")
Back to Journals » International Medical Case Reports Journal » Volume 10
Drug-induced dyspnea versus cystic fibrosis exacerbation: a diagnostic dilemma
Authors Walayat S , Hussain N, Patel J, Hussain F, Patel P, Dhillon S, Aulakh B , Chittivelu S
Received 6 April 2017
Accepted for publication 28 June 2017
Published 19 July 2017 Volume 2017:10 Pages 243—246
DOI https://doi.org/10.2147/IMCRJ.S139022
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ronald Prineas
Video presented by Saqib Walayat
Views: 576
Saqib Walayat,1 Nooreen Hussain,1 Jaymon Patel,1 Faiz Hussain,2 Preeti Patel,1 Sonu Dhillon,1 Bhagat Aulakh,3,4 Subramanyam Chittivelu3
1Department of Internal Medicine, University of Illinois College of Medicine at Peoria, Peoria, 2Department of Internal Medicine, West Suburban Medical Center, Oak Park, 3Department of Pulmonary, 4Department of Critical Care, University of Illinois College of Medicine at Peoria, Peoria, IL, USA
Abstract: Cystic fibrosis (CF) is a disease caused by a mutation in the cystic fibrosis transmembrane conductance regulator protein in the epithelial membrane, and affects at least 30,000 people in the USA. There are between 900 and 1000 new cases diagnosed every year. Traditionally, CF has been treated symptomatically with pancreatic enzymes, bronchodilators, hypertonic saline, and pulmozyme. In July 2015, the US Food and Drug Administration approved Orkambi (lumacaftor/ivacaftor), a combination drug that works on reversing the effects of the defective cystic fibrosis transmembrane conductance regulator protein. Orkambi and mucolytics decrease the viscosity of mucous secretions, leading to an accumulation of hypoviscous fluid in the alveoli, resulting in dyspnea. This presentation can be mistaken for an infective exacerbation. We present a case in which a young female with CF recently started on Orkambi therapy presented to her primary care physician with dyspnea and increased respiratory secretions and was admitted to the hospital for 2 weeks of intravenous and inhaled antibiotic therapy for a presumed CF exacerbation. We highlight this case to bring awareness and educate patients and clinicians of the side-effect profile of Orkambi therapy with an intent to avoid unnecessary hospitalizations, inpatient antibiotics, and other costly medical services.
Keywords: cystic fibrosis, lumacaftor/ivacaftor, dyspnea, CFTR protein, pulmozyme, hypertonic saline, pancrelipase
Introduction
Cystic fibrosis (CF) is caused by a mutation in the gene that codes for cystic fibrosis transmembrane conductance regulator (CFTR) protein, which is most commonly present in the epithelial membrane (Figure 1). It is a disease known for multisystem dysfunction of organs such as the lungs and the pancreas, leading to premature death in affected patients. The most common CFTR mutation is Phe508del gene, which is present in almost 70% of patients, while about 45% of patients with CF are homozygous for this gene mutation.1,2 Traditionally, CF has been treated symptomatically with pancreatic enzymes for digestion, and bronchodilators, mucolytics, and DNase for airway congestion. In July 2015, the US Food and Drug Administration (FDA) approved Orkambi (lumacaftor/ivacaftor), a combination drug that works to help restore the function of the defective CFTR protein in CF patients.3
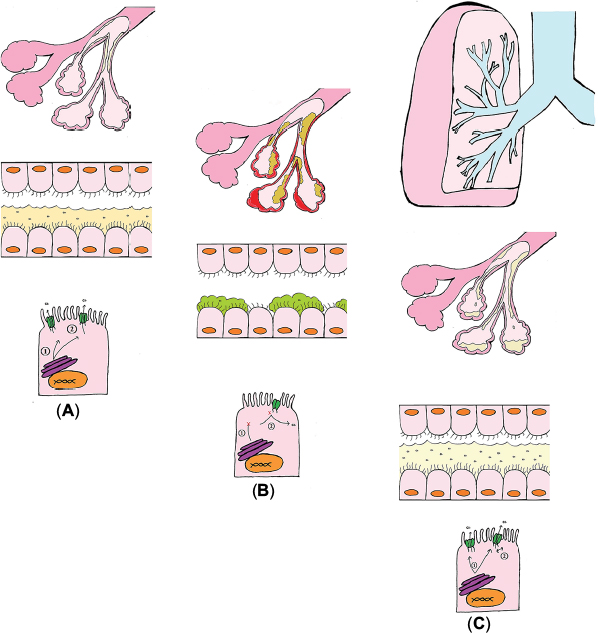 | Figure 1 Alveoli and respiratory epithelium of (A) normal lung (B) Cystic Fibrosis lung, and (C) Orkambi Therapy lung. Notes: (A) CFTR is expressed at the apical membrane and allows the flow of chloride and water in and out of the cell. (1) Normally it is tranlated, folded, and processed, then sent to the apical membrane. (2) Normal CFTR allows for the efflux of chloride and water to produce a thin mucus gel layer. (B) Cystic fibrosis is caused by mutations which result in a deficiency in and dysfunctional CFTR. (1) The F508del mutation results in impaired cellular processing. (2) Other mutations affect the conductance of the channel. (C) Orkambi is a combination drug that has a corrector and a potentiator. (1) Lumacaftor acts as a chaperone and aids in cellular processing. (2) Ivacaftor potentiates CFTR that is at the membrane. It keeps the channel open and allows chloride to flow through. |
Lumacaftor is an FDA-approved drug that corrects the processing of Phe508del gene, subsequently increasing CFTR protein availability at the cell surface, while ivacaftor increases the duration of the opening of the CFTR channels.4 While the two drugs individually have not been shown to improve clinical outcomes, when these two medications were combined they have been shown to improve forced expiratory volume in one second (FEV1), reduce the rate of pulmonary exacerbations, increase body mass index, and decrease sweat chloride in patients with CF who were homozygous for the Phe508del gene mutation.5
Previous studies have shown that Orkambi therapy has a side-effect profile that has generally been well tolerated by patients.1 Adverse effects include chest tightness, dyspnea, diarrhea, nausea, upper respiratory tract infection symptoms, and elevated liver enzymes.1 While long-term side effects of the drug remain unknown at this time, it has been suggested that these drugs may counteract each other, and eventually the benefit may be lost.1 We present a case of a patient with a homozygous Phe508del CF mutation on Orkambi therapy with a reflection on management of her symptoms, and recommendations to improve clinical outcomes.
Case report
A 31-year-old female with a history of CF (genotype homozygous ΔF508) who had been started by her primary care provider on Orkambi (lumacaftor/ivacaftor) six weeks earlier, presented to our hospital with complaints of experiencing increased fatigue and feeling “more winded”. At baseline, the patient stated that she was an active individual who ran up to a mile comfortably. More recently, she noticed an increase in sputum and sinus secretions, and felt as if she were “drowning” in her secretions. She was admitted to the hospital for dyspnea and a concern for a CF exacerbation as she grew multidrug resistant (MDR) Pseudomonas aeruginosa in previous sputum cultures. New pulmonary function studies showed a decreased FEV1/FVC (forced vital capacity) of 56% of predicted from 61% from 3 months prior. Her FEV1 also significantly decreased to 0.99 L, 34% of predicted from 58%. A computed tomography scan of her chest showed bilateral upper, right middle and lower lobe bronchiectasis, mucous plugging, cavitary lesions, fibrous scarring, and an atrophic pancreas. The patient was treated with meropenem (2 g q 8 h) and tobramycin (IV 700 mg qd and inhaled 300 mg bid) for 14 days as repeat sputum cultures during the admission grew three strains of MDR P. aeruginosa sensitive only to meropenem. Her CF exacerbation was also treated with albuterol and hypertonic saline nebulization, Advair, and Orkambi daily along with aggressive pulmonary rehabilitation. Pulmonary function tests showed serial improvement in FEV1 from 34% predicted to 42% predicted. Her respiratory status improved to near baseline and she was discharged after 2 weeks of inpatient therapy.
Written informed consent was obtained from the patient for the case details to be published.
Discussion and review
The advancement in therapy for CF is truly one of the success stories in medicine. This disease was first described in 1938 by Dr Dorothy Andersen. At that time, life expectancy with CF was only around 6 months, while in 2013, this had increased to 40.7 years.5,6 This success is attributed to a multidisciplinary approach in the management of CF. This approach focuses on nutrition repletion, relieving airway obstruction, infection management, and suppression of inflammation. Nutrition repletion is crucial, as more than 85% of affected patients have pancreatic insufficiency at birth.7 This has been addressed with the advent of pancreatic enzymes, which have been shown to significantly improve nutrition status in patients.6 Mucous plugging is a key feature of CF, and with the biotechnology of human DNase, hypertonic saline, B-agonist inhalers, and chest vest therapies (high frequency chest wall oscillation), there have been improvements in mucous clearing, thus relieving airway obstruction.6 Infections are managed by the use of oral, IV, and inhaled antibiotics chronically and acutely with a goal of treatment and prevention of respiratory tract infections.6 The final approach deals with suppression of inflammation, and this is done with steroids and nonsteroidal anti-inflammatory drugs; however therapy may often be limited due to side effects of these drugs.6
Orkambi therapy may now be thought of as another crucial approach under the umbrella of CF management. This is the first drug that has been FDA approved that works on the actual mechanism of action of the disease.8 This tablet contains fixed doses of lumacaftor and ivacaftor. Defects in CFTR chloride channel in CF impairs transport of salt and water, thus resulting in defective gland fluid secretion and reduced secreted fluid volume, increased protein concentration, and a viscous dehydrated air surface liquid, which causes reduced oxygen diffusion and a relatively hypoxic environment.9 The submucosal glands become hypertrophied as the disease progresses, with mucosal plugging of the airways.9 These hyperviscous secretions reduce airway mucociliary clearance and normal defense to antimicrobial infections.9 Ivacaftor has been studied to bind directly to and open the CFTR chloride channel via ATP-dependent mechanisms, while lumacaftor increases the quantity of CFTR protein present on the cell surface.10,11 This, in turn, increases the chloride flux through these channels, resulting in an increased amount of thin, watery fluid secretions.12 We believe that Orkambi and mucolytics work together in these patients to cause a decrease in viscosity of secretions, leading to an accumulation of fluid in the alveoli that results in respiratory distress and shortness of breath, mirroring a congestive heart failure exacerbation. This is an interesting observation that we have noticed at our CF clinic and more research needs to be done to validate this hypothesis. Dyspnea and respiratory chest tightness have been reported in 14% and 9.8% of the patients on Orkambi in a 24-week trial period.2
Our patient historically felt more on the dry side with regard to respiratory secretions. She was an active individual who liked to walk and run outdoors, and on a good day she was able to run a mile without difficulty. However, when started on Orkambi, she reported malaise, respiratory distress, and feeling that “she was drowning in her secretions”. Orkambi was continued along with aggressive pulmonary rehab and pulmonary toilet using the multidisciplinary approach, and her symptoms eventually improved.
In the PROGRESS trial (which extended for a 96-week period), the most common reported adverse effects were cough, increased sputum production, and infective pulmonary exacerbations.13 The classification of “infective pulmonary exacerbations” was based on the Modified Fuchs Criteria in which a patient is treated with parenteral antibiotics if they have 4 of 12 identified respiratory signs or symptoms.14 However, these respiratory symptoms can be nonspecific, and we believe that an individual presenting with just four of these symptoms may not actually have an active infection, as seen in our patient, who experienced dyspnea, malaise, change in sinus discharge, and a decrease in pulmonary function tests (PFTs) (4 of the 12 modified Fuchs Criteria). In reality, the individual may actually be experiencing a drug side effect that can be mistaken as an infective pulmonary exacerbation.
Thus, from our clinical experience, we would like to emphasize that patients started on Orkambi should be educated about this side-effect profile, and they should continue to be involved in pulmonary rehab and pulmonary toilet. The dyspnea they may be experiencing, we believe, is primarily due to increased secretions from the therapy itself, and not an exacerbation of the disease. However, due to these presenting symptoms, they will be hospitalized unnecessarily, and treated for a presumed “CF exacerbation”, while in reality what is being treated is a “drug-induced dyspnea” with unnecessary IV and inhaled antibiotics. Many of these individuals are chronically colonized with Pseudomonas and other bacteria in their respiratory cultures and may get exposed to more unnecessary antibiotics.
While the mean annual health cost for CF patients is based on severity and progression of the disease, the majority of costs are accounted for by hospitalized inpatients (58%), medications (29%), medical services (10%), complications (2%), and diagnostic testing (1%).15 Our patient underwent aggressive pulmonary rehab and toilet along with education about the expected side-effect profile. She was subsequently continued on Orkambi with a favorable outcome. Her PFTs and respiratory status improved when continuing Orkambi along with Pulmozyme, albuterol, and Advair daily. We bring this case up to highlight the importance of awareness of the side-effect profile of Orkambi therapy. Doing so will allow clinicians and patients to maintain the multidisciplinary approach to CF management in the setting of possible adverse side effects of Orkambi therapy, and at the same time, avoid unnecessary hospitalizations, inpatient antibiotics, and other costly medical services.
Another point to be made with regard to management in these patients is to be aware of the effect of Orkambi in conjugation with Pulmozyme and hypertonic saline nebulization therapy. We believe that starting Orkambi while already on the other two drugs can actually cause an excessive amount of thin watery secretions, rather than its intent to break up hyperviscous secretions. Patients on Orkambi therapy may benefit from Pulmozyme alone, rather than a combination of Orkambi, Pulmozyme, and hypertonic saline. This, in turn, may limit these hypoviscous secretions. Further studies of this regimen may be of benefit for patients on Orkambi therapy.
More studies are underway to assess the effects of Orkambi on dyspnea and exercise performance (Clinical trials.gov).
Author contributions
All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Wainwright CE, Stuart Elborn J, Ramsey BW, et al. Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med. 2015;373(3):220–231. | ||
Schneider EK, Reyes‐Ortega F, Li J, Velkov T. Can cystic fibrosis patients finally catch a breath with Orkambi? Clin Pharmacol Ther. 2016;101:130–141. | ||
Veit G, Avramescu RG, Chiang AN, et al. From CFTR biology toward combinatorial pharmacotherapy: expanded classification of cystic fibrosis mutations. Mol Biol Cell. 2016;27(3):424–433. | ||
Wainwright CE, Elborn JS, Ramsey BW, et al; TRAFFIC Study Group; TRANSPORT Study Group. Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med. 2015;373(3):220–231. | ||
Spoonhower KA, Davis PB. Epidemiology of cystic fibrosis. Clin Chest Med. 2016;37(1):1–8. | ||
Davis PB. Cystic fibrosis since 1938. Am J Respir Crit Care Med. 2006; 173(5):475–482. | ||
Mascarenhas MR. Treatment of gastrointestinal problems in cystic fibrosis. Curr Treat Options Gastroenterol. 2003;6(5):427–441. | ||
Deeks ED. Lumacaftor/Ivacaftor: a review in cystic fibrosis. Drugs. 2016;76(12):1191–1201. | ||
Verkman AS, Song Y, Thiagarajah JR. Role of airway surface liquid and submucosal glands in cystic fibrosis lung disease. Am J Physiol Cell Physiol. 2003;284(1):C2–C15. | ||
Eckford PD, Li C, Ramjeesingh M, Bear CE. Cystic fibrosis transmembrane conductance regulator (CFTR) potentiator VX-770 (ivacaftor) opens the defective channel gate of mutant CFTR in a phosphorylation-dependent but ATP-independent manner. J Biol Chem. 2012;287(44):36639–36649. | ||
Ren HY, Grove DE, De La Rosa O, et al. VX-809 corrects folding defects in cystic fibrosis transmembrane conductance regulator protein through action on membrane-spanning domain 1. Mol Biol Cell. 2013;24(19):3016–3024. | ||
Boyle MP, De Boeck K. A new era in the treatment of cystic fibrosis: correction of the underlying CFTR defect. Lancet Respir Med. 2013;1(2):158–163. | ||
Konstan MW, McKone EF, Moss RB, et al. Assessment of safety and efficacy of long-term treatment with combination lumacaftor and ivacaftor therapy in patients with cystic fibrosis homozygous for the F508del-CFTR mutation (PROGRESS): a phase 3, extension study. Lancet Respir Med. 2017;5(2):107–118. | ||
Fuchs HJ, Borowitz DS, Christiansen DH, et al. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The Pulmozyme Study Group. N Engl J Med. 1994;331(10):637–642. | ||
Van Gool K, Norman R, Delatycki MB, Hall J, Massie J. Understanding the costs of care for cystic fibrosis: an analysis by age and health state. Value Health. 2013;16(2):345–355. | ||
Elkins MR, Bye PT. Mechanisms and applications of hypertonic saline. J R Soc Med. 2011;104(Suppl 1):S2–S5. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.