")
Back to Journals » International Journal of Nanomedicine » Volume 12
Double-receptor-targeting multifunctional iron oxide nanoparticles drug delivery system for the treatment and imaging of prostate cancer
Authors Ahmed MSU , Salam AB, Yates C, Willian K, Jaynes J, Turner T, Abdalla MO
Received 6 April 2017
Accepted for publication 21 June 2017
Published 19 September 2017 Volume 2017:12 Pages 6973—6984
DOI https://doi.org/10.2147/IJN.S139011
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Md Shakir Uddin Ahmed,1 Ahmad Bin Salam,1 Clayton Yates,1 Kyle Willian,2 Jesse Jaynes,3 Timothy Turner,4 Mohamed O Abdalla5
1Department of Biology, Tuskegee University, Tuskegee, 2Department of Chemistry and Biochemistry, Auburn University, Auburn, 3Department of Environmental Sciences, Tuskegee University, Tuskegee, AL, 4Department of Biology, Jackson State University, Jackson, MS, 5Department of Chemistry, Tuskegee University, Tuskegee, AL, USA
Abstract: As an alternative therapeutic treatment to reduce or eliminate the current side effects associated with advanced prostate cancer (PCa) chemotherapy, a multifunctional double-receptor-targeting iron oxide nanoparticles (IONPs) (luteinizing hormone-releasing hormone receptor [LHRH-R] peptide- and urokinase-type plasminogen activator receptor [uPAR] peptide-targeted iron oxide nanoparticles, LHRH-AE105-IONPs) drug delivery system was developed. Two tumor-targeting peptides guided this double-receptor-targeting nanoscale drug delivery system. These peptides targeted the LHRH-R and the uPAR on PCa cells. Dynamic light scattering showed an increase in the hydrodynamic size of the LHRH-AE105-IONPs in comparison to the non-targeted iron oxide nanoparticles (NT-IONPs). Surface analysis showed that there was a decrease in the zeta potential values for drug-loaded LHRH-AE105-IONPs compared to the NT-IONPs. Prussian blue staining demonstrated that the LHRH-AE105-IONPs were internalized efficiently by the human PCa cell line, PC-3. In vitro, magnetic resonance imaging (MRI) results confirmed the preferential binding and accumulation of LHRH-AE105-IONPs in PC-3 cells compared to normal prostate epithelial cells (RC77N/E). The results also showed that LHRH-AE105-IONPs significantly maintained T2 MRI contrast effects and reduced T2 values upon internalization by PC-3 cells. These paclitaxel-loaded double-receptor-targeting IONPs also showed an approximately twofold reduction in PC-3 cell viability compared to NT-IONPs.
Keywords: LHRH-R, uPAR, prostate cancer, iron oxide nanoparticles, targeted drug delivery
Introduction
Prostate cancer (PCa) is considered the most commonly caused non-skin malignancy and the second leading cause of cancer death in men.1 Although current chemotherapeutics including docetaxel have shown promise for the treatment of hormone-refractory PCa, toxicity has been a serious limitation associated with systemic chemotherapy. Thus, novel strategies including targeted therapies are gaining importance and are being intensively investigated.2–5 Targeted nanoparticle (NP) therapies reduce the toxic side effects of anticancer drugs in normal cells and tissues by targeting overexpressed cell surface receptors unique to cancer, directly or indirectly killing cancer cells while sparing noncancer cells.6–9
One of the most commonly studied targeted drug delivery systems is the peptide-guided nanocarrier which is intended to interact specifically with a unique cancer-specific surface protein. Upon interaction with the cancer-specific surface protein, the peptide-guided nanocarrier is internalized by the cancer cells.10–12 However, a major drawback of the majority of these targeted drug delivery systems is that they suffer from the singularity of their target. PCa is a highly heterogeneous tumor; like most solid tumors, it undergoes temporal changes in cancer cell surface receptor profile, cellular behavior, and phenotype throughout tumor progression.13–17 Thus, to capture the variety of molecular features of advanced PCa tumors, a more complex targeted drug delivery system is warranted. Consequently, the objective of this study was to test the feasibility of developing a novel double-receptor-targeting nanoscale drug delivery system.
The developed system was designed to target two overexpressed cell surface proteins on PCa cells. The overexpressed cell surface proteins targeted were luteinizing hormone-releasing hormone receptor (LHRH-R) and urokinase-type plasminogen activator receptor (uPAR).8,18–30 This system utilized a combination of two different targeting peptides: a modified LHRH peptide and an AE105 peptide that targets LHRH-R and uPAR, respectively. The modified LHRH peptide has a single amino acid alternation at the tyrosine position of naturally occurring luteinizing hormone-releasing hormone, which has been replaced by tryptophan.31,32 AE105 is a potent nine-mer peptide antagonist of uPAR binding that has been developed by affinity maturation and combinatorial chemistry.33,34 The peptides were conjugated to an amphiphilic polymer-coated iron oxide nanoparticles (IONPs) core. An inherent benefit of the developed drug delivery system is found in the IONPs which provide an ideal condition for imaging functionality. These superparamagnetic IONPs are used as contrast agents for clinical diagnostics with magnetic resonance imaging (MRI).35–37
Here, we report the development of a multifunctional drug delivery system that can be tracked by MRI. This system utilizes the overexpression of LHRH-R and uPAR on PCa cells to selectively and preferentially deliver the chemotherapeutic paclitaxel (PTX) into the tumor. This novel delivery approach allows for enhanced intracellular drug accumulation to selectively cause significant growth inhibition and cell death in PCa cells.
Materials and methods
Materials
Amphiphilic polymer-coated IONPs terminated with carboxylic acid groups and the conjugation kit were purchased from Ocean Nanotech (San Diego, CA, USA). PTX, ethyl-3-dimethyl amino propyl carbodiimide (EDAC), and N-hydroxysulfosuccinimide (NHS) were acquired from Sigma-Aldrich (St Louis, MO, USA). The peptides LHRH and AE105 were acquired from GenScript (Piscataway, NJ, USA). All other chemicals were obtained from Sigma-Aldrich and were of analytical grade.
Cell culture
Human prostate carcinoma PC-3 cells were purchased from ATCC (Manassas, VA, USA). PC-3 cells were cultured in RPMI 1640 medium (Mediatech, Manassas, VA, USA) supplemented with 10% fetal bovine serum (FBS) (Sigma-Aldrich) and 1% penicillin/streptomycin (Mediatech). Immortalized PCa cell lines RC77T/E and RC77N/E were obtained by radical prostatectomy according to the protocol of the Internal Review Board at the Walter Reed Army Medical Center, the Uniformed Services University of the Health Sciences, and Tuskegee University.38 RC77T/E cells were maintained in Gibco Keratinocyte Serum-Free Medium (SFM; Thermo Fisher Scientific, Waltham, MA, USA) supplemented with human recombinant epidermal growth factor (rEGF) and bovine pituitary extract (25 mg) and 1% penicillin/streptomycin.38 RC77N/E cells were grown on plates coated with FNC Coating Mix (Athens Enzyme Systems).38 RC77N/E cells were grown in Keratinocyte-SFM, supplemented with bovine pituitary extract and rEGF. All cell lines were grown in sterile 100-mm2 cell culture dishes (Corning Costar Corp., Corning, NY, USA) in a fully humidified incubator containing 95% air and 5% CO2 at 37°C.
Conjugation protocol
The conjugation of the peptides to IONPs was achieved by amide bond formation between the carboxylic group of the IONPs and the free amine group in the N-terminus in the peptides. Briefly, IONPs were activated by using an EDAC/NHS mixture in the activation buffer. The activated IONPs were reacted with an equal amount of LHRH and AE105 peptides in the coupling buffer for 2 hours. Then, the peptides-conjugated IONPs (LHRH-AE105-IONPs) were separated from the reaction mixer, washed with storage buffer, and kept at 4°C for future experiments. Single-targeted IONPs (LHRH-IONPs and AE105-IONPs) were prepared using the same protocol.
NPs characterization
The hydrodynamic size and zeta potential of the non-targeted iron oxide nanoparticles (NT-IONPs), LHRH-AE105-IONPs, and single-targeted IONPs (LHRH-IONPs and AE105-IONPs) were determined using dynamic light scattering (DLS) (Malvern Instruments, Malvern, UK). The samples were prepared by diluting 20 μL of the non-targeted, single-targeting, and double-targeting IONP formulations using 800 μL of double-distilled water. All measurements were made at a scattering angle of 90° at 25°C.
Drug-loading efficiency
PTX loading was performed using a 1:4 ratio of drug to IONPs for both targeted IONPs and NT-IONPs. The mixture was mixed at room temperature for 4 hours and then overnight at 4°C. The resulting mixture was filtered through a 100 K nanosep centrifugal column (VWR, Radnor, PA, USA), and the free drug in the flow-through was analyzed by high-performance liquid chromatography (HPLC; HP Agilent 1200 HPLC; Agilent Technologies, Santa Clara, CA, USA). The encapsulation efficiency of PTX was calculated as the ratio of the amount of drug entrapped in IONPs to the initial amount of PTX used in drug loading.
pH-dependent drug release
To determine the physiological conditions that can be effective for releasing the PTX encapsulated within the IONPs, drug release analysis was performed using a series of different pH buffers. PTX-containing IONPs were incubated in buffers of different pHs (4, 5, 6, and 7) for 4 hours. Free PTX molecules in the buffer were then separated from the NT-IONPs and LHRH-AE105-IONPs by using a Nanosep 100 K centrifugal column (VWR). The amount of PTX released was determined by HPLC (Agilent 1200 HPLC; Agilent Technologies).
Binding and internalization of NPs by PC-3 cells
Prussian blue staining
Prussian blue staining was used to confirm the presence of iron in cells treated with IONPs. PC-3 cells were cultured, treated, and incubated for 4 hours with 30 pmol/mL of LHRH-AE105-IONPs, NT-IONPs, LHRH-IONPs, and AE105-IONPs. After incubation, the cells were gently washed three times with 1X PBS and fixed using 4% formaldehyde solution. Prussian blue staining was done by incubating the fixed cells for 30 minutes at 25°C in a solution containing a 1:1 mixture of 5% potassium ferrocyanide and 5% HCl. The cells were then examined under an inverted microscope. For each treatment group, bright-field optical images were obtained, and blue-stained cells were quantified and presented as a percentage of the total number of cells.
Intracellular iron concentration
To determine the intracellular iron concentration in PC-3 cells, a colorimetric method was used.39 PC-3 cells were treated with 40 μg/mL of IONPs and incubated for 24 hours. Following incubation, the cells were gently washed with 1X PBS, trypsinized, and collected. The collected cell pellets were dispensed in 100 μL of 12% HCl solution and incubated at 60°C for an additional 4 hours. The suspension was then centrifuged, and the supernatants were collected. A volume of 50 μL of supernatant was mixed with 50 μL of 1% ammonium persulfate and 100 μL of 0.1 M potassium thiocyanate into a 96-well plate, and incubated for 5 minutes to allow the formation of iron thiocyanate indicated by a red color. The absorption was read by a microplate reader (Model 3550; Bio-Rad, Hercules, CA, USA) at a wavelength of 490 nm. The iron content was then determined by using the standard curve of iron.
MRI scanning
PC-3 and RC77N/E cells were grown at a density of 2.2×106 cells/mL. The cells were then treated for 2 hours with LHRH-AE105-IONPs, LHRH-IONPs, AE105-IONPs, and NT-IONPs at concentrations of 5 and 10 pmol/mL. Untreated cells for each cell type were used as a control. After 2 hours, the cells were gently washed three times using 1X PBS, trypsinized, centrifuged, and collected. The collected cells were resuspended in 1.5 mL eppendorf tubes and embedded in 1% agarose gel. All samples were scanned in a 3-T MRI Scanner (Siemens Verio Open-Bore 3T Scanner; Auburn University, AL, USA) using fresh water as a control and multiecho T2-weighted fast-spin echo imaging sequences. T2 values for each sample were calculated from obtained multiecho (TEi, i =20, range from 10 to 200ms, interval =10ms).
Cell cytotoxicity analysis (MTT assay)
Cell lines were seeded at a density of 2.5×103 cells per well into 96-well plates (Corning). The cells were treated with SFM containing free PTX or PTX-loaded NT-IONPs, LHRH-IONPs, AE105-IONPs, and LHRH-AE105-IONPs. Cells without any treatment were used as a control. After 24 hours, the treated and control cells were incubated with 100 μL of MTT (Sigma-Aldrich) reagent for an additional 3 hours. After incubation, the MTT reagent was removed, and 100 μL of DMSO was added to each well to dissolve the formazan crystal (MTT metabolic product). After shaking for 5 minutes, the absorbance was taken at 570 nm on a Thermomax microplate reader (Molecular Devices, Sunnyvale, CA, USA). The percentage of cell death was determined from the difference between the absorbance of control and treated wells.
Statistical analysis
Data are presented as mean ± SEM of at least three separate experiments performed in triplicate. Statistical analysis was performed by analysis of variance. P<0.05 was considered statistically significant.
Results and discussion
Preparation and characterization of peptide-conjugated IONPs
A double-receptor-targeted multifunctional IONPs drug delivery system was designed, developed, and optimized to deliver the chemotherapeutic agent PTX with the ultimate goal of improving the efficiency of tumor drug delivery and thus decreasing the side effects. Figure 1 shows the interaction of double-receptor-targeting IONPs with a cancer cell. The two targeting peptides, one for LHRH-R and one for uPAR, were coupled to the carboxyl ends of the amphiphilic polymer coating on the IONPs to form the LHRH-AE105-IONPs conjugate.
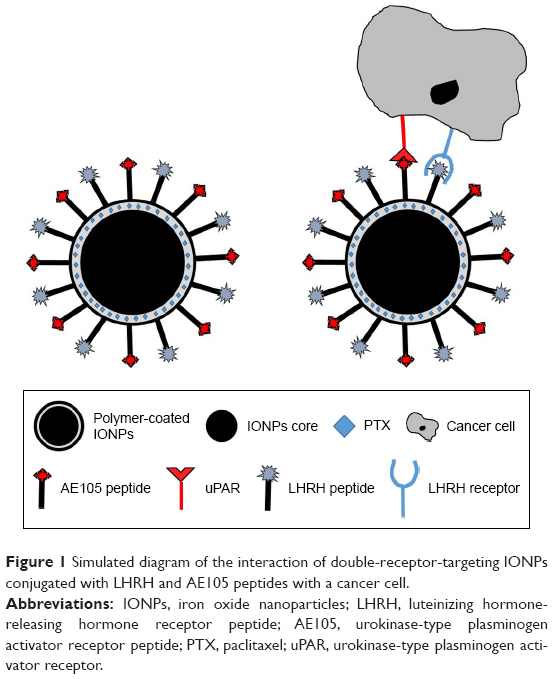 | Figure 1 Simulated diagram of the interaction of double-receptor-targeting IONPs conjugated with LHRH and AE105 peptides with a cancer cell. |
Hydrodynamic size of NPs
Effective drug delivery to tumor tissue requires appropriately sized NPs that can circulate in the bloodstream for necessary time to selectively release their payload at the tumor site.40 Since elevated levels of vascular permeability characterize most solid tumors, it is pertinent for the NPs to possess a compact size to steer through immature and leaky tumor vessels and proficiently enter the tumor mass.41 Thus, ideally, NPs should be large enough (>10 nm) to prevent their rapid leakage into blood capillaries but small enough (<100 nm) to escape capture by macrophages that are lodged in the reticuloendothelial system.19,40 To obtain an accurate measure of the hydrodynamic size and size distribution of the prepared peptide–IONP conjugates, we analyzed the samples using DLS. The size distribution is fundamentally significant to examine as it will reveal if the magnetic NPs have formed any aggregates after conjugation of the peptide and loading of the drug.42 Table 1 illustrates the average hydrodynamic size of NT-IONPs and targeted IONPs (LHRH-AE105-IONPs, LHRH-IONPs, and AE105-IONPs). The results also show that the targeted IONPs retained their narrow size distribution after conjugation of the peptides. The average hydrodynamic size of LHRH-AE105-IONPs (15.74±0.44 nm) was greater than the NT-IONPs (12.33±0.23 nm) which is expected due to conjugated peptides on the IONPs surface. The size of the LHRH-AE105-IONPs was within the expected size of IONPs, between 10 and 100 nm, which allowed them to deliver drug efficiently to the cancer cells as well as avoid clearance by the body immune system.43
 | Table 1 Hydrodynamic size of targeted IONPs and NT-IONPs |
Surface charge of NPs
The surface charge of NPs determines their circulation time in the bloodstream and is a measure of NP stability in aqueous solution and of their interaction with the cell membrane.44 The zeta potential values of the IONPs used in these experiments are presented in Table 2. The NT-IONPs had a negative surface charge of −70.44±2.18 mV, which was attributed to the carboxyl groups on the polymer coating on the IONPs. Upon conjugation of the peptides, the negative surface charge of LHRH-AE105-IONPs decreased to −58.06±1.72 mV; the negative surface charge of the AE105-IONPs and LHRH-IONPs was −51.3±1.23 and −54.3±0.88 mV, respectively. The peptides conjugation to the carboxyl ends on IONPs resulted in a decrease in the zeta potential of targeted IONPs because of the elimination of these negatively charged groups. Despite the reduction in the surface charge, the peptides-conjugated IONPs were well dispersed in the aqueous storage buffer and displayed no visible precipitation over a period of several months.
 | Table 2 Zeta potential of NT-IONPs and targeted IONPs |
Efficiency of PTX encapsulation
To be an effective therapeutic treatment, it is very important to incorporate a sufficient amount of drugs onto the IONPs coating. We took advantage of the interactions between the hydrophobic inner layer of IONPs and hydrophobic drug PTX. Table 3 presents the efficiency of PTX encapsulation on NT-IONPs and peptide-conjugated IONPs. Using a ratio of 1 mg drug to 4 mg IONPs, we were able to encapsulate 85.6% of PTX onto the NT-IONPs as determined by HPLC analysis. We observed that there was approximately a 7.0% decrease in efficiency of drug encapsulation when PTX was loaded into LHRH-AE105-IONPs. There was also 3.0% decrease in PTX loading in the case of single-targeted IONPs. Several studies reported the use of PTX-loaded IONPs in targeted drug delivery systems for various cancer treatments, including PCa.45–47 The PTX encapsulation in biodegradable, nontoxic, nanoscale drug delivery systems is very useful for drug delivery because this type of system can protect PTX from degradation during circulation. This characteristic lowers the toxicity of PTX by increasing drug circulation time, showing significant pharmacokinetic efficiency and better patient outcome.48
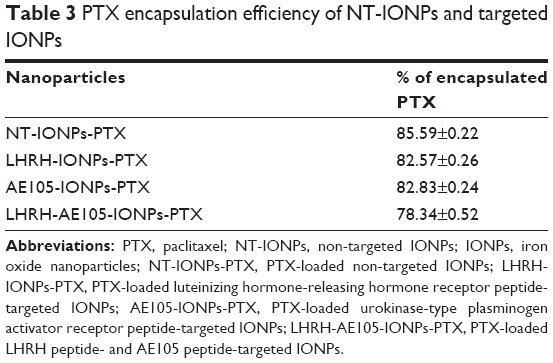 | Table 3 PTX encapsulation efficiency of NT-IONPs and targeted IONPs |
pH-dependent drug release from IONPs
It is recognizable that the efficiency of any drug delivery vehicle is best evaluated by its ability to release the loaded drug into cells and not systemically in the patient’s circulation, which would exacerbate the condition of the patient.42,49 Thus, we investigated the pH-dependent PTX release from LHRH-AE105-IONPs by placing the drug-loaded NPs in four buffers of different pHs. To simulate physiological conditions, we examined the amount of PTX that was released from the NPs at pH 7. Slightly acidic pH conditions (pH 5 and 6) simulated the environment of tumor interstitium, while more acidic pH (pH 4) simulated highly acidic conditions in the intracellular vesicles such as endosomes and lysosomes. Figure 2A shows the overlays of HPLC chromatograms to depict the amount of PTX detected in the supernatant of PTX-loaded LHRH-AE105-IONPs (LHRH-AE105-IONPs-PTX) at various pHs. The results indicated that the amount of drug released increased with a decrease in the pH of the buffer. The maximum amount of PTX was released at pH 4 ((≈60%), and a small amount was released at pH 7 (≈10%). There are variations in the physical environment in the cell and tissue compartments with a slightly acidic environment in cancer tissue and a more acidic pH in endosomes (5.5–6.8) and in lysosomes (4.0–5.0).45 The area under the peak was used to calculate the percentage of drug release from the LHRH-AE105-IONPs-PTX and NT-IONPs (Figure 2B). The peptide-conjugated IONPs, LHRH-AE105-IONPs-PTX, showed a slightly lower percentage of drug release because of the peptides conjugated on the surface of the NPs. This can be attributed to that the conjugated peptides may play a temporary barrier for the drug release from the surface of the IONPs. For the LHRH-AE105-IONPs-PTX, 10% of drug was detected at pH 7, and ~20% at pH 6. At pH 5, the percentage of drug released increased to ~40% and to over ~60% at pH 4. These results indicated strong adsorption of drug onto the IONPs surface, thus facilitating the drug release at an acidic pH. Enhancement of drug release may be due to the onset of polymer degradation at lower pH or the breakage of hydrophobic interactions between polymer and drug molecules.50 Therefore, the drug loading and release study suggested that the LHRH-AE105-IONPs can serve as an efficient vehicle for drug delivery in acidic environments that exist particularly in the hypoxic tumor milieu.
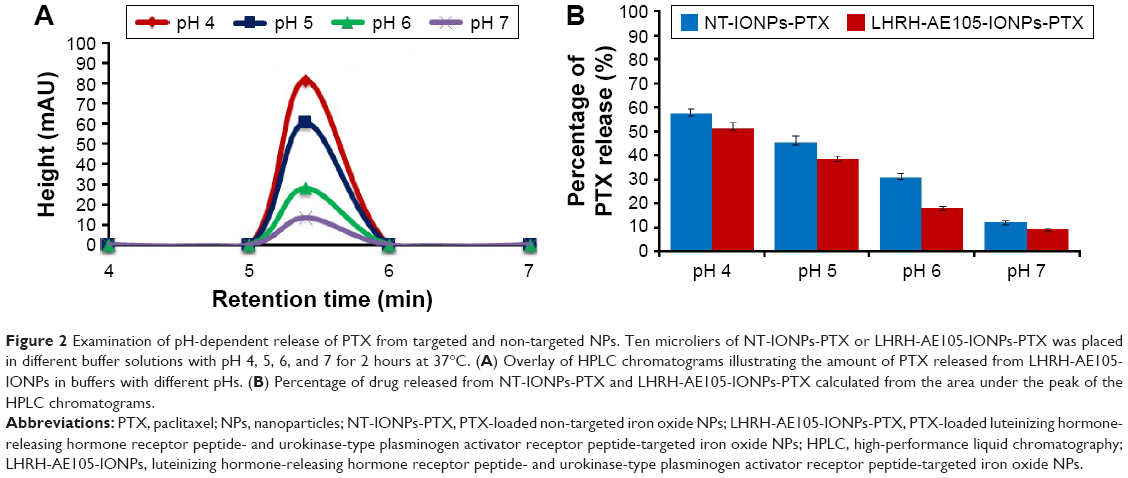 | Figure 2 Examination of pH-dependent release of PTX from targeted and non-targeted NPs. Ten microliers of NT-IONPs-PTX or LHRH-AE105-IONPs-PTX was placed in different buffer solutions with pH 4, 5, 6, and 7 for 2 hours at 37°C. (A) Overlay of HPLC chromatograms illustrating the amount of PTX released from LHRH-AE105-IONPs in buffers with different pHs. (B) Percentage of drug released from NT-IONPs-PTX and LHRH-AE105-IONPs-PTX calculated from the area under the peak of the HPLC chromatograms. |
Binding and internalization efficiency of receptor-targeted IONPs
The in vitro NPs binding and internalization was investigated by Prussian blue staining, chemical analysis of intracellular iron concentration, and MRI.
Prussian blue staining
Figure 3A shows the bright-field images of PC-3 cells treated with NT-IONPs. These cells exhibited very low levels of blue staining, suggesting nonspecific iron staining. Figure 3B presents the staining images of PC-3 cells incubated with the double-receptor-targeting NPs, LHRH-AE105-IONPs, where an intense blue staining is shown. PC-3 cells treated with single-targeted IONPs, LHRH-IONPs, and AE105-IONPs also showed a very good level of iron staining (Figure 3C and D). Figure 3E illustrates the quantification of the percentage of blue-stained cells by Prussian blue stain. PC-3 cells incubated with LHRH-AE105-IONPs demonstrated a high level of stained cells (>75% stained cells) compared to PC-3 cells incubated with NT-IONPs which showed a very low level of stained cells. In addition to that, PC-3 cells incubated with either single-targeted IONPs (LHRH-IONPs and AE105-IONPs) has a lower level of iron staining compared with LHRH-AE105-IONPs. This observation suggests that double-receptor-targeting NPs can bind and internalize more efficiently than others. Both LHRH-R and uPAR proteins are highly overexpressed in PC-3 cells with virtually no expression detectable in normal prostate cells.20,21,23,30,51 Therefore, these staining patterns suggested that high LHRH-R and uPAR expression facilitated specific binding and internalization of LHRH-AE105-IONPs upon ligand–receptor binding in PC-3 cells.
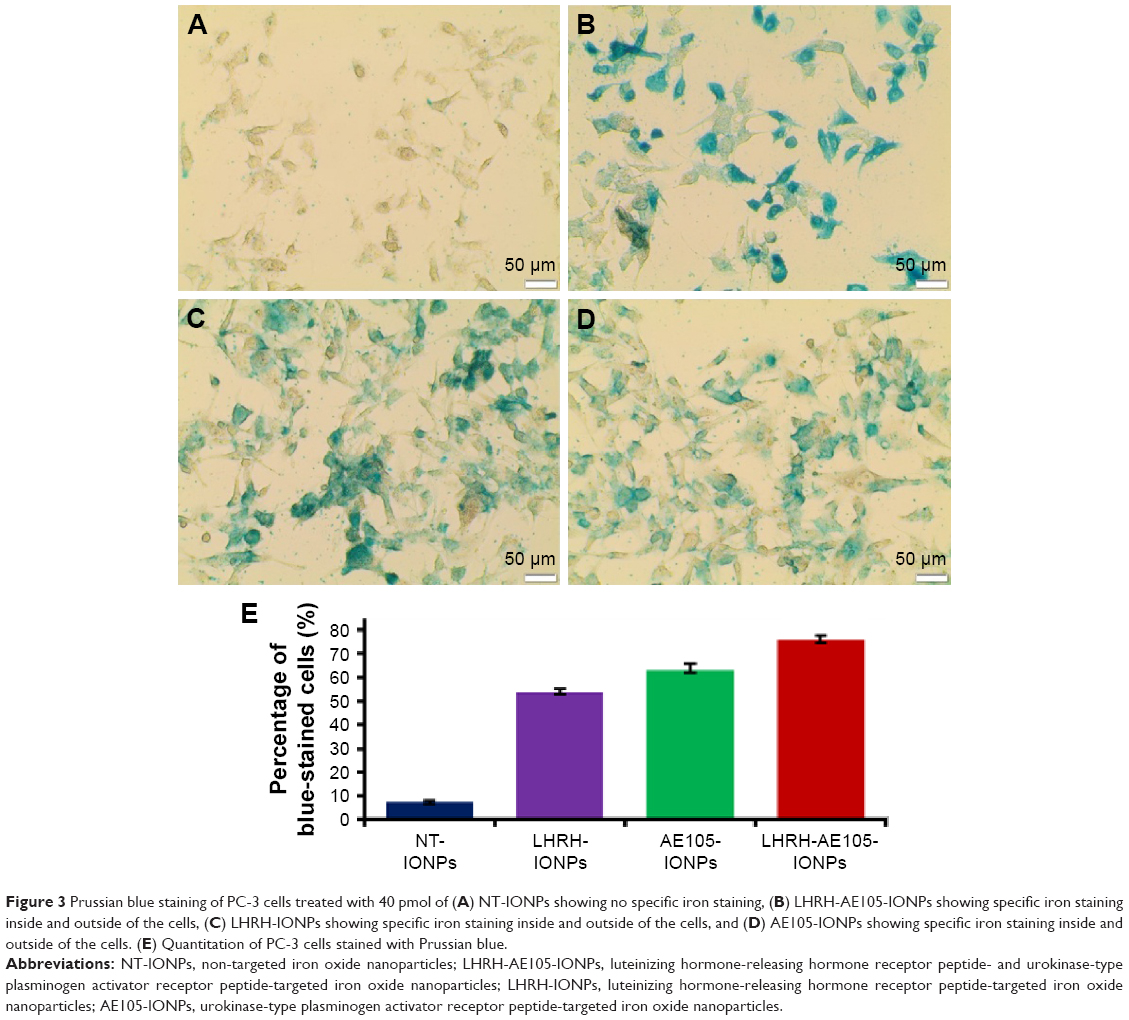 | Figure 3 Prussian blue staining of PC-3 cells treated with 40 pmol of (A) NT-IONPs showing no specific iron staining, (B) LHRH-AE105-IONPs showing specific iron staining inside and outside of the cells, (C) LHRH-IONPs showing specific iron staining inside and outside of the cells, and (D) AE105-IONPs showing specific iron staining inside and outside of the cells. (E) Quantitation of PC-3 cells stained with Prussian blue. |
Chemical analysis of intracellular iron concentration
The concentration of intracellular iron was measured to test the efficiency of LHRH-AE105-IONPs to corroborate receptor binding and internalization further. For the LHRH-AE105-IONPs, the intracellular iron concentration was found to be approximately 10 μg, whereas in the case of NT-IONPs, approximately 2 μg of intracellular iron was found. Thus, the amount of iron delivered by the LHRH-AE105-IONPs inside the cells was approximately five times higher than that of the NT-IONPs (Figure 4). The difference in iron concentration in the case of LHRH-AE105-IONPs was significant when compared to NT-IONPs (P<0.002). In addition to that, the iron content for LHRH-AE105-IONPs was also significant when compared to LHRH-IONPs (P<0.005) and AE105-IONPs (P<0.038). It is worthy to note the point that the results indicated that the iron content was higher in the case of LHRH-AE105-IONPs than any of the single-targeted IONPs. These results further support the receptor-mediated internalization of targeting IONPs by cells.52–54
 | Figure 4 Intracellular iron content after treating PC-3 cells with 40 μg/mL of NT-IONPs, LHRH-IONPs, AE105-IONPs, and LHRH-AE105-IONPs. The highest iron concentration was found in the cells treated with the double-targeting LHRH-AE105-IONPs compared to the single-targeting LHRH-IONPs and AE105-IONPs and NT-IONPs. |
In vitro MRI
Multifunctional NPs which can serve to simultaneously deliver anticancer drugs and allow tumor imaging are being increasingly investigated for their improved therapeutic efficacy. Superparamagnetic IONPs are widely used as contrast agents in MRI because of their negative enhancement effect on T2-weighted sequences. The double-targeted multifunctional NPs (LHRH-AE105-IONPs) included the iron oxide core which typically induces reduction of transverse relaxation time and T2-weighted contrast change. The signal drops in MRI allow for tracking the targeted drug delivery along with the noninvasive assessment of cancer, using clinically applicable MRI systems.55,56 Thus, in vitro MRI scanning was used to evaluate the targeting specificity and suitability of the LHRH-AE105-IONPs as a contrast agent. Figure 5A and B shows the MRI scans of PC-3 cells treated with two different concentrations (5 and 10 pmol) of NT-IONPs, LHRH-IONPs, AE105-IONPs, and LHRH-AE105-IONPs. The MR images demonstrated significant target specificity. The largest noticeable reduction of T2 values was detected in PC-3 cells treated with the LHRH-AE105-IONPs compared to the single-targeted IONPs (LHRH-IONPs and AE105-IONPs) and NT-IONPs. This indicated that large amounts of IONPs are bound and internalized by the PC-3 cells due to the higher affinity of the double-receptor-targeting LHRH-AE105-IONPs compared to the single-targeted IONPs. This finding furthermore proved our hypothesis that targeting two receptors will significantly increase the chances of the binding and internalization of IONPs by the cancer cells. Figure 5C shows the quantification of the reduction in the T2 for both concentrations used, and the LHRH-AE105-IONPs showed the lowest T2 values at both concentrations. These results suggested the enhanced binding ability of double-receptor-targeting LHRH-AE105-IONPs to PC-3 cells compared to single-peptide-guided IONPs.
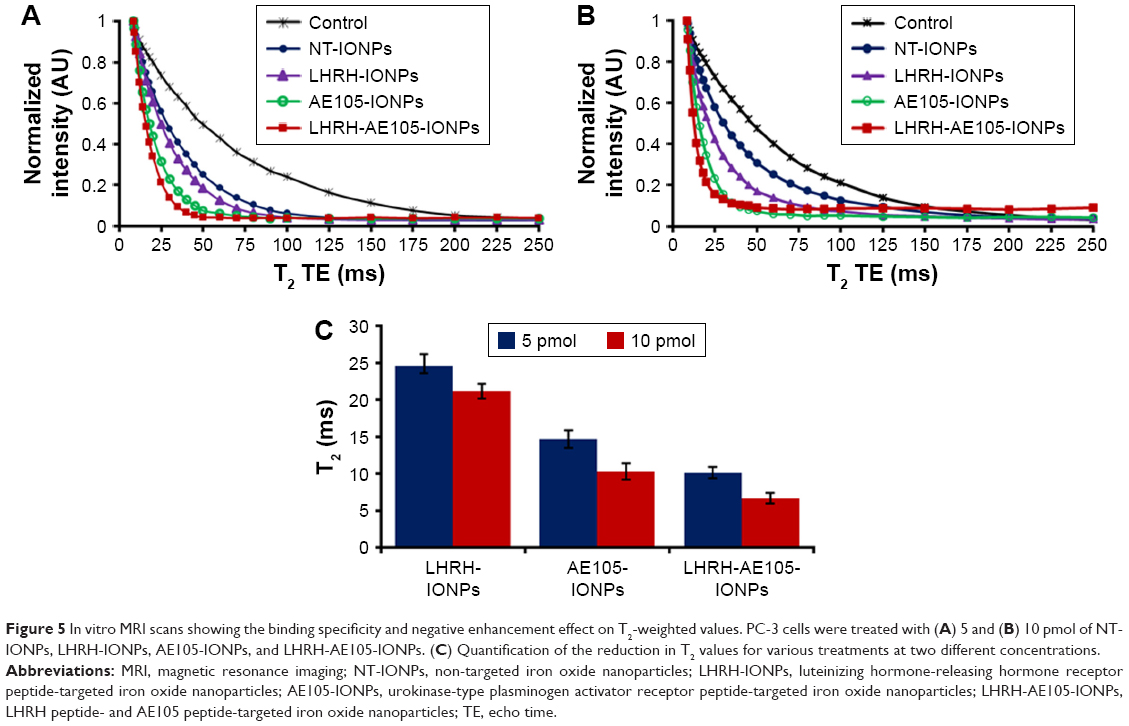 | Figure 5 In vitro MRI scans showing the binding specificity and negative enhancement effect on T2-weighted values. PC-3 cells were treated with (A) 5 and (B) 10 pmol of NT-IONPs, LHRH-IONPs, AE105-IONPs, and LHRH-AE105-IONPs. (C) Quantification of the reduction in T2 values for various treatments at two different concentrations. |
To further confirm the target specificity, normal prostate RC77N/E cells which have low expression of LHRH-R and uPAR were used as a control to corroborate the data on the reduction of T2 values. Figure 6A and B shows the overlaid curves of T2 values for PC-3 and RC77N/E cells treated with 5 and 10 pmol of NT-IONPs, LHRH-IONPs, AE105-IONPs, and LHRH-AE105-IONPs. Compared to the PC-3 cells, RC77N/E cells did not show a reduction in the T2 values at both concentrations. Figure 6C shows the quantification of the T2 values reduction for the PC-3 cells and RC77N/E cells treated with LHRH-AE105-IONPs. At both 5 and 10 pmol IONP concentrations, the PC-3 cells showed a significant reduction in the T2 values compared to the RC77N/E cells. These results proved that the overexpression of the LHRH-R and uPAR is essential for the successful binding and internalization of the targeting IONPs. The MRI signal variances of different samples in T2-weighted images correlated well with the reduction in the T2 values of different specimens. Figure 6D shows that the PC-3 cells treated with LHRH-IONPs, AE105-IONPs, and LHRH-AE105-IONPs all appeared blue in color which indicated the binding of the IONPs to the cells. PC-3 cells treated with AE105-IONPs and LHRH-AE105-IONPs showed a solid dark blue in agreement with the samples having the lowest T2 values. On the contrary, untreated PC-3, and PC-3 and RC77N/E cells treated with NT-IONPs remained bright and indistinguishable due to the absence of iron. RC77N/E cells treated with single-targeted or double-receptor-targeting IONPs also remained bright and indistinguishable due to the lack of iron. The inefficiency of T2 reduction and T2-weighted color maps indicated that RC77N/E cells were not able to internalize any of the targeted IONPs because they do not significantly express LHRH-R and uPAR.
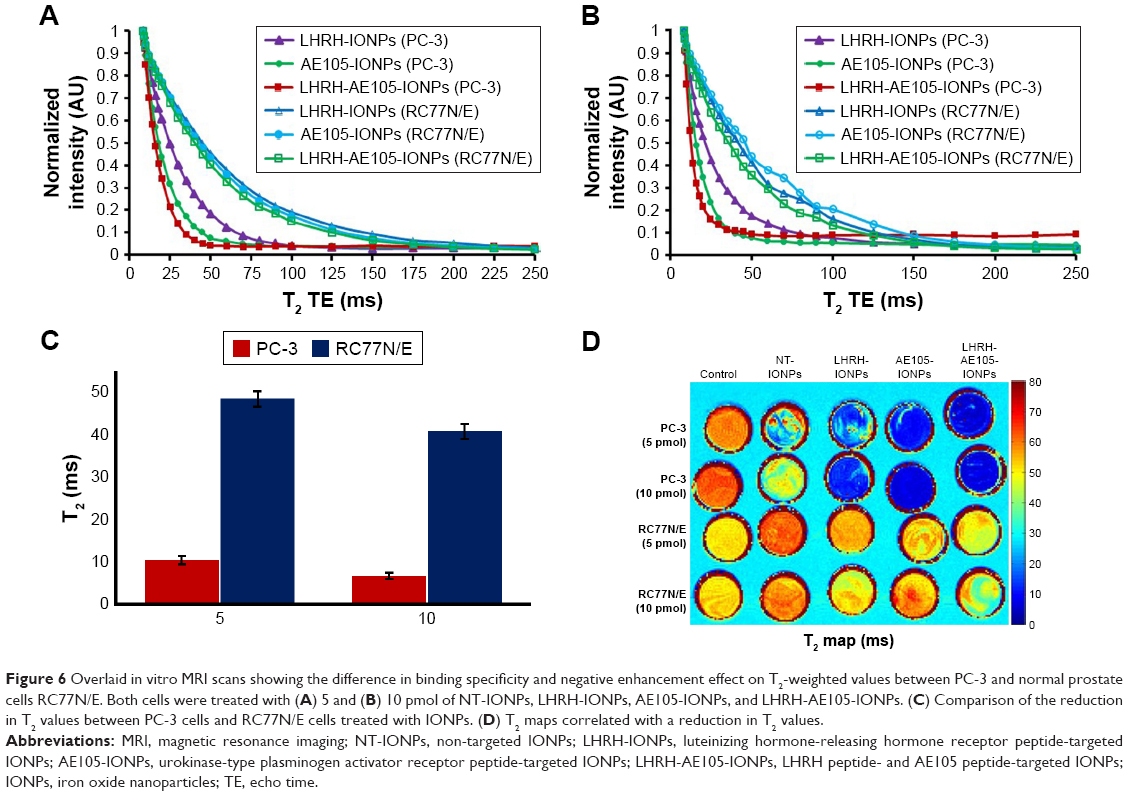 | Figure 6 Overlaid in vitro MRI scans showing the difference in binding specificity and negative enhancement effect on T2-weighted values between PC-3 and normal prostate cells RC77N/E. Both cells were treated with (A) 5 and (B) 10 pmol of NT-IONPs, LHRH-IONPs, AE105-IONPs, and LHRH-AE105-IONPs. (C) Comparison of the reduction in T2 values between PC-3 cells and RC77N/E cells treated with IONPs. (D) T2 maps correlated with a reduction in T2 values. |
Cell cytotoxicity analysis
To evaluate the selective advantage of double-receptor-targeting IONPs to enhance PTX delivery compared to single-receptor-targeting IONPs and free drug, we examined the cytotoxic effect of PTX-loaded IONPs on PC-3. PC-3 cells were treated with double-receptor-targeting IONPs (LHRH-AE105-IONPs-PTX), PTX-loaded single-receptor-targeting IONPs (LHRH-IONPs-PTX and AE105-IONPs-PTX), NT-IONPs, and free PTX. All treatments were done at a fixed PTX concentration of 10 ng/mL. Figure 7A shows that PC-3 cells treated with LHRH-AE105-IONPs-PTX showed an approximately twofold reduction in cell viability compared to LHRH-IONPs-PTX and AE105-IONPs-PTX. This experiment proved the enhanced efficiency of the double-receptor-targeting IONPs as a drug delivery system compared to the single-receptor-targeting IONPs. Moreover, the LHRH-AE105-IONPs-PTX showed approximately threefold better cell cytotoxicity compared to the NT-IONPs and free PTX. Figure 7B shows the amount of free drug required to achieve the 85% cell death caused by LHRH-AE105-IONPs-PTX treatment on PC-3 cells. Interestingly, while the concentration of the drug on the LHRH-AE105-IONPs-PTX was only 10 ng/mL, 100 ng/mL of free PTX was needed to achieve comparable cytotoxic effects of the double-receptor-targeting IONPs. This result indicated the highly enhanced capability (~10 times) of the double-receptor-targeting IONPs in killing cancer cells by delivering the maximum amount of drug inside the cells. To show the enhanced selectivity in killing cancer cells of receptor-targeted drug delivery systems,57,58 the normal prostate epithelial cell line, RC77N/E, was treated with LHRH-AE105-IONPs-PTX at a drug concentration of 10 ng/mL. As shown in Figure 7C, RC77N/E cells did not show a pronounced cell death which occurred in the PC-3 cells treated with LHRH-AE105-IONPs-PTX at the same drug concentration. Even increasing the concentration of PTX-loaded LHRH-AE105-IONPs from 10 to 20 ng/mL failed to raise the level of cytotoxicity in RC77N/E cells. This experiment confirmed the targeted delivery of PTX via the two targeting peptides (AE105 and LHRH) to the overexpressed LHRH-R and uPAR in PC-3 cells.
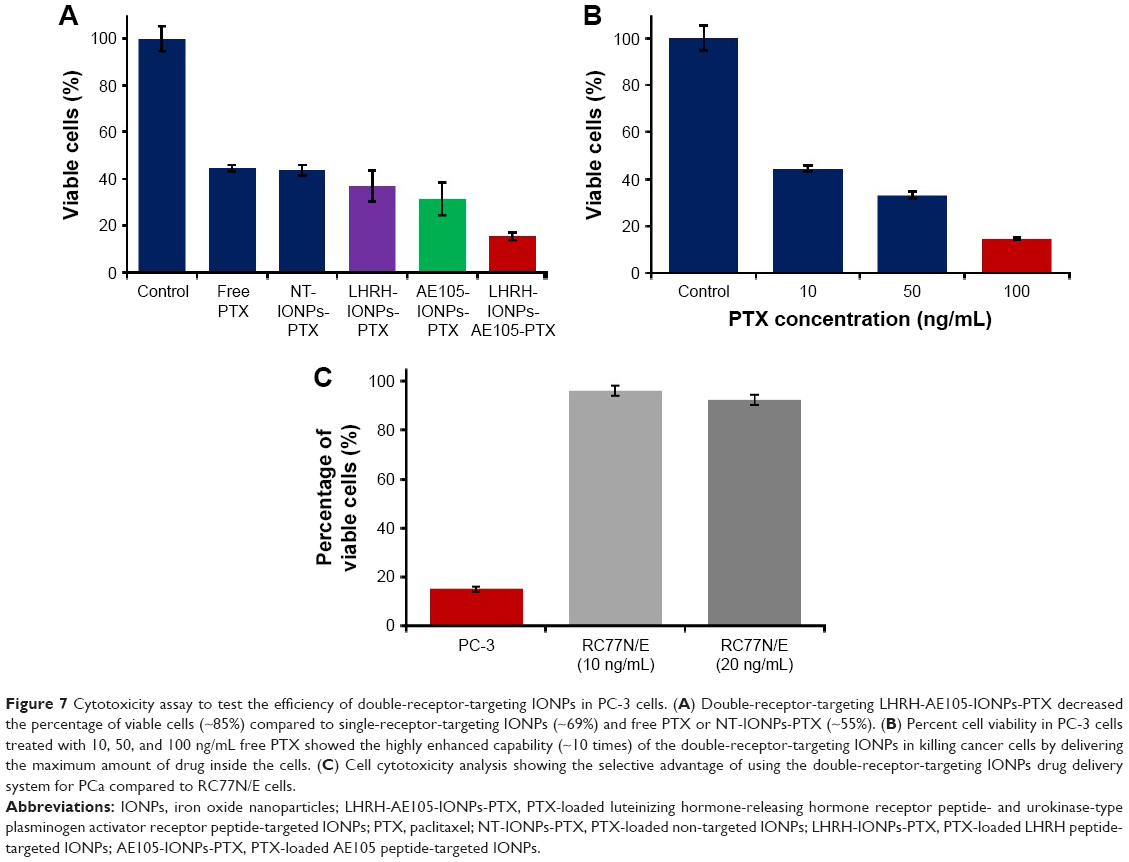 | Figure 7 Cytotoxicity assay to test the efficiency of double-receptor-targeting IONPs in PC-3 cells. (A) Double-receptor-targeting LHRH-AE105-IONPs-PTX decreased the percentage of viable cells (~85%) compared to single-receptor-targeting IONPs (~69%) and free PTX or NT-IONPs-PTX (~55%). (B) Percent cell viability in PC-3 cells treated with 10, 50, and 100 ng/mL free PTX showed the highly enhanced capability (~10 times) of the double-receptor-targeting IONPs in killing cancer cells by delivering the maximum amount of drug inside the cells. (C) Cell cytotoxicity analysis showing the selective advantage of using the double-receptor-targeting IONPs drug delivery system for PCa compared to RC77N/E cells. |
Conclusion
Based on our knowledge, this is the first report of a multifunctional nanoscale drug delivery system possessing double-receptor-targeting ability and imaging capability using PCa cells. We have demonstrated via Prussian blue staining and in vitro MRI scan that the developed double-receptor-targeting nanoscale drug delivery system was successfully bound and internalized by LHRH-R- and uPAR-overexpressing PC-3 cells. We were able to successfully load and release the anticancer drug PTX which was physically adsorbed on the polymeric coating of the IONPs. The LHRH-AE105-IONPs exhibited a significant MRI contrast effect because of the superparamagnetic iron oxide NPs which served as the core of the drug delivery system. The enhanced efficiency of double-receptor-targeting LHRH-AE105-IONPs-PTX resulted in approximately two times greater cell cytotoxicity compared to single-receptor-targeting IONPs. Moreover, the LHRH-AE105-IONPs-PTX resulted in 10 times reduction in the concentration of PTX required to achieve similar cytotoxic effect produced by the free drug. Based on the results of this study, double-receptor-targeting IONPs drug delivery system may offer a potential novel therapeutic alternative for PCa to enhance the quality of life and longevity of the patients.
Acknowledgments
This work was supported by grant funding from NIH/RCMI G12MD00758 and NIH/NCI U54 CA118623. The abstract of this paper was presented as a poster with interim findings at the 106th Annual Meeting of the American Association for Cancer Research, held on April 18–22, 2015, in Philadelphia. The abstract was published in the meeting proceedings, and cited as Cancer Res. 2015;75(15 Suppl):abstract nr 3675. doi: 10.1158/1538-7445.AM2015-3675.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11–30. | ||
Petrylak DP. The treatment of hormone-refractory prostate cancer: docetaxel and beyond. Rev Urol. 2006;8 Suppl 2:S48–S55. | ||
Sajja HK, East MP, Mao H, Wang YA, Nie S, Yang L. Development of multifunctional nanoparticles for targeted drug delivery and noninvasive imaging of therapeutic effect. Curr Drug Discov Technol. 2009;6(1):43–51. | ||
Kim K, Kim JH, Park H, et al. Tumor-homing multifunctional nanoparticles for cancer theragnosis: simultaneous diagnosis, drug delivery, and therapeutic monitoring. J Control Release. 2010;146(2):219–227. | ||
Xie J, Huang J, Li X, Sun S, Chen X. Iron oxide nanoparticle platform for biomedical applications. Curr Med Chem. 2009;16(10):1278–1294. | ||
Wu J, Joseph SO, Muggia FM. Targeted therapy: its status and promise in selected solid tumors part I: areas of major impact. Oncology (Williston Park). 2012;26(10):936–943. | ||
Joseph SO, Wu J, Muggia FM. Targeted therapy: its status and promise in selected solid tumors. Part II: impact on selected tumor subsets, and areas of evolving integration. Oncology (Williston Park). 2012;26(11):1021–1030, 1035. | ||
Brannon-Peppas L, Blanchette JO. Nanoparticle and targeted systems for cancer therapy. Adv Drug Deliv Rev. 2012;64:206–212. | ||
Glasgow MD, Chougule MB. Recent developments in active tumor targeted multifunctional nanoparticles for combination chemotherapy in cancer treatment and imaging. J Biomed Nanotechnol. 2015;11(11):1859–1898. | ||
Huang J, Li Y, Orza A, et al. Magnetic nanoparticle facilitated drug delivery for cancer therapy with targeted and image-guided approaches. Adv Funct Mater. 2016;26(22):3818–3836. | ||
Li Z, Ye E, David, Lakshminarayanan R, Loh XJ. Recent advances of using hybrid nanocarriers in remotely controlled therapeutic delivery. Small. 2016;12(35):4782–4806. | ||
Zhu L, Zhou Z, Mao H, Yang L. Magnetic nanoparticles for precision oncology: theranostic magnetic iron oxide nanoparticles for image-guided and targeted cancer therapy. Nanomedicine (Lond). 2017;12(1):73–87. | ||
Sun XX, Yu Q. Intra-tumor heterogeneity of cancer cells and its implications for cancer treatment. Acta Pharmacol Sin. 2015;36(10):1219–1227. | ||
Seguin L, Desgrosellier JS, Weis SM, Cheresh DA. Integrins and cancer: regulators of cancer stemness, metastasis, and drug resistance. Trends Cell Biol. 2015;25(4):234–240. | ||
Boyd LK, Mao X, Lu YJ. The complexity of prostate cancer: genomic alterations and heterogeneity. Nat Rev Urol. 2012;9(11):652–664. | ||
Zhang L, Wu C, Hoffman RM. Prostate cancer heterogeneous high-metastatic multi-organ-colonizing chemo-resistant variants selected by serial metastatic passage in nude mice are highly enriched for multinucleate giant cells. PLoS One. 2015;10(11):e0140721. | ||
Lalonde E, Ishkanian AS, Sykes J, et al. Tumour genomic and microenvironmental heterogeneity for integrated prediction of 5-year biochemical recurrence of prostate cancer: a retrospective cohort study. Lancet Oncol. 2014;15(13):1521–1532. | ||
Yang L, Mao H, Wang YA, et al. Single chain epidermal growth factor receptor antibody conjugated nanoparticles for in vivo tumor targeting and imaging. Small. 2009;5(2):235–243. | ||
Cho K, Wang X, Nie S, Chen ZG, Shin DM. Therapeutic nanoparticles for drug delivery in cancer. Clin Cancer Res. 2008;14(5):1310–1316. | ||
Limonta P, Dondi D, Moretti RM, Maggi R, Motta M. Antiproliferative effects of luteinizing hormone-releasing hormone agonists on the human prostatic cancer cell line LNCaP. J Clin Endocrinol Metab. 1992;75(1):207–212. | ||
Limonta P, Dondi D, Moretti R, Fermo D, Garattini E, Motta M. Expression of luteinizing hormone-releasing hormone mRNA in the human prostatic cancer cell line LNCaP. J Clin Endocrinol Metab. 1993;76(3):797–800. | ||
Dondi D, Limonta P, Moretti RM, Marelli MM, Garattini E, Motta M. Antiproliferative effects of luteinizing hormone-releasing hormone (LHRH) agonists on human androgen-independent prostate cancer cell line DU 145: evidence for an autocrine-inhibitory LHRH loop. Cancer Res. 1994;54(15):4091–4095. | ||
Limonta P, Moretti RM, Marelli MM, Dondi D, Parenti M, Motta M. The luteinizing hormone-releasing hormone receptor in human prostate cancer cells: messenger ribonucleic acid expression, molecular size, and signal transduction pathway. Endocrinology. 1999;140(11):5250–5256. | ||
Goessl C, Müller M, Heicappell R, et al. DNA-based detection of prostate cancer in urine after prostatic massage. Urology. 2001;58(3):335–338. | ||
Halmos G, Arencibia JM, Schally AV, Davis R, Bostwick DG. High incidence of receptors for luteinizing hormone-releasing hormone (LHRH) and LHRH receptor gene expression in human prostate cancers. J Urol. 2000;163(2):623–629. | ||
Tieva A, Stattin P, Wikström P, Bergh A, Damber JE. Gonadotropin-releasing hormone receptor expression in the human prostate. Prostate. 2001;47(4):276–284. | ||
Fekete M, Zalatnai A, Comaru-Schally AM, Schally AV. Membrane receptors for peptides in experimental and human pancreatic cancers. Pancreas. 1989;4(5):521–528. | ||
Gründker C, Völker P, Griesinger F, et al. Antitumor effects of the cytotoxic luteinizing hormone-releasing hormone analog AN-152 on human endometrial and ovarian cancers xenografted into nude mice. Am J Obstet Gynecol. 2002;187(3):528–537. | ||
Zhang J, Sud S, Mizutani K, Gyetko MR, Pienta KJ. Activation of urokinase plasminogen activator and its receptor axis is essential for macrophage infiltration in a prostate cancer mouse model. Neoplasia. 2011;13(1):23–30. | ||
Li Y, Cozzi PJ. Targeting uPA/uPAR in prostate cancer. Cancer Treat Rev. 2007;33(6):521–527. | ||
Folkers K, Bowers CY, Tang P, Kubota M. Decapeptides as effective agonists from L-amino acids biologically equivalent to the luteinizing hormone-releasing hormone. Proc Natl Acad Sci U S A. 1986;83(4):1070–1074. | ||
Dharap SS, Wang Y, Chandna P, et al. Tumor-specific targeting of an anticancer drug delivery system by LHRH peptide. Proc Natl Acad Sci U S A. 2005;102(36):12962–12967. | ||
Ploug M, Gårdsvoll H, Jørgensen TJ, Lønborg Hansen L, Danø K. Structural analysis of the interaction between urokinase-type plasminogen activator and its receptor: a potential target for anti-invasive cancer therapy. Biochem Soc Trans. 2002;30(2):177–183. | ||
Ploug M. Structure-driven design of radionuclide tracers for non-invasive imaging of uPAR and targeted radiotherapy. The tale of a synthetic peptide antagonist. Theranostics. 2013;3(7):467–476. | ||
Sundaresan V, Menon JU, Rahimi M, Nguyen KT, Wadajkar AS. Dual-responsive polymer-coated iron oxide nanoparticles for drug delivery and imaging applications. Int J Pharm. 2014;466(1–2):1–7. | ||
Bates D, Abraham S, Campbell M, Zehbe I, Curiel L. Development and characterization of an antibody-labeled super-paramagnetic iron oxide contrast agent targeting prostate cancer cells for magnetic resonance imaging. PLoS One. 2014;9(5):e97220. | ||
Ramachandra Kurup Sasikala A, Thomas RG, Unnithan AR, et al. Multifunctional nanocarpets for cancer theranostics: remotely controlled graphene nanoheaters for thermo-chemosensitisation and magnetic resonance imaging. Sci Rep. 2016;6:20543. | ||
Theodore S, Sharp S, Zhou J, et al. Establishment and characterization of a pair of non-malignant and malignant tumor derived cell lines from an African American prostate cancer patient. Int J Oncol. 2010;37(6):1477–1482. | ||
Gupta AK, Gupta M. Cytotoxicity suppression and cellular uptake enhancement of surface modified magnetic nanoparticles. Biomaterials. 2005;26(13):1565–1573. | ||
Gaumet M, Vargas A, Gurny R, Delie F. Nanoparticles for drug delivery: the need for precision in reporting particle size parameters. Eur J Pharm Biopharm. 2008;69(1):1–9. | ||
Veiseh O, Gunn JW, Zhang M. Design and fabrication of magnetic nanoparticles for targeted drug delivery and imaging. Adv Drug Deliv Rev. 2010;62(3):284–304. | ||
Yang L, Cao Z, Sajja HK, et al. Development of receptor targeted magnetic iron oxide nanoparticles for efficient drug delivery and tumor imaging. J Biomed Nanotechnol. 2008;4(4):439–449. | ||
Longmire M, Choyke PL, Kobayashi H. Clearance properties of nano-sized particles and molecules as imaging agents: considerations and caveats. Nanomedicine (Lond). 2008;3(5):703–717. | ||
Gupta AK, Wells S. Surface-modified superparamagnetic nanoparticles for drug delivery: preparation, characterization, and cytotoxicity studies. IEEE Trans Nanobioscience. 2004;3(1):66–73. | ||
Hua MY, Yang HW, Chuang CK, et al. Magnetic-nanoparticle-modified paclitaxel for targeted therapy for prostate cancer. Biomaterials. 2010;31(28):7355–7363. | ||
Yang HW, Hua MY, Liu HL, et al. Cooperative dual-activity targeted nanomedicine for specific and effective prostate cancer therapy. ACS Nano. 2012;6(2):1795–1805. | ||
Aina OH, Sroka TC, Chen ML, Lam KS. Therapeutic cancer targeting peptides. Pept Sci. 2002;66(3):184–199. | ||
Ma P, Mumper RJ. Paclitaxel nano-delivery systems: a comprehensive review. J Nanomed Nanotechnol. 2013;4(2):1000164. | ||
Abdalla MO, Karna P, Sajja HK, et al. Enhanced noscapine delivery using uPAR-targeted optical-MR imaging trackable nanoparticles for prostate cancer therapy. J Control Release. 2011;149(3):314–322. | ||
Meng F, Zhong Z, Feijen J. Stimuli-responsive polymersomes for programmed drug delivery. Biomacromolecules. 2009;10(2):197–209. | ||
Lee DE, Koo H, Sun IC, Ryu JH, Kim K, Kwon IC. Multifunctional nanoparticles for multimodal imaging and theragnosis. Chem Soc Rev. 2012;41(7):2656–2672. | ||
Zhu XM, Wang YX, Leung KC, et al. Enhanced cellular uptake of aminosilane-coated superparamagnetic iron oxide nanoparticles in mammalian cell lines. Int J Nanomedicine. 2012;7:953–964. | ||
Accardo A, Aloj L, Aurilio M, Morelli G, Tesauro D. Receptor binding peptides for target-selective delivery of nanoparticles encapsulated drugs. Int J Nanomedicine. 2014;9:1537–1557. | ||
Sabnis S, Sabnis NA, Raut S, Lacko AG. Superparamagnetic reconstituted high-density lipoprotein nanocarriers for magnetically guided drug delivery. Int J Nanomedicine. 2017;12:1453–1464. | ||
Huang J, Zhong X, Wang L, Yang L, Mao H. Improving the magnetic resonance imaging contrast and detection methods with engineered magnetic nanoparticles. Theranostics. 2012;2(1):86–102. | ||
Korchinski DJ, Taha M, Yang R, Nathoo N, Dunn JF. Iron oxide as an MRI contrast agent for cell tracking. Magn Reson Insights. 2015;8 Suppl 1:15–29. | ||
Srinivasarao M, Galliford CV, Low PS. Principles in the design of ligand-targeted cancer therapeutics and imaging agents. Nat Rev Drug Discov. 2015;14(3):203–219. | ||
Barve A, Jin W, Cheng K. Prostate cancer relevant antigens and enzymes for targeted drug delivery. J Control Release. 2014;187:118–132. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.