Back to Journals » Journal of Inflammation Research » Volume 18
Downregulation of IFIT3 Relieves the Inflammatory Response in Ulcerative Colitis via Selectively Regulating Macrophage M1 Polarization and the STAT1/2 Signaling Pathway
Authors Du M, Tao Y, Yang K, Liu J, Yang X ![]()
Received 22 May 2025
Accepted for publication 30 August 2025
Published 6 September 2025 Volume 2025:18 Pages 12295—12309
DOI https://doi.org/10.2147/JIR.S542033
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Meiling Du,1,* Yiran Tao,2,* Ke Yang,3 Jin Liu,1 Xia Yang1
1Department of Gastroenterology, Fudan University Affiliated Shanghai Fifth People’s Hospital, Shanghai, People’s Republic of China; 2Department of General Medicine, Zhoupu Community Health Service Center, Shanghai, People’s Republic of China; 3Department of Pathology, Fudan University Affiliated Shanghai Fifth People’s Hospital, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xia Yang; Jin Liu, Email [email protected]; [email protected]
Purpose: Ulcerative colitis (UC) is an inflammatory condition of the colon. Interferon-induced protein with tetratricopeptide repeat 3 (IFIT3), a member of the IFIT family, is known to be associated with antiviral immunity and cellular regulation. However, the functional and molecular mechanisms by which IFIT3 affects the occurrence and development of UC have not been reported.
Patients and Methods: Intestinal mucosa samples were collected from 22 UC patients and 20 healthy controls. After immunohistochemical staining and image analysis, the correlation between IFIT3 expression and clinical characteristics of UC patients was analyzed. Subsequently, we applied a dextran sulfate sodium (DSS)-induced colitis model in adeno-associated virus 9 -mediated IFIT3 silencing mice. Inflammatory cytokines and signal pathway molecules were examined. Moreover, THP-1 cells, lipopolysaccharide (LPS) and upadacitinib (UPA) were used in vitro experiments, and various assays were performed to evaluate IFIT3 expression, cellular responses, and signaling pathways.
Results: First, our results demonstrated that IFIT3 was upregulated in colon tissues of UC patients and was positively correlated with the Mayo clinical score and fecal calprotectin levels. Second, knockdown of IFIT3 significantly attenuated the intestinal inflammatory response and reduced phosphorylated signal transducer and activator of transcription 1 and 2 (pSTAT1 and pSTAT2) protein levels in DSS-induced UC mice. Further mechanistic studies revealed that IFIT3 regulated LPS-induced M1 polarization in THP-1 macrophages by modulating the STAT1 signaling pathway. In addition, UPA exerted anti-inflammatory effects in vitro, to some extent, through the inhibition of IFIT3 expression.
Conclusion: Our findings highlight the role of IFIT3 in inflammatory responses and macrophage polarization in colitis, suggesting that IFIT3 may be a promising target for UC treatment.
Keywords: interferon-induced protein with tetratricopeptide repeat 3, ulcerative colitis, inflammatory response, macrophage, signal transducer and activator of transcription 1/2
Introduction
Ulcerative colitis (UC) is a chronic non-specific intestinal inflammation condition of unknown etiology. The pathogenesis of UC is closely associated with genetics, immunity, intestinal microbiota, and environmental factors.1 The disorder starts in the rectum and generally extends proximally in a continuous manner through the entire colon, presenting with clinical symptoms such as recurrent abdominal pain, diarrhea, and bloody stools containing mucus or pus and other manifestations. It affects 5 million people globally, with an increasing incidence annually, particularly in Northern Europe and North America.2 Compared with conventional drugs (such as 5-aminosalicylic acid, corticosteroids and immunomodulators), biological agents (such as tumor necrosis factor-alpha (TNF-α) inhibitors, integrin inhibitors and interleukin (IL)-12/23 inhibitors) and small molecule inhibitors (such as janus kinase (JAK) inhibitors, PDE4 inhibitors and S1P receptor modulator) have emerged as a significant treatment option for moderate to severe active UC, with fewer side effects and more accurate targeting.3,4 However, they are not effective for all UC patients. Therefore, it is imperative to further investigate gene targeting strategies for inhibiting inflammatory or cellular signaling pathways against UC, to provide more treatment options for UC patients.
The interferon (IFN)-induced proteins with tetratricopeptide repeats (IFIT) family consists of four members: IFIT1, IFIT2, IFIT3, and IFIT5. Previous studies have focused primarily on the antiviral immune response and innate immune response to these proteins, including IFIT3. Specifically, after a virus invades, it activates the body’s innate immune system and produces a large amount of IFN, which in turn stimulates the production of a variety of antiviral genes, including the IFIT family, through the JAK/signal transducer and activator of transcription (STAT) pathway.5,6 IFIT proteins directly bind to viral RNA7 or indirectly regulate the interaction of related cytokines, such as TBK1/IRF38,9 and MITA,10 effectively inhibiting viral replication and transcription and exerting powerful antiviral effects.
Moreover, the role of IFIT3 varies in different tumors, either promoting tumor progression (eg, oral squamous cell carcinoma11 and pancreatic ductal carcinoma12) or suppressing it (eg, hepatocellular carcinoma13). Recently, IFIT3 was shown to be involved in inflammatory responses in diseases such as multiple sclerosis,14 myocardial infarction,15 and liver ischemia‒reperfusion injury.16 Consequently, targeted modulation of IFIT3 expression in mouse models could significantly mitigate the inflammatory response observed in these diseases. In Yang’s study on berberine,17 IFIT3 was reported to be highly expressed in colon tissues of dextran sulfate sodium (DSS)-induced UC mice, but the role and potential mechanism of IFIT3 in UC were not further explored. Therefore, further exploration of IFIT3 in UC is necessary, with the hope that the findings could provide new evidence for the potential clinical application of IFIT3 in UC treatment.
Materials and Methods
Patients and Samples
All 42 intestinal mucosa samples were collected from Shanghai Fifth People’s Hospital in 2024, including samples from 22 UC patients and 20 healthy controls (HCs). Mayo clinical score (MCS) and fecal calprotectin (FC) levels were collected, both of which are widely used in clinical trials to assess the clinical status of UC patients. The MCS consists of four subscores, with each of the following scored 0, 1, 2 and 3: stool frequency, rectal bleeding, endoscopy findings, and physician’s global assessment.18 The study was approved by the Research Ethics Committee of the hospital, and written informed consent was obtained from all the subjects.
Immunohistochemistry (IHC)
According to the instructions of the ultrasensitive SP detection kit (#KIT-9730, Maxim, Fuzhou, China), IHC staining was performed using a primary antibody against IFIT3 (1:400; #15201-1-AP, Proteintect, Rosemont, USA). Immunohistochemical staining results were evaluated using a semiquantitative method, which was calculated by multiplying the staining intensity (graded as 0: negative; 1: weak; 2: moderate; and 3: strong) by the percentage of positive cells (graded as 0: negative; 1: ≤10%; 2: 11–50%; 3: 51–80%; and 4: ≥81%).19 IFIT3-positive cells were counted in five different areas of each section by two pathologists under double-blind conditions.
IFIT3 Silencing in DSS-Induced UC Mice
This study was approved by Shanghai Fifth People’s Hospital. The animal experimental procedures were carried out in strict accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Seven-week-old C57BL/6 male mice were purchased from SPF Biotechnology Co., Ltd. (Permission No. SCXK 2024–0001) and were maintained on a 12-hour light/12-hour dark cycle in a temperature- and humidity-controlled environment (23 °C, 50% humidity) for one week prior to experimentation. Seventy-two hours before UC induction, mice were administered 100 µL of either adeno-associated virus 9 containing short hairpin small interfering RNA (shRNA) targeting IFIT3 (AAV9-shIFIT3, designed and synthesized by Obio, Shanghai, China) or control AAV9 (AAV9-shNC) at a concentration of 1×1012 ng/mL via intraperitoneal injection,20 with 5 mice in each group. DSS (#9011-18-1, MP Biomedicals, California, USA) was subsequently added to the daily drinking water of mice at a final concentration of 2.5% for one week. Weight changes of mice were recorded daily. Upon induction of UC, mice were euthanized, and colon samples were obtained for further analysis. The target sequences of shIFIT3 and shNC were as follows: 5′-GCCTACATAAAGCACCTAGAT-3′ and 5′-CCTAAGGTTAAGTCGCCCTCG-3′. It was important to note that the efficacy of colonic transduction mediated by AAV9 (carrying eGFP under the control of the ubiquitous CMV promoter) was visualized and analyzed prior to the study by using paraffin sections and the IVIS Lumina III imaging system (provided by PerkinElmer in Massachusetts, USA), through intraperitoneal injection of AAV9 and saline.
Hematoxylin and Eosin (H&E) Staining
Tissues from the transverse colon of mice were fixed in 4% paraformaldehyde overnight, dehydrated in graded ethanol, embedded in paraffin, and sectioned. The tissue sections were then stained with H&E for microscopic examination and evaluated with a tissue damage index, which was derived from a combination of the epithelial barrier injury score (0–4) and the infiltration leukocyte score (0–4).21
Immunofluorescence Staining
To visualize the macrophages, double immunofluorescence staining was conducted. Following antigen retrieval and blocking, the tissue sections were incubated with primary antibodies against CD68 (1:500; #28058-1-AP, Proteintect) and IFIT3 (1:500; #15201-1-AP, Proteintech) at 4 °C overnight. The sections were subsequently incubated with Cy3-conjugated (1:300; #GB 21303, Servicebio, Wuhan, China) or HRP-conjugated (1:500; #GB23303, Servicebio) secondary antibodies for 1 hour at room temperature. Finally, the sections were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; #G1012, Servicebio). Fluorescence images were captured using a microscope (Olympus, Tokyo, Japan).
Cell Culture and Differentiation
THP-1 cells were obtained from the cell bank at the Chinese Academy of Sciences and cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum and 0.05 mM β-mercaptoethanol at 37 °C in a humidified atmosphere of 5% CO2. Routine medium changes were performed during passaging, and cells in the logarithmic growth phase were used for experiments.
Subsequently, THP-1 cells were seeded into 6-well plates at a density of 1×106 cells per well and differentiated into macrophages with 100 ng/mL phorbol-12-myristate-13-acetate (#S1819, Beyotime, Shanghai, China) for 24 hours. Successfully differentiated macrophages were then treated with 100 ng/mL lipopolysaccharide (LPS; #S1732, Beyotime) alone or in combination with 20 ng/mL IFN‐γ (#C104, Novoprotein, Suzhou, China) for the M1 phenotype, 20 ng/mL IL-4 (#P5129, Beyotime) for the M2 phenotype, or left untreated (M0) as a control for 48 hours.
Cell Transfection and Upadacitinib (UPA) Treatment
To silence IFIT3, M1 THP-1 macrophages were infected with either two siRNAs targeting IFIT3 (siIFIT3-1 and siIFIT3-2) or a control siRNA (siNC) using a siRNA transfection kit (#P09410, LeapWal, Changsha, China). Additionally, M0 THP-1 macrophages were pretreated with 100 nM and 400 nM UPA (#HY-19569, Medchemexpress, New Jersey, USA) for 1 hour prior to stimulation with 100 ng/mL LPS. Cells were subsequently cultured for 48 hours to collect RNA and for 96 hours to collect protein or perform flow cytometry. The siRNA sense sequences were as follows: 5′-GGAAGAGAUCAAAGACCAATT-3′ (siIFIT3-1) and 5′-GCACUAAAGCAAUAUGCUATT-3′. (siIFIT3-2), and 5′-UUCUCCGAACGUGUCACGUTT-3′ (siNC).
Flow Cytometry
Macrophage polarization was assessed using flow cytometry. Briefly, THP-1 cells were harvested from 6-well plates, resuspended in PBS, and incubated with the following antibodies on ice for 20 minutes in the dark: FITC-conjugated anti-human CD68 (#333805, Biolegend, San Diego, USA) and PE-conjugated anti-human CD86 (#374206, Biolegend). Cells were subsequently analyzed using a flow cytometer (DAKEWE, Shenzhen, China).
Quantitative Real-Time PCR (qRT‒PCR)
Total RNA was isolated from transverse colon tissues or THP-1 cells using an RNA extraction kit (#RK30120, ABclonal, Wuhan, China) following the manufacturer’s instructions. The RNA was subsequently reverse transcribed into cDNA using Primescript™ RT Master Mix (#RR036A, Takara, Shiga, Japan). Quantitative real-time PCR (qRT‒PCR) was then conducted using TB Green® Premix Ex Taq™ II (#RR820A, Takara) in a QuantStudio™ 6 Flex Real‒Time PCR instrument (Applied Biosystems, USA) The primers used in the study are listed in Table 1.
 |
Table 1 Primers Used in This Study |
RNA-Seq Analysis
The purity, concentration, and integrity of the total RNA samples were assessed prior to RNA-seq analysis. Library preparation, cluster generation, and sequencing were subsequently conducted by Novogene Bioinformatics Technology. Gene expression analysis of different samples was performed using the DESeq2 R package. Genes with “|log2FC| (the absolute log2 value of the fold change in gene expression) > 2” and “P value < 0.05” were identified as differentially expressed genes (DEGs). For the functional enrichment analysis of the identified DEGs, we employed the clusterProfiler package in R 4.2.2. This comprehensive approach facilitated the exploration of significantly enriched Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways associated with DEGs.
Western Blot
Total proteins from transverse colon samples or THP-1 cells were extracted with RIPA lysis buffer containing 1% phosphatase inhibitors. After quantification using the BCA protein assay kit (#P0012, Beyotime), equal amounts of proteins were separated by 10% SDS‒PAGE, transferred to PVDF membranes, and blocked with TBST containing 5% BSA for 1 hour. The PVDF membranes were incubated with primary antibodies against IFIT3 (1:3000; #15201-1-AP, Proteintect), STAT1 (1:1000; #R25799, Zenbio, Chengdu, China), pSTAT1 (1:1000; #bsm-52209R, Bioss, Beijing, China), STAT2 (1:1000; #R381419 Zenbio), pSTAT2 (1:1000; #bs-3428R, Bioss), and GAPDH (1:1000; #GB15002-100, Servicebio) overnight at 4 °C and then incubated with HRP-labeled secondary antibodies at room temperature for 1 hour. Finally, protein bands were observed via an enhanced chemiluminescence developer (Tanon, Shanghai, China).
Statistical Analysis
Statistical analysis was performed using the GraphPad Prism software (version 9.0). Each experiment was reproduced at least 3 times. The data obtained from this study are presented as the means ± standard deviations (SDs). Statistical significance was calculated by 2-tailed Student’s t test or one-way analysis of variance with Dunnett’s multiple-comparison test. Moreover, Pearson analysis was used for correlation analysis. A value of p < 0.05 was considered statistically significant (*p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001).
Results
IFIT3 Was Upregulated in the Colon Tissues of UC Patients
IHC results revealed that the mean expression level of IFIT3 in the colon mucosa of UC patients was 7.98 ± 2.65, which was significantly greater than that in HCs (2.18 ± 1.76, p < 0.0001) (Figure 1A). Specifically, IFIT3 expression was found to be abundant in the epithelium and localized to the cytoplasm.
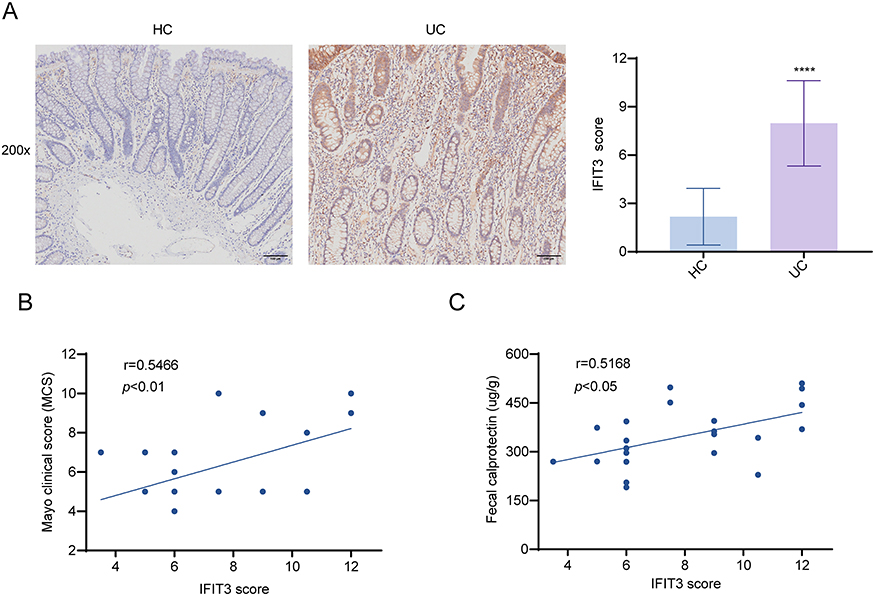 |
Figure 1 IFIT3 was upregulated in colon tissues of UC patients by IHC results. (A) Representative images and score of IHC staining for IFIT3 from 20 HCs and 22 UC samples. Scale bar = 100 μm. (B) The correlation between colonic IFIT3 score and MCS in 22 UC patients. (C) The correlation between colonic IFIT3 score and FC levels in 22 UC patients. ****p < 0.0001. Abbreviations: IFIT3, interferon-induced protein with tetratricopeptide repeat 3; UC, ulcerative colitis; IHC, immunohistochemistry; HC, healthy control; MCS, Mayo clinical score; FC, fecal calprotectin. |
As MCS and FC levels are commonly used to describe the clinical status of UC patients, we investigated the correlation between MCS/FC levels and colonic IFIT3 expression in these patients. Correlation analysis revealed that both the MCS and FC levels were positively associated with the IFIT3 score in colon tissues (Figure 1B and C). These findings suggest that IFIT3 might be a potential biomarker for colonic injury in UC patients.
IFIT3 Was Upregulated in DSS-Induced UC Mice
M1 macrophages play a crucial role in the pathogenesis of UC. Therefore, we induced colitis in mice using DSS to investigate the expression of IFIT3 in inflamed colon tissues (Figure 2A). Our results revealed that both the mRNA and protein levels of IFIT3, as well as the mRNA levels of the M1 macrophage-related genes TNF-α, IL-1β, and IL-6, were significantly elevated in the colon tissues of mice with colitis compared with those in the control group (Figure 2B and C). Moreover, immunofluorescence revealed that co-expression level of IFIT3 and CD68 (a macrophage surface marker) was higher in the colon tissues of mice with colitis (Figure 2D). These findings suggest that IFIT3 may be involved in the pathogenesis of UC through its interaction with M1 macrophages.
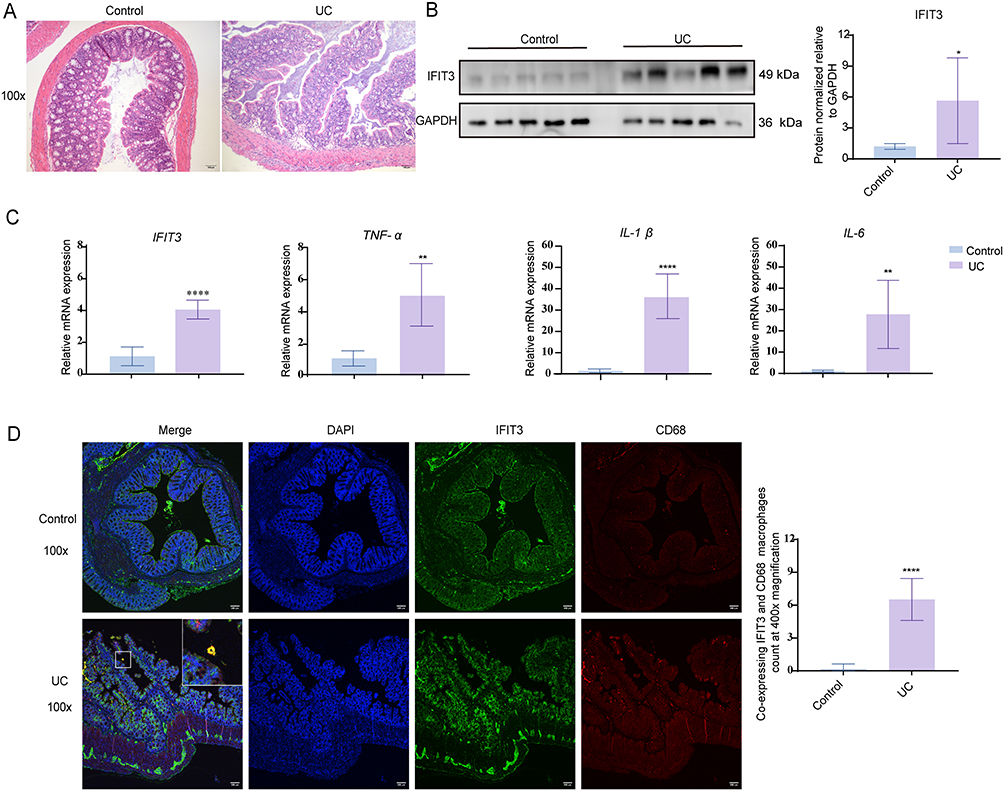 |
Figure 2 IFIT3 was upregulated in colon tissues of DSS-induced UC mice. (A) Representative images of H&E-stained colon tissues from control and DSS-induced UC mice. Scale bar = 100 μm. (B) Protein levels of IFIT3 in colon tissues from the two mouse groups. (C) mRNA levels of IFIT3, TNF-α, IL-1β, and IL-6 in colon tissues from the two mouse groups. (D) Representative images of double immunofluorescence staining for IFIT3 and CD68 in colon tissues from the two mouse groups, and quantification of co-expressing IFIT3 and CD68 macrophages at 400x magnification. Scale bar = 100 μm. Data was presented as mean ± SD (n = 5). * p < 0.05, ** p < 0.01, **** p < 0.0001. Abbreviations: IFIT3, interferon-induced protein with tetratricopeptide repeat 3; DSS, dextran sulfate sodium; UC, ulcerative colitis; H&E, hematoxylin and eosin; TNF-α, tumor necrosis factor-alpha; IL, interleukin; SD, standard deviation. |
IFIT3 Knockdown Relieved the Intestinal Inflammatory Response and Inhibited the Activation of the STAT1/2 Pathway in DSS-Induced UC Mice
Compared with saline, AAV9-mediated colon transduction efficacy following intraperitoneal injection was more potent, as visualized in paraffin sections and in the IVIS Lumina III imaging system (Figure 3A and B). Subsequently, to determine the role of IFIT3 in UC, we compared the pathological changes in DSS-induced UC mice that were intraperitoneally injected with AAV9-shIFIT3 or AAV9-shNC. As shown in Figure 3C, the average colon length of the AAV9-shIFIT3-treated UC mice was not significantly greater than that of the AAV9-shNC-treated UC mice. Consistently, slight weight gain was observed after 3 days of virus treatment and 2 days of DSS treatment in both groups. However, from the sixth day onward, both groups of mice gradually decreased in weight; the weight of the AAV9-shIFIT3-treated mice decreased less than that of the AAV9-shNC-treated mice from days 8 to 10 (Figure 3D).
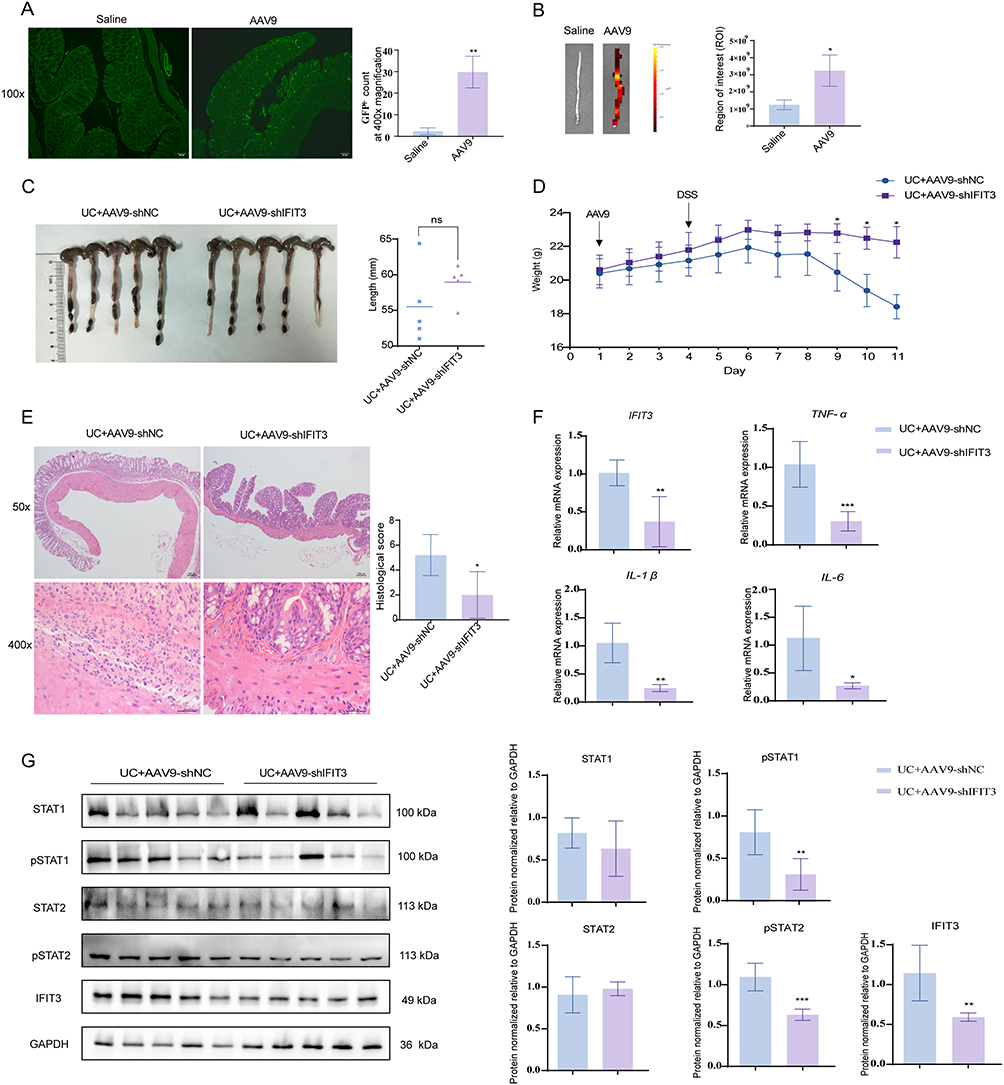 |
Figure 3 IFIT3 knockdown relieved the intestinal inflammatory response and inhibited the activation of the STAT1/2 pathway in DSS-induced UC mice. (A and B) Comparison of colon transduction efficacy in mice between intraperitoneal injection of saline and AAV9 using paraffin sections (A) and the IVIS Lumina III imaging system (B). Scale bar = 100 µm. (C) Comparison of colon length between AAV9-shNC- and AAV9-shIFIT3-treated UC mice. (D) Weight changes in the two mouse groups. (E) Representative images and histological score of H&E staining in colon tissues from the two mouse groups. Scale bar = 100 µm (top) and 25 µm (bottom). (F) mRNA levels of IFIT3, TNF-α, IL-1β and IL-6 in colon tissues from the two mouse groups. (G) Protein expressions of STAT1, pSTAT1, STAT2, pSTAT2 and IFIT3 in colon tissues from the two mouse groups. Data was presented as mean ± SD (n = 5). * p < 0.05, ** p < 0.01, *** p < 0.001. Abbreviations: IFIT3, interferon-induced protein with tetratricopeptide repeat 3; UC, ulcerative colitis; AAV9, adeno-associated virus 9; H&E, hematoxylin and eosin; pSTAT, phosphorylated signal transducer and activator of transcription; TNF-α, tumor necrosis factor-alpha; IL, interleukin; SD, standard deviation. |
Moreover, histological analysis of H&E-stained colonic tissues revealed that, compared with the AAV9-shNC-treated mice, the AAV9-shIFIT3-treated mice presented less damage, with incomplete loss of the epithelial barrier and less infiltration of inflammatory cells in the colon, with lower histological score after 7 days of DSS treatment (5.20 ± 1.64 vs 2.00 ± 1.87, p < 0.05; Figure 3E). Consequently, the relative mRNA levels of TNF-α, IL-1β, and IL-6 in the colon tissues of the AAV9-shIFIT3-treated mice were markedly lower than those in the colon tissues of the AAV9-shNC-treated mice (Figure 3F). To further explore the underlying mechanism of the role of IFIT3 in UC, we measured the activation of the STAT1/2 pathway in the colon tissues of both groups using Western blot analysis. Our findings indicated that knockdown of IFIT3 inhibited the activation of the STAT1/2 pathway, as evidenced by the decreased phosphorylated STAT1 and STAT2 (pSTAT1 and pSTAT2), with no significant change observed in the total protein levels of STAT1 and STAT2 (Figure 3G).
Furthermore, we performed RNA-Seq to assess the effect of IFIT3 on global gene expression in the colons of AAV9-shIFIT3- and AAV9-shNC-treated mice. A total of 974 upregulated and 1364 downregulated genes were identified between the two sample groups (Figure 4A). Among the downregulated genes, IFIT3 and several M1 macrophage-related genes, including TNF-α, IL-1β, and IL-6, were identified in AAV9-shIFIT3-treated mice (Figure 4B). GO analysis returned 8 enriched pathways, including the humoral immune response, acute inflammatory response, and chemokine-mediated signaling pathway. In addition, KEGG pathway analysis demonstrated that these DEGs were enriched in several M1-related pathways, such as cytokine‒cytokine receptor interaction and the PI3K‒Akt signaling pathway (Figure 4C and D).
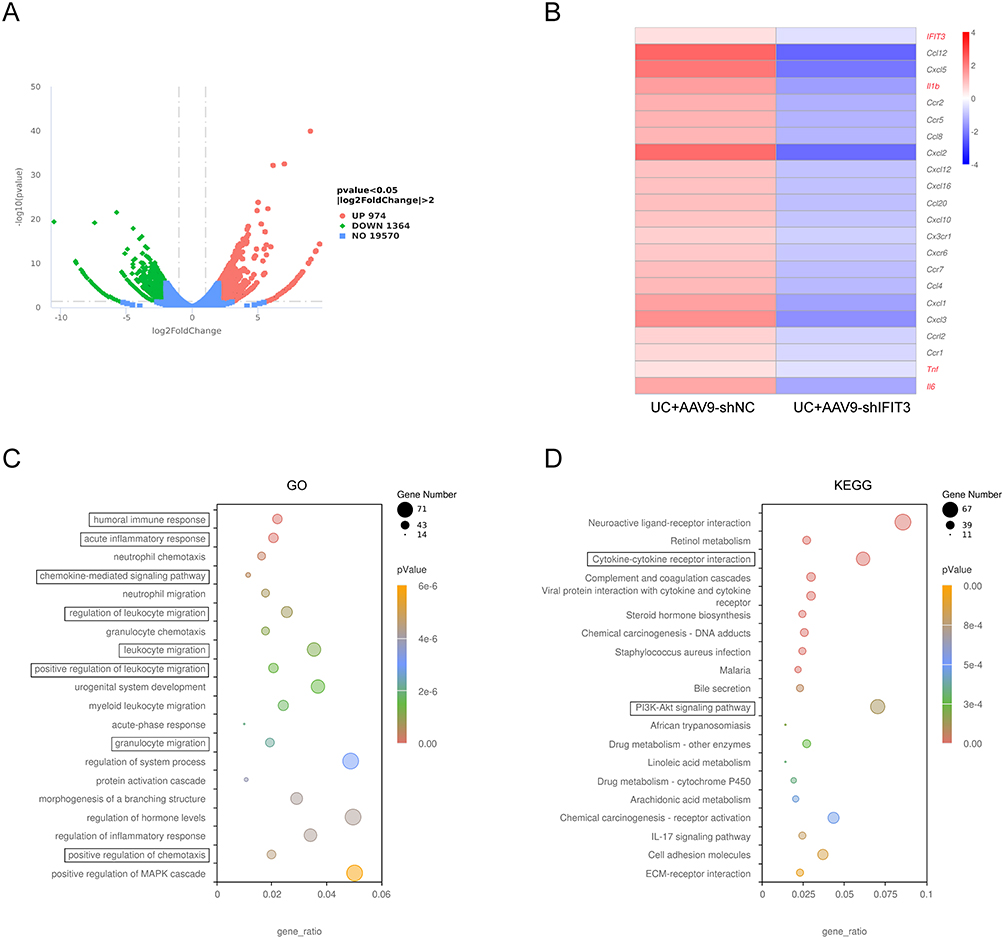 |
Figure 4 IFIT3 knockdown relieved the intestinal inflammatory response in DSS-induced UC mice by RNA-Seq analysis. (A) Volcano plot showed DEGs in colon tissues between AAV9-shNC- and AAV9-shIFIT3-treated UC mice (n=1). (B) Heatmap showed IFIT3 and differentially expressed macrophage polarization–related genes, including TNF-α, IL-1β, and IL-6, in colon tissues from the two mouse groups. (C and D) Significantly enriched GO terms (C) and KEGG pathways (D) of DEGs in colon tissues from the two mouse groups. Abbreviations: IFIT3, interferon-induced protein with tetratricopeptide repeat 3; UC, ulcerative colitis; DEG, differentially expressed gene; GO, gene ontology; KEGG, kyoto encyclopedia of genes and genomes. |
IFIT3 Expression Was Upregulated in M1 THP-1 Macrophages
To examine the expression of IFIT3 in different types of macrophages, we stimulated THP-1 cells with LPS alone or LPS + IFN-γ simultaneously to induce M1 polarization and with IL-4 to induce M2 polarization. Compared with those in the control group (M0), the relative mRNA and protein levels of IFIT3 were significantly greater in the LPS- and LPS + IFN-γ-stimulated macrophages, especially in the latter, but not in the IL-4-stimulated macrophages (Figure 5A and B). These findings suggest a close correlation between IFIT3 expression and M1 polarization.
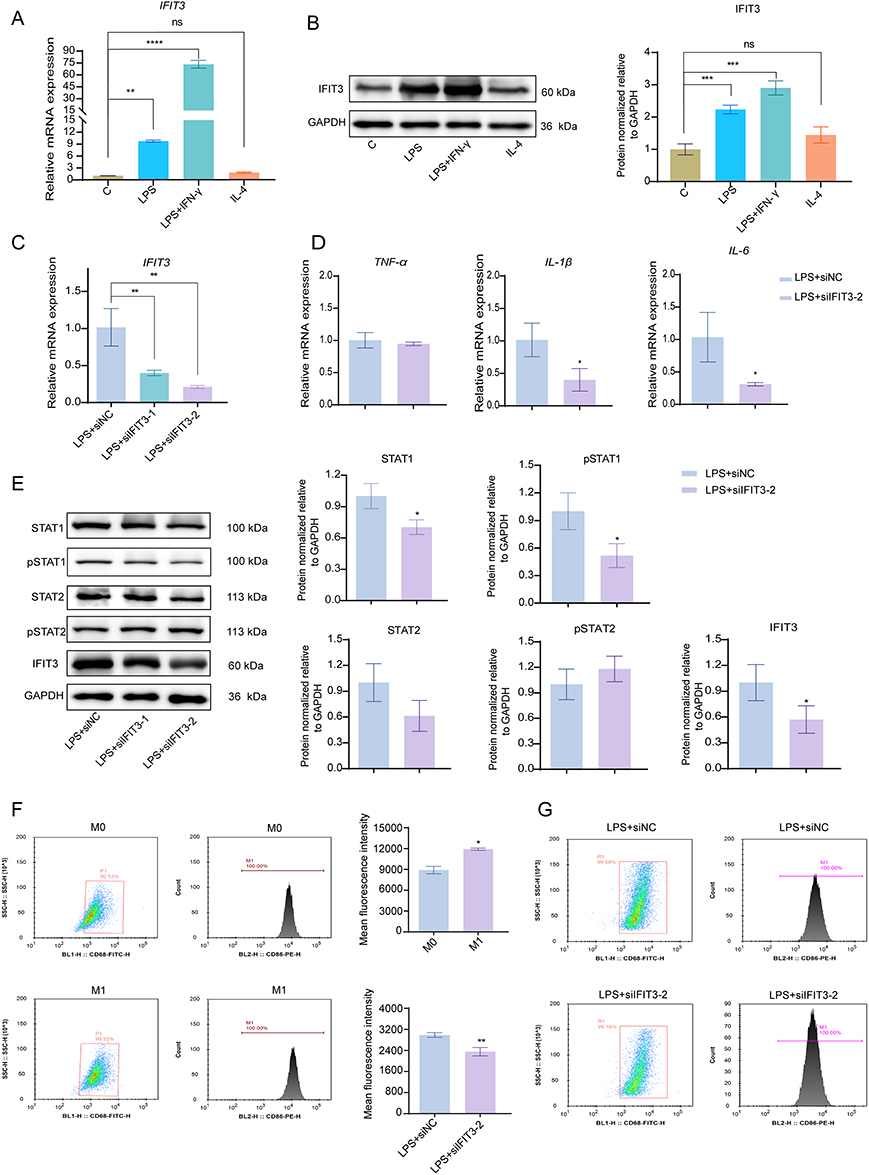 |
Figure 5 IFIT3 knockdown repressed macrophage M1 polarization by inhibiting the STAT1 pathway in vitro. (A and B) mRNA and protein levels of IFIT3 in untreated THP‐1 macrophages or treated with 100 ng/mL LPS or 100 ng/mL LPS + 20 ng/mL IFN‐γ or 20 ng/mL IL‐4. (C) mRNA levels of IFIT3 in 100 ng/mL LPS-stimulated macrophages treated with siNC or two siRNAs targeting IFIT3 (siIFIT3-1 and siIFIT3-2). (D) mRNA levels of TNF-α, IL-1β and IL-6 in 100 ng/mL LPS-stimulated macrophages treated with siNC and siIFIT3-2. (E) Protein levels of STAT1, pSTAT1, STAT2, pSTAT2 and IFIT3 in each group. (F) Representative gating and the co-expression of CD86 and CD68 in untreated (M0) and 100 ng/mL LPS-stimulated macrophages (M1) by flow cytometry. (G) Representative gating and the co-expression of CD68 and CD86 in 100 ng/mL LPS-stimulated macrophages treated with siNC and siIFIT3-2 by flow cytometry. Data was presented as mean ± SD from 3 independent experiments. * p < 0.05, ** p < 0.01, ***p < 0.001, **** p < 0.0001. Abbreviations: IFIT3, interferon-induced protein with tetratricopeptide repeat 3; LPS, lipopolysaccharide; IFN‐γ, interferon‐γ; IL, interleukin; pSTAT, phosphorylated signal transducer and activator of transcription; TNF-α, tumor necrosis factor-alpha. |
IFIT3 Knockdown Repressed LPS-Induced M1 Polarization in THP-1 Macrophages by Inhibiting the STAT1 Pathway
As depicted in Figure 5C, successful knockdown of IFIT3 by small interfering RNA was observed in LPS-stimulated macrophages, particularly those treated with siIFIT3-2, but this effect was not evident in LPS + IFN-γ-stimulated macrophages. Subsequently, an in-depth investigation was carried out to elucidate the role of IFIT3 in LPS-stimulated macrophages. Knockdown of IFIT3 in LPS-stimulated macrophages resulted in decreased mRNA levels of IL-1β and IL-6 (Figure 5D) and protein levels of STAT1 and pSTAT1 compared with those in the control group (Figure 5E). However, no significant difference in STAT2 or pSTAT2 protein levels was detected between the two cell groups (Figure 5E). Moreover, flow cytometry analysis revealed that CD68 and CD86, which are cell-surface molecules associated with M1 polarization, were both highly expressed in LPS-stimulated macrophages. Conversely, the co-expression was significantly reduced in siIFIT3-2-treated macrophages (Figure 5F and G). These findings suggest that the STAT1 pathway is involved in the IFIT3-induced proinflammatory response in the LPS-induced M1 polarization of THP-1 macrophages.
Furthermore, according to RNA-Seq, a total of 172 genes were upregulated and 280 genes were downregulated in LPS-stimulated macrophages with siIFIT3-2 interference compared with those in the control group (Figure 6A), among which the expression of IFIT3 and M1-related genes (such as IL-1a and IL-1b) was reduced in siIFIT3-2-treated macrophages (Figure 6B). These DEGs were associated with cytokine activity and chemotaxis according to GO analysis and enriched in inflammatory bowel disease (IBD) and the JAK/STAT signaling pathway, as indicated by KEGG analysis (Figure 6C and D).
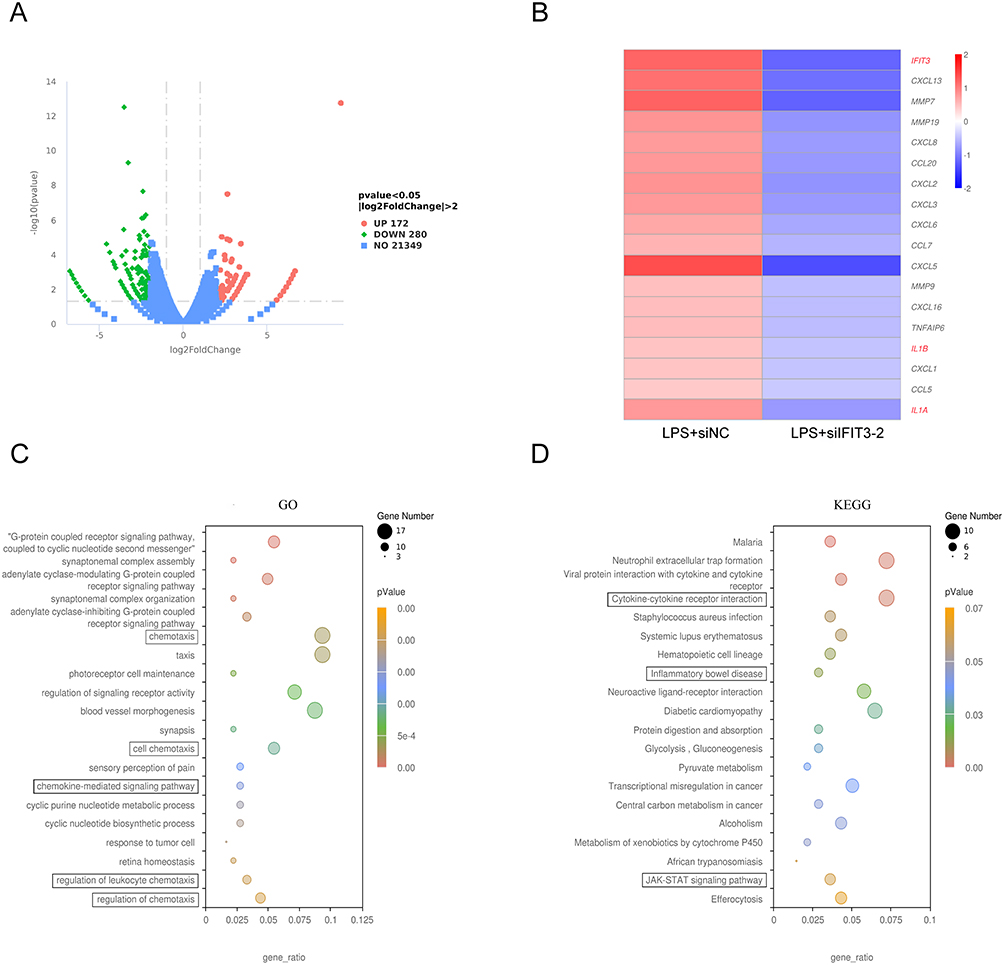 |
Figure 6 IFIT3 knockdown inhibited macrophage M1 polarization in vitro by RNA-Seq analysis. (A) Volcano plot showed DEGs in 100 ng/mL LPS-stimulated macrophages treated with siNC and siIFIT3-2 (n=1). (B) Heatmap showed IFIT3 and differentially expressed macrophage polarization–related genes (such as IL-1a and IL-1b) in LPS-stimulated macrophages treated with siIFIT3-2 compared to the control. (C and D) Significantly enriched GO terms (C) and KEGG pathways (D) of DEGs between the two sample groups. Abbreviations: IFIT3, interferon-induced protein with tetratricopeptide repeat 3; DEG, differentially expressed gene; LPS, lipopolysaccharide; GO, gene ontology; KEGG, kyoto encyclopedia of genes and genomes. |
UPA Inhibits Inflammation by Silencing IFIT3 in LPS-Stimulated Macrophages
UPA, a JAK1 inhibitor, was utilized to investigate whether its anti-inflammatory effect was associated with IFIT3 levels in vitro. The results revealed a significant decrease in both IFIT3 mRNA and protein levels in LPS-stimulated macrophages treated with 100 nM and 400 nM UPA compared with those in the untreated group, with more pronounced effects observed at the higher concentration of 400 nM. Meanwhile, the mRNA levels of TNF-α, IL-1β, and IL-6 were notably reduced in both experimental cell groups (Figure 7A). And there was a significant decrease in the protein levels of STAT1, STAT2, pSTAT1 and pSTAT2 (Figure 7B).
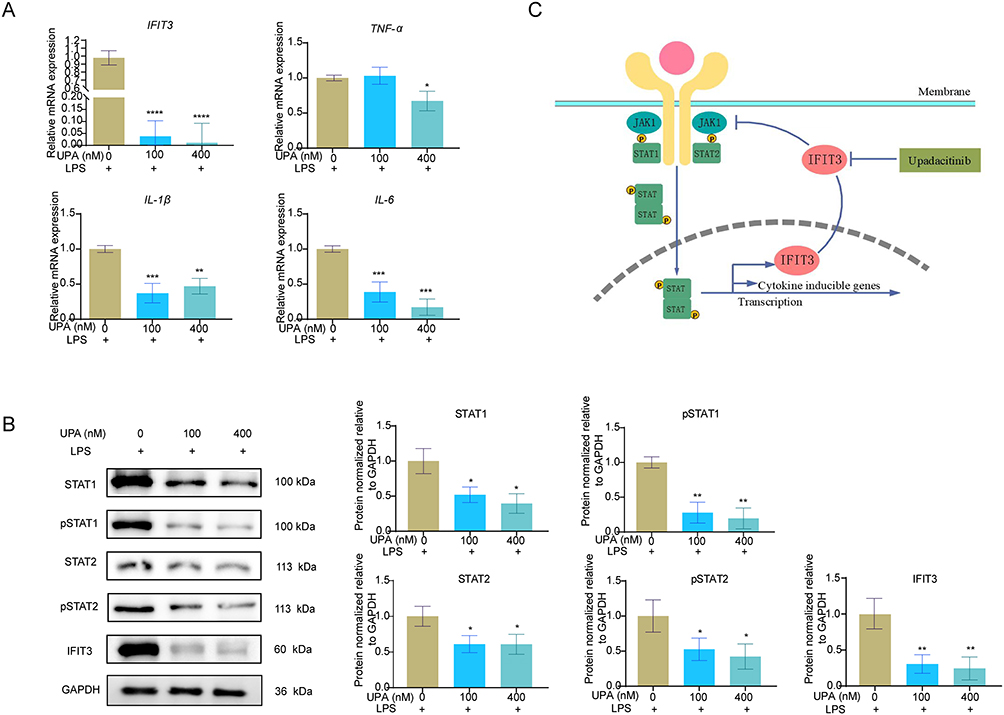 |
Figure 7 UPA inhibits inflammation by silencing IFIT3 in LPS-stimulated macrophages. (A) mRNA levels of IFIT3, TNF-α, IL-1β and IL-6 in 100 ng/mL LPS-stimulated macrophages with untreated or treated with 100 nM and 400 nM UPA. (B) Protein levels of STAT1, pSTAT1, STAT2, pSTAT2 and IFIT3 among cell-groups. (C) Schematic diagram of relation among UPA, IFIT3 and STAT1/2 pathways. Data was presented as mean ± SD from 3 independent experiments. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. Abbreviations: UPA, upadacitinib; IFIT3, interferon-induced protein with tetratricopeptide repeat 3; LPS, lipopolysaccharide; TNF-α, tumor necrosis factor-alpha; IL, interleukin; pSTAT, phosphorylated signal transducer and activator of transcription. |
Discussion
In this study, our results demonstrated for the first time that IFIT3 plays a pivotal role in the pathophysiological process of UC, particularly in the inflammatory response. Knocking down IFIT3 delayed UC progression in an in vivo mouse model and inhibited macrophage M1 polarization in vitro, by suppressing the STAT1/2 signaling pathway. Additionally, UPA exerted anti-inflammatory effects in vitro, partly through the inhibition of IFIT3 expression (Figure 7C). Overall, our findings may enhance the understanding of molecular interventions targeting IFIT3 for UC treatment.
UC is a common intestinal inflammatory disease with limited therapeutic options. Identifying related genes and exploring the mechanisms of this disease offer more opportunities for the treatment of UC. IFIT3 was found to be upregulated in UC patients and DSS-induced UC mice, indicating that IFIT3 may play a role in the development of UC. The pathogenesis of UC involves the response of macrophages to colonic antigenic immune attacks. Specifically, during the active phase of UC, many activated M1 macrophages colonize the colon mucosal lamina propria and produce many proinflammatory cytokines, such as TNF-α, IL-1β, and IL-6, resulting in epithelial barrier damage, epithelial cell apoptosis, and exacerbation of the disease.22–24 In this study, IFIT3 was highly expressed in CD68+ macrophages in the colons of mice with colitis. Knocking down IFIT3 ameliorated clinical symptoms and decreased inflammatory infiltration in UC model mice, revealing that IFIT3 might contribute to UC by enhancing the inflammatory response.
To further understand the role of IFIT3 in macrophages, we initially stimulated THP-1 cells with LPS, LPS + IFN-γ, or IL-4 to polarize them into M1 or M2 phenotypes. The expression of IFIT3 was found to be increased in M1 macrophages induced by LPS or LPS + IFN-γ, particularly in the latter case, which aligns with the findings of previous study.25 Subsequent silencing of IFIT3 in LPS-stimulated macrophages resulted in decreased co-expression level of M1 phenotype markers (CD68 and CD86) and mRNA levels of M1-related genes (IL-1β and IL-6), further confirming the pivotal role of IFIT3 in M1 polarization of macrophages. Reactive macrophages exhibit two phenotypes with a continuous spectrum of functional heterogeneity, namely, the M1 phenotype and the M2 phenotype. In contrast to proinflammatory macrophages (M1) in UC, M2 macrophages are anti-inflammatory and play a role in tissue repair and inflammation resolution.26,27 Notably, the expression of IFIT3 in macrophages remained unchanged under M2-polarizing condition. The predominance of M1 macrophages (not the M2 phenotype), which was mediated by certain genes, such as Mef2c, was also typically observed during the active phases of UC, contributing to sustained inflammation.28
IFIT3 is involved in the JAK/STAT signaling pathway, which is activated by various cytokines.5,6 In this study, we silenced IFIT3 in mice and observed a decrease in pSTAT1 and pSTAT2 protein levels, whereas total STAT1 and STAT2 protein levels remained unchanged. Interestingly, targeted silencing of IFIT3 in LPS-stimulated macrophages resulted in decreased expression of the STAT1 and pSTAT1 proteins but no changes in the STAT2 and pSTAT2 proteins. Unlike STAT2/pSTAT2, STAT1/pSTAT1 typically mediate the signaling pathway activated by Type II IFNs such as IFN-γ, a key cytokine that drives M1 polarization.29,30 Increased expression of STAT1 has been linked to increased M1 macrophage activation in UC.22,31,32 In certain autoimmune diseases, such as multiple sclerosis, STAT1 expression was significantly upregulated, whereas STAT2 expression was downregulated,14 indicating their differential regulation under pathological conditions. In addition, Aniska Chikhalya et al reported that the antiviral effect of IFIT3 involved the activation of STAT1 rather than STAT2.8 These findings suggest that, compared with STAT2/pSTAT2, STAT1/pSTAT1 may play a more prominent role in inflammatory responses.
UPA, a selective inhibitor of JAK1, effectively disrupts the downstream signaling cascades initiated by inflammatory factors, leading to reduced inflammation and amelioration of symptoms in conditions such as IBD and rheumatoid arthritis.33,34 Our study demonstrated that UPA can effectively suppress the expression of IFIT3 in LPS-stimulated macrophages, exerting anti-inflammatory effects. Notably, low concentration of UPA and IFIT3 silencing did not reduce TNF-α expression. Yuya Fujita et al explored the effects of various types of JAK inhibitors, including UPA, by selectively monitoring IL-6 expression in GM-CSF-stimulated THP-1 cells.35 In line with these findings, our experiments revealed that, regardless of whether IFIT3 was silenced or UPA was applied in vitro, the reduction in IL-6 expression was pronounced.
Certainly, our research has some inevitable limitations. First, due to the degradation of some RNA samples, there were not enough RNA-Seq samples of tissues or cells. However, both heatmap results revealed that IFIT3 silencing effectively reduced the expression of M1-related inflammatory factors, and DEGs enriched in the JAK/STAT signaling pathway were identified via KEGG analysis via cell sequencing. Second, owing to the interference efficiency of the siRNAs, we explored the effect of IFIT3 in LPS-induced M1 macrophages. Lentiviral vector-mediated IFIT3 silencing in IFN-γ + LPS-induced M1 macrophages needs to be studied in the future. In addition, IFIT3 seemed to be expressed not only in macrophages but also in other cells (such as intestinal epithelial cells) in the UC model, and it is speculated that IFIT3 would affect other cells as well, which in turn interfered with UC through different mechanisms. This hypothesis needs to be verified in future studies.
Conclusion
In summary, we provided evidence that IFIT3 was upregulated in inflamed colon tissues and that the knockdown of IFIT3 exerted potent protective effects against inflammation, possibly through the suppression of macrophage M1 polarization and the STAT1/2 signaling pathway. In addition, UPA attenuated inflammation, to some extent, by decreasing the level of IFIT3. These findings suggest that IFIT3 may be proposed as a therapeutic target for the treatment of UC.
Data Sharing Statement
The datasets used and/or analyzed in the current study are available from the corresponding author on reasonable request.
Ethics Approval and Informed Consent
All experiments involving clinical samples and mice were conducted in accordance with the protocol approved by the Ethics Review Committee of Shanghai Fifth People’s Hospital (Approval No. 2022JSWY-046 and 2024-118). All participants provided written consent to take part in the research. The experiments were carried out in compliance with the Declaration of Helsinki.
Acknowledgments
The authors are grateful to the support and assistance in terms of technological help provided by Pathology Department of Shanghai Fifth People’s Hospital. We would like to thank the all participants involved in the study.
Funding
This work was supported by Natural Science Fund of Minhang District (NO. 2024MHZ010) and Scientific Research Project Fund of Shanghai Fifth People’s Hospital (NO. 2022WYZT01).
Disclosure
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1. Borowitz SM. The epidemiology of inflammatory bowel disease: clues to pathogenesis? Front Pediatr. 2022;10:1103713. doi:10.3389/fped.2022.1103713
2. Le Berre C, Honap S, Peyrin-Biroulet L. Ulcerative colitis. Lancet. 2023;402(10401):571–584. doi:10.1016/S0140-6736(23)00966-2
3. Liu J, Di B, Xu LL. Recent advances in the treatment of IBD: targets, mechanisms and related therapies. Cytokine Growth Factor Rev. 2023;71-72:1–12. doi:10.1016/j.cytogfr.2023.07.001
4. Stallmach A, Atreya R, Grunert PC, Stallhofer J, de Laffolie J, Schmidt C. Treatment strategies in inflammatory bowel diseases. Dtsch Arztebl Int. 2023;120(45):768–778. doi:10.3238/arztebl.m2023.0142
5. Zhang W, Li Y, Xin S, et al. The emerging roles of IFIT3 in antiviral innate immunity and cellular biology. J Med Virol. 2023;95(1):e28259. doi:10.1002/jmv.28259
6. Zhou X, Michal JJ, Zhang L, et al. Interferon induced IFIT family genes in host antiviral defense. Int J Biol Sci. 2013;9(2):200–208. doi:10.7150/ijbs.5613
7. Fleith RC, Mears HV, Leong XY, et al. IFIT3 and IFIT2/3 promote IFIT1-mediated translation inhibition by enhancing binding to non-self RNA. Nucleic Acids Res. 2018;46(10):5269–5285. doi:10.1093/nar/gky191
8. Chikhalya A, Dittmann M, Zheng Y, Sohn SY, Rice CM, Hearing P. Human IFIT3 protein induces interferon signaling and inhibits adenovirus immediate early gene expression. mBio. 2021;12(6):e0282921. doi:10.1128/mBio.02829-21
9. Liu XY, Chen W, Wei B, Shan YF, Wang C. IFN-induced TPR protein IFIT3 potentiates antiviral signaling by bridging MAVS and TBK1. J Immunol. 2011;187(5):2559–2568. doi:10.4049/jimmunol.1100963
10. Li Y, Li C, Xue P, et al. ISG56 is a negative-feedback regulator of virus-triggered signaling and cellular antiviral response. Proc Natl Acad Sci U S A. 2009;106(19):7945–7950. doi:10.1073/pnas.0900818106
11. Pidugu VK, Wu MM, Yen AH, et al. IFIT1 and IFIT3 promote oral squamous cell carcinoma metastasis and contribute to the anti-tumor effect of gefitinib via enhancing p-EGFR recycling. Oncogene. 2019;38(17):3232–3247. doi:10.1038/s41388-018-0662-9
12. Wang Z, Qin J, Zhao J, et al. Inflammatory IFIT3 renders chemotherapy resistance by regulating post-translational modification of VDAC2 in pancreatic cancer. Theranostics. 2020;10(16):7178–7192. doi:10.7150/thno.43093
13. Yang Y, Zhou Y, Hou J, et al. Hepatic IFIT3 predicts interferon-alpha therapeutic response in patients of hepatocellular carcinoma. Hepatology. 2017;66(1):152–166. doi:10.1002/hep.29156
14. Sun R, Wang YF, Yang X. Knockdown of IFIT3 ameliorates multiple sclerosis via selectively regulating M1 polarization of microglia in an experimental autoimmune encephalomyelitis model. Int Immunopharmacol. 2024;128:111501. doi:10.1016/j.intimp.2024.111501
15. Sun J, Zhang Q, Liu X, Shang X. Downregulation of interferon-induced protein with tetratricopeptide repeats 3 relieves the inflammatory response and myocardial fibrosis of mice with myocardial infarction and improves their cardiac function. Exp Anim. 2021;70(4):522–531. doi:10.1538/expanim.21-0060
16. Guan G, Shen Y, Yu Q, et al. Down-regulation of IFIT3 protects liver from ischemia-reperfusion injury. Int Immunopharmacol. 2018;60:170–178. doi:10.1016/j.intimp.2018.04.045
17. Yang T, Ma X, Wang R, et al. Berberine inhibits IFN-gamma signaling pathway in DSS-induced ulcerative colitis. Saudi Pharm J. 2022;30(6):764–778. doi:10.1016/j.jsps.2022.03.015
18. Kawakatsu S, Zhu R, Zhang W, et al. A longitudinal model for the Mayo Clinical Score and its sub-components in patients with ulcerative colitis. J Pharmacokinet Pharmacodyn. 2022;49(2):179–190. doi:10.1007/s10928-021-09789-2
19. Du M, Feng J, Tao Y, Pan Q, Chen F. GNAO1 as a novel predictive biomarker for late relapse in hepatocellular carcinoma. J Healthc Eng. 2021;2021:7631815. doi:10.1155/2021/7631815
20. Ma LT, Lian JX, Bai Y, et al. Adeno-associated virus vector intraperitoneal injection induces colonic mucosa and submucosa transduction and alters the diversity and composition of the faecal microbiota in rats. Front Cell Infect Microbiol. 2022;12:1028380. doi:10.3389/fcimb.2022.1028380
21. Zhang T, Jiang J, Liu J, et al. MK2 is required for neutrophil-derived ROS production and inflammatory bowel disease. Front Med. 2020;7:207. doi:10.3389/fmed.2020.00207
22. Zhang M, Li X, Zhang Q, Yang J, Liu G. Roles of macrophages on ulcerative colitis and colitis-associated colorectal cancer. Front Immunol. 2023;14:1103617. doi:10.3389/fimmu.2023.1103617
23. Dharmasiri S, Garrido-Martin EM, Harris RJ, et al. Human intestinal macrophages are involved in the pathology of both ulcerative colitis and crohn disease. Inflamm Bowel Dis. 2021;27(10):1641–1652. doi:10.1093/ibd/izab029
24. Chen Y, Chen D, Zhou C, Cao X, He J. Correlation of Macrophages with inflammatory reaction in ulcerative colitis and influence of curcumin on macrophage chemotaxis. Altern Ther Health Med. 2023;29(2):97–103.
25. Huang C, Lewis C, Borg NA, et al. Proteomic identification of interferon-induced proteins with tetratricopeptide repeats as markers of M1 macrophage polarization. J Proteome Res. 2018;17(4):1485–1499. doi:10.1021/acs.jproteome.7b00828
26. Hong S, Wang H, Chan S, et al. Identifying macrophage-related genes in ulcerative colitis using weighted coexpression network analysis and machine learning. Mediators Inflamm. 2023;2023:4373840. doi:10.1155/2023/4373840
27. Yao Y, Xu T, Li X, et al. Selenoprotein S maintains intestinal homeostasis in ulcerative colitis by inhibiting necroptosis of colonic epithelial cells through modulation of macrophage polarization. Theranostics. 2024;14(15):5903–5925. doi:10.7150/thno.97005
28. Zhao X, Di Q, Liu H, et al. MEF2C promotes M1 macrophage polarization and Th1 responses. Cell Mol Immunol. 2022;19(4):540–553. doi:10.1038/s41423-022-00841-w
29. Chen S, Saeed A, Liu Q, et al. Macrophages in immunoregulation and therapeutics. Signal Transduct Target Ther. 2023;8(1):207. doi:10.1038/s41392-023-01452-1
30. Zhou F. Molecular mechanisms of IFN-gamma to up-regulate MHC class I antigen processing and presentation. Int Rev Immunol. 2009;28(3–4):239–260. doi:10.1080/08830180902978120
31. Wang H, Lou J, Liu H, et al. TRIM59 deficiency promotes M1 macrophage activation and inhibits colorectal cancer through the STAT1 signaling pathway. Sci Rep. 2024;14(1):16081. doi:10.1038/s41598-024-66388-0
32. Zhang K, Guo J, Yan W, Xu L. Macrophage polarization in inflammatory bowel disease. Cell Commun Signal. 2023;21(1):367. doi:10.1186/s12964-023-01386-9
33. Dignass A, Esters P, Flauaus C. Upadacitinib in Crohn’s disease. Expert Opin Pharmacother. 2024;25(4):359–370. doi:10.1080/14656566.2024.2333964
34. Mohamed MF, Bhatnagar S, Parmentier JM, Nakasato P, Wung P. Upadacitinib: mechanism of action, clinical, and translational science. Clin Transl Sci. 2024;17(1):e13688. doi:10.1111/cts.13688
35. Fujita Y, Matsuoka N, Temmoku J, et al. JAK inhibitors impair GM-CSF-mediated signaling in innate immune cells. BMC Immunol. 2020;21(1):35. doi:10.1186/s12865-020-00365-w
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.