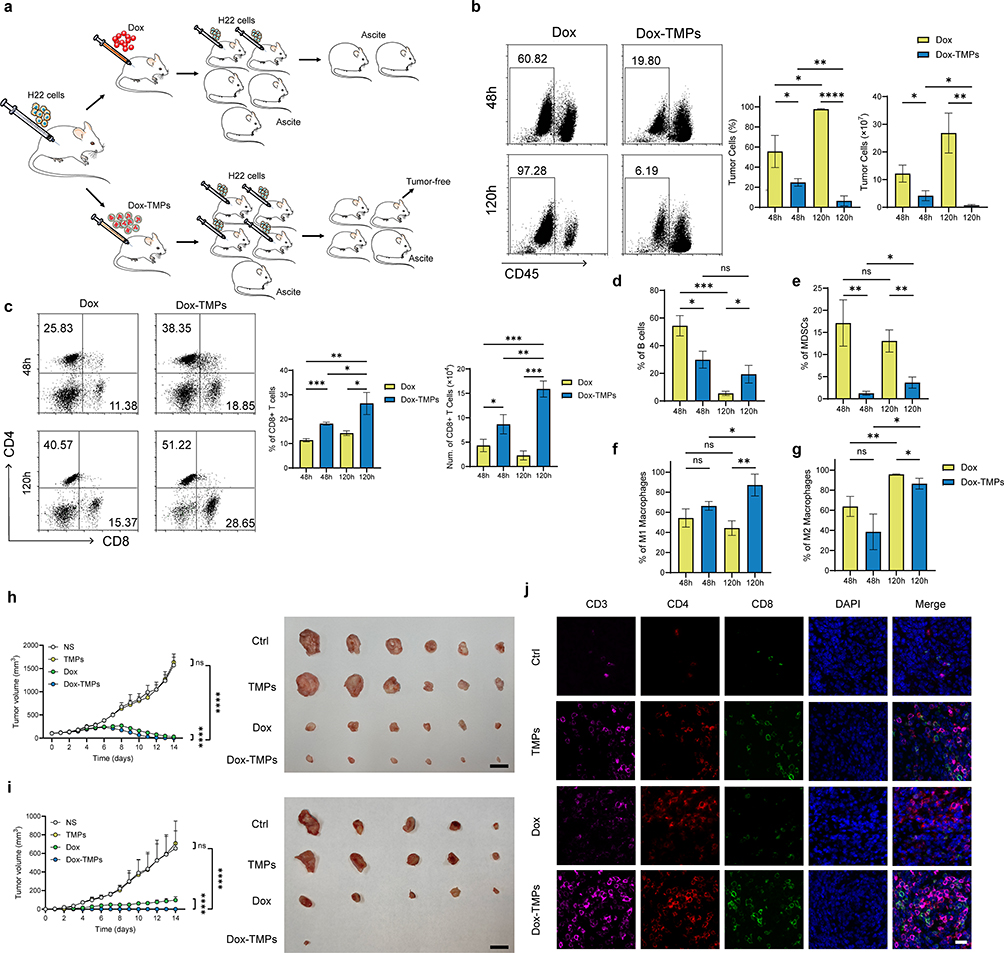 |
Figure 3 Modulation of the Tumor Immune Microenvironment by Dox-TMPs. (a) Schematic Diagram of H22-ascite mice re-challenged with H22 cells after Dox and Dox-TMPs treatment. A portion of mice after Dox-TMPs treatment were tumor-free while re-challenged with H22 cells. H22-ascite mice that did not develop ascites after Dox treatment, when re-challenged with H22 cells, all subsequently developed ascites. (b–g) While mice were treated with the final dose of Dox or Dox-TMPs (as in Figure 1e), and at 48h and 120h thereafter, peritoneal lavage fluid was collected for flow cytometry analysis to determine CD45- cells (b), CD4+/CD8+ T cells (c), B cells (d), DMSCs (e) and M1/M2-like macrophage (f and g). (h and i) Subcutaneous tumor model was established by injecting 2×10^5 H22 cells subcutaneously into the right dorsal side of each mouse. When the tumor volume reached approximately 100 mm3, intratumoral injections of PBS, TMPs, Dox, or Dox-TMPs were administered consecutively for five days. Tumor size was measured daily (h, left). On the 14th day, the subcutaneous tumors were completely excised (h, right). H22 cells (same quantity as before) were injected subcutaneously into the opposite dorsal side, and subcutaneous tumor volume was measured daily (i, left). On the 14th day, the second subcutaneous tumors were completely excised (i, right). (j) Fluorescence images of T cells infiltration in the excised subcutaneous tumors in (h). T cells were labeled with Cy5-conjugated CD3 antibody (purple), Cy3-conjugated CD4 antibody (red) and FITC-conjugated CD8 antibody (green). Cell nuclei were stained with DAPI (blue). Scale bars: 10μm. Data are representative of three independent experiments and presented as mean ± SEM. ns, not statistically significant; *P<0.05; **P<0.01; ***P<0.001, ****P<0.0001. |
Furthermore, we validated the therapeutic and preventive effects of Dox-TMPs in a subcutaneous tumor model. H22 cells were inoculated into the right flank of mice, and after treatment with NS, TMPs, Dox, and Dox-TMPs, the control group and TMPs group exhibited progressively increasing subcutaneous tumor volumes. In contrast, the tumors treated with Dox and Dox-TMPs gradually shrank, with the Dox-TMPs group showing a greater reduction in tumor size (Figure 3h). After the complete removal of the subcutaneous tumors on the right flank of mice, H22 cells were reinoculated into the left flank. We observed that the control group and TMPs group mice still developed subcutaneous tumors at the injection site. In the Dox group, the subcutaneous tumors were relatively smaller. In contrast, the Dox-TMPs group mice exhibited minimal subcutaneous tumor formation, with only a few showing small, palpable nodules (Supplementary Figure 1a, Figure 3i). The first-time resected subcutaneous tumors, when subjected to immunofluorescence staining, displayed a significant infiltration of lymphocytes within the tumor tissue of the Dox-TMPs group (Figure 3j). In contrast, samples from the control group, TMPs group, and Dox group, when analyzed using HE staining following the second resection, all exhibited a widespread presence of tumor cells. However, the sole sample obtained from the Dox-TMPs group showed only a limited number of tumor cells, indicating a striking contrast (Supplementary Figure 1b). The results above provide evidence that in the liver cancer metastasis model, Dox-TMPs modulate the tumor immune microenvironment to generate an intrinsic anti-tumor immune response.
Combination of Dox-TMPs with Anti-PD-1 Effectively Activated CTLs, Leading to an Anti-Tumor Effect
The persistence of responses to anti-PD-1/PD-L1 therapy in cancer treatment remains low, primarily due to the lack of infiltrating T lymphocytes within tumors and the presence of an immunosuppressive tumor microenvironment12. Since we have already confirmed that Dox-TMPs can enhance T cell infiltration in the tumor microenvironment and modulate the immunosuppressive tumor microenvironment, we combined anti-PD-1 with Dox-TMPs for the treatment of H22 ascites mice (as in Figure 4a). K-M survival analysis curve demonstrates that the use of anti-PD-1 alone is not effective in inhibiting ascites formation. When the number of H22 cell injected was increased and the start of treatment was delayed, Dox-TMPs did not demonstrate a significant advantage over Dox or Dox combined with anti-PD-1 in terms of therapeutic efficacy. When Dox-TMPs were used in combination with anti-PD-1, 90% of the mice survived without ascites (Figure 4b). We next investigated the activation status of peritoneal T lymphocytes in mice treated with Dox and Dox-TMPs, either alone or in combination with anti-PD-1. On the fifth day after the end of treatment, mice were rechallenged with H22 cells at a dose ten times higher than the initial injection. Two days later, peritoneal lavage fluid was collected and analyzed using flow cytometry to assess T lymphocyte activation. The results revealed that the CD4+/CD8+ T cell ratio in the Dox-TMPs+anti-PD-1 group and the Dox-TMPs group was significantly higher than that in the Dox and Dox+anti-PD-1 groups. The combination of Dox-TMPs with anti-PD-1 further increased the T cell ratio (Figure 4c). In the Dox-TMPs+anti-PD-1 group, CD4+ T cells exhibited significantly higher IL-2 production compared to the Dox and TMPs groups, but there was no significant difference compared to the Dox+anti-PD-1 group (Figure 4d). The TMPs group showed higher production of IFN-γ, TNF-α, Ki-67, and Granzyme B in CD8+ T cells compared to the Dox and Dox+anti-PD-1 groups. When anti-PD-1 was combined, the production of these cytokines further increased (Figure 4e–h). In vitro experiments also yielded similar results. CD8+ T cells were extracted from the mesenteric lymph nodes of mice in each group and co-cultured with H22 cells at different E:T ratios. The results showed that T cells extracted from the Dox group exhibited no specific cytotoxic effect against H22 cells in vitro. In contrast, T cells from the Dox-TMPs+anti-PD-1 group displayed highly efficient cytotoxic effects against H22 compared to the Dox and Dox+anti-PD-1 groups. Furthermore, as the E:T ratio increased, the cytotoxic effect became more pronounced (Figure 4i). This indicates that Dox-TMPs+anti-PD-1 can effectively induce the specific activation of CTL cells, exerting an anti-tumor effect.
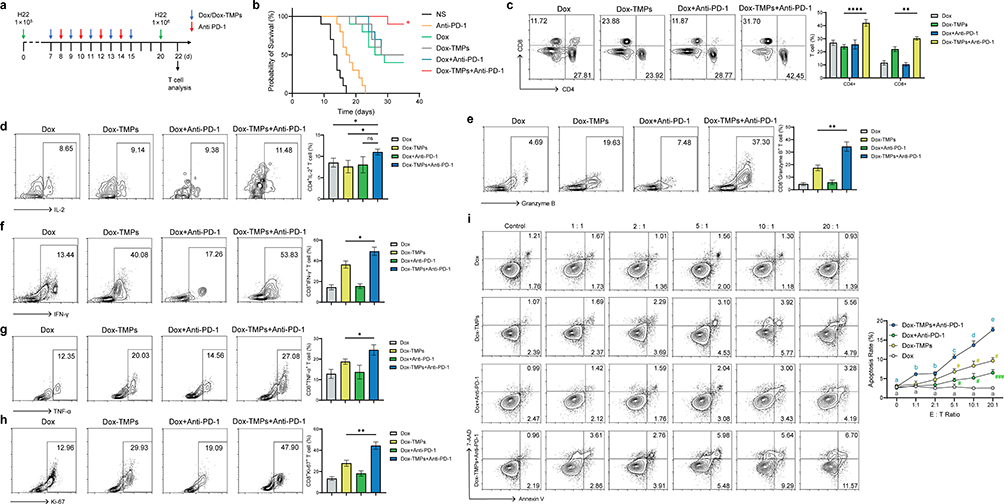 |
Figure 4 Combination of Dox-TMPs with Anti-PD-1 effectively activated CTLs, leading to an anti-tumor effect. (a) Schematic diagram of Dox or Dox-TMPs combined or not with anti-PD-1 treatment in H22-ascite mice. (b) Kaplan–Meier survival plot of H22-ascitic mice re-challenged with H22 cells after intraperitoneal injection of NS, anti-PD-1, Dox or Dox-TMPs combined or not with anti-PD-1 at the time points indicated in (a) (n=10). (c–h) On the second day (day 22) after re-challenged with H22 cells in mice (as in a), peritoneal lavage fluid was collected and the CD4+/CD8+ T cell ratio (c) as well as the activation status of CD4+/ CD8+ T cells (e–h) were assessed using flow cytometry. IL-2 expression was used to assess the activation status of CD4+ T cells, while IFN-γ, TNF-α, Ki67 and Granzyme B was used to evaluate the activation status of CD8+ T cells. i T cells were isolated from mesenteric lymph nodes of mice from each group and were co-cultured in vitro with H22 cells at different E:T ratios. After 24 hours of co-culture, CD45- cell death was assessed using flow cytometry. Data are representative of three independent experiments and presented as mean ± SEM. ns, not statistically significant; *P<0.05; **P<0.01; ****P<0.0001; #P<0.05; ###P<0.001; The same letters represent no statistical difference, while different letters indicate statistical differences. |
Discussion
TMPs as the drug carriers can reverse tumor drug resistance, efficiently target and eliminate cancer stem cells, thus reducing the risk of tumor recurrence and metastasis.18,21 Furthermore, TMPs have the ability to mobilize immune cells to target and eliminate tumor cells, thereby reshaping the tumor immune microenvironment.19,22–26 They recruit neutrophils from peripheral blood, inducing their transformation into cytotoxic N1 neutrophils.24 Drug-loaded TMPs can also target M2 macrophages within the TME, converting part of them into anti-tumor M1 macrophages, working collaboratively to eliminate tumor cells.25,27 Additionally, TMPs can function as tumor vaccines by activating dendritic cells, effectively initiating specific T cell immune responses, thereby reshaping the immune microenvironment and establishing lasting anti-tumor immunity.19,26 In this study, we utilized Dox-TMPs to treat H22 ascites-bearing mice and confirmed their highly effective anti-tumor properties both in vitro and in vivo. Moreover, we demonstrated their potential as tumor vaccines, eliciting lasting anti-tumor immune responses. We have provided evidence that Dox-TMPs augment tumor-infiltrating T lymphocytes, resulting in the elimination of tumor cells, and upon re-exposure to tumor cells, CD8+ T lymphocytes become specifically activated, effectively targeting and eradicating tumor cells.
In the ascites model, mice treated with Dox-TMPs displayed increased infiltration of T lymphocytes compared to the other groups, and results regarding MDSCs and macrophages indicated an improved status of the TME. Nevertheless, the roles of other immune cells such as Treg cells and monocyte-derived macrophages in this context require further investigation. In the subcutaneous tumor model, extensive lymphocyte infiltration was observed, which undoubtedly addresses two significant challenges in the current field of low durable responses to anti-PD-1/PD-L1 therapy. The results of Dox-TMPs in combination with anti-PD-1 also indicated a synergistic effect, leading to a significant increase in T lymphocyte proportions and enhanced secretion of cytokines, thereby exerting a robust anti-tumor effect. Co-culture experiments with T lymphocytes and tumor cells demonstrated that CD8+ T cells from mice treated with Dox-TMPs were capable of directly and specifically killing H22 tumor cells. This suggests the development of endogenous anti-H22 tumor immunity, which effectively targets tumor cells upon re-exposure, thereby reducing the risk of tumor recurrence. Of particular interest, in vitro experiments revealed that H22-derived Dox-TMPs exhibited high cytotoxic effects not only against H22 cells but also against different cell lines, such as Hepa1-6, and even human HCC cell lines HepG2 and Huh7. Previous studies have also demonstrated that H22-derived TMPs, when used as a tumor vaccine, could provide protection against both H22 and Hepa1-6,19 indicating shared antigens among TMPs of the same tumor type. These antigens can be cross-presented by host antigen-presenting cells, thereby expanding the potential applications of Dox-TMPs.
Currently, liver cancer vaccines are primarily categorized as preventive and therapeutic types, with the main goal of initiating or enhancing the human body’s immune response against cancer.28 Some preventive vaccines, such as DNA vaccines,29 live-attenuated Listeria vaccines30 and peptide vaccine31 have shown effective preventive results. On the other hand, personalized peptide vaccines have demonstrated efficacy in postoperative liver cancer prevention,32 but their effectiveness varies due to individual patient characteristics and the heterogeneity of HCC. Moreover, the immune microenvironment in HCC often exhibits immune-suppressive characteristics, which can impact the immune response to vaccines. Our research suggests that Dox-TMPs can improve the immune-suppressive microenvironment. Additionally, they possess characteristics of a tumor vaccine, making them more advantageous in generating long-term antitumor immunity and preventing tumor recurrence compared to traditional tumor vaccines.
Although Dox-TMPs have demonstrated promising results in preventing the recurrence of liver cancer in murine models, thus offering a new perspective for clinical translation, there are still several challenges when transitioning from the laboratory to human clinical trials. Firstly, this study verified the preventive effects of Dox-TMPs using peritoneal metastasis and subcutaneous tumor models, but the efficacy in preventing intrahepatic recurrence and lung metastasis has not been investigated and may require further research in subsequent studies. Secondly, an important decision to be made in practical applications is whether to use patient-derived tumor cells or immortalized cell lines. Currently, MPs derived from immortalized cell lines have been utilized in clinical applications, but they lack “self” characteristics to evade recognition by the immune system. On the other hand, using patient-specific tumor cells presents challenges such as lengthy preparation periods and the need for personalized therapies.
In summary, Dox-TMPs demonstrate effective treatment for liver cancer by increasing T lymphocyte infiltration within the tumor tissue and improving the tumor immune microenvironment. Furthermore, their combination with antiPD-1 drugs enhances T lymphocyte infiltration and anti-cancer activity, resulting in long-term T cell-mediated memory immune responses. These findings suggest that Dox-TMPs can serve as an effective approach for the treatment of liver cancer and prevention of its recurrence.
Abbreviations
HCC, Hepatocellular Carcinoma; Dox, Doxorubicin; MPs, Microparticles; TMPs, Tumor-derived microparticles; Dox-TMPs, Doxorubicin-loaded tumor-derived microparticles; TME, Tumor immune microenvironment; PD-1/PD-L1, Programmed cell death 1/PD-1 ligand; PS, Penicillin-streptomycin; CFSE, 5(6)-Carboxyfluorescein diacetate N-succinimidyl ester; DAPI, 4’,6-Diamidino-2-Phenylindole; NTA, tracking analysis nanoparticle; HPLC, High Performance Liquid Chromatography.
Acknowledgments
The Figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license. Graphical Abstract were drawn by Figdraw.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Ferlay J, Colombet M, Soerjomataram I, et al. Cancer statistics for the year 2020: an overview. Intl J Cancer. 2021;149(4):778–789. doi:10.1002/ijc.33588
2. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA a Cancer J Clinicians. 2021;71(3):209–249. doi:10.3322/caac.21660
3. Lan T, Li H, Zhang D, et al. KIAA1429 contributes to liver cancer progression through N6-methyladenosine-dependent post-transcriptional modification of GATA3. Mol Cancer. 2019;18(1):186. doi:10.1186/s12943-019-1106-z
4. Kulik L, Heimbach JK, Zaiem F, et al. Therapies for patients with hepatocellular carcinoma awaiting liver transplantation: a systematic review and meta‐analysis. Hepatology. 2018;67(1):381–400. doi:10.1002/hep.29485
5. Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet. 2018;391(10127):1301–1314. doi:10.1016/S0140-6736(18)30010-2
6. Bruix J, Takayama T, Mazzaferro V, et al. Adjuvant sorafenib for hepatocellular carcinoma after resection or ablation (STORM): a Phase 3, randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2015;16(13):1344–1354. doi:10.1016/S1470-2045(15)00198-9
7. Lu LC, Cheng AL, Poon R. recent advances in the prevention of hepatocellular carcinoma recurrence. Semin Liver Dis. 2014;34(04):427–434. doi:10.1055/s-0034-1394141
8. Kassel R, Cruise MW, Iezzoni JC, Taylor NA, Pruett TL, Hahn YS. Chronically inflamed livers up-regulate expression of inhibitory B7 family members. Hepatology. 2009;50(5):1625–1637. doi:10.1002/hep.23173
9. Llovet JM, Castet F, Heikenwalder M, et al. Immunotherapies for hepatocellular carcinoma. Nat Rev Clin Oncol. 2022;19(3):151–172. doi:10.1038/s41571-021-00573-2
10. El-Khoueiry AB, Sangro B, Yau T, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, Phase 1/2 dose escalation and expansion trial. Lancet. 2017;389(10088):2492–2502. doi:10.1016/S0140-6736(17)31046-2
11. Zhang J, Dang F, Ren J, Wei W. Biochemical aspects of pd-l1 regulation in cancer immunotherapy. Trends Biochem Sci. 2018;43(12):1014–1032. doi:10.1016/j.tibs.2018.09.004
12. Binnewies M, Roberts EW, Kersten K, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24(5):541–550. doi:10.1038/s41591-018-0014-x
13. Tominaga N, Yoshioka Y, Ochiya T. A novel platform for cancer therapy using extracellular vesicles. Adv Drug Delivery Rev. 2015;95:50–55. doi:10.1016/j.addr.2015.10.002
14. Colombo M, Raposo G, Biogenesis TC. Secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. 2014;30(1):255–289. doi:10.1146/annurev-cellbio-101512-122326
15. D’Souza-Schorey C, Clancy JW. Tumor-derived microvesicles: shedding light on novel microenvironment modulators and prospective cancer biomarkers. Genes Dev. 2012;26(12):1287–1299. doi:10.1101/gad.192351.112
16. Lee TH, D’Asti E, Magnus N, Al-Nedawi K, Meehan B, Rak J. Microvesicles as mediators of intercellular communication in cancer—the emerging science of cellular ‘debris. Semin Immunopathol. 2011;33(5):455–467. doi:10.1007/s00281-011-0250-3
17. Quesenberry PJ, Aliotta JM. Cellular phenotype switching and microvesicles. Adv Drug Delivery Rev. 2010;62(12):1141–1148. doi:10.1016/j.addr.2010.06.001
18. Tang K, Zhang Y, Zhang H, et al. Delivery of chemotherapeutic drugs in tumour cell-derived microparticles. Nat Commun. 2012;3(1):1282. doi:10.1038/ncomms2282
19. Zhang H, Tang K, Zhang Y, et al. Cell-free tumor microparticle vaccines stimulate dendritic cells via cgas/sting signaling. Cancer Immunol Res. 2015;3(2):196–205. doi:10.1158/2326-6066.CIR-14-0177
20. Ortiz-Bonilla CJ, Uccello TP, Gerber SA, Lord EM, Messing EM, Lee YF. Bladder cancer extracellular vesicles elicit a CD8 T cell-mediated antitumor immunity. IJMS. 2022;23(6):2904. doi:10.3390/ijms23062904
21. Ma J, Zhang Y, Tang K, et al. Reversing drug resistance of soft tumor-repopulating cells by tumor cell-derived chemotherapeutic microparticles. Cell Res. 2016;26(6):713–727. doi:10.1038/cr.2016.53
22. Guo M, Wu F, Hu G, et al. Autologous tumor cell–derived microparticle-based targeted chemotherapy in lung cancer patients with malignant pleural effusion. Sci Transl Med. 2019;11(474):eaat5690. doi:10.1126/scitranslmed.aat5690
23. Wan C, Sun Y, Tian Y, et al. Irradiated tumor cell–derived microparticles mediate tumor eradication via cell killing and immune reprogramming. Sci Adv. 2020;6(13):eaay9789. doi:10.1126/sciadv.aay9789
24. Gao Y, Zhang H, Zhou N, et al. Methotrexate-loaded tumour-cell-derived microvesicles can relieve biliary obstruction in patients with extrahepatic cholangiocarcinoma. Nat Biomed Eng. 2020;4(7):743–753. doi:10.1038/s41551-020-0583-0
25. Wei K, Zhang H, Yang S, et al. Chemo-drugs in cell microparticles reset antitumor activity of macrophages by activating lysosomal P450 and nuclear hnRNPA2B1. Sig Transduct Target Ther. 2023;8(1):22. doi:10.1038/s41392-022-01212-7
26. Ma J, Wei K, Zhang H, et al. Mechanisms by which dendritic cells present tumor microparticle antigens to cd8+ t cells. Cancer Immunol Res. 2018;6(9):1057–1068. doi:10.1158/2326-6066.CIR-17-0716
27. Sun Y, Zheng Z, Zhang H, et al. Chemotherapeutic tumor microparticles combining low-dose irradiation reprogram tumor-promoting macrophages through a tumor-repopulating cell-curtailing pathway. OncoImmunology. 2017;6(6):e1309487. doi:10.1080/2162402X.2017.1309487
28. Palmer DH, Midgley RS, Mirza N, et al. A Phase II study of adoptive immunotherapy using dendritic cells pulsed with tumor lysate in patients with hepatocellular carcinoma. Hepatology. 2009;49(1):124–132. doi:10.1002/hep.22626
29. Meko’o JLD, Xing Y, Zhang H, Lu Y, Wu J, Cao R. immunopreventive effects against murine h22 hepatocellular carcinoma in vivo by a DNA vaccine targeting a gastrin-releasing peptide. Asian Pac J Cancer Prev. 2014;15(20):9039–9043. doi:10.7314/APJCP.2014.15.20.9039
30. Hochnadel I, Hoenicke L, Petriv N, et al. Safety and efficacy of prophylactic and therapeutic vaccine based on live-attenuated Listeria monocytogenes in hepatobiliary cancers. Oncogene. 2022;41(14):2039–2053. doi:10.1038/s41388-022-02222-z
31. Kwon S, Kim D, Park BK, et al. Induction of immunological memory response by vaccination with TM4SF5 epitope-CpG-DNA-liposome complex in a mouse hepatocellular carcinoma model. Oncol Rep. 2013;29(2):735–740. doi:10.3892/or.2012.2130
32. Cai Z, Su X, Qiu L, et al. Personalized neoantigen vaccine prevents postoperative recurrence in hepatocellular carcinoma patients with vascular invasion. Mol Cancer. 2021;20(1):164. doi:10.1186/s12943-021-01467-8
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.