Back to Journals » International Journal of Nanomedicine » Volume 13
Doxorubicin-loaded protease-activated near-infrared fluorescent polymeric nanoparticles for imaging and therapy of cancer
Authors Yildiz T, Gu R ![]() , Zauscher S, Betancourt T
, Zauscher S, Betancourt T
Received 22 May 2018
Accepted for publication 24 July 2018
Published 31 October 2018 Volume 2018:13 Pages 6961—6986
DOI https://doi.org/10.2147/IJN.S174068
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Video abstract presented by Tugba Yildiz
Views: 1537
Tugba Yildiz,1 Renpeng Gu,2 Stefan Zauscher,2 Tania Betancourt1,3
1Materials Science, Engineering, and Commercialization Program, Texas State University, San Marcos, TX, 2Department of Mechanical Engineering and Materials Science, Duke University, Durham, NC, 3Department of Chemistry and Biochemistry, Texas State University, San Marcos, TX, USA
Introduction: Despite significant progress in the field of oncology, cancer remains one of the leading causes of death. Chemotherapy is one of the most common treatment options for cancer patients but is well known to result in off-target toxicity. Theranostic nanomedicines that integrate diagnostic and therapeutic functions within an all-in-one platform can increase tumor selectivity for more effective chemotherapy and aid in diagnosis and monitoring of therapeutic responses.
Material and methods: In this work, theranostic nanoparticles were synthesized with commonly used biocompatible and biodegradable polymers and used as cancer contrast and therapeutic agents for optical imaging and treatment of breast cancer. These core–shell nanoparticles were prepared by nanoprecipitation of blends of the biodegradable and biocompatible amphiphilic copolymers poly(lactic-co-glycolic acid)-b-poly-L-lysine and poly(lactic acid)-b-poly(ethylene glycol). Poly-L-lysine in the first copolymer was covalently decorated with near-infrared fluorescent Alexa Fluor 750 molecules.
Results: The spherical nanoparticles had an average size of 60–80 nm. The chemotherapeutic drug doxorubicin was encapsulated in the core of nanoparticles at a loading of 3% (w:w) and controllably released over a period of 30 days. A 33-fold increase in near-infrared fluorescence, mediated by protease-mediated cleavage of the Alexa Fluor 750-labeled poly-L-lysine on the surface of the nanoparticles, was observed upon interaction with the model protease trypsin. The cytocompatibility of drug-free nanoparticles and growth inhibition of drug-loaded nanoparticles on MDA-MB-231 breast cancer cells were investigated with a luminescence cell-viability assay. Drug-free nanoparticles were found to cause minimal toxicity, even at high concentrations (0.2–2,000 µg/mL), while doxorubicin-loaded nanoparticles significantly reduced cell viability at drug concentrations >10 µM. Finally, the interaction of the nanoparticles with breast cancer cells was studied utilizing fluorescence microscopy, demonstrating the potential of the nanoparticles to act as near-infrared fluorescence optical imaging agents and drug-delivery carriers.
Conclusion: Doxorubicin-loaded, enzymatically activatable nanoparticles of less than 100 nm were prepared successfully by nanoprecipitation of copolymer blends. These nanoparticles were found to be suitable as controlled drug delivery systems and contrast agents for imaging of cancer cells.
Keywords: nanomedicine, theranostics, drug delivery, fluorescence imaging, enzymatic activation, nanoprecipitation, block copolymers, PLGA, PLA, PEG, poly-L-lysine, nanoparticles
Introduction
Despite great progress in prevention, detection, and treatment modalities, cancer remains one of the most common and deadly diseases worldwide and is the second-leading cause of mortality of all Americans.1 Breast cancer is the most commonly diagnosed cancer in women and the second-leading cause of cancer deaths in women worldwide.2 Specifically, breast cancer represents 30% of all new cancer cases in the US.3 The American Cancer Society estimated that in the US in 2017 there would be over 250,000 new cases and over 40,000 people would die of this disease. The most common cancer treatment methods for breast cancer are surgery, chemotherapy, radiation therapy, hormone therapy, or antibody-based therapy, with the preferred treatment being surgery followed by chemotherapy. The most common chemotherapeutic drugs used for early breast cancer are anthracyclines (doxorubicin and epirubicin) and taxanes (paclitaxel and docetaxel).4 Chemotherapy often causes systemic toxicity-related side effects, due to aspecific biodistribution and activity. Further, half of all chemotherapy patients develop drug resistance, a situation that also leads to treatment failure.5 For this reason, efforts to develop alternative chemotherapy treatments have focused on designing nanoscale agents that are more specific against cancer cells to minimize toxic side effects and improve therapeutic efficacy.6–13 The use of nanoscale agents enhances tumor selectivity via passive and active targeting.14
The combination of therapeutic and diagnostic agents within a single nanoscale “theranostic” platform offers significant potential to make personalized nanomedicine a clinical reality for cancer patients.15 The biodistribution, pharmacokinetics, and tumor accumulation of theranostic nanoparticles (NPs) can be monitored while at the same time enabling a therapeutic response and obtaining information about the biological characteristics of the tumor tissue (eg, protease overexpression, which is known to be related to tumor aggression and therapeutic outcome). Clinicians can then make decisions about dosage, frequency, drug choice, and treatment strategies by tracking therapeutic responses. Theranostic nanomedicines can be monitored by noninvasive molecular imaging techniques, including those based on optical imaging.16 Optical imaging offers many advantages over other imaging techniques, because it is generally safer and considerably simpler, and it can be used to monitor the progression of cancer and the effectiveness of treatment over time. Optical imaging using fluorescent probes has been preclinically and clinically used for imaging of superficial tissue, tissue that can be accessed endoscopically or laparoscopically,17–20 internal tissue imaging during surgical procedures (intraoperative fluorescence imaging or fluorescence-guided surgery),21,22 and ex vivo analysis of diseased tissue after in vivo probe administration.23,24 Specifically, clinical fluorescence imaging has been readily used for imaging of tissue perfusion, fluorescence-guided surgery,21,22 sentinel lymph-node mapping and general lymphography,20,25–27 detection of premalignant/malignant lesions (ClinicalTrials.gov; NCT02113202 and NCT01972373), and tumor identification and margin detection,24,28–30 among other applications. Several fluorescence-imaging systems, including the SPY system from Novadaq Technologies, and fluorescent contrast agents, including indocyanine green, methyl blue, and fluorescein, have been US Food and Drug Administration-approved for clinical use, boding well for the translational future of fluorescence-based theranostic nanomedicines.
Theranostic NPs are considered the future of medicine, allowing for less invasive imaging, detection, and therapy. These NPs can be used for diagnosis or investigation of disease characteristics, followed by therapy with personalized strategies likely to be effective to a given patient, or for administering therapy and monitoring early response to treatment. Theranostic NPs have found wide application not only in cancer treatment but also inflammatory/autoimmune disease,31–34 neurodegenerative diseases,35,36 diabetes,37,38 and lung39 and cardiovascular40,41 afflictions. Various types of theranostic NPs based on organic and inorganic materials have been developed over the last decade for imaging and treating cancer,42 including quantum dots, carbon nanotubes, and gold, silica, iron oxide, and polymer NPs. Due to their biocompatibility and diversity, polymer-based theranostic NPs have become some of the most intensely studied platforms for image-guided therapy. Polymer-based theranostic systems developed to date include polymer conjugates, nanocapsules, polymersomes, dendrimers, hydrogels, micelles, and NPs.43
Doxorubicin (Dox) has been used for treatment of several different types of cancer, such as leukemia, lung, breast, stomach, ovarian, and soft-tissue sarcoma.44 Dox intercalates into base pairs of DNA, and localizes in the minor groove of the double helix. The progression of the enzyme topoisomerase II, which winds DNA for transcription, is inhibited by Dox intercalation.45,46 To reduce the systemic toxicity of Dox caused by its conventional administration, it has been encapsulated within PEGylated liposomes to make it more targetable for tumors.47,48 Dox is inherently fluorescent, due to its central anthracycline chromophore group. Dox distribution in tissue or cells can thus be viewed under fluorescence-imaging systems in the visible range.44 However, absorption and emission interference between Dox and biological chromophores, such as hemoglobin, oxyhemoglobin, and melanin, hamper Dox’s efficiency as an imaging agent for tumor diagnosis and monitoring, especially in vivo. This is the main reason that other imaging agents, such as those with near-infrared (NIR) capabilities, need to be used to monitor tumor location and response to Dox treatment.
In this work, we prepared theranostic NPs for NIR imaging and Dox delivery to the highly invasive MDA-MB-231 ductal carcinoma cells. This cell line is known as triple-negative, because both estrogen and progesterone hormone receptors and HER2 show minimal–no expression. Triple-negative cancer cells do not respond to hormonal or HER2-receptor-targeted therapies, because these hormones do not support their growth. This cell line thereby represents a more aggressive, higher-grade, and more likely recurrent tumor type than other types of breast cancer.49,50
Our theranostic NPs are designed in such a way that NIR-fluorescence contrast is activated by proteolytic enzymes that are overexpressed in tumor tissue. High levels of proteases are involved in progression and tumor growth at both primary and metastatic sites. Tumor margins secrete high amounts of proteases to degrade the basement membrane and extracellular matrix and open up more space for tumor growth.51–54 Peptide-based NIR-fluorescence-imaging probes have been studied broadly as targeting optical probes for image-guided drug delivery and for monitoring of drug pharmacokinetics, distribution, and tumor accumulation in real time.55 These probes can interact with proteases overexpressed by tumors, such as cathepsins, MMPs, and trypsin.56–60 Serine proteases, such as trypsin, are associated with such diseases as cancer and atherosclerosis. Trypsin specifically is known to participate in colorectal carcinogenesis, where it is critical for cancer-cell proliferation and metastasis.61,62 Highly invasive metastatic breast carcinoma cell lines secrete overexpressed trypsin enzymes, leading to autocrine activation of cell-surface PAR2.63 PAR2 is overexpressed in a variety of breast cancer cells, such as MDA-MB-231, and causes cell growth, migration, and invasion.64–66
Here, we focus on the development and evaluation of theranostic NPs that can be easily synthesized with commonly used, biocompatible, and biodegradable polymers, and that can be controlled to provide imaging contrast and exert a therapeutic effect to treat breast cancer. Based on our prior work on the synthesis of enzymatically activated NIR fluorescent nanoprobes,67 in this work NPs with a core–shell structure (Figure 1) were prepared by nanoprecipitation of amphiphilic block-copolymer blends. The hydrophobic core was used to load the chemotherapeutic agent Dox free base (Dox-FB) for drug delivery. The core was made of the biodegradable and biocompatible polymers poly(lactic-co-glycolic acid) (PLGA) and poly(lactic acid) (PLA). NP surfaces contained the hydrophilic polymers poly(ethylene glycol) (PEG) and protease-cleavable polypeptide poly-L-lysine (PLL). The polypeptide was used as a site for conjugation of NIR fluorescent Alexa Fluor 750 (AF750) for imaging of tumor cells. The densely decorated NIR-fluorescent molecules self-quench on the NP surface, providing a means for enzymatically controlled fluorescence development in cancerous tissue through the action of tumor proteases. The synthesis and characterization of copolymers, conjugation of NIR dye on polypeptide, drug loading and release, and characterization of NPs, as well as in vitro evaluation of the function of the NPs as imaging and therapeutic agents, are described.
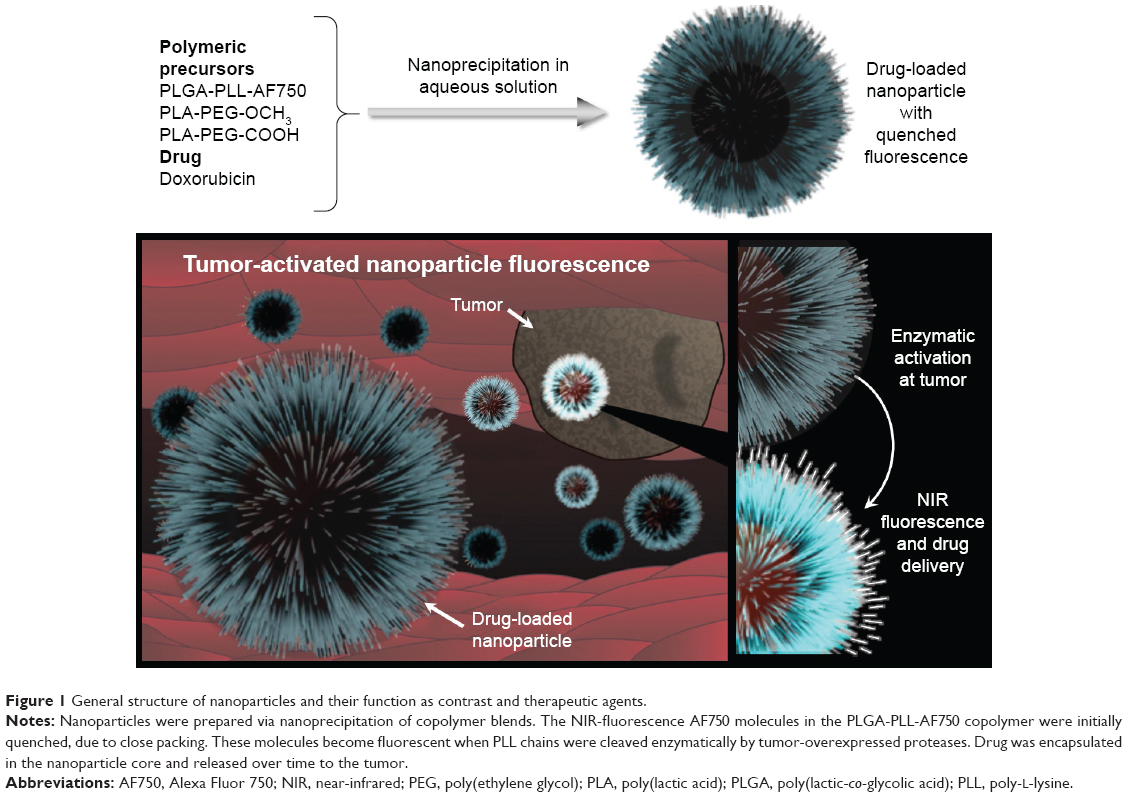 | Figure 1 General structure of nanoparticles and their function as contrast and therapeutic agents. |
Materials and methods
Materials
Carboxylic acid-terminated PLGA copolymer (PLGA-COOH) was bought from Corbion (Amsterdam, the Netherlands). The PLGA-COOH was 17 kDa in molecular weight (MW) and composed of 50:50 D,L-lactide:glycolide molar ratio. Heterofunctional PEG (OH-PEG-COOH) of 5 kDa molecular weight was obtained from Laysan Bio (Arab, AL, USA). Heterofunctional PEG methyl ether (OH-PEG-OCH3, MW 5 kDa), PLL hydrobromide (MW 3.8 kDa), poly-D-lysine (PDL) hydrobromide (MW 3.8 kDa), N-α-tosyl-L-lysine chloromethyl ketone hydrochloride (TLCK), N,N′-dicyclohexylcarbodiimide (DCC), trypsin solution from porcine pancreas (10×, 25 g/L solution in 0.9% sodium chloride), and BSA were acquired from Sigma-Aldrich (St Louis, MO, USA). N-hydroxysuccinimide (NHS) was purchased from Thermo Fisher Scientific (Waltham, MA, USA). 3,6-Dimethyl-1,3-dioxane-2,5-dione (D,L-lactide), N-(dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (HCl), tin(II) 2-ethylhexanoate (stannous octoate, Sn[Oct]2), Dox HCl, triethylamine (TEA), and MTT were obtained from Alfa Aesar (Haverhill, MA, USA). AF750 succinimidyl ester was obtained from Thermo Fisher Scientific. CellTiter-Glo Luminescent cell-viability assay was obtained from Promega (Madison, WI, USA). Toluene, methanol, acetone, dimethyl sulfoxide (DMSO), dimethyl formamide (DMF), and diethyl ether solvents were American Chemical Society grade. Ultrapure deionized water was obtained from a Millipore Direct Q system.
Instrumentation
The structure of copolymers was characterized using proton nuclear magnetic resonance (1H-NMR) spectroscopy with a 400 MHz Bruker Avance. Fourier-transform IR (FTIR) spectra of copolymers were acquired with a Bruker Tensor 27. Samples were prepared for FTIR spectroscopy by depositing copolymer solutions onto polished sodium chloride crystal optic disks (Alfa Aesar 38415) and fully drying them prior to analysis. Ultraviolet-visible NIR-luminescence measurements were obtained using a BioTek Synergy 4 hybrid microplate reader. Scanning electron microscopy (SEM) images were obtained using an FEI Helios NanoLab 400 (10 kV and 0.34 nA) at 80,000× magnification. SEM samples were coated with 2 nm iridium using an EMS 150T turbo-pumped sputter-coater. Transmission electron microscopy (TEM) images were obtained using a JEOL JEM 1200 EXII (beam current 80 μA) with a Gatan Orius charge-coupled-device digital camera at 150,000× magnification. Tapping-mode atomic force microscopy (AFM) images were acquired with a Bruker Multimode AFM system using tapping-mode-air silicon cantilevers (kF 20–80 N/m, fres 230–410 kHz, Rtip <12 nm). NanoScope Analysis software was used to analyze the images and develop the average height and diameter of the features. Dynamic light-scattering (DLS) measurements were obtained using a Malvern Zetasizer Nano ZS using a 173° backscatter-measurement angle. A VWR Symphony Ultrasonic Cleaner was used for resuspension of NP pellets. A Beckman Coulter Avanti J-26 XPI centrifuge was used to purify polymers and NPs.
Cell culture
MDA-MB-231 human mammary adenocarcinoma cells were obtained from the American Type Culture Collection. Cells were grown in DMEM supplemented with 10% fetal bovine serum, 1% HEPES, and 1% penicillin–streptomycin in a 37°C humidified incubator with a 5% CO2 and 95% air atmosphere.
Synthesis of PLA-PEG-COOH and PLA-PEG-OCH3
PLA-PEG copolymers were synthesized via ring-opening polymerization of lactide. D,L-Lactide and carboxylated or methoxylated PEG were reacted in the presence of stannous octoate as a catalyst and toluene as a solvent at 110°C for 16 hours, as previously described.68 Copolymers were recovered by precipitation in diethyl ether. For further purification, polymers were redissolved in acetone and precipitated in diethyl ether twice more. The polymer was then freeze-dried and stored desiccated at −25°C for later use. The synthesis scheme is shown in Figure 2.
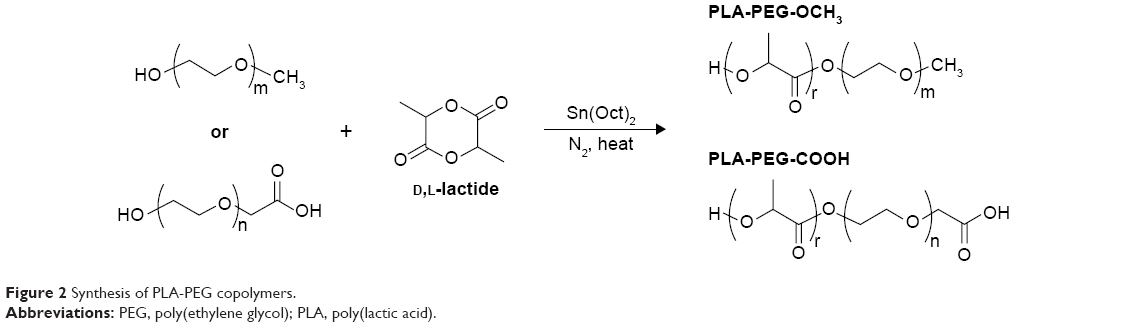 | Figure 2 Synthesis of PLA-PEG copolymers. |
Synthesis of PLGA-PLL
PLGA-PLL was prepared by carbodiimide-mediated conjugation of PLGA-COOH and PLL. PLGA-COOH (0.5 g), DCC (0.062 g), and NHS (0.035 g) (PLGA:NHS:DCC molar ratio of 1:10:10) were dissolved in 4 mL DMF and reacted for 4 hours while stirring under argon to activate PLGA-COOH into the amine-reactive NHS ester. Activated PLGA-NHS was reacted with 0.121 g PLL in methanol in the presence of 8.43 μL TEA (PLGA:PLL:TEA molar ratio of 1:1:2) for 24 hours at room temperature. Figure 3 details the conjugation of PLGA-PLL. An identical method was utilized for the preparation of PLGA-PDL, which was used as a non-protease-labile control in this work. Copolymers were recovered by precipitation in deionized (DI) water and centrifuging. The polymer pellet was then dissolved in acetone, precipitated in water, and centrifuged twice more to remove unbound water-soluble PLL and remaining byproducts. The resulting polymers were then freeze-dried and stored at −25°C.
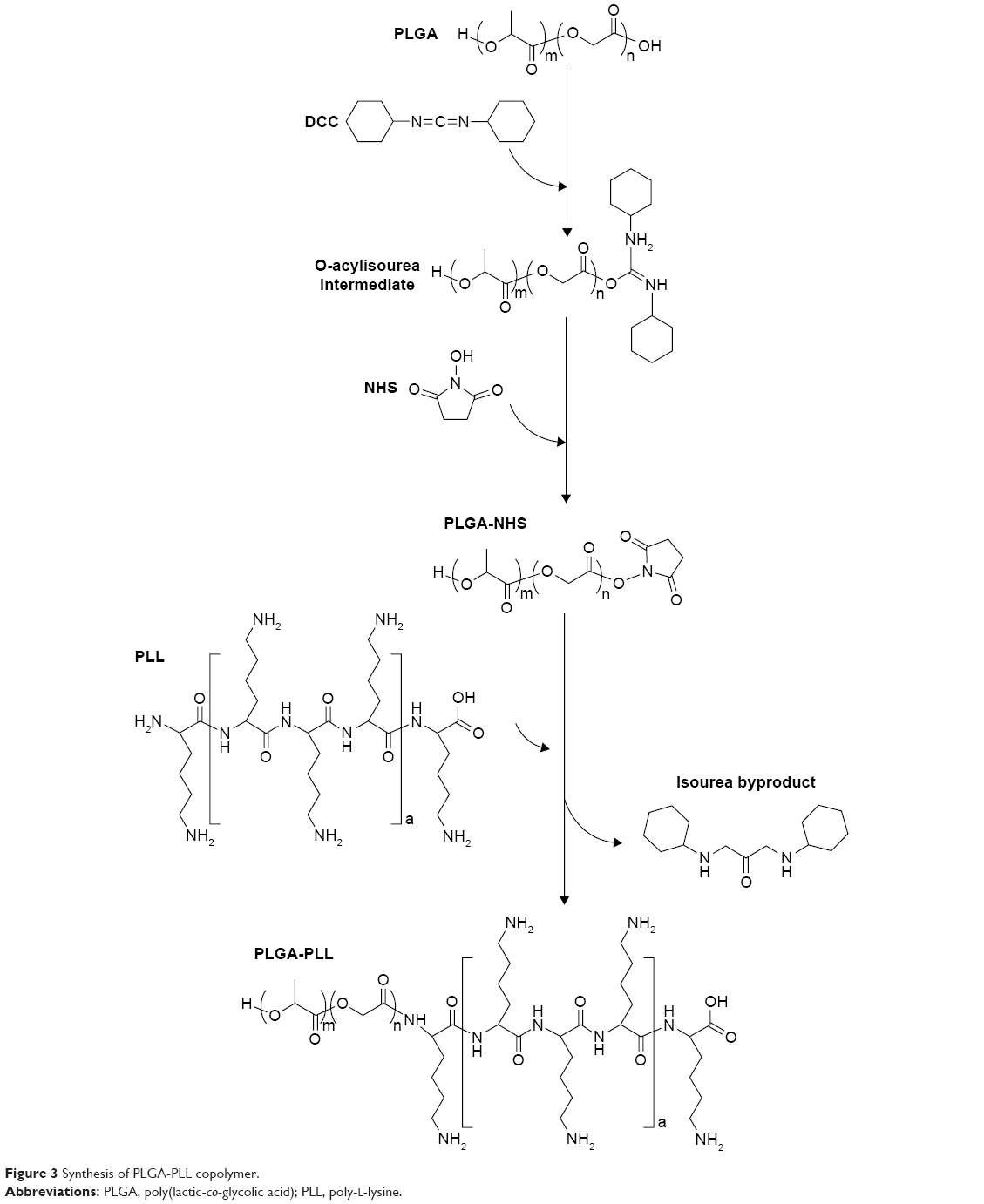 | Figure 3 Synthesis of PLGA-PLL copolymer. |
Conjugation of fluorescent dye to PLGA-PLL copolymer
PLGA-PLL copolymer (6.25 mg) was dissolved in 125 μL DMF. A mass of 0.3 mg AF750 NIR fluorescent dye in 300 μL DMF was added to the copolymer solution in the presence of 13.25 μL TEA for 12 hours in the dark at room temperature. The theoretical feed molar ratio of AF750/PLL is 2.5. AF750 NHS ester reacts with primary amines on PLL, forming stable amide bonds. PLGA-PLL-AF750 polymer conjugate was purified by three cycles of precipitation in ice-cold methanol and dissolution in acetone to remove unbound AF750, and then dried under vacuum. An identical method was used for fluorescence labeling of PLGA-PDL, which acted as a inactivable fluorescence control in our studies.
Preparation of NPs
For preparation of drug-loaded NPs, Dox HCl was converted to its hydrophobic FB form utilizing a method adopted from the literature.69 Dox HCl was dispersed in 12.5% v:v methanol in acetone and deprotonated using TEA (1:5 molar ratio of Dox to TEA) at 3.5 mg/mL. After overnight reaction, the resulting clear-orange solution of Dox-FB was evaporated and freeze-dried.
NPs loaded with Dox-FB and labeled with AF750 (Dox-FB-AF750 NPs) were prepared by nanoprecipitation of blends of PLGA-PLL-AF750/PLGA-PDL-AF750, PLA-PEG-COOH, PLA-PEG-OCH3, and Dox-FB. Dox-FB was dissolved in DMSO at 3.5 mg/mL. Each of the three copolymers were dissolved in acetone separately at 50 mg/mL. The polymer blend (187.5 μL, including 125 μL PLGA-PLL-AF750, 31.25 μL PLA-PEG-COOH, and 31.25 μL PLA-PEG-OCH3) and Dox-FB solution (141 μL) were combined with acetone (500 μL) and methanol (86 μL). The final polymer concentration was 10.25 mg/mL in the organic phase. The mixed solution (mass ratio of PLGA-PLL-AF750:PLA-PEG-COOH:PLA-PEG-OCH3 4:1:1) was then added dropwise to 3.75 mL stirring aqueous solution of 10 mg/mL BSA to enable NP formation via nanoprecipitation. Dox-FB-only NPs (no AF750) and blank NPs (no Dox-FB, no AF750) were also prepared as described, but utilizing PLGA-PLL (or PLGA-PDL for inactivable controls) instead of the fluorescently labeled copolymer and replacing Dox-FB solution with the equivalent volume of DMSO for the latter case.
NPs were centrifuged at 74,200 g for an hour at 10°C and washed five times with 10 mg/mL BSA in DI water. BSA was used as a stabilizer to minimize NP aggregation during centrifugation.70 The NP pellet was resuspended with PBS after the last centrifugation for further studies. The size of NPs was determined using DLS with appropriate dispersant parameters (viscosity and refractive index of 10 mg/mL BSA, 1 mg/mL BSA, or PBS for unwashed and washed/redispersed NPs). The stability of NPs over time in PBS and in serum was evaluated by DLS. For this purpose, NPs were suspended in PBS and then 100 μL NP suspension was diluted in PBS and/or serum as appropriate to prepare samples with final concentrations of 0, 10%, 20%, and 90% FBS. Samples were then stored at room temperature (22°C) or 37°C for 4 days.
The ζ-potential of the NPs was determined using laser Doppler microelectrophoresis with the Malvern Zetasizer Nano ZS. This technique is used to measure the movement of charged particles in an electric field. Particle mobility is determined from the known applied electric field and measured particle velocity. The ζ-potential is then calculated from mobility using the Smoluchowski model.71 For ζ-potential measurements, a few drops of aqueous NP suspension were added to 1 mL 1 mM potassium chloride (KCl). A dip-cell electrode was then used to determine the ζ-potential of NPs.
The size and morphology of NPs were examined by TEM, SEM, and AFM. For TEM imaging, NP suspensions were dried on carbon-coated 200-mesh copper grids and stained with 2% aqueous uranyl acetate. For SEM imaging, a drop of NP suspension was dried on a silicon wafer. NPs were then coated with iridium (2 nm thickness) before SEM imaging. Samples were prepared for AFM by placing a drop of the NP suspension onto freshly cleaved mica.
Loading percentage of Dox within NPs
NPs were first frozen at −86°C and freeze-dried with a Labconco FreeZone lyophilizer. Drug loading and entrapment efficiency of Dox-FB in NPs were determined via fluorescence spectroscopy using a BioTek H4 multimode plate reader (λEx 500 nm, λEm 600 nm). A known mass of freeze-dried NPs was dissolved in a known volume of DMSO. For determination of drug-loading percentage, the concentration and mass of Dox in NPs were determined based on a standard curve of Dox in DMSO. Drug loading was determined utilizing Equation 1:
|
|
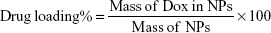
Enzymatic activation of NP fluorescence
Dox-loaded AF750-labeled NPs were diluted in PBS to a tenth the concentration of the NP stock (0.2 mg/mL, value based on the known mass of NPs after freeze-drying). The BioTek H4 multimode plate reader was used to determine the fluorescent enhancement of the NPs (λEx 740 nm, λEm 780 nm). Four repetitions were done for each sample. A volume of 50 μL 0.2 mg/mL NPs was mixed with 40 μL PBS and 30 μL 10× trypsin (25 mg/mL). Fluorescence development was recorded at specific intervals for up to 4 hours. Controls included NPs that were incubated with PBS buffer (10 mM) in the absence of a protease, as well as NPs exposed to trypsin together with the protease inhibitor TLCK (1:1 molar ratio of trypsin to TLCK). Fluorescence readings were normalized with respect to the control consisting of NPs exposed to PBS only. The same study was carried out with NPs contained PLGA-PDL instead of PLGA-PLL as a control to show lack of fluorescent development based on trypsin’s lack of activity on PDL. Paired two-tailed t-tests were used to determine statistical differences between individual conditions and the control (NPs exposed to PBS).
In vitro drug-release study
Drug (Dox) release from NPs was studied using a dialysis method. A D-Tube dialyzer mini (MW cutoff 12–14 kDa; EMD Millipore) tubes were filled with 180 μL 2 mg/mL NP suspension (actual mass of Dox in NPs was 0.28 mg), based on loading and determination of the known mass of NPs after freeze-drying. Dialyzer tubes were immersed in 5 mL PBS buffer solution (release medium, pH 7.4) at 37°C and stirred continuously at 200 rpm using an OptiMag Plus hot-plate stirrer with a round dry block for 20 mL scintillation vials. Four independent replicates were used for each sample. Dox HCl solution in DI water at the same concentration of Dox-FB in NPs was used as control to show the rapid diffusion of a dissolved (unencapsulated) molecule of similar size and properties to Dox-FB across the dialysis membranes utilized. Aliquots (120 μL) of the release medium were collected for analysis at different time intervals and replaced with 120 μL PBS buffer at 37°C. Dox-FB NP-release runs were continued for 30 days, while those of Dox HCl were stopped at 24 hours, when complete dialysis was visually observed.
Release samples were stored at −25°C until the end of the study. At the end of the study, the samples remaining in the dialyzer tubes were freeze-dried and dissolved in DMSO. The fluorescence of Dox in the release samples and NPs in dialyzer tubes was read with the BioTek H4 multimode plate reader at λEx 500 nm and λEm 600 nm. The mass released at each time point was determined using a standard calibration curve.
Determination of NP cytocompatibility
The CellTiter-Glo luminescence assay was used for determination of NP cytocompatibility. MDA-MB-231 breast cancer cells were cultivated in complete DMEM (including 10% FBS) at 37°C and 5% CO2. For these studies, cells were seeded in black clear-bottom 96-well plates (Falcon 353219) at a density of 7,500 cells/well with 100 μL complete medium. After 36 hours of incubation to provide sufficient time for the cells to adhere to the plates, the growth medium was replaced with suspensions of blank NPs (NPs free of Dox or AF750) in the concentration range of 0.02–2,000 μg/mL and cells incubated for 72 hours. Cells cultured in complete media without NPs represented the positive viability control (+), and cell-free wells with serum-free medium were used as the negative control (−). After the period of exposure, media were replaced with 100 μL CellTiter-Glo luminescence mixture. This mixture was prepared by dissolving the lyophilized CellTiter-Glo substrate (one vial) in CellTiter-Glo Buffer (100 mL), as specified by the manufacturer. The mixture was diluted 1:5 with Tris buffer (250 mM Tris, 27 mM KCl, 1.37 M NaCl, 10% glycerol, 1% Triton X-100) before use in the cells. The 96-well plate was mixed for 2 minutes on a shaker, and after a 10-minute incubation at 37°C, the emitted luminescence was detected using the BioTek H4. Luminescence (λEm 540/35 nm) of six repeat wells for each concentration were read. Paired two-tailed t-tests were used to determine statistical differences between individual conditions and the control (cells incubated in full media).
Determination of NP-induced growth inhibition
Cell-growth inhibition induced by Dox-NPs was determined with the CellTiter-Glo luminescence assay. MDA-MB-231 cells were plated at 7,500 cells/well in black clear-bottom 96-well plates with 100 μL complete cell-culture medium. After 36 hours of incubation, the time necessary to enable cells to adhere fully to the surface of these plates, the growth medium was replaced with suspensions of Dox-FB NPs in a drug-concentration range of 0.001–100 μM and cells incubated for 72 hours. Dox-FB (1% DMSO) and Dox HCl solutions in a concentration range of 0.001–100 μM were also used as controls for the growth-inhibition study. For dilution of Dox-FB, 1% DMSO in media was used to maintain DMSO content constant at different concentrations, since this form of the drug is not directly soluble in aqueous solutions. Cells cultured without NPs/drug in complete media and cells without NPs/drug in complete media containing 1% DMSO were the positive (+) controls for the Dox HCl and Dox-FB solution studies, respectively. Cell-free wells with serum-free medium were used as negative control (−). Viability was determined with luminescence assays. Data were fit to a four-parameter dose–response curve in GraphPad Prism 7.0 software. The equations of the regression curves were utilized to estimate GI50.
Investigation of NP–cell interaction
MDA-MB-231 cells were plated on eight-well Lab-Tek chambered coverslips at a density of 50,000 cells per well in 0.3 mL media. Cells grew under normal culture conditions for 24 hours prior to beginning the studies. Cell media were replaced with the Dox-FB-AF750 NP suspensions at 0.2 mg/mL. After incubation for specific time periods (24 hours, 48 hours, 72 hours, and 96 hours), cells were washed three times with DPBS and fixed using 3.7% formaldehyde in PBS. The interaction of NPs with cells was monitored over time with an Evos FL LED-based fluorescence microscope utilizing a Cy7 filter cube (λEx 710/40 nm, λEm 775/46) for imaging of AF750 fluorescence development. The delivery of Dox-FB to cells was also monitored with fluorescence microscopy utilizing an RFP filter cube (λEx 531/40, λEm 593/40). Dox-FB-AF750 NPs containing PLGA-PDL copolymer instead of PLGA-PLL were used as control for determining the influence of enzymatic fluorescence development on imaging studies.
Fluorescence quantification was carried out using ImageJ utilizing raw fluorescence images of the AF750 channel only. Images were processed by assigning a fluorescence threshold to differentiate background from cell fluorescence, determining fluorescence density for each cell event, and subtracting background fluorescence. The resulting fluorescence densities were averaged between three independent images for each condition.
Results
Synthesis of PLGA-PLL, PLA-PEG-COOH, and PLA-PEG-OCH3 copolymers
PLGA-PLL and PLGA-PDL copolymers were synthesized by carbodiimide-mediated coupling of acid-terminated PLGA and PLL. Figure 4 shows the 1H-NMR spectra of PLGA-PLL in deuterated DMSO (DMSO-d6). NMR spectra confirmed the presence of PLGA and PLL in the copolymer. Proton peaks for PLL were assigned as previously reported:67 β, γ, and δ protons at 1.2 ppm (6 hours), ε protons at 2.8 ppm (2 hours), α proton at 4.0–4.5 ppm (1 hour), PLGA proton peaks corresponding to lactide methane quartets and methyl doublets at 5.2 ppm (1 hour) and 1.6 ppm (3 hours), respectively, and glycolide methane protons at 4.8 ppm (2 hours).
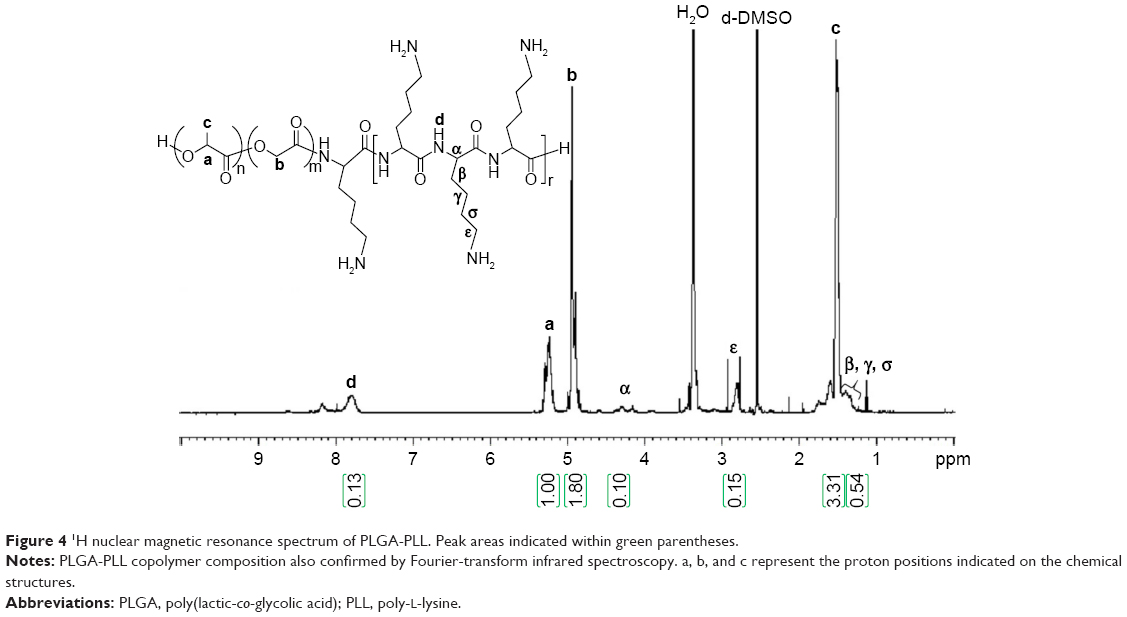 | Figure 4 1H nuclear magnetic resonance spectrum of PLGA-PLL. Peak areas indicated within green parentheses. |
Figure 5 shows FTIR spectra of PLGA, PLL, and PLGA-PLL copolymers. The spectrum of PLGA (red curve) shows and alkyl C−H stretch peak at 2,950–2,850 cm−1, ester C=O stretch peak at 1,750–1,735 cm−1, and C−O−C stretch peak at 1,250–1,050 cm−1, respectively. The spectrum of PLL (blue curve) shows a broad medium band for primary amines N−H stretch peak at 3,500–3,350 cm−1, characteristic strong amide C=O stretch peak (amide I band) at 1,690–1,630 cm−1, and broad and strong N−H bend (amide II band) peak at 1,510–1,580 cm−1, respectively. Finally, the spectrum of PLGA-PLL (green curve) shows PLL amide I and II bands, as well as alkyl, C=O, and C−O−C stretch peaks, confirming the presence of both blocks in the copolymer.
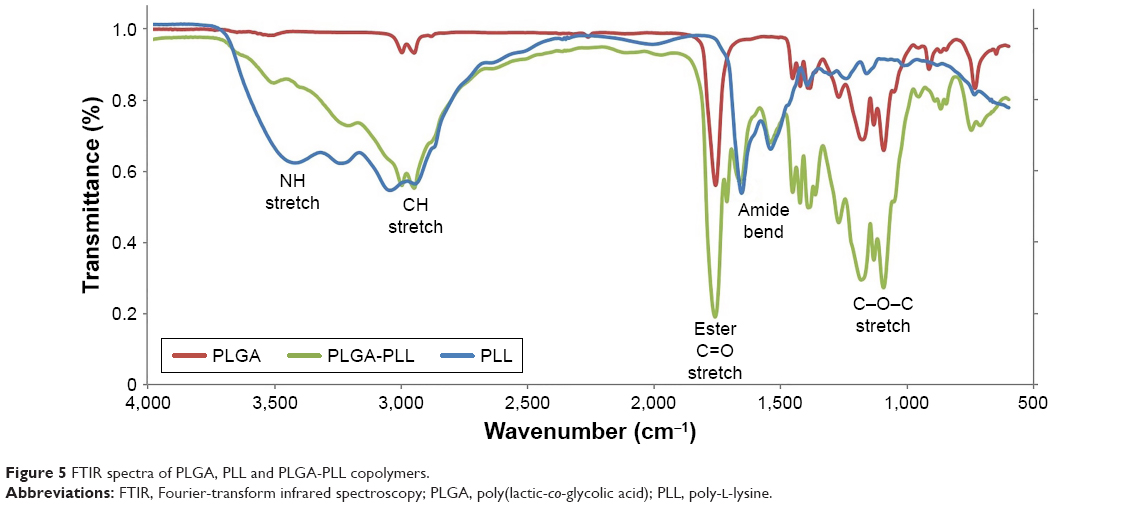 | Figure 5 FTIR spectra of PLGA, PLL and PLGA-PLL copolymers. |
PLA-PEG copolymers were synthesized via the ring-opening polymerization of lactide initiated by terminal hydroxyl groups of heterofunctional PEG. Polylactide protons at 1.6 (3 H) and 5.2 (1 H) ppm and PEG protons at 3.6 ppm (4 hours) on the NMR spectra confirmed the synthesis of the copolymers, as shown in Figure 6. The MW of PLA-PEG copolymers was calculated by comparing the peak area of PEG protons with that of polylactide peaks. The MW of polymers was determined to be 21,200 Da for PLA-PEG-COOH and 14,066 Da for PLA-PEG-OCH3 from NMR results.
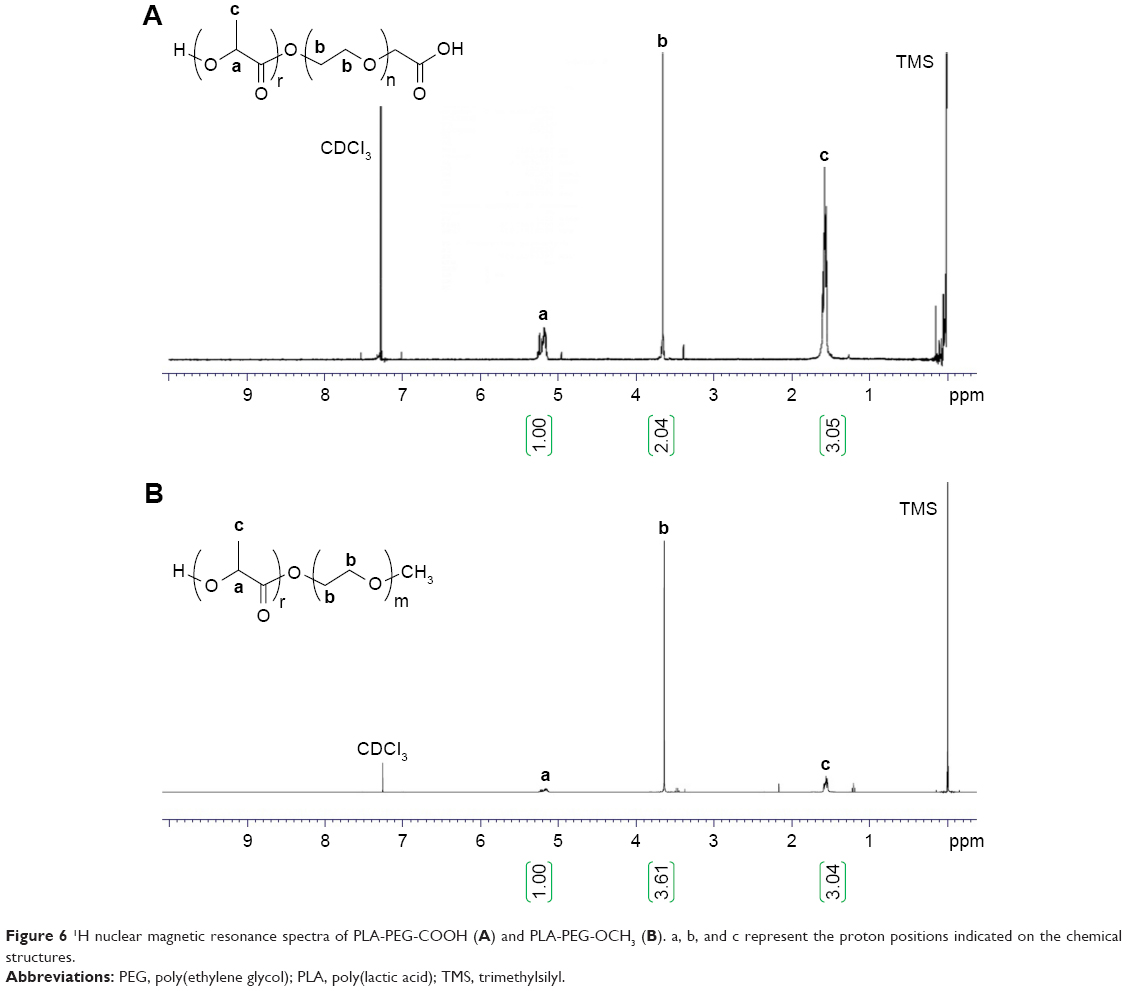 | Figure 6 1H nuclear magnetic resonance spectra of PLA-PEG-COOH (A) and PLA-PEG-OCH3 (B). a, b, and c represent the proton positions indicated on the chemical structures. |
The structure of PLA-PEG-COOH and PLA-PEG-OCH3 copolymers was also confirmed by FTIR (Figure 7). The copolymers presented peaks for C−H bonds of alkyl groups at 2,800–2,950 cm−1 and ether C−O−C stretch at ~1,100 cm−1 associated with the PEG block of PLA-PEG. A characteristic sharp C=O stretch peak at ~1,755 cm−1 of the PLA polyester block and alkyl absorption peaks at 2,950–3,000 cm−1 were also observed. As expected, no significant difference was observed in FTIR spectra of these copolymers, as the terminal carboxylic acid of PLA-PEG-COOH could not be observed from the spectrum as a result of the high MW of the polymer (225 carbonyls from the PLA polyester section vs one carbonyl from the carboxylic acid).
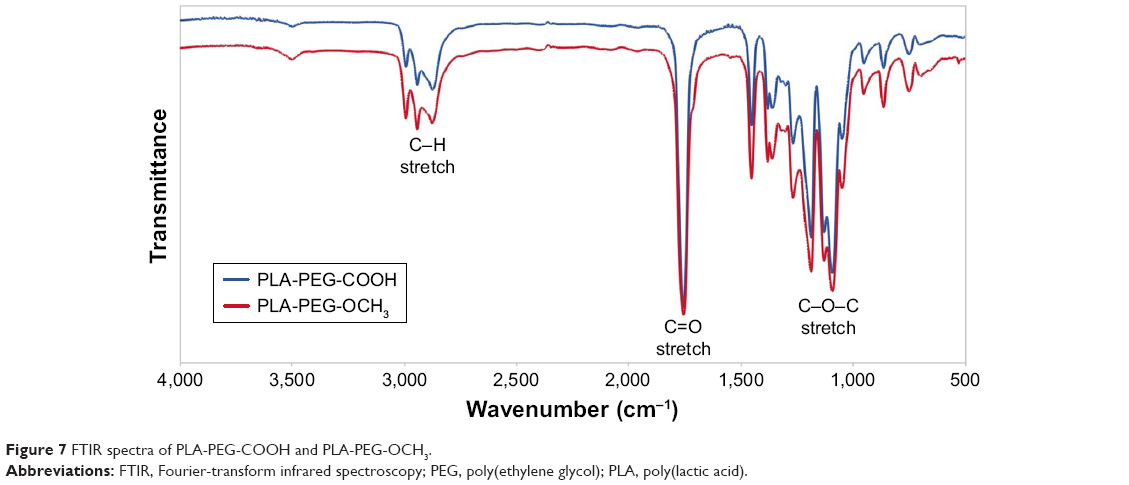 | Figure 7 FTIR spectra of PLA-PEG-COOH and PLA-PEG-OCH3. |
Conjugation of AF750 fluorescent dye to PLGA-PLL copolymers
PLL and PDL moieties of PLGA-PLL and PLGA-PDL were covalently decorated with AF750. Figure 8 shows the visual difference between AF750-labeled and unlabeled PLGA-PLL copolymers after purification. The dark blue of the AF750-PLL-PLGA copolymer provided clear indication of successful conjugation reaction between AF750-NHS ester and polylysine primary amine groups.
 | Figure 8 Appearance of PLGA-PLL (A) vs PLGA-PLL-AF750 (B) copolymers. |
Preparation of NPs
Dox was first converted into its FB form to enhance entrapment within the NPs by increasing its hydrophobicity. Figure 9 shows images of the initial cloudy Dox HCl dispersion in methanol–acetone (12.5%:87.5% v:v) mixture and then of the clear solution that results after reaction in the presence of TEA, confirming the conversion of Dox into the more hydrophobic Dox-FB form now fully soluble in the organic cosolvent mixture.
 | Figure 9 Conversion of Dox HCl dispersion (A) in acetone/methanol into a Dox-FB solution (B) after overnight reaction with TEA. |
NPs were prepared via nanoprecipitation of a blend of amphiphilic copolymers in an aqueous solution. Figure 10 shows the different types of NP suspensions: blank NPs (no drug, no AF750), Dox-FB NPs (no AF750), and Dox-FB-AF750 NPs.
 | Figure 10 Images of concentrated nanoparticle suspensions: (A) Blank, (B) Dox-FB NP, and (C) Dox-FB-AF750 NPs. |
Figure 11A shows the ultraviolet-visible-NIR absorbance spectrum of Dox-FB-AF750 NP suspension in water. While Dox-FB absorbs and fluoresces at 500 nm and 600 nm, AF750 absorbs and fluoresces at 740 nm and 780 nm, respectively. The spectral separation between Dox-FB and AF750 is sufficient to ensure that there is no optical interference during spectroscopic quantification of the drug or dye or during fluorescence microscopy. Figure 11B shows Dox-FB-AF750 NPs pelleted after centrifugation (left) and the NP suspension in PBS (right), confirming the strong association of both Dox-FB and AF750 with the polymer, yet the ability of the particles to be resuspended easily in aqueous media.
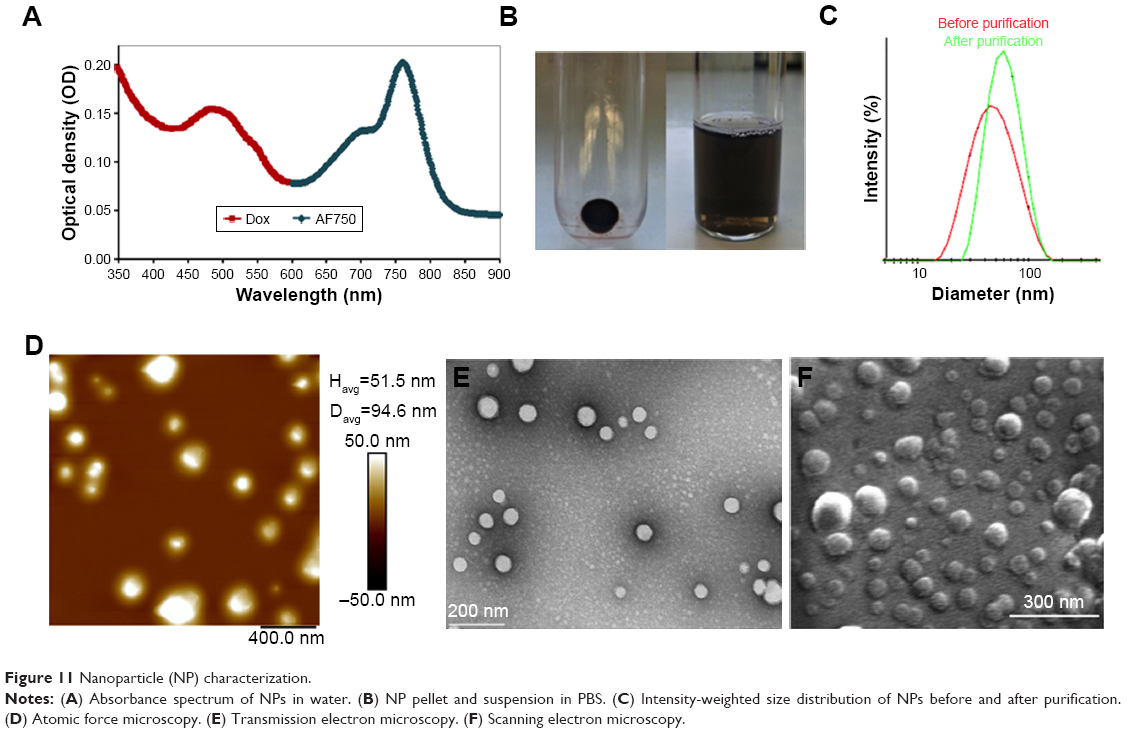 | Figure 11 Nanoparticle (NP) characterization. |
The size of the NPs in PBS was determined by DLS. Figure 11C shows the intensity-weighted size distribution of Dox-FB-AF750 NPs before and after purification. The change in size distribution observed in Figure 11C was likely due to the fact that after further purifications of NPs via centrifugation, smaller NPs stayed in the supernatant, as we have previously observed with PLA-PEG NP formulations.68 The size and polydispersity-index (the square of the standard deviation divided by the square of the mean particle size) values of the three types of NPs are listed in Table 1. Polydispersity-index values of NPs decreased after purification, providing further evidence that smaller NPs were lost during centrifugation.
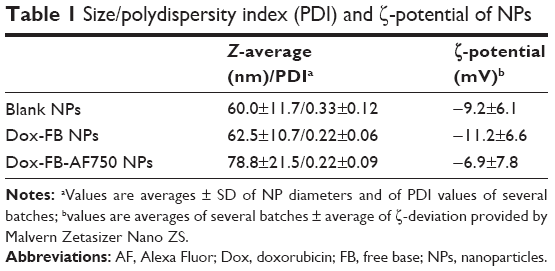 | Table 1 Size/polydispersity index (PDI) and ζ-potential of NPs |
To confirm size and morphology of the NPs, AFM, TEM, and SEM were used (Figure 11D–F). Spherical NPs were all smaller than 200 nm, with an average size of 60–80 nm in their dry state. The colloidal stability of the NPs was measured over time at 22°C and 37°C in various dilutions of serum. Figure S1 in summarizes the data. Samples incubated in PBS or in 10%, 20%, or 90% serum at room temperature remained stable, presenting changes (increases or decreases) in diameter <10% over 96 hours (Figure S1A). At 37°C, NPs remained stable in PBS throughout the 96-hour study, as did those suspended in all dilutions of serum for the first 48 hours. Samples suspended in 10%, 20%, and 90% serum increased in size >10% of initial size after 72 hours (Figure S1B).
The charge on a colloidal can be ascertained by measuring the particle’s electrokinetic potential (ζ-potential), which is a beneficial measure for understanding and predicting interactions among particles in suspension. The ζ-potential of NPs depends on the end groups of charged polymers, such as PLL (positively charged), PEG-COOH (neutral polymer with negatively charged end group at pH 7.4), the ratio of polymers in NPs, the type of drug encapsulated, and the type of surfactant (BSA, negatively charged) utilized during their preparation and purification. The surface charge of the NPs was found to be between −6.9 and −11.2 mV. As can be seen in Table 1, there were no significant ζ-potential differences between blank, AF750-labeled, and Dox-loaded NPs.
The smallness of the spherical NPs is expected to allow them to extravasate easily into the leaky vasculature of tumor vessels upon intravenous administration, resulting in passive accumulation into tumors. NP sizes of about 10–200 nm are believed to favor prolonged circulation and passive targeting of carriers, thanks to the enhanced permeability and retention effect.72,73
The amount of entrapped drug in the NP formulations was determined from the fluorescence intensity of Dox in dissolved NP solutions using fluorescence spectroscopy. The analysis was carried out in triplicate for each batch of drug-loaded NPs. The target loading of Dox based on feed ratios was 5% (mg Dox/mg NP × 100). We achieved 3%±0.4% (w:w) drug loading.
Drug-loading studies were carried out with Dox HCl (water-soluble form) as well. With this form of the drug, <1% drug loading was achieved. Since Dox HCl is highly soluble in water, the hydrophobic core of NPs could not entrap the drug efficiently, and furthermore Dox HCl was also released very rapidly from the NPs. For this reason, Dox HCl was converted into its water-insoluble FB form.
Enzymatic activation of NP fluorescence
PLL can be degraded by intracellular proteases, including cathepsins B and L and trypsin.60,74–76 When AF750 molecules are bound to PLL on the surface of the NPs in close proximity to one another, they are able to transfer their energy to one another and self-quench. This quenched fluorescence signal can, however, be recovered after proteolytic degradation of the PLL backbone. The optimal loading ratio that provided the appropriate quenching/dequenching effect and protease accessibility was determined from a series of polymer–dye conjugates with differing amounts of fluorescent molecules (data not shown). From these studies and considering MW, structure, excitation/emission wavelengths, molar absorption, quantum yield, and cost of AF750, an AF750:PLL ratio of 2.5 was selected for preparation of Dox-FB-AF750 NPs.
Trypsin was used as a model protease for activation of NP fluorescence. As shown in Figure 12A, the fluorescence intensity of NP suspensions increased nearly instantaneously 2.6-fold and reached a 32.7-fold increase within 240 minutes of exposure to trypsin in vitro, while only a slight increase (2.9-fold) in fluorescence was observed in the same period in the presence of trypsin and its inhibitor TLCK. As expected, there was no increase in fluorescence in the absence of trypsin. This novel “off/on” switch of fluorescence enhancement demonstrates the capabilities of our NPs to function as enzymatically triggered contrast agents ultimately triggered by tumor-overexpressed enzymes. For comparison, the fluorescence signal of AF750-labeled uncleavable PDL NPs did not increase significantly (2.2-fold vs >30-fold) upon interaction with trypsin (Figure 12B), confirming that proteolysis is required to activate the quenched fluorescence signal.
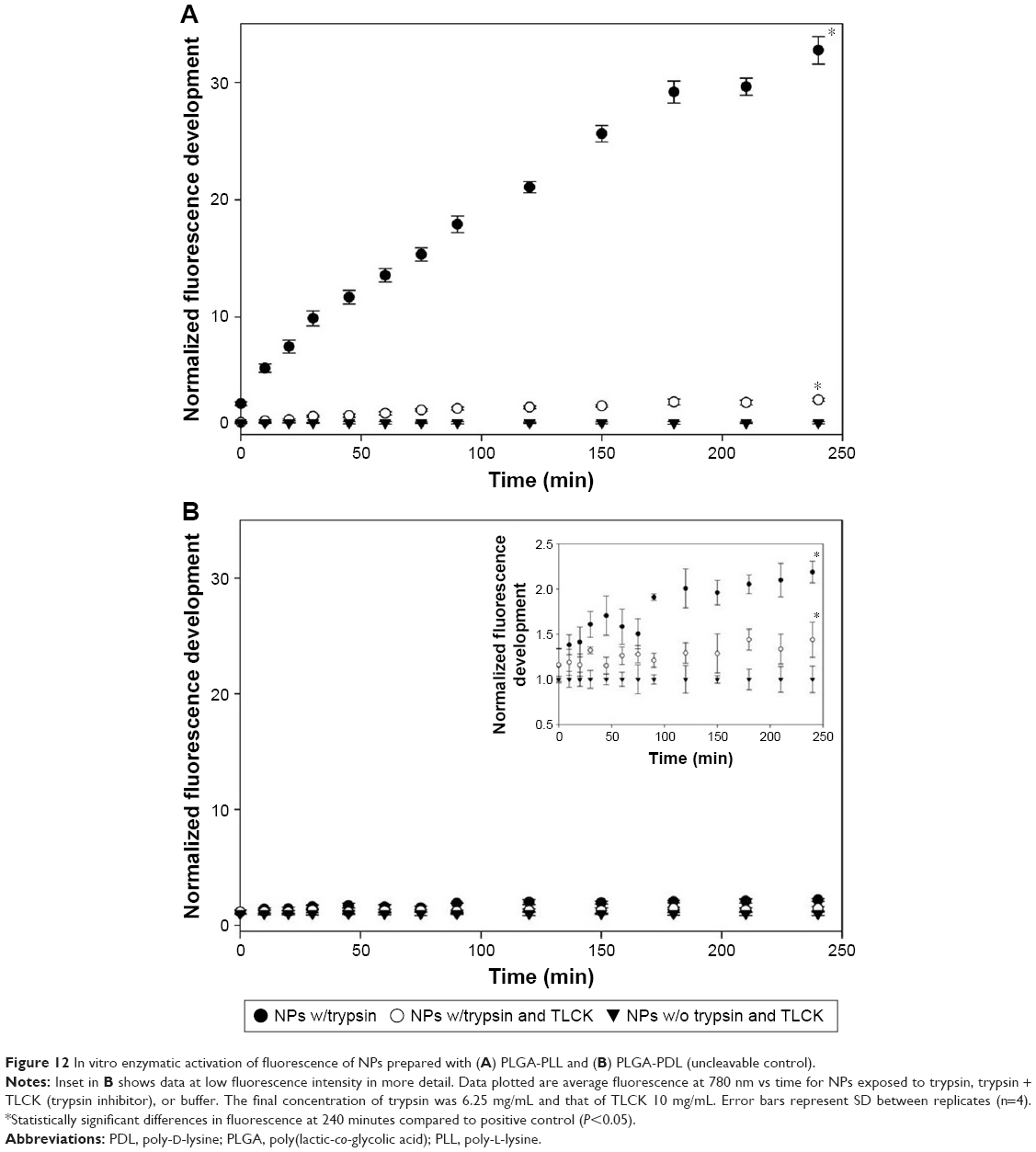 | Figure 12 In vitro enzymatic activation of fluorescence of NPs prepared with (A) PLGA-PLL and (B) PLGA-PDL (uncleavable control). |
In vitro drug-release study
Dox HCl solutions were used as controls in the release studies to confirm the lack of diffusion barrier imposed by the dialysis membrane onto this low-MW drug. The cumulative diffusion of Dox HCl and cumulative in vitro release of Dox-FB from NPs at pH 7.4 is shown in Figure 13. Fast diffusion of Dox HCl across the dialysis membrane was observed, as expected, releasing 99.3%±0.2% of drug in the first 18 hours.
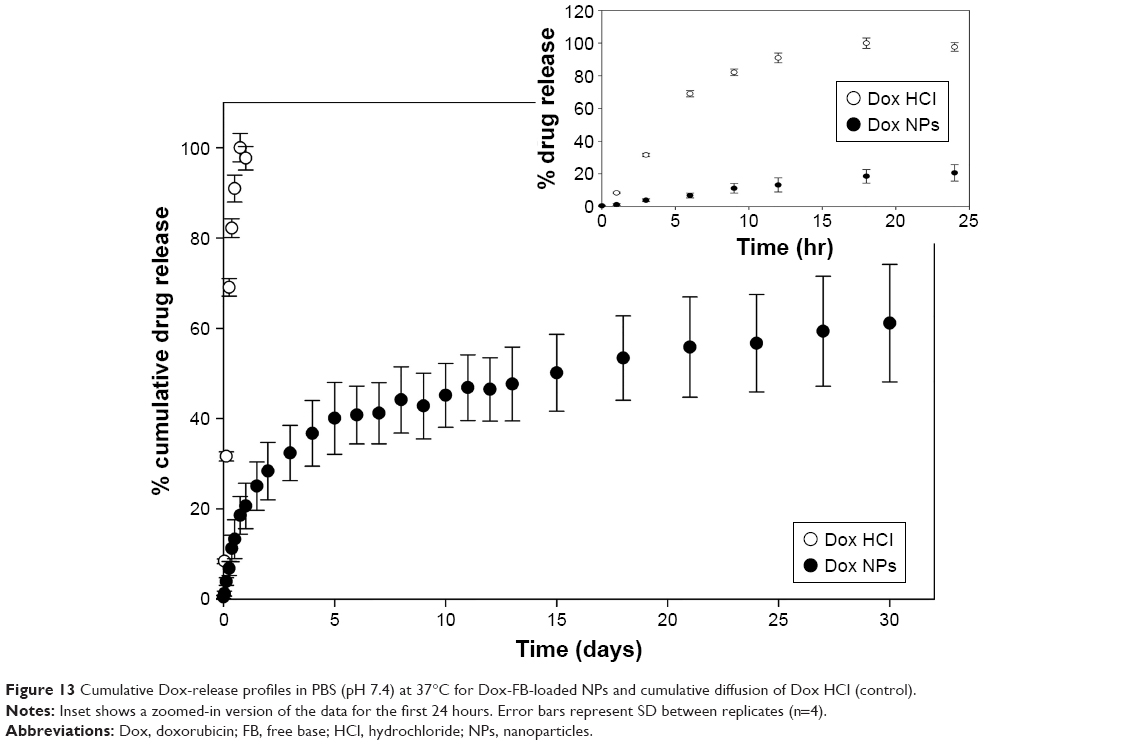 | Figure 13 Cumulative Dox-release profiles in PBS (pH 7.4) at 37°C for Dox-FB-loaded NPs and cumulative diffusion of Dox HCl (control). |
Dox-FB release from the NPs occurred in a controlled manner. An initial fast release in the first day associated with drug adsorbed or entrapped near the surface of the NPs was followed by a slow release rate over the rest of the release period. In fact, the cumulative percentage of Dox-FB released from NPs at pH 7.4 was 20.6% within 24 hours and 61% within 30 days. The remaining drug would have continued to release at a slow rate until polymer degradation was such that there was no longer a diffusional barrier for drug dissolution. Figure S2 shows the concentration of Dox-FB measured at each time point. As shown, therapeutically active micromolar concentrations were readily obtained during the release study.
We used a simple power-law expression (Equation 2), originally proposed by Peppas et al,77,78 to determine the release kinetics of Dox-FB from the NPs utilizing our in vitro drug-release data:
|
|
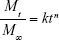
where Mt/M∞ is the fraction of drug release, k a constant, t the release time, and n the diffusional exponent that provides information about the mechanism associated with drug release from the particles. For spherical particles, n<0.43 represents quasi-Fickian diffusion, n=0.43 purely Fickian diffusion, n<0.43–<0.85 anomalous (non-Fickian) transport, n=0.85 represents case II transport, and n=1 zero-order release.77–79 Equation 2 was used to analyze the first 60% of the drug release of the total released in 30 days. We determined a diffusional exponent of n=0.5 (Figure S3), which indicates a non-Fickian diffusion behavior for drug release and suggests that the process was likely influenced not only by diffusion but also by drug dissolution and polymer relaxation.
Determination of NP cytocompatibility
The cytocompatibility of the blank NPs (no Dox, no AF750) was determined with MDA-MB-231 cancer cells using the CellTiter-Glo luminescence assay. Cell-free wells with serum-free medium were used as negative control, in accordance with previous reports.80 Figure 14 shows the viability of cells exposed to blank NPs for 72 hours. The presence of the NPs did not affect cell viability, except at very high concentrations (2 mg/mL), where viability decreased to ~80% upon 72 hours of continuous exposure. Similar studies were conducted with an MTT assay in a narrower concentration range (1–7.8 mg/mL), revealing similar results (data not shown).
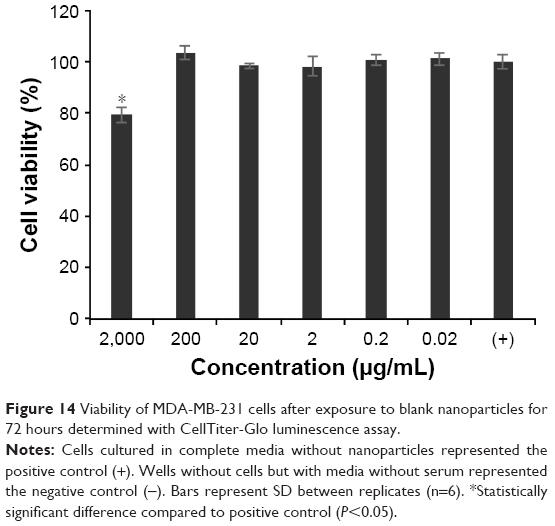 | Figure 14 Viability of MDA-MB-231 cells after exposure to blank nanoparticles for 72 hours determined with CellTiter-Glo luminescence assay. |
In addition to quantitative measures of cell viability, cells were monitored via microscopy during the studies, and no visual differences in cell morphology or growth were observed between NP-treated cells compared to cells in the positive viability control set (data not shown). Taken together, these studies overall suggest that the blank NPs are biocompatible.
Determination of NP-induced growth inhibition
In vitro growth inhibition induced by Dox-loaded NPs (no AF750) was evaluated with the CellTiter-Glo luminescence assay in MDA-MB-231 breast cancer cells. Figure 15 shows the viability of cells exposed to Dox-FB, Dox HCl, and Dox-FB NPs at drug concentrations of 0–100 μM. Lower cell viability represents a higher level of growth inhibition and thereby cytotoxic, cytostatic, and/or antiadhesive effects.81
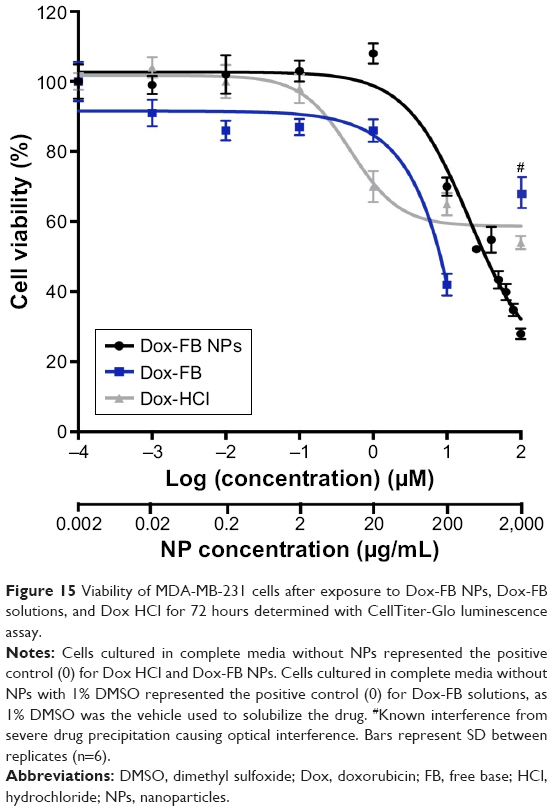 | Figure 15 Viability of MDA-MB-231 cells after exposure to Dox-FB NPs, Dox-FB solutions, and Dox HCl for 72 hours determined with CellTiter-Glo luminescence assay. |
GI50 of Dox-FB NPs and Dox-FB was estimated at 33 and 8 μM, respectively, from the nonlinear regression curve of the data presented in Figure 15. Overall, cells treated with low concentrations of Dox-FB NPs showed low growth inhibition. This was confirmed through visual observation of the samples. The reason for this is most likely the fact that Dox released slowly to cells from NPs, thereby exposing the cells to very low effective concentrations within the 72-hour exposure. However, significant growth inhibition was seen with Dox-FB NPs at concentrations >1 μM. It should be noted that no precipitation of NPs or Dox-FB delivered from Dox-FB NPs was observed at any of the concentrations tested in this study, demonstrating that the NPs are readily able to “solubilize” the hydrophobic Dox-FB in aqueous media. On the other hand, Dox-FB solutions readily precipitated during the study, despite being added to DMSO as a vehicle. In fact, the viability of cells treated with Dox-FB in solution (not NPs) at 100 μM appears higher than that of cells exposed to 10 μM Dox-FB as a result of Dox-FB precipitation, which stains the well (Figure S4), causing optical interference, light scattering, and background that leads to erroneous readings. This is in fact the reason that the luminescent assay was utilized, as the effect was much more pronounced with colorimetric MTT assays. For this reason, this data point was not included in the regression analysis. Because of precipitation, Dox-FB could not be utilized in vivo directly. Finally, while Dox HCl is highly soluble and readily able to enter cells, concentrations >1 μM did not further inhibit cell growth. In fact, the GI50 of Dox HCl could not be determined, as cell growth was not inhibited <58%.
With Dox-FB NPs at a drug concentration of 100 μM, cell viability was reduced significantly more than that of cells treated with Dox HCl. While the reason for the enhancement in growth inhibition by Dox-FB NPs at high concentrations relative to that of Dox HCl is not completely understood, it is possible that the NPs were better able to solubilize and deliver the drug intracellularly at high levels compared to the unencapsulated drug. It is also possible that the NPs enabled avoidance of drug efflux by transmembrane transporters, such as the P-glycoprotein and other multidrug-resistance-related proteins, leading to better cell-growth inhibition than Dox HCl. Enhanced therapeutic efficacy by several other Dox-nanomedicine formulations has been attributed to the delivery of high concentrations of drugs intracellularly to the cytoplasm or endosomal compartments, which avoids recognition and excretion of the drugs by transmembrane-efflux pumps.82–84
Finally, due to the long-release capabilities of the formulation, it is expected that cancer-cell viability could be further decreased with longer exposure, not feasible with an in vitro assay. In addition, in vivo use of the NPs is expected to increase their localization at the tumors by passive targeting via the enhanced permeability and retention effect, thereby providing a further mechanism for specificity and long-term localized treatment.
Microscopy study of NP interaction with cells
Fluorescence microscopy was used to investigate the ability of the NPs to mediate NIR fluorescence and Dox delivery to MDA-MB-231 cells. As shown in Figure 16, an increase in AF750-fluorescence intensity was observed throughout the 4-day study, suggesting that the fluorescence was in fact being activated by the cells. On the other hand, the fluorescence intensity of Dox did not change significantly over time. These results suggest that the NPs were already internalized in the cells by day 1 and that the NIR fluorescence of AF750 was “developed” by cell proteases. Decreased cell density was observed over time, due to the growth inhibition induced by Dox.
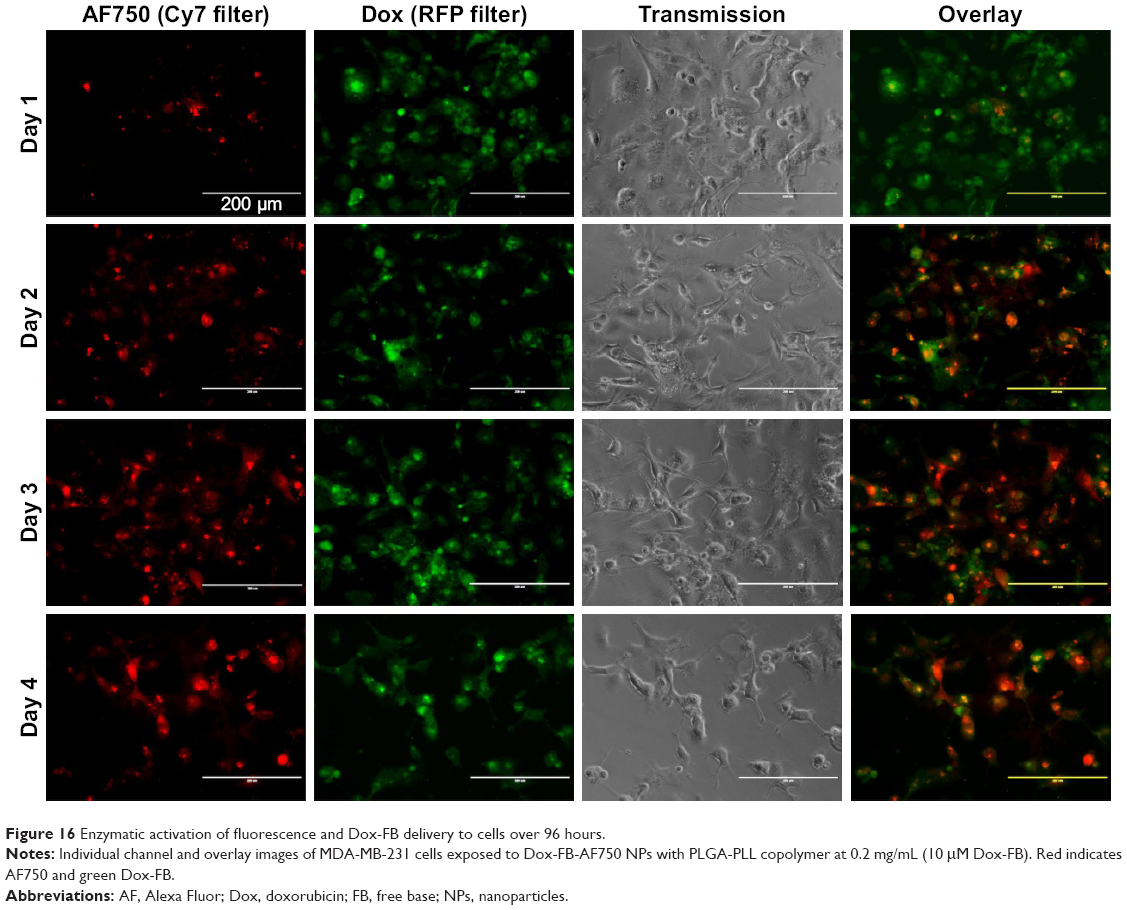 | Figure 16 Enzymatic activation of fluorescence and Dox-FB delivery to cells over 96 hours. |
To confirm that NIR-fluorescence development was in fact caused by cell activation of AF750 fluorescence and not simply a result of differences in AF750 and Dox diffusion into the cells, studies were then conducted with NPs prepared with uncleavable PLGA-PDL. As shown in Figure 17, NPs containing inert PDL polypeptide showed very low and constant AF750 fluorescence during the 4-day study. These data agree with the enzymatic activation study of PDL-NPs, in which we observed very low fluorescence. As in the study with PLGA-PLL NPs, steady fluorescence intensity of Dox was observed over time and cell density decreased during the study.
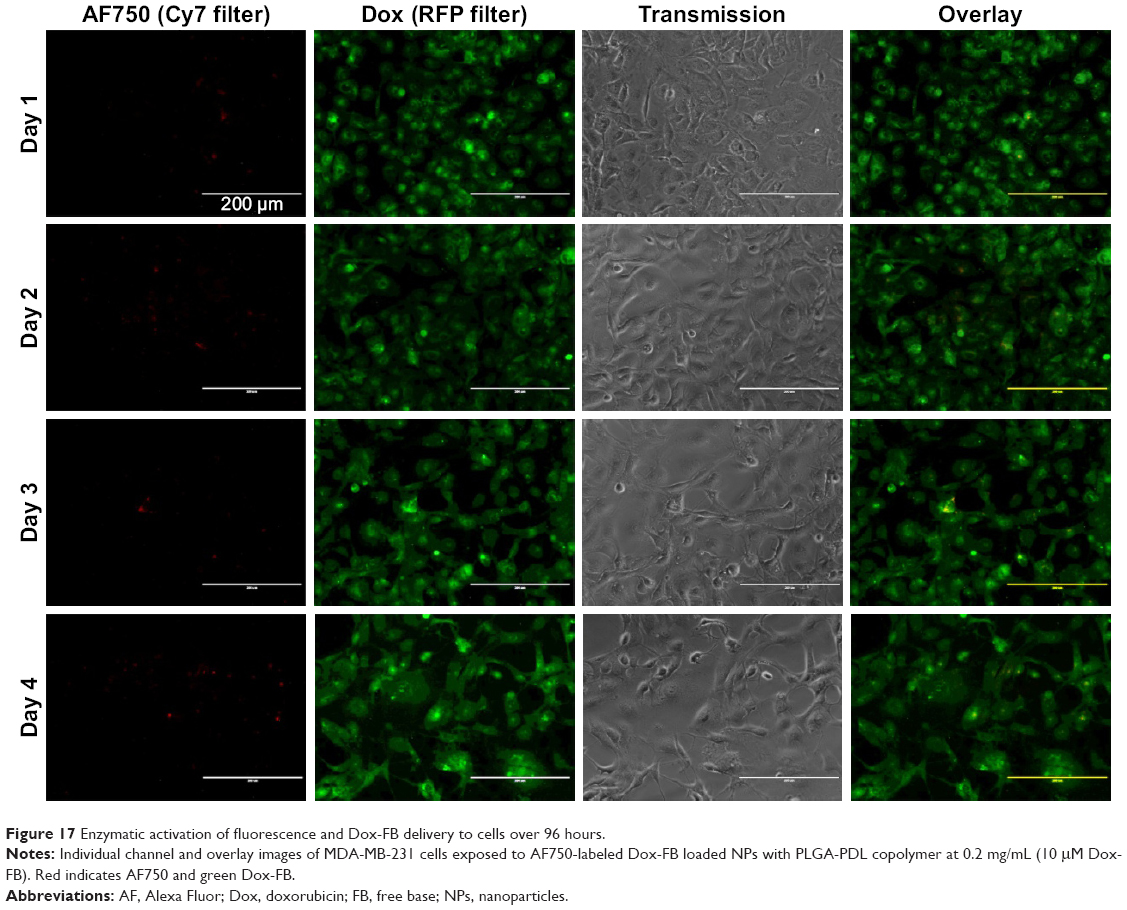 | Figure 17 Enzymatic activation of fluorescence and Dox-FB delivery to cells over 96 hours. |
Quantification of AF750-fluorescence development with incubation time for both PLGA-PLL and PLGA-PDL NPs is provided in Figure S5. A statistically significant increase in fluorescence relative to the 24-hour time point of the PLGA-PLL and PLGA-PDL NPs was observed over time for PLGA-PLL NPs. The increase was lower than that observed in NP suspensions exposed to trypsin, since protease levels in the cells were lower than those used in our experiments.
To confirm that cell viability was maintained during the study in the absence of Dox, a no-treatment control was included. Bright-field images of these cells are shown in Figure S6. No changes in cell morphology or growth were observed, and confluence was reached at 72–96 hours, as expected, in the absence of Dox-FB-AF750 NPs. Cell-media color was monitored for changes in pH, and no changes were observed during the study.
Discussion
Early detection of and determination of suitable treatments of cancer are critical to improve patient outcomes. Novel nanoscale drug-delivery and imaging systems improve the pharmacological behavior of chemotherapeutic drugs, enable the visualization of target tissue, and provide the ability to evaluate and adjust treatment protocols over time. Theranostic nanomedicines have significant potential as tools for personalized medicine. In this work, biodegradable polymeric NPs were developed and evaluated for imaging and treatment of breast cancer (Figure S7). These NPs have a core–shell architecture. The hydrophobic core, made of PLGA and PLA polyesters, acts as a suitable compartment for encapsulation of the chemotherapeutic drug Dox. The hydrophilic shell contains PEG and the protease-cleavable polypeptide PLL, which was decorated with AF750 for optical imaging. NPs were prepared by the nanoprecipitation of blends of amphiphilic PLGA-PLL and PLA-PEG copolymers synthesized by carbodiimide conjugation and ring-opening polymerization, respectively. The size of the spherical NPs was in the range of 60–80 nm.
The encapsulation of Dox-FB in the core of NPs was efficient and reproducible. While nanoencapsulation of Dox HCl is well established, fewer formulations of Dox-FB have been reported in the literature. This less water-soluble form of the drug enables long-term drug release. Missirlis et al reported the preparation of Dox-FB-loaded NPs composed of PEG and poloxamer 407 by inverse emulsion photopolymerization.85 After an initial burst release accounting for approximately 10% of the encapsulated drug, approximately 50% of the drug released at a slower rate over a week.85 Kataoka et al prepared Dox-FB-loaded micelles composed of a PEG–poly(β-benzyl-L-aspartate) block copolymer by an oil:water emulsion method.86 The release profile from these micelles was characterized by a rapid release in the initial stage (first 10 hours), followed by a slow and sustained release over a prolonged period (phase II, up to 80 hours).87 The drug-release kinetic studies of Missirlis and Kataoka support the results of the present study, in which the drug was observed to release approximately 20.6% of the drug in the first day, followed by a slow release rate over a month. The slow release kinetics of hydrophobic Dox-FB base may provide an advantage in vivo. If NPs are targeted to specific types of cells, they will have enough time to reach the targeted site and release drug into cells with minimal aspecific release in the circulation.
The imaging agent AF750 was covalently conjugated to protease-cleavable PLL to provide a mechanism for fluorescence activation. This mechanism is based on Förster resonance energy transfer (FRET) between donor and acceptor chromophores. FRET is a dynamic energy-transfer (and quenching in this case) mechanism in which energy transfers while the donor is in the excited state. FRET depends strongly on donor–acceptor distance.88 The efficiency of FRET is inversely proportional to the sixth power of the distance between donor and acceptor. FRET can typically occur over distances of up to 1–10 nm. Proteases overexpressed in tumors, including trypsin and cathepsins, can activate fluorescence by cleaving PLL into small fluorescently labeled peptide fragments that can diffuse away from NPs, thereby increasing the distance between chromophores and inhibiting FRET. Increased fluorescence of almost 33-fold for NP suspensions exposed to trypsin was observed over time in the in vitro studies. The inactive form of polylysine D-enantiomer was used as a control. Only L-amino acids are produced in cells and incorporated into proteins. D-Isomers of amino acids are of a biologically inert nature. As expected, NPs prepared with AF750-labeled PDL did not show significant fluorescence development.
In our previous work demonstrating enzymatically activable nanoprobes, fluorescent agents were conjugated to PLL present on the surface of NPs after their preparation by nanoprecipitation of polymer blends into BSA solutions.67 BSA was used as surfactant and stabilizer for NP preparation. BSA is a serum protein that consists of 60 lysines;89 consequently, covalent attachment of fluorophores to PLL after NP preparation carries the risk of resulting in labeling of BSA adsorbed onto the surface of the NPs. In the current work, AF750 was conjugated with PLGA-PLL copolymer and purified prior to NP preparation. The modified NP-labeling method prevents AF750 from binding to BSA, and thereby provides better control of NP function.
A luminescence-based viability assay was used for determination of cell viability upon exposure to NPs. This assay enables determination of cell viability culture indirectly by detection of ATP, which signals the presence of metabolically active cells with a luciferase-catalyzed bioluminescence reaction. The luminescence assay was specifically utilized, as opposed to more common MTT assays, to minimize interference from the chromophores present in the NPs. The absorbance of MTT reagent (λAbs 550 nm peak in DMSO) and Dox (λAbs 450–570 nm) overlap significantly. Consequently, Dox-FB within the cells or precipitated onto the well surfaces can result in artificially high viability values with the MTT assay for samples tested with high drug concentrations. The luminescence assay minimizes this interference. With this assay, high viability was observed in cells exposed to blank NPs, even at very high concentrations. These data confirm that blank NPs (in the absence of Dox and AF750) are biocompatible. Dox-FB-NPs, on the other hand, showed dose-dependent decreases in cell viability, as expected, especially in the range of 10–100 μM. Ultimately, the potential of these NPs to enhance the therapeutic effect of the drug should be evaluated in vivo, where nanoencapsulation is expected to provide improved tumor accumulation and long-term drug release.
Microscopy studies showed an increase in NIR AF750-fluorescence intensity over time that was associated with enzymatic activation of the PLL-AF750 conjugates, as confirmed with uncleavable PDL controls. A significant change was not observed on Dox-fluorescence intensity, suggesting that NPs were taken into the cells rapidly, after which the drug was able to release and AF750 fluorescence be developed inside the cells. Therefore, the multifunctional theranostic NPs allowed imaging of the cancer cells by NIR-fluorescence imaging and at the same time delivery of anticancer drug. We anticipate that theranostic NPs may be utilized in a distinct way to develop combined diagnosis and therapy for cancer treatment.
Theranostic nanomedicines hold significant promise in cancer management. Alternative polymeric nanomedicines with theranostic properties have been reported in the literature. For example, multifunctional micelles of poly(caprolactone)-b-PEG were prepared for single-photon-emission computed tomography and NIR-fluorescence imaging by inclusion of 188Re and the fluorescent dye IR780, respectively. These micelles also served as agents for photothermal therapy by virtue of the NIR-absorbing dye.90 PEG-coated phospholipid micelles loaded with paclitaxel and hydrophobic superparamagnetic iron oxide NPs were recently demonstrated to be effective magnetic resonance-imaging (MRI) agents and drug-delivery systems, both in vitro and in vivo.91 Blends of poly(hydroxyethyl methacrylate-co-histidine)-g-PLA and PLA-PEG loaded with Dox and labeled with Cy5.5 have been used for dual chemotherapy and imaging.92 Similarly, theranostic NPs have been prepared from a hydroxypropyl methyl acrylamide polymer covalently functionalized with Cy5.5 and gadolinium for optical imaging and MRI, respectively.93 In this system, paclitaxel was covalently attached to the polymer through a peptide linker that enabled enzymatic drug release. Paclitaxel-loaded theranostic micelles have also been similarly prepared from triblock PLA-PEG-PLL copolymers that were decorated with diethylenetriamine pentaacetic acid for chelation of gadolinium. These micelles were used for MRI and drug delivery to hepatocellular carcinoma.94 The same group also prepared theranostic NPs that contained a pH-sensitive poly(L-histidine)–PEG–biotin copolymer for pH-controlled release of sorafenib.95 Nanocapsules of triblock copolymers of PEG-PLGA-PLL have also been used as carriers for adriamycin and siRNA.96 Encapsulation of Cy5 within these nanocapsules enabled monitoring of hepatic carcinoma targeting. Fluorescent NPs have also been prepared by functionalizing poly(hydroxyethyl)aspartamide with rhodamine, PLA, and a PEG–galactose conjugate.97 These NPs enabled in vitro imaging of hepatocellular carcinoma cells. Finally, Cy5.5-labeled theranostic self-assembled supramolecular NPs stabilized by PEGylated phospholipids and including two chemotherapeutic agents in the form of prodrugs have been demonstrated to be effective imaging and therapeutic agents in a colorectal carcinoma mouse model.13
All these systems demonstrate the potential of theranostic nanomedicines. Our system, however, provides the means for imaging contrast to be activated at the tumor site, which is expected to provide significant increase in tumor signal:background ratio. Because of this, we believe that our system has significant potential in the field of cancer theranostics.
Conclusion
This work describes the preparation of biodegradable and biocompatible polymeric NPs as theranostic agents for cancer. NPs were prepared by nanoprecipitation blends of PLGA-PLL and two types of PLA-PEG block copolymers. This led to the formation of spherical core–shell NPs <100 nm in size. In vitro studies demonstrated the ability of the NPs to deliver Dox-FB over an extended period, as well as fluorescence activation upon NP interaction with proteolytic enzymes. Cell studies demonstrated the cytocompatibility of the NPs and their theranostic properties as contrast agents for fluorescence imaging and anticancer agents. Future work will investigate the behavior of these NPs in vivo and investigate the effect of targeting agents on NP selectivity toward cancer cells.
Acknowledgments
This work was funded by the National Science Foundation PREM Center for Interfaces (DMR-1205670) and the National Science Foundation Research Triangle Materials Research Science and Engineering Center: Programmable Assembly of Soft Matter (DMR-1121107).
Disclosure
The authors report no conflicts of interest in this work.
References
Jemal A, Tiwari RC, Murray T, et al. Cancer statistics, 2004. CA Cancer J Clin. 2004;54(1):8–29. | ||
WCRF. Data for cancer frequency by country: World Cancer Research Fund International. World Cancer Res Fund. 2017. | ||
American Cancer Society. Cancer Facts & Figures 2016. Atlanta: American Cancer Society; 2016. Available from: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2016/cancer-facts-and-figures-2016.pdf. Accessed October 4, 2018. | ||
DeSantis C, Ma J, Bryan L, Jemal A, Statistics Bcancer. Breast cancer statistics, 2013. CA Cancer J Clin. 2014;64(1):52–62. | ||
Giaccone G, Pinedo HM, Resistance D. Drug Resistance. Oncologist. 1996;1(1 & 2):82–87. | ||
Xu X, Xie K, Zhang XQ, et al. Enhancing tumor cell response to chemotherapy through nanoparticle-mediated codelivery of siRNA and cisplatin prodrug. Proc Natl Acad Sci U S A. 2013;110(46):18638–18643. | ||
du Y, Ren W, Li Y, et al. The enhanced chemotherapeutic effects of doxorubicin loaded PEG coated TiO2 nanocarriers in an orthotopic breast tumor bearing mouse model. J Mater Chem B. 2015;3(8):1518–1528. | ||
Chen AM, Zhang M, Wei D, et al. Co-delivery of doxorubicin and Bcl-2 siRNA by mesoporous silica nanoparticles enhances the efficacy of chemotherapy in multidrug-resistant cancer cells. Small. 2009;5(23):2673–2677. | ||
Wang H, Zhao Y, Wu Y, et al. Enhanced anti-tumor efficacy by co-delivery of doxorubicin and paclitaxel with amphiphilic methoxy PEG-PLGA copolymer nanoparticles. Biomaterials. 2011;32(32):8281–8290. | ||
Wang H, Lu Z, Wang L, et al. New generation nanomedicines constructed from self-assembling small-molecule prodrugs alleviate cancer drug toxicity. Cancer Res. 2017;77(24):6963–6974. | ||
Wang H, Wu J, Xie K, et al. Precise Engineering of Prodrug Cocktails into Single Polymeric Nanoparticles for Combination Cancer Therapy: Extended and Sequentially Controllable Drug Release. ACS Appl Mater Interfaces. 2017;9(12):10567–10576. | ||
Wang H, Xie H, Wu J. Structure-Based Rational Design of Prodrugs to Enable Their Combination with Polymeric Nanoparticle Delivery Platforms for Enhanced Antitumor Efficacy. Angew Chemie Int Ed. 2014;53:11532–11537. | ||
Wang H, Chen J, Xu C, et al. Cancer nanomedicines stabilized by π-π stacking between heterodimeric prodrugs enable exceptionally high drug loading capacity and safer delivery of drug combinations. Theranostics. 2017;7(15):3638–3652. | ||
Taurin S, Greish K. Enhanced Vascular Permeability in Solid Tumors: A Promise for Anticancer Nanomedicine. Tight Junctions in Cancer Metastasis. In: Martin TA, Jiang WG, editors. Vol Dordrecht: Springer Netherlands; 2013:81–118. | ||
Lammers T, Kiessling F, Hennink WE, Storm G, Nanotheranostics SG. Nanotheranostics and image-guided drug delivery: current concepts and future directions. Mol Pharm. 2010;7(6):1899–1912. | ||
Weissleder R, Pittet MJ. Imaging in the era of molecular oncology. Nature. 2008;452(7187):580–589. | ||
Tobis S, Knopf J, Silvers C, et al. Near infrared fluorescence imaging with robotic assisted laparoscopic partial nephrectomy: initial clinical experience for renal cortical tumors. J Urol. 2011;186(1):47–52. | ||
Schlottmann F, Patti MG. Evaluation of Gastric Conduit Perfusion During Esophagectomy with Indocyanine Green Fluorescence Imaging. J Laparoendosc Adv Surg Tech A. 2017;27(12):1305–1308. | ||
Boni L, David G, Mangano A, et al. Clinical applications of indocyanine green (ICG) enhanced fluorescence in laparoscopic surgery. Surg Endosc. 2015;29(7):2046–2055. | ||
Currie AC, Brigic A, Thomas-Gibson S, et al. A pilot study to assess near infrared laparoscopy with indocyanine green (ICG) for intraoperative sentinel lymph node mapping in early colon cancer. Eur J Surg Oncol. 2017;43(11):2044–2051. | ||
Bernard ME, Glaser SM, Gill BS, Nagaya T, Nakamura YA, Choyke PL, et al. Results of a Single Institution Experience with Dose-Escalated Chemoradiation for Locally Advanced Unresectable Non-Small Cell Lung Cancer. Front Oncol. 2017;7(314):1–16. | ||
Tipirneni KE, Warram JM, Moore LS, et al. Oncologic Procedures Amenable to Fluorescence-guided Surgery. Ann Surg. 2017;266(1):36–47. | ||
de Boer E, Warram JM, Tucker MD, et al. In Vivo Fluorescence Immunohistochemistry: Localization of Fluorescently Labeled Cetuximab in Squamous Cell Carcinomas. Sci Rep. 2015;5:10169. | ||
Whitley MJ, Cardona DM, Lazarides AL, et al. A mouse-human phase 1 co-clinical trial of a protease-activated fluorescent probe for imaging cancer. Sci Transl Med. 2016;8320(320):ra4. | ||
Peek MC, Charalampoudis P, Anninga B, Baker R, Douek M. Blue dye for identification of sentinel nodes in breast cancer and malignant melanoma: a systematic review and meta-analysis. Future Oncol. 2017;13(5):455–467. | ||
Ogata F, Narushima M, Mihara M, Azuma R, Morimoto Y, Koshima I. Intraoperative lymphography using indocyanine green dye for near-infrared fluorescence labeling in lymphedema. Ann Plast Surg. 2007;59(2):180–184. | ||
Chang DW, Suami H, Skoracki R. A prospective analysis of 100 consecutive lymphovenous bypass cases for treatment of extremity lymphedema. Plast Reconstr Surg. 2013;132(5):1305–1314. | ||
Aldave G, Tejada S, Pay E, et al. Prognostic value of residual fluorescent tissue in glioblastoma patients after gross total resection in 5-aminolevulinic Acid-guided surgery. Neurosurgery. 2013;72(6):915–921. | ||
Unkart JT, Chen SL, Wapnir IL, González JE, Harootunian A, Wallace AM. Intraoperative Tumor Detection Using a Ratiometric Activatable Fluorescent Peptide: A First-in-Human Phase 1 Study. Ann Surg Oncol. 2017;24(11):3167–3173. | ||
Tummers QR, Hoogstins CE, Gaarenstroom KN, et al. Intraoperative imaging of folate receptor alpha positive ovarian and breast cancer using the tumor specific agent EC17. Oncotarget. 2016;7(22):32144–32155. | ||
Patel SK, Janjic JM. Macrophage targeted theranostics as personalized nanomedicine strategies for inflammatory diseases. Theranostics. 2015;5(2):150–172. | ||
Singh AV, Khare M, Gade WN, Zamboni P. Theranostic implications of nanotechnology in multiple sclerosis: a future perspective. Autoimmune Dis. 2012;2012:1–12. | ||
McCarthy JR, Korngold E, Weissleder R, Jaffer FA. A light-activated theranostic nanoagent for targeted macrophage ablation in inflammatory atherosclerosis. Small. 2010;6(18):2041–2049. | ||
Crielaard BJ, Lammers T, Schiffelers RM, Storm G. Drug targeting systems for inflammatory disease: one for all, all for one. J Control Release. 2012;161(2):225–234. | ||
Aulić S, Bolognesi ML, Legname G. Small-molecule theranostic probes: a promising future in neurodegenerative diseases. Int J Cell Biol. 2013;2013:150952. | ||
Bolognesi ML, Gandini A, Prati F, Uliassi E. From Companion Diagnostics to Theranostics: A New Avenue for Alzheimer’s Disease? J Med Chem. 2016;59(17):7759–7770. | ||
Wang P, Moore A. Theranostic magnetic resonance imaging of type 1 diabetes and pancreatic islet transplantation. Quant Imaging Med Surg. 2012;2(3):151–162. | ||
Wang P, Moore A. Theranostic MRI: the future for Type 1 diabetes management? Imaging Med. 2014;6(1):25–39. | ||
Howell M, Wang C, Mahmoud A, Hellermann G, Mohapatra SS, Mohapatra S. Dual-function theranostic nanoparticles for drug delivery and medical imaging contrast: perspectives and challenges for use in lung diseases. Drug Deliv Transl Res. 2013;3(4):352–363. | ||
Mccarthy JR. Multifunctional agents for concurrent imaging and therapy in cardiovascular disease. Adv Drug Deliv Rev. 2010;62(11):1023–1030. | ||
Wang D, Lin B, Ai H. Theranostic nanoparticles for cancer and cardiovascular applications. Pharm Res. 2014;31(6):1390–1406. | ||
Xie J, Lee S, Chen X. Nanoparticle-based theranostic agents. Adv Drug Deliv Rev. 2010;62(11):1064–1079. | ||
Zhu X, Anquillare ELB, Farokhzad OC, Shi J. Polymer- and Protein-Based Nanotechnologies for Cancer Theranostics. In: Cancer Theranostics. 2014:419–436. | ||
Mohan P, Rapoport N. Doxorubicin as a molecular nanotheranostic agent: effect of doxorubicin encapsulation in micelles or nanoemulsions on the ultrasound-mediated intracellular delivery and nuclear trafficking. Mol Pharm. 2010;7(6):1959–1973. | ||
Cummings J, Smyth JF. DNA topoisomerase I and II as targets for rational design of new anticancer drugs. Ann Oncol. 1993;4(7):533–543. | ||
Fornari FA, Randolph JK, Yalowich JC, Ritke MK, Gewirtz DA. Interference by doxorubicin with DNA unwinding in MCF-7 breast tumor cells. Mol Pharmacol. 1994;45(4):649–656. | ||
Gabizon A, Shmeeda H, Barenholz Y. Pharmacokinetics of pegylated liposomal Doxorubicin: review of animal and human studies. Clin Pharmacokinet. 2003;42(5):419–436. | ||
Gabizon AA. Liposomal drug carrier systems in cancer chemotherapy: current status and future prospects. J Drug Target. 2002;10(7):535–538. | ||
Holliday DL, Speirs V. Choosing the right cell line for breast cancer research. Breast Cancer Res. 2011;13(4):215. | ||
Tuzlali S. Pathology of Breast Cancer. In: Breast Disease: Diagnosis and Pathology. Vol. 1. 2015;1:241–266. | ||
Hilger I, Hergt R, Kaiser WA. Use of magnetic nanoparticle heating in the treatment of breast cancer. IEE Proc Nanobiotechnol. 2005;152(1):33–39. | ||
Onda N, Kemmochi S, Morita R, Ishihara Y, Shibutani M. In vivo imaging of tissue-remodeling activity involving infiltration of macrophages by a systemically administered protease-activatable probe in colon cancer tissues. Transl Oncol. 2013;6(6):628–IN4. | ||
Mito JK, Ferrer JM, Brigman BE, et al. Intraoperative detection and removal of microscopic residual sarcoma using wide-field imaging. Cancer. 2012;118(21):5320–5330. | ||
Mieog JS, Hutteman M, van der Vorst JR, et al. Image-guided tumor resection using real-time near-infrared fluorescence in a syngeneic rat model of primary breast cancer. Breast Cancer Res Treat. 2011;128(3):679–689. | ||
Reubi JC. Peptide receptors as molecular targets for cancer diagnosis and therapy. Endocr Rev. 2003;24(4):389–427. | ||
Weissleder R, Tung CH, Mahmood U, Bogdanov A. In vivo imaging of tumors with protease-activated near-infrared fluorescent probes. Nat Biotechnol. 1999;17(4):375–378. | ||
Tung CH, Bredow S, Mahmood U, Weissleder R. Preparation of a cathepsin D sensitive near-infrared fluorescence probe for imaging. Bioconjug Chem. 1999;10(5):892–896. | ||
Lee S, Park K, Lee SY, et al. Dark quenched matrix metalloproteinase fluorogenic probe for imaging osteoarthritis development in vivo. Bioconjug Chem. 2008;19(9):1743–1747. | ||
Bremer C, Tung CH, Weissleder R. In vivo molecular target assessment of matrix metalloproteinase inhibition. Nat Med. 2001;7(6):743–748. | ||
Lee SK, Han MS, Tung CH. Layered nanoprobe for long-lasting fluorescent cell label. Small. 2012;8(21):3315–3320. | ||
Rakash S. Role of proteases in cancer: A review. Biotechnol Mol Biol Rev. 2012;7(4):90–101. | ||
Soreide K, Janssen EA, Körner H, Baak JP. Trypsin in colorectal cancer: molecular biological mechanisms of proliferation, invasion, and metastasis. J Pathol. 2006;209(2):147–156. | ||
Ge L, Shenoy SK, Lefkowitz RJ, Defea K. Constitutive protease-activated receptor-2-mediated migration of MDA MB-231 breast cancer cells requires both beta-arrestin-1 and -2. J Biol Chem. 2004;279(53):55419–55424. | ||
Su S, Li Y, Luo Y, et al. Proteinase-activated receptor 2 expression in breast cancer and its role in breast cancer cell migration. Oncogene. 2009;28(34):3047–3057. | ||
Matěj R, Mandáková P, Netíková I, Poucková P, Olejár T. Proteinase-activated receptor-2 expression in breast cancer and the role of trypsin on growth and metabolism of breast cancer cell line MDA MB-231. Physiol Res. 2007;56(4):475–484. | ||
Morris DR, Ding Y, Ricks TK, Gullapalli A, Wolfe BL, Trejo J. Protease-activated receptor-2 is essential for factor VIIa and Xa-induced signaling, migration, and invasion of breast cancer cells. Cancer Res. 2006;66(1):307–314. | ||
Özel T, White S, Nguyen E, et al. Enzymatically activated near infrared nanoprobes based on amphiphilic block copolymers for optical detection of cancer. Lasers Surg Med. 2015;47(7):579–594. | ||
Hamon CL, Dorsey CL, Özel T, Barnes EM, Hudnall TW, Betancourt T. Near-infrared fluorescent aza-BODIPY dye-loaded biodegradable polymeric nanoparticles for optical cancer imaging. J Nanoparticle Res. 2016;18(7):207. | ||
Foy SP, Stine A, Jain TK, Labhasetwar V. Magnetic Nanoparticles for Drug Delivery. Methods in Bioengineering: Nanoscale Bioengineering and Nanomedicine. In: Rege K, Medintz IgorL, editors. Vol Boston: Artech House; 2009:123–136. | ||
Betancourt T, Brown B, Brannon-Peppas L. Doxorubicin-loaded PLGA nanoparticles by nanoprecipitation: preparation, characterization and in vitro evaluation. Nanomedicine. 2007;2(2):219–232. | ||
Kaszuba M, Corbett J, Watson FM, Jones A. High-concentration zeta potential measurements using light-scattering techniques. Philos Trans A Math Phys Eng Sci. 2010;368(1927):4439–4451. | ||
Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5(4):505–515. | ||
Ledet G, Mandal TK. Nanomedicine: Emerging therapeutics for the 21st century. US Pharm. 2012;37:7–11. | ||
Olson ES, Jiang T, Aguilera TA, et al. Activatable cell penetrating peptides linked to nanoparticles as dual probes for in vivo fluorescence and MR imaging of proteases. Proc Natl Acad Sci U S A. 2010;107(9):4311–4316. | ||
Funovics M, Weissleder R, Tung CH. Protease sensors for bioimaging. Anal Bioanal Chem. 2003;377(6):956–963. | ||
Choi Y, Weissleder R, Tung CH. Selective antitumor effect of novel protease-mediated photodynamic agent. Cancer Res. 2006;66(14):7225–7229. | ||
Peppas NA. Analysis of Fickian and non-Fickian drug release from polymers. Pharm Acta Helv. 1985;60(4):110–111. | ||
Siepmann J, Peppas NA. Derivation Higuchi Equation: applications, use and misuse. Int J Pharm. 2011;418(1):6–12. | ||
Ritger PL, Peppas NA. A simple equation for description of solute release. J Control Release. 1987;5:23–36. | ||
Wang P, Henning SM, Heber D, Limitations Of MTT. Limitations of MTT and MTS-based assays for measurement of antiproliferative activity of green tea polyphenols. PLoS One. 2010;5(4):e10202. | ||
Ciavatta ML, Lefranc F, Carbone M, et al. Marine Mollusk-Derived Agents with Antiproliferative Activity as Promising Anticancer Agents to Overcome Chemotherapy Resistance. Med Res Rev. 2017;37(4):702–801. | ||
Wong HL, Rauth AM, Bendayan R, et al. A new polymer-lipid hybrid nanoparticle system increases cytotoxicity of doxorubicin against multidrug-resistant human breast cancer cells. Pharm Res. 2006;23(7):1574–1585. | ||
Lee ES, Na K, Bae YH. Doxorubicin loaded pH-sensitive polymeric micelles for reversal of resistant MCF-7 tumor. J Control Release. 2005;103(2):405–418. | ||
Torchilin VP. Strategies and Means for Drug Targeting: An Overview. Biomedical Aspects of Drug Targeting. In: Muzykantov V, Torchilin V, editors. Boston MA: Springer; 2002:3–26. | ||
Missirlis D, Kawamura R, Tirelli N, Hubbell JA. Doxorubicin encapsulation and diffusional release from stable, polymeric, hydrogel nanoparticles. Eur J Pharm Sci. 2006;29(2):120–129. | ||
Kataoka K, Matsumoto T, Yokoyama M, et al. Doxorubicin-loaded poly(ethylene glycol)-poly(β-benzyl-L-aspartate) copolymer micelles: Their pharmaceutical characteristics and biological significance. Journal of Controlled Release. 2000;64:143–153. | ||
Kataoka K, Matsumoto T, Yokoyama M, et al. Doxorubicin-loaded poly(ethylene glycol)-poly(beta-benzyl-L-aspartate) copolymer micelles: their pharmaceutical characteristics and biological significance. J Control Release. 2000;64(1–3):143–153. | ||
Saini S, Singh H, Bagchi B. Fluorescence resonance energy transfer (FRET) in chemistry and biology: Non-Förster distance dependence of the FRET rate. J Chem Sci. 2006;118(1):23–35. | ||
Huang BX, Kim HY, Dass C. Probing three-dimensional structure of bovine serum albumin by chemical cross-linking and mass spectrometry. J Am Soc Mass Spectrom. 2004;15(8):1237–1247. | ||
Peng CL, Shih YH, Lee PC, Hsieh TM, Luo TY, Shieh MJ. Multimodal image-guided photothermal therapy mediated by 188Re-labeled micelles containing a cyanine-type photosensitizer. ACS Nano. 2011;5(7):5594–5607. | ||
Upponi JR, Jerajani K, Nagesha DK, et al. Polymeric micelles: Theranostic co-delivery system for poorly water-soluble drugs and contrast agents. Biomaterials. 2018;170:26–36. | ||
Tsai HC, Chang WH, Lo CL, et al. Graft and diblock copolymer multifunctional micelles for cancer chemotherapy and imaging. Biomaterials. 2010;31(8):2293–2301. | ||
Cai H, Wang X, Zhang H, et al. Enzyme-sensitive biodegradable and multifunctional polymeric conjugate as theranostic nanomedicine. Appl Mater Today. 2018;11:207–218. | ||
Liu Y, Li J, Liu F, et al. Theranostic polymeric micelles for the diagnosis and treatment of hepatocellular carcinoma. J Biomed Nanotechnol. 2015;11(4):613–622. | ||
Liu Y, Feng L, Liu T, et al. Multifunctional pH-sensitive polymeric nanoparticles for theranostics evaluated experimentally in cancer. Nanoscale. 2014;6(6):3231–3242. | ||
Liu P, Yu H, Sun Y, Zhu M, Duan Y. A mPEG-PLGA-b-PLL copolymer carrier for adriamycin and siRNA delivery. Biomaterials. 2012;33(17):4403–4412. | ||
Craparo EF, Licciardi M, Conigliaro A, et al. Hepatocyte-targeted fluorescent nanoparticles based on a polyaspartamide for potential theranostic applications. Polymer. 2015;70:257–270. |
Supplementary materials
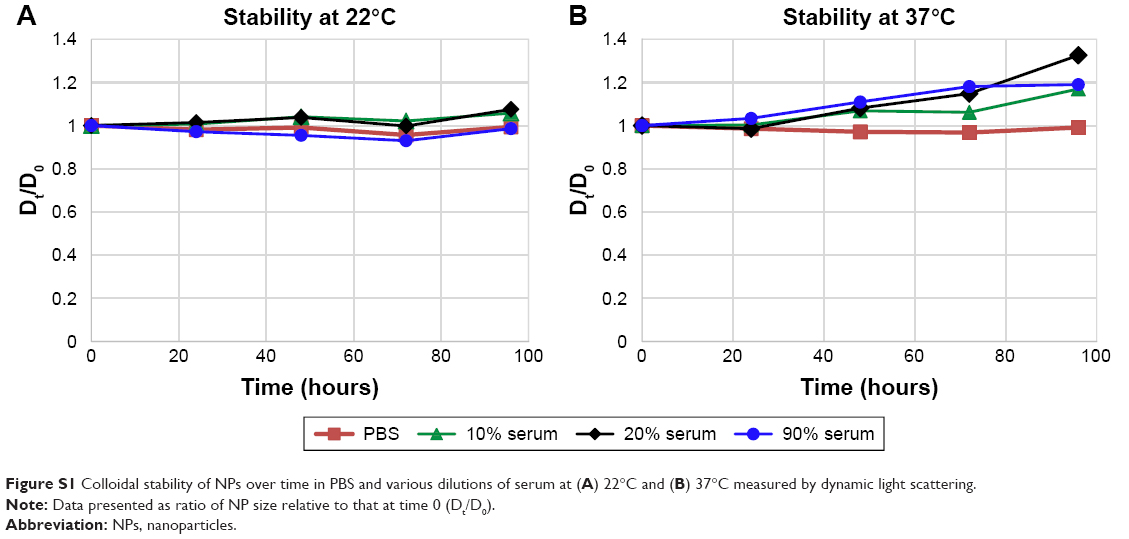 | Figure S1 Colloidal stability of NPs over time in PBS and various dilutions of serum at (A) 22°C and (B) 37°C measured by dynamic light scattering. |
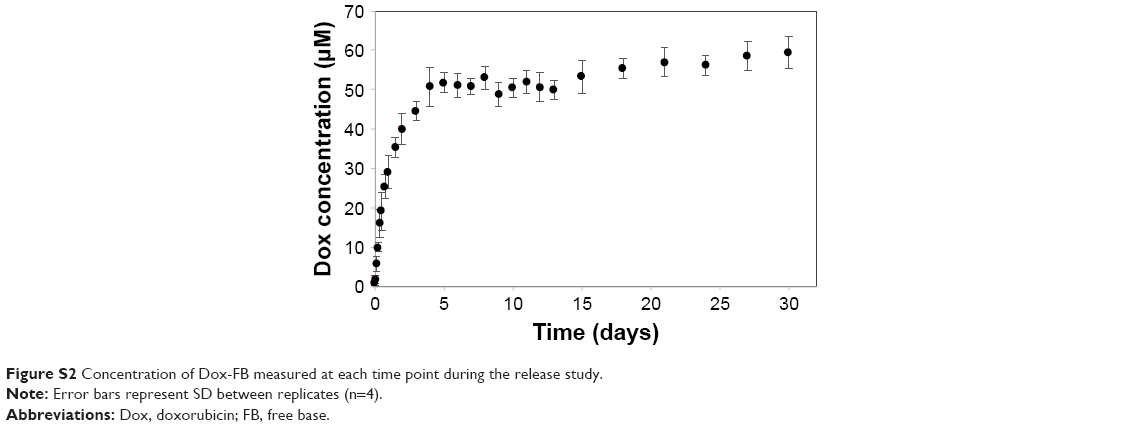 | Figure S2 Concentration of Dox-FB measured at each time point during the release study. |
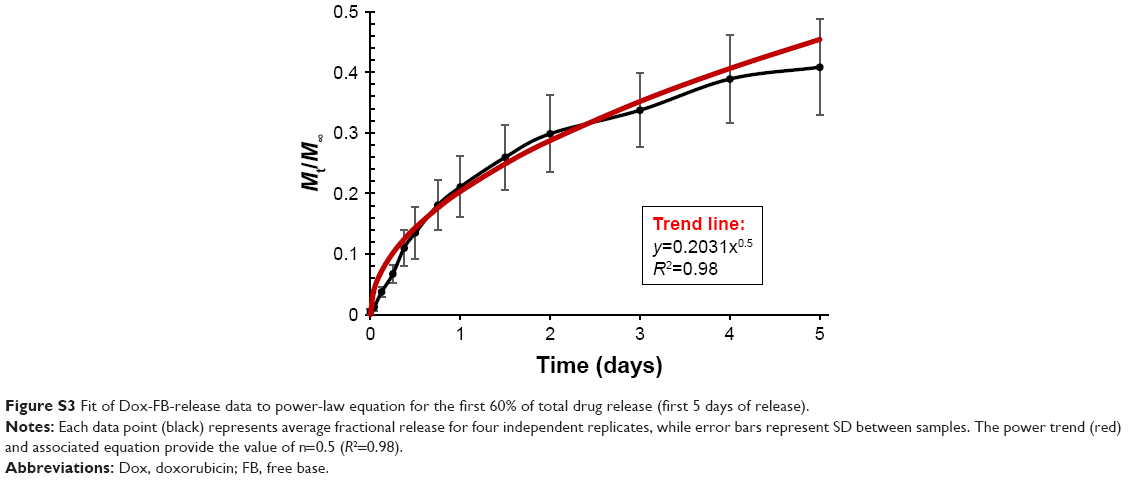 | Figure S3 Fit of Dox-FB-release data to power-law equation for the first 60% of total drug release (first 5 days of release). |
 | Figure S4 Appearance of 96-well plate after cells had been incubated with Dox-FB solution for 72 hours and washed with DPBS. |
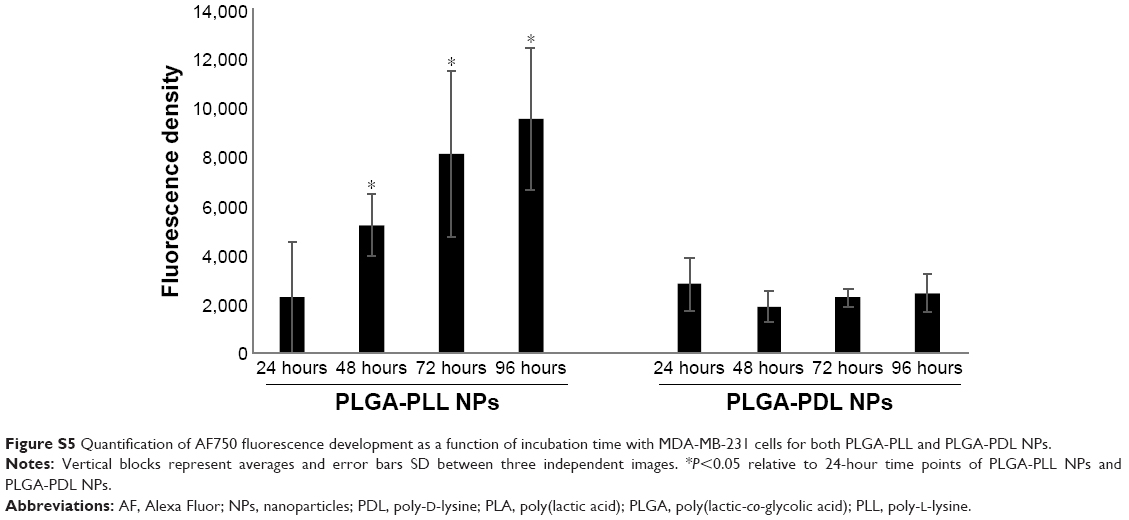 | Figure S5 Quantification of AF750 fluorescence development as a function of incubation time with MDA-MB-231 cells for both PLGA-PLL and PLGA-PDL NPs. |
 | Figure S6 Bright-field images of cells seeded in eight-well Lab-Tek chambered coverslips at 50,000 cells/well in 0.3 mL medium at 24-hour time points before study start. |
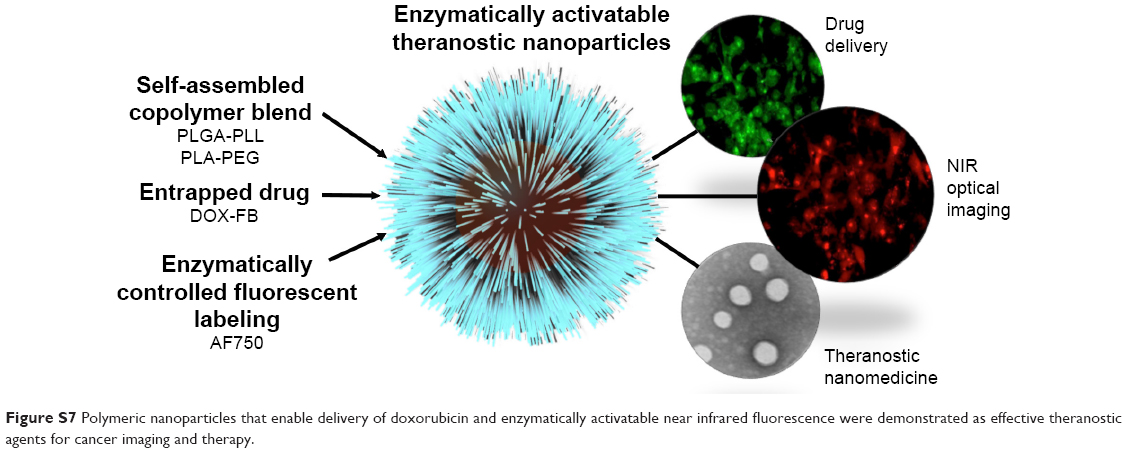 | Figure S7 Polymeric nanoparticles that enable delivery of doxorubicin and enzymatically activatable near infrared fluorescence were demonstrated as effective theranostic agents for cancer imaging and therapy. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.