Back to Journals » International Journal of Nanomedicine » Volume 12
Docetaxel-loaded PLGA and PLGA-PEG nanoparticles for intravenous application: pharmacokinetics and biodistribution profile
Received 8 September 2016
Accepted for publication 29 November 2016
Published 27 January 2017 Volume 2017:12 Pages 935—947
DOI https://doi.org/10.2147/IJN.S121881
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Pedram Rafiei, Azita Haddadi
Division of Pharmacy, College of Pharmacy and Nutrition, University of Saskatchewan, Saskatoon, SK, Canada
Abstract: Docetaxel is a highly potent anticancer agent being used in a wide spectrum of cancer types. There are important matters of concern regarding the drug’s pharmacokinetics related to the conventional formulation. Poly(lactide-co-glycolide) (PLGA) is a biocompatible/biodegradable polymer with variable physicochemical characteristics, and its application in human has been approved by the United States Food and Drug Administration. PLGA gives polymeric nanoparticles with unique drug delivery characteristics. The application of PLGA nanoparticles (NPs) as intravenous (IV) sustained-release delivery vehicles for docetaxel can favorably modify pharmacokinetics, biofate, and pharmacotherapy of the drug in cancer patients. Surface modification of PLGA NPs with poly(ethylene glycol) (PEG) can further enhance NPs’ long-circulating properties. Herein, an optimized fabrication approach has been used for the preparation of PLGA and PLGA–PEG NPs loaded with docetaxel for IV application. Both types of NP formulations demonstrated in vitro characteristics that were considered suitable for IV administration (with long-circulating sustained-release purposes). NP formulations were IV administered to an animal model, and docetaxel’s pharmacokinetic and biodistribution profiles were determined and compared between study groups. PLGA and PEGylated PLGA NPs were able to modify the pharmacokinetics and biodistribution of docetaxel. Accordingly, the mode of changes made to pharmacokinetics and biodistribution of docetaxel is attributed to the size and surface properties of NPs. NPs contributed to increased blood residence time of docetaxel fulfilling their role as long-circulating sustained-release drug delivery systems. Surface modification of NPs contributed to more pronounced docetaxel blood concentration, which confirms the role of PEG in conferring long-circulation properties to NPs.
Keywords: poly(lactide-co-glycolide), poly(lactide-co-glycolide)–poly(ethylene glycol), polymeric nanoparticles, docetaxel, emulsification solvent evaporation, sustained release, pharmacokinetics, biodistribution
Introduction
Medically known as malignant neoplasm, cancer includes a wide spectrum of various diseases that generally involve a circumstance of unregulated cell growth.1 Cancer cells proliferate uncontrollably and potentially affect nearby organs, resulting in disorders. Although new diagnostic and therapeutic approaches have improved the level of success of therapy and the quality of life of patients, cancer is still a leading cause of death all around the world.2 Accordingly, currently applied and well-established treatments for cancer (eg, chemotherapy, radiotherapy, and surgery) have proven to be variably effective depending on the type of cancer.3 For example, primary solid tumors being localized in the body can usually be removed by surgical interventions or subjected to radiation therapy to shrink the cancer tissue. However, cases with spreading features and tumor metastasis require extensive chemotherapy.4 Chemotherapy uses highly cytotoxic agents that interfere with normal cell activities such as cell division and growth upon entering the body and might also activate cell death pathways, promoting tumor shrinkage.
After administration, most anticancer agents demonstrate widespread distribution throughout the body and therefore can easily reach nontumor tissues. Furthermore, most chemotherapeutic agents possess a fairly rapid elimination rate after administration to the body. The widespread distribution of chemotherapeutic agents along with their fast elimination from the body results in a pronounced drop in the systemic concentration of anticancer agents. Therefore, to reach the required systemic therapeutic levels in the body, anticancer agents are required to be administered at high and frequent doses. However, most types of anticancer agents do not discriminate between normal cells and cancer cells.
Nonspecific activity and widespread distribution of anticancer agents in the body lead to systemic toxicity and adverse effects. Accordingly, a delicate balance must be maintained between the therapeutic and toxic effectiveness of anticancer agents. Otherwise, underdosing results in lack of therapeutic outcomes and overdosing can lead to immoderate toxicity. Toxicity due to anticancer agents and extensive adverse effects are common and fairly important in patients undergoing chemotherapy. They are of great importance because depending on the type of cancer and anticancer drug regimen, patients can experience a wide range of adverse effects. This can potentially lead to extreme patient suffering and discomfort that secondarily contributes to delays, reduction of dose, and cessation of chemotherapy.
Anticancer agents often have variable physicochemical characteristics (eg, variable water solubility). Incorporation of such active agents into formulations suitable for administration to the body requires the use of nonactive ingredients (ie, excipients) such as cosolvents and vehicles in the formulation. Interestingly, these excipients can cause serious side effects on their own (eg, hypersensitivity reaction, nephrotoxicity, and neurotoxicity) to a level that would also contribute to cessation of chemotherapy.5–8 Therefore, development of strategies that can modify cancer chemotherapy by intervening the mechanisms that lead to toxicity would help patients tolerate the adverse effects, comply with cancer treatment regimen, and ultimately have a better quality of life.
Docetaxel is a member of the taxane family (antineoplastic agents), which exert their cytotoxic effects on microtubules.9 It is derived semisynthetically from 10-deacetyl-baccatin III that is isolated from trees of the Taxus family (eg, T. baccata and T. brevifolia).10 Docetaxel inhibits the proliferation of cells by inducing a sustained block at the metaphase–anaphase boundary by disrupting the microtubular network necessary for mitotic cellular function.11 It inhibits the disassembly of tubulin leading to inhibited cell division and cell death.12 Docetaxel has demonstrated antitumor activity in patients with recurrent ovarian cancer, non-small-cell lung cancer, metastatic and adjuvant breast cancer, squamous cell head and neck cancer, and gastric cancer. In addition to these indications, docetaxel has demonstrated activity in previously treated patients with carcinomas of endometrial cancer, esophageal cancer, bladder cancer, prostate cancer, small cell lung cancer, as well as lymphomas and other neoplasms.12 The drug has a high level of plasma protein binding (>90%) and possesses a relatively large volume of distribution (Vd) accompanied with binding to a wide range of tissues.11 However, its peak plasma concentrations generally exceed levels required to induce relevant biologic effects.12 Docetaxel is insoluble in water, and hence a combination of ethanol and tween 80 (polysorbate 80) has been used in the commercial docetaxel formulation (taxoter®) to increase its solubility.10 Issues such as hypersensitivity reactions, decreased uptake by tumor tissue, and increased exposure of other body compartments to the drug have been accompanied with the formulation and are believed to be due to formulation ingredients (ethanol/tween 80).13–15
Accordingly, alternate drug delivery systems that help circumvent problems accompanied with docetaxel pharmacotherapy are needed. “Particulate drug carrier systems” have long been used to address such problems with anticancer drug pharmacokinetics.16–20 In particular, poly(lactide-co-glycolide) (PLGA) polymer whose application in human has been approved by the United States Food and Drug Administration has been widely used to prepare spectacular nanoparticles (NPs) as drug delivery vehicles.21 NP–drug association can provide favorable impacts on pharmacokinetics of the parent drug, leading to favorable pharmacodynamics and biological responses.22,23
PLGA has a variable nature that gives different physicochemical characteristics to the polymer. Other than the polymer itself, there are several NP fabrication methods introduced to the field. Therefore, any PLGA NP formulation is uniquely distinct from another formulation prepared differently. This brings attraction and novelty to the field of drug delivery using PLGA NP and leaves more space to explore. Herein, an optimized fabrication approach (data not shown) has been used for the preparation of PLGA NPs loaded with docetaxel for intravenous (IV) application. Drug-loaded NPs were prepared and characterized as long-circulating sustained-release delivery vehicles of docetaxel. Surface-modified PLGA NPs with poly(ethylene glycol) (PEG) have also been prepared using the copolymer of PLGA and PEG to have a NP formulation with enhanced long-circulating features. Then, PLGA and PLAG–PEG NPs were intravenously administered to our animal model, and the pharmacokinetic and biodistribution profiles of docetaxel were determined and compared between study groups.
Materials and methods
Materials
PLGA polymer with acid terminal group, lactide:glycolide monomer ratio of 50:50, and low-molecular-weight (ie, 6,700 Da, PLGA inherent viscosity of 0.15–0.25 dL/g in hexafluoroisopropanol) was purchased from Absorbable Polymers International (Pelham, AL, USA). PLGA–PEG diblock copolymer (50:50 PLGA attached to mPEG 5000, 15%. wt) was obtained from Evonik Degussa Corporation (Birmingham, AL, USA). Docetaxel was purchased from LC Laboratories (Woburn, MA, USA). BALB/c mice were purchased from Charles River Laboratories Inc. (Saint Constant, QC, Canada). All procedures were conducted in accordance with the Canadian Council of Animal Care guidelines for the care and use of laboratory animals and were approved by the Animal Care and Supply Committee of the University of Saskatchewan. All reagents were analytical grade or above and used as received unless otherwise stated.
Methods
Preparation of NPs
Docetaxel-loaded NPs were prepared from PLGA or PLGA–PEG diblock copolymer using a modified emulsification solvent evaporation technique as established in our laboratory. Briefly, the polymer and proper amount of drug were dissolved in ethyl acetate to give a solution of 10% (w/v) PLGA or PLGA–PEG and 1.5 mg/mL (w/v) docetaxel, which was then added to a 2.2% (w/v) poly(vinyl alcohol) (PVA) solution. The mixture was then vigorously shaken and subjected to sonication. The resulting nanosuspension was then left to stir for 2 h to let the organic solvent evaporate. The NPs were ultimately obtained after consecutive ultracentrifugation/washing steps with distilled water. The obtained nanoparticles were finally resuspended in 1% sucrose aqueous solution and freeze-dried. The freeze-dried nanoparticles were then kept at −20°C for further use.
NP characterization
NPs’ characteristics, including mean diameter/size distribution profile, polydispersity index (PDI), and zeta potential, were measured using Zetasizer (Nano-series, Nano ZS, model ZEN3600; Malvern Instruments, Worcestershire, UK). Nanoparticles were suspended in water prior to the measurements at 25°C. The drug loading and encapsulation efficiency of the preparation method were evaluated through extraction of docetaxel from freeze-dried PLGA or PLGA–PEG NP formulations. Briefly, acetone was added to 5 mg of NPs to dissolve both polymer and drug. The mixture was vortexed for 30 s and subjected to bath sonication for 30 min. Then, it was centrifuged for 20 min at 1,700× g. The supernatant was separated and preserved. Acetone was added to the precipitate, and the same procedure was repeated. The obtained supernatants from the first and second centrifugation steps were mixed and evaporated. Methanol was added to the residue to precipitate the polymer, vortexed for 30 s, and centrifuged for 20 min at 1,700× g. Docetaxel was then quantified in the supernatant using the mass spectrometry method described later.
Encapsulation efficiency of the preparation method and NPs’ drug loading were then calculated as follows:
|
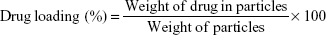
|
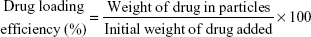
Docetaxel release profile from NPs
To have an estimate of the rate and pattern of release of docetaxel encapsulated inside NPs, in vitro drug release test was performed on docetaxel-loaded PLGA and PLGA–PEG NP formulations. Briefly, 20 mg of NPs was resuspended in 4 mL of phosphate-buffered saline (PBS) and placed at 37°C in a shaker incubator. At designated time points (1, 6, and 12 h, followed by 1, 2, 3, 4, and 5 days), the suspension was centrifuged at 12,000× g for 10 min and the supernatant was harvested. NPs were resuspended in fresh PBS in the original tube for further incubation. The supernatant was then extracted twice, and the extract was subjected to quantitative analysis by mass spectrometer.
Mass spectrometric method for rapid quantification of docetaxel
A mass spectrometry (MS/MS) method was developed and validated in our laboratory and used to quantitatively determine docetaxel in NPs24 and biological samples. Fragmentation pattern of docetaxel was determined as follows. The solution of docetaxel and paclitaxel (internal standard [IS]) in methanol containing 0.1% (v/v) formic acid was directly infused into the ionization source using a Model 11 Plus Syringe Pump (Harvard Apparatus, Holliston, MA, USA) at a flow rate of 10 μL/min into a Hybrid Triple Quadrupole/Linear Ion trap mass spectrometer (4000 QTRAP MS/MS System; AB Sciex, Framingham, MA, USA). Heater gas, nebulizer, and curtain gas pressure were set to 40, 40, and 30 psi, respectively. Turbo Ion Spray Source was adjusted to 5,500 V. Paclitaxel’s declustering potential (DP) and docetaxel’s DP were 55 and 46, respectively. The collision energy of paclitaxel and docetaxel was 93 and 21, respectively. For both docetaxel and paclitaxel, the collision cell exit potential was 18. Other parameters of the instrument were as follows: exit potential =10, collisionally activated dissociation =5, and interface heater = ON 150°C. Using positive electrospray ionization (+ESI), product-ion scans (MS/MS) were finally performed with appropriate set mass (parent m/z).
Based on the obtained fragmentation pattern of docetaxel and paclitaxel, four docetaxel fragments were selected for application in multiple reaction monitoring (MRM) transitions and further docetaxel quantitation. A G1329A (1200 Series) Autosampler (Agilent Technologies, Santa Clara, CA, USA) and a Quaternary Pump (1200 Series) were used to inject samples into the mass spectrometer. The flow rate of mobile phase, solution of 0.1% formic acid in methanol (isocratic elution), was set to 200 μL/min through a precolumn guard (Eclipse XDB-Rapid resolution, C18, 2.1 mm×30 mm, 3.5 μm; Agilent Technologies). Analyst software (version 1.6) was used for the quantitation step of corresponding docetaxel and paclitaxel MRM transitions.
Animal experiments
Animal experiments were conducted using 8-week-old female BALB/c mice after a 7-day period acclimatization in the Laboratory Animal Services Unit (LASU) of Health Sciences Building (University of Saskatchewan). A total of 96 mice were randomly divided into three groups with free access to food and water. Each group received 5 mg/kg of docetaxel formulation in the form of docetaxel-loaded PLGA NPs, docetaxel-loaded PLGA–PEG NPs, or docetaxel solution. Docetaxel formulations (200 μL) were injected through mouse tail vein. Then, at various time points (0.5, 1, 2, 4, 6, 12, 18, and 24 h) after dosing, mice were anesthetized and sacrificed by cardiopuncture for collection of blood. Organs, including the lung, heart, liver, and kidneys, were harvested and preserved in a freezer for later docetaxel extraction and analysis. Each time point was done in four replicates.
Biodistribution and pharmacokinetic analysis
For biodistribution studies, mouse organs were subjected to extraction procedure, and mass spectrometric analysis was performed to determine the concentration of docetaxel in each tissue. Then, the relationship between docetaxel concentration and time was determined by plotting the corresponding graphs. Pharmacokinetic analysis was performed using the collected mouse serum at different time points as follows. Briefly, the obtained blood was centrifuged for 5 min at 10,600× g. Serum was then separated from the rest of blood components and subjected to extraction procedure and further analyzed by mass spectrometer. Using GraphPad Prism software (Version 5.04), the plot of docetaxel concentration versus time was prepared. Noncompartmental pharmacokinetic analysis method was used to determine pharmacokinetically important parameters in mouse serum and other organs. Then, results from each study group were compared with each other. Pharmacokinetic parameters were calculated as follows:
- Elimination half-life (T1/2): Graphical relationship between docetaxel concentration at y-axis and time at x-axis was established. Docetaxel concentration at y-axis was then transformed to natural logarithm (ln) to convert the data into straight-line relationship. Then, data points from terminal elimination phase were used in nonlinear regression to plot a straight line (Y = −KeX + B). Half-life was calculated as
where Ke is the slope of plotted line from nonlinear regression analysis, Y is the natural logarithm (ln) of concentration, X is time, and B is the intercept of terminal elimination phase line.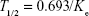
- Concentration at time zero (C0): Residuals were calculated between the plotted line and data points at distribution phase. Using residual data points and regression analysis, a second line was plotted (Y = −αX + A). C0 was then calculated as
where A and B are the intercepts from residual line and terminal elimination phase line, respectively, Y is the natural logarithm (ln) of concentration, X is time, and α is the slope of residual line.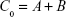
- Area under the serum concentration versus time curve (AUC): AUC from time zero (C0) until 24 h was calculated by software. AUC from t=24 h to infinity was calculated as
Therefore, total AUC was calculated as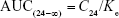
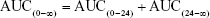
- Clearance (Cl): Docetaxel Cl was calculated as
where, dose is the amount of docetaxel injected intravenously and AUC(0–8) is the total AUC.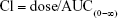
- Volume of distribution (Vd): Vd was calculated as
Area under the moment curve (AUMC): The product of concentration (including C0) multiplied by time at each time point was calculated (ie, concentration × time). Then, the graphical relationship between docetaxel concentration × time at y-axis and time at x-axis was established. AUMC from time zero until t=24 h was calculated by software. AUMC from 24 h to infinity was calculated as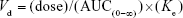
Therefore, total AUMC was calculated as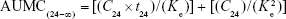
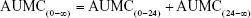
- Mean residence time (MRT): The MRT of docetaxel was calculated as
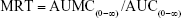
Statistical analysis
One-way analysis of variance (ANOVA) was used to compare the mean of different treatment groups. A P-value of <0.05 was considered statistically significant, unless otherwise stated.
Results and discussion
In vitro NP characteristics
Table 1 exhibits the various characteristics of fabricated NP formulations. Docetaxel-loaded PLGA NPs had an average diameter of 123.6±9.5 nm, a PDI of 0.245±0.041, and a zeta potential of −28.3±1.2 mV. However, PLGA–PEG nanoparticles exhibited a unidistributed population (ie, PDI 0.103±0.100) with 186.7±2.9 nm average diameter and −25.9±3.5 mV zeta potential. Drug loading of PLGA and PLGA–PEG NPs was calculated to be 5.58±0.24 μg/mg NP (ie, 37.25%±1.60% drug-loading efficiency) and 8.89±0.10 μg/mg NP (ie, 59.30%±0.70% drug-loading efficiency), respectively.
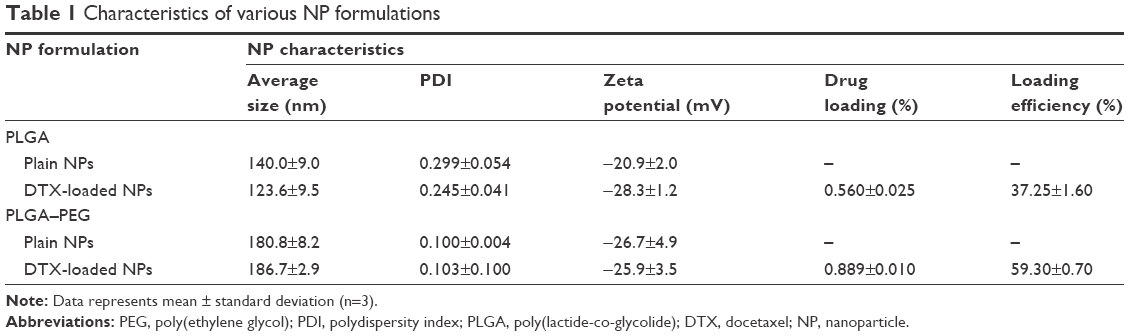 | Table 1 Characteristics of various NP formulations |
Both types of NP formulations demonstrated size ranges that were considered to be suitable for IV administration for long-circulating sustained-release properties. Size plays an important part in determining the biofate of NPs upon administration to the body.25,26 It is believed that a size range of <10 nm results in the removal of NPs by renal filtration.27,28 However, it is shown that NPs with hydrodynamic radii of >200 nm demonstrate a higher rate of Cl compared to the ones with lower size range.29 In other words, very small particles can be rapidly cleared from the systemic circulation via renal filtration, while very big particles have the potential to become entrapped inside the organs of reticuloendothelial system (RES), such as liver, spleen, and bone marrow.25
The negative zeta potential values of fabricated docetaxel-loaded PLGA and PLGA–PEG NPs suit IV administration as well. Surface characteristics such as zeta potential affect the fate of nanoparticles in the systemic circulation.30 Positively charged NPs have a higher rate of cell uptake and Cl when compared to neutral and negatively charged formulations.31 The presence of negatively charged serum proteins can lead to formation of aggregates after IV administration of positively charged NPs.32 These aggregates can secondarily be entrapped inside RES organs or result in embolism in blood capillaries.29,33–35
The fate of NPs in the body has also been correlated to the surface properties. The surface decoration of nanoparticles with PEG, a flexible, neutral, and hydrophilic polymer, forms a barrier-like layer that sterically hinders the surface of the original nanoparticle.36,37 Other than steric hindrance, it confers a hydrophilic neutral attribute to the surface of nanoparticles that works as a layer opposing the interaction of the NP surface and other materials (eg, opsonins and proteins). This provides efficient protection to NPs and gives long-circulation properties to PEGylated NPs.29,38,39
Docetaxel release profile from NPs
Release pattern of docetaxel from PLGA and PLGA–PEG NP formulations are shown in Figure 1. As exhibited, both formulations demonstrate an initial burst release during the first 24 h of incubation. This initial burst can be attributed to the drug molecules embedded at outermost layers of the NP and those adsorbed to the surface of the particles. The obtained profile after the initial burst is in favor of a sustained-release pattern for both types of NPs. This has resulted in an overall 25% and 49% drug depletion from PLGA and PLGA–PEG NPs, respectively. Diffusion of the drug, erosion, swelling of the polymer matrix, and degradation of the polymer are considered to be the main mechanisms for drug release.40 This sustained-release behavior is attributed to the slow degradation rate of polymers,41 and therefore release of docetaxel from the NPs is dependent mainly on drug diffusion and matrix erosion.42
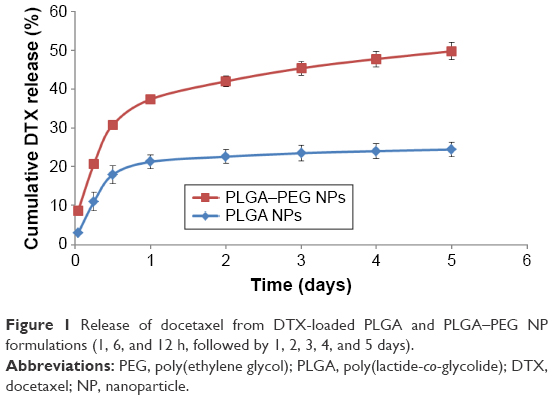 | Figure 1 Release of docetaxel from DTX-loaded PLGA and PLGA–PEG NP formulations (1, 6, and 12 h, followed by 1, 2, 3, 4, and 5 days). |
It is evident that PLGA–PEG NPs demonstrate higher and faster drug release profile compared to PLGA NPs. This difference could be accounted for the role of PEG chain moieties at the surface of PLGA–PEG NPs. PEG molecules can attract water to the surface of NPs, result in more pronounced wetting/hydration of PLGA–PEG NPs;43 and ultimately bring about higher release percentages compared to naked PLGA NPs. Other than surface properties, differences in characteristics such as particle size, hardness, and porosity can potentially result in varied drug release profiles.44 However, other studies have demonstrated the biphasic release behavior of docetaxel from PLGA45–48 and PLGA–PEG NPs.45,46,49
Pharmacokinetics and biodistribution of docetaxel
Docetaxel in mouse serum
Docetaxel serum concentration versus time curve for drug-loaded NPs and solution of free drug after IV injection to mice is demonstrated in Figure 2. The concentration of docetaxel in all formulations demonstrates a gradual declining trend. Drug concentration due to docetaxel solution is significantly lower than that of NP formulations throughout the study period being detectable in serum only up to 12 h. This is attributed to the large Vd of docetaxel and its binding affinity to various tissues.50–52 In fact, although docetaxel is highly bound to proteins of plasma, such as albumin and acid glycoproteins,53 docetaxel can escape the blood and get distributed to various organs immediately after IV injection of the drug solution.54 This is why docetaxel concentrations in serum have dropped fairly rapid compared to NP formulations during the first few time points followed by a rapid elimination phase. However, docetaxel nanoparticle formulations maintained the serum concentration of the drug up to 24 h postinjection. This is attributed to the role of NPs as long-circulating sustained-release drug delivery vehicles. Several studies have demonstrated the contribution of docetaxel-loaded nanoparticles in maintaining docetaxel concentration levels in the blood compared to the free solution of drug.47,55–58 Up to 4 h after injection, drug concentration due to PLGA NPs is higher than that of PLGA–PEG NPs. This is while after 4 h, drug concentration due to PLGA–PEG NPs becomes dominant. This is considered to be due to the differences in size (ie, larger average size of PLGA–PEG NPs) and surface properties (ie, hydrophilic surface of PEGylated NPs) between nanoparticle formulations that ultimately contribute to different biodistribution and entrapment levels in other organs.
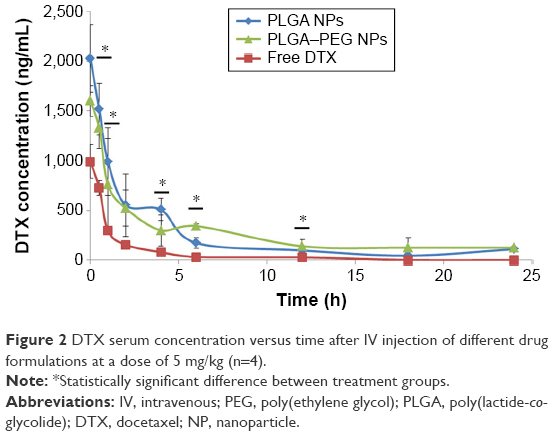 | Figure 2 DTX serum concentration versus time after IV injection of different drug formulations at a dose of 5 mg/kg (n=4). |
Table 2 demonstrates docetaxel’s important pharmacokinetic parameters in mouse serum. Compared to free docetaxel formulation, both types of NPs have resulted in a decrease in elimination rate (Ke) of drug from serum (P<0.05), which is reflected in their corresponding half-lives. This is particularly evident in case of PLGA–PEG nanoparticles increasing docetaxel’s elimination half-life (T1/2) by ~3.7-fold compared to free drug solution. This change is attributed to the presence of PEG moiety on the surface of PLGA NPs and its role in conferring characteristics to nanoparticles to evade the Cl mechanisms of the body.21,57,58 As a result, the level of serum exposure of docetaxel (AUC) is increased in case of PLGA–PEG NPs followed by PLGA NPs compared to that obtained from free drug solution (P<0.05). This is in agreement with the results obtained by other studies evaluating the effect of docetaxel loading in PLGA NPs on docetaxel pharmacokinetics.46,58 In turn, this has contributed to the pronounced reduction in levels of docetaxel Cl from serum when NP formulations have been used. Compared to free drug solution, docetaxel Cl demonstrates a significant decrease (ie, 3.6- and 5-fold for PLGA and PLGA–PEG NPs, respectively) when NP formulations were used (P<0.05). Both NP formulations have lowered docetaxel’s Vd (P<0.05). Vd of docetaxel decreased from 383 mL in case of free drug solution to 150 and 290 mL in case of PLGA and PLGA–PEG NPs, respectively. This reduction is more pronounced in case of PLGA NPs than in case of PLGA–PEG NPs. The reason is that PLGA–PEG NPs have both increased the serum exposure to docetaxel (AUC) and decreased the serum elimination rate of docetaxel in parallel (P<0.05). Furthermore, the MRT of docetaxel demonstrates statistically significant difference (P<0.05) between treatment groups. MRTs exhibited ~4.8- and 2.4-fold increases in case of PLGA–PEG (18.46 h) and PLGA (9.29 h) NPs, respectively, compared to the solution of docetaxel (3.81 h). It means that PLGA NPs and particularly PLGA–PEG NPs (as delivery vehicles) have increased the average transit time of docetaxel molecules through the body.
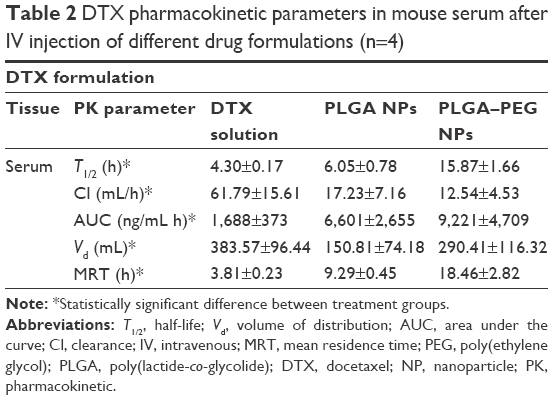 | Table 2 DTX pharmacokinetic parameters in mouse serum after IV injection of different drug formulations (n=4) |
Docetaxel in mouse liver
Figure 3 exhibits concentration versus time profile of docetaxel in mouse liver after IV administration of various drug formulations. The gradual declining trend in docetaxel concentration over time is evident for all drug formulations. Docetaxel from free drug solution is seen in liver only up to 12 h. This is while in case of PLGA and PLGA–PEG NPs, docetaxel is seen in the liver up to 18 and 24 h, respectively. Although docetaxel concentration due to PLGA–PEG NPs is lower than that of free drug solution and PLGA NPs during the first four time points (ie, 0.5, 1, 2, and 4 h), it becomes dominant over the last four time points (ie, 6, 12, 18, and 24 h). This can generally be attributed to the size distribution of PLGA–PEG NPs. Compared to PLGA NPs, PLGA–PEG NPs possess a higher particle size range and therefore can better come into contact with Kupffer cells of hepatic lobules as well as liver sinusoidal endothelial cells, get entrapped more easily in liver sinusoids, and ultimately remain in the liver for a longer time.59 Accordingly, Park et al60 demonstrated the association of liver and hepatic cells in liver filtering PLGA NPs from systemic circulation. In their study, PLGA NPs were highly localized in sinusoidal area, while Kupffer cells largely retained administered NPs. Therefore, we can conclude that, over time, PLGA NPs because of their lower particle size ranges would have less chance of entrapment in the liver.61,62 Consequently, PLGA–PEG NPs tend to remain in the liver for a longer period of time. That is the main reason why the elimination half-life of docetaxel in liver due to PLGA–PEG NPs is significantly higher than that of free docetaxel solution and PLGA NPs (P<0.05). Accordingly, the AUC of docetaxel concentration versus time for different formulations was as follows: PLGA–PEG NPs > PLGA NPs > free docetaxel solution (no statistically significant difference). This justifies the contribution of PLGA–PEG NPs to lower liver Cl levels compared to PLGA NPs and free solution of drug (P<0.05). Throughout the study period, drug concentration from PLGA NPs was higher than that from free docetaxel solution.
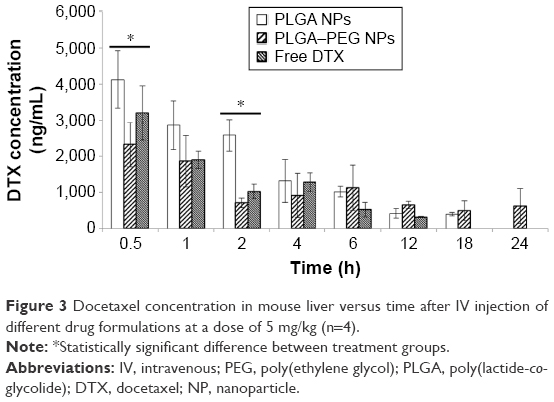 | Figure 3 Docetaxel concentration in mouse liver versus time after IV injection of different drug formulations at a dose of 5 mg/kg (n=4). |
Pharmacokinetic parameters of docetaxel in mouse liver are listed in Table 3. T1/2 of docetaxel in liver increased from 6.15 h in case of free drug solution to 8.15 and 19.03 h in case of PLGA and PLGA–PEG NPs (P<0.05). Parallel to T1/2, the Cl of docetaxel tends to decrease when NP formulations are used (ie, 1.5- and 1.9-fold for PLGA and PLGA–PEG NPs, respectively) (P<0.05). However, MRT of docetaxel is increased from 7.31 h (free drug solution) to 9.29 and 28.3 h when PLGA and PLGA–PEG NPs were used, respectively. The role of PLGA–PEG NPs in increasing docetaxel’s MRT in liver can potentially be used to target localized cancer tissues in liver.59
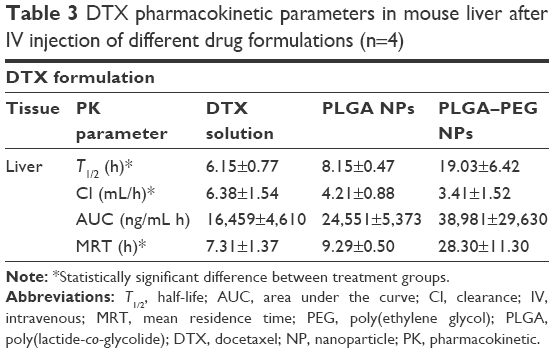 | Table 3 DTX pharmacokinetic parameters in mouse liver after IV injection of different drug formulations (n=4) |
Docetaxel in mouse kidney
Figure 4 demonstrates docetaxel concentration in mouse kidney at various time points after IV injection of three different drug formulations to mice. Docetaxel concentration demonstrates a declining trend throughout the study period and is detectable up to 24 h in all formulations except PLGA–PEG NPs, which is detectable up to 18 h. PLGA NPs demonstrate a significant contribution to docetaxel concentration in mouse kidney compared to other formulations (P<0.05). The average size of PLGA NPs is ~120 nm, while PLGA–PEG NPs possess a larger average size range (ie, 180 nm). Average size is an important determinant of the fate and biodistribution of particles.63 However, it is generally considered that particles with an average size of ≤10 nm are filtered in the kidneys.30,64 Therefore, the difference in NP size is not considered to be responsible for the different biodistribution behaviors between PLGA and PLGA–PEG NPs. Both types of NPs can potentially get entrapped in kidney capillary bed.26,65 Rather, PLGA nanoparticles with unmodified surface can get entrapped more. This is while PLGA–PEG NPs due to the presence of PEG moiety on the surface tend to remain in the blood circulation.66
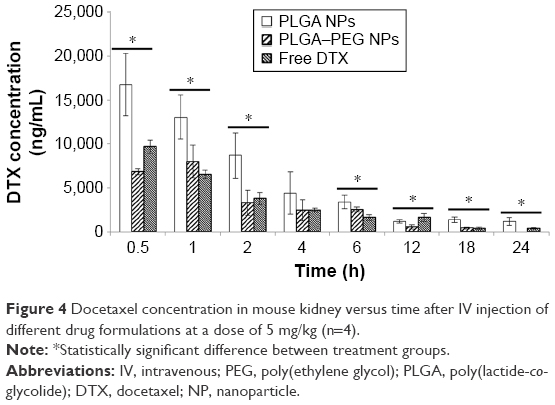 | Figure 4 Docetaxel concentration in mouse kidney versus time after IV injection of different drug formulations at a dose of 5 mg/kg (n=4). |
Table 4 exhibits docetaxel’s pharmacokinetic parameters in kidney subsequent to IV administration of various drug formulations. Docetaxel’s T1/2 in kidney tends to decrease (P<0.05) when loaded into PLGA–PEG NPs compared to free solution of drug (ie, from 7.54 to 4.91 h). This is while loading of NPs in PLGA NPs has contributed to 1.5-fold increase in docetaxel T1/2 (P<0.05). This is again attributed to the difference in surface properties between NP formulations. The Cl of docetaxel in kidney is almost the same for PLGA–PEG NP and free drug formulation (2.55 and 2.13 mL/h, respectively) (P>0.05) compared to that for PLGA NPs (1.09 mL/h) (P<0.05). In addition, MRT of docetaxel in kidney is increased and decreased in case of PLGA (13.10 h) and PLGA–PEG NPs (5.72 h), respectively, than in case of free docetaxel solution (8.46 h). PLGA–PEG nanoparticles have contributed to a lower AUC. It means that PLGA–PEG NPs demonstrate less kidney exposure compared to other formulations (P<0.05).
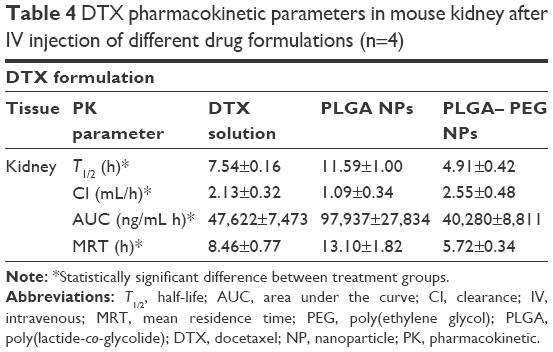 | Table 4 DTX pharmacokinetic parameters in mouse kidney after IV injection of different drug formulations (n=4) |
Docetaxel in mouse heart
Changes in docetaxel concentration in mouse heart versus time after IV administration of drug-loaded PLGA NPs, PLGA–PEG NPs, and solution of free drug are demonstrated in Figure 5. In contrast to PLGA–PEG NPs showing a relatively constant level of docetaxel in heart during the first few time points, docetaxel concentration due to PLGA NPs and free drug solution demonstrates a declining trend throughout the study time (P>0.05). In addition, the concentration of docetaxel provided by PLGA–PEG NPs is significantly lower (P<0.05) than that obtained from other drug formulations particularly during the first few time points. Docetaxel concentration from PLGA–PEG NPs demonstrates minimal variations during the distribution phase.
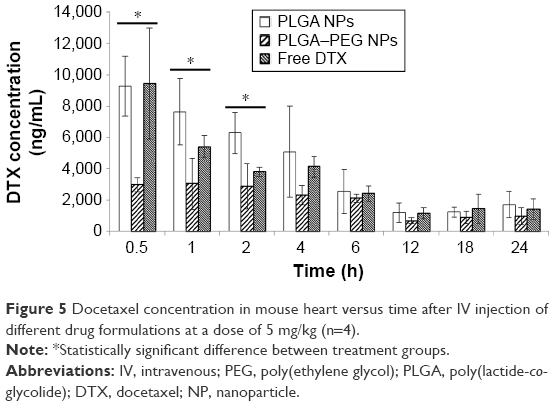 | Figure 5 Docetaxel concentration in mouse heart versus time after IV injection of different drug formulations at a dose of 5 mg/kg (n=4). |
Generally, after injection of drug formulations into the systemic circulation, they are taken to the mouse heart.67 During this visit, a high portion of NPs can get trapped there and result in high levels of docetaxel detected in the heart. In contrast to PLGA NPs, PLGA–PEG NPs (with surface modification) tend to demonstrate lower levels of entrapment in the heart. Later, blood flow can wash away trapped NPs from capillary bed of the heart. Microtubules are the major component of cytoskeleton of myocytes that contribute to structural integrity of cardiac cells.68 Docetaxel has a high affinity for microtubules.9 The first pass of docetaxel formulation from heart (being a microtubule-rich tissue69) and the high affinity that docetaxel has for microtubules result in the accumulation of docetaxel in heart. This might be the reason why free solution of docetaxel has resulted in high concentrations in heart tissue compared to NP formulations.
Loading of docetaxel in NPs has modified the pharmacokinetic parameters of the drug in mouse heart (Table 5). Half-life of docetaxel in heart demonstrates a decrease from 21.74 h (free drug solution) to 11.34 and 14.00 h in case of PLGA and PLGA–PEG NP formulations, respectively (P<0.05). It is due to high levels of docetaxel tissue accumulation in the heart after the injection of free docetaxel solution due to drug’s high affinity for cardiac tissue. This is while for heart, only a small increment is evident in Cl of docetaxel when NP formulations are used, that is, 1.28 and 2.14 mL/h for PLGA and PLGA–PEG NPs, respectively, and 1.12 mL/h for free solution of docetaxel (P>0.05). However, surface modification of PEGylated NPs can potentially contribute to less entrapment and therefore less AUC of docetaxel concentration versus time for PLGA–PEG NP formulation. However, PLGA NPs with unmodified surface demonstrate higher entrapment and consequently higher AUC. However, the difference between the AUC of treatment groups was not statistically significant (P>0.05). This is while MRT demonstrates a decrement in case of both NP formulations (17.36 and 20.711 h for PLGA and PLGA–PEG NPs, respectively) compared to the free docetaxel solution (28.08 h) (P>0.05). Theoretically, NP formulations have the chance to be gradually washed away from the heart after initial entrance to heart tissue postinjection. This could have influenced the T1/2 of docetaxel due to nanoparticles compared to free solution of drug.
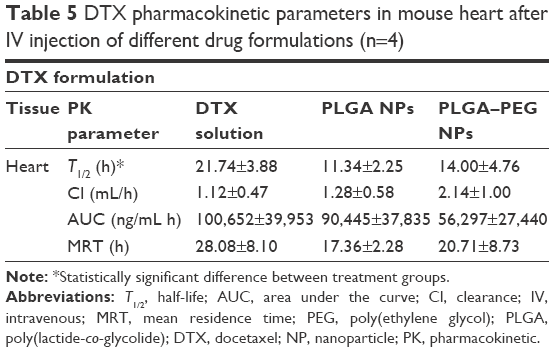 | Table 5 DTX pharmacokinetic parameters in mouse heart after IV injection of different drug formulations (n=4) |
Docetaxel in mouse lung
Figure 6 exhibits concentration versus time profile of docetaxel in mouse lung after IV administration of various docetaxel formulations. Docetaxel levels in the lung were high during the first time points (ie, up to 2 h) particularly for PLGA NPs and free drug solution. Later, docetaxel levels showed a significant decrease from time point 4 to 24 h. Drug formulations are considered to enter mouse heart and lung immediately postinjection.67 In other words, after injection, significant influx of a large portion of injected material happens to the lung. Consequently, there is the chance that docetaxel from free drug solution accumulates in the lung. Later, with further recirculation of blood in the lungs, docetaxel concentration tends to decrease gradually. However, NPs can get trapped in the lung immediately after IV injection being a result of lung capillary filtration effects.70 NPs can become physically entrapped in the lung or get recognized by the lung phagocytic macrophages. PLGA NPs appear to contribute to higher docetaxel concentrations in lung compared to PLGA–PEG NPs (during the first few time points) (P<0.05). Considering their lower size range (compared to PLGA–PEG NPs), surface-unmodified PLGA NPs are easily taken up in the lung. Esmaeili et al47 reported a significant amount of NP entrapment in mice lung after IV injection of PLGA NPs. In other words, based on docetaxel concentration–time profile, PLGA–PEG NPs demonstrate less accumulation in lung.
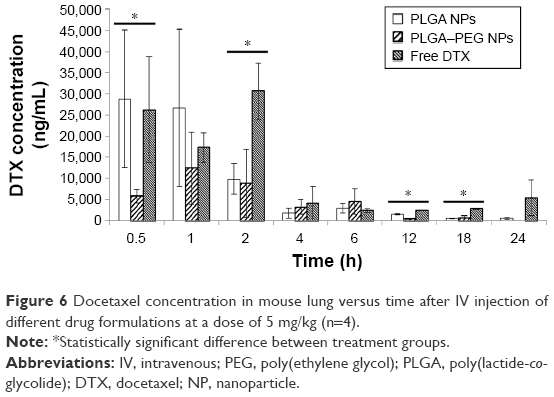 | Figure 6 Docetaxel concentration in mouse lung versus time after IV injection of different drug formulations at a dose of 5 mg/kg (n=4). |
Docetaxel pharmacokinetic parameters in mouse lung after IV injection of different drug formulations are summarized in Table 6. PLGA–PEG NPs show the lowest half-life (4.12 h) for docetaxel in mouse lung compared to other formulations (12.57 and 7.40 h for free drug solution and PLGA NPs, respectively) (P<0.05). Tendency of PLGA–PEG NPs to remain in the systemic circulation might be the reason for this. The Cl of docetaxel tends to increase from 0.53 mL/h in case of free drug solution to 1.12 mL/h (~2-fold) in case of PLGA NPs and 2.29 mL/h (~4.3-fold) in case of PLGA–PEG NPs. NP formulations demonstrate to decrease the MRT of docetaxel to 5.95 h (2.88-fold) (PLGA NPs) and 5.37 h (3.2-fold) (PLGA–PEG NPs) compared to the free solution of drug (17.19 h) (P<0.05). All these changes indicate the contribution of PLGA and PLGA–PEG NPs to lower delivery, distribution, retention, and residence of docetaxel in lungs compared to the free solution of docetaxel (P<0.05). This is potentially helpful in deviating the delivery of docetaxel away from the lung.
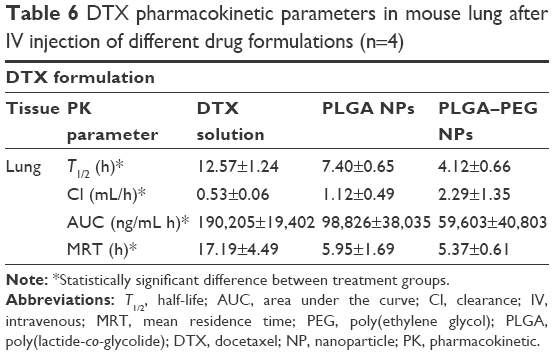 | Table 6 DTX pharmacokinetic parameters in mouse lung after IV injection of different drug formulations (n=4) |
Conclusion
The method of NP fabrication used here provided NPs with characteristics suitable for an IV sustained-release drug delivery system. The average particle sizes (~120 nm for PLGA and 180 nm for PEGylated PLGA nanoparticles) of <200 nm were considered to be at a range suitable to evade both filtration in RES organs and filtration in kidneys. The negative surface zeta potential values of NPs were in favor of long-circulating NPs in the blood. Fabricated NPs demonstrated a biphasic in vitro release profile. It was in fact favorable because the burst release phase potentially provides a loading dose of docetaxel followed by the release of drug in a sustained manner.
Association of docetaxel with PLGA and PEGylated PLGA NPs modified the pharmacokinetics and biodistribution of docetaxel. Accordingly, the mode of changes made to pharmacokinetics and biodistribution of docetaxel is attributed to the size and surface properties of NPs. Loading of docetaxel in NPs contributed to an increased blood residence time of docetaxel fulfilling NP’s role as a long-circulating sustained-release drug delivery system. Surface modification of NPs, however, contributed to more pronounced blood concentrations of docetaxel, confirming the role of PEG in helping NPs evade from Cl mechanisms present in the systemic circulation and the body. The role of NP’s average size in modifying the biodistribution is also evident. The obtained data from pharmacokinetics and biodistribution studies provide valuable insight about the relationship between NPs’ characteristics and their in vivo behaviors, which can be used to further manipulate PLGA nanoparticles for cancer chemotherapy.
Acknowledgments
The authors thank Dr Jane Alcorn for her significant contribution to this study. Also, kind assistance of Dr Mehran Yarahmadi during animal experiments is deeply appreciated. This work was funded by a research grant from the Natural Sciences and Engineering Research Council of Canada (NSERC).
Disclosure
The authors report no conflicts of interest in this work.
References
Cooper GM. Chapter 2: classification and development of neoplasms. Elements of Human Cancer. Burlington, MA: Jones and Bartlett Publishers Inc.; 1992:15–30. | ||
Canadian Cancer Society. Canadian Cancer Statistics. Toronto, ON: Canadian Cancer Society; 2015. | ||
Chidambaram M, Manavalan R, Kathiresan K. Nanotherapeutics to overcome conventional cancer chemotherapy limitations. J Pharm Pharm Sci. 2011;14(1):67–77. | ||
Maeda H, Bharate GY, Daruwalla J. Polymeric drugs for efficient tumor-targeted drug delivery based on EPR-effect. Eur J Pharm Biopharm. 2009;71(3):409–419. | ||
Feng S-S, Chien S. Chemotherapeutic engineering: application and further development of chemical engineering principles for chemotherapy of cancer and other diseases. Chem Eng Sci. 2003;58(18):4087–4114. | ||
Gelderblom H, Verweij J, Nooter K, Sparreboom A. Cremophor EL: the drawbacks and advantages of vehicle selection for drug formulation. Eur J Cancer. 2001;37(13):1590–1598. | ||
Singla AK, Garg A, Aggarwal D. Paclitaxel and its formulations. Int J Pharm. 2002;235(1–2):179–192. | ||
Bergh M, Magnusson K, Nilsson JL, Karlberg AT. Contact allergenic activity of Tween 80 before and after air exposure. Contact Dermatitis. 1997;37(1):9–18. | ||
Herbst RS, Khuri FR. Mode of action of docetaxel – a basis for combination with novel anticancer agents. Cancer Treat Rev. 2003;29(5):407–415. | ||
Zhao P, Astruc D. Docetaxel nanotechnology in anticancer therapy. ChemMedChem. 2012;7(6):952–972. | ||
Takimoto C, Beeram M. Microtubule stabilizing agents in clinical oncology, the taxanes. In: Fojo T, editor. The Role of Microtubules in Cell Biology, Neurobiology, and Oncology. Totowa, NJ: Human Press; 2008:395–419. | ||
Hitt R, Rodríguez C. Docetaxel. In: Schwab M, editor. Encyclopedia of Cancer. Berlin: Springer-Verlag; 2011:1148–1150. | ||
van Zuylen L, Verweij J, Sparreboom A. Role of formulation vehicles in taxane pharmacology. Invest New Drugs. 2001;19(2):125–141. | ||
ten Tije AJ, Verweij J, Loos WJ, Sparreboom A. Pharmacological effects of formulation vehicles: implications for cancer chemotherapy. Clin Pharmacokinet. 2003;42(7):665–685. | ||
Sanna V, Roggio AM, Posadino AM, et al. Novel docetaxel-loaded nanoparticles based on poly(lactide-co-caprolactone) and poly(lactide-co-glycolide-co-caprolactone) for prostate cancer treatment: formulation, characterization, and cytotoxicity studies. Nanoscale Res Lett. 2011;6(1):260. | ||
Blanco E, Hsiao A, Mann AP, Landry MG, Meric-Bernstam F, Ferrari M. Nanomedicine in cancer therapy: innovative trends and prospects. Cancer Sci. 2011;102(7):1247–1252. | ||
Haley B, Frenkel E. Nanoparticles for drug delivery in cancer treatment. Urol Oncol. 2008;26(1):57–64. | ||
Conti M, Tazzari V, Baccini C, Pertici G, Serino LP, De Giorgi U. Anticancer drug delivery with nanoparticles. In vivo. 2006;20(6A): 697–701. | ||
Cho K, Wang X, Nie S, Chen ZG, Shin DM. Therapeutic nanoparticles for drug delivery in cancer. Clin Cancer Res. 2008;14(5):1310–1316. | ||
Jabir NR, Tabrez S, Ashraf GM, Shakil S, Damanhouri GA, Kamal MA. Nanotechnology-based approaches in anticancer research. Int J Nanomedicine. 2012;7:4391–4408. | ||
Dinarvand R, Sepehri N, Manoochehri S, Rouhani H, Atyabi F. Polylactide-co-glycolide nanoparticles for controlled delivery of anticancer agents. Int J Nanomedicine. 2011;6:877–895. | ||
Hamidi M, Azadi A, Rafiei P, Ashrafi H. A pharmacokinetic overview of nanotechnology-based drug delivery systems: an ADME-oriented approach. Crit Rev Ther Drug Carrier Syst. 2013;30(5):435–467. | ||
Song X, Zhao X, Zhou Y, Li S, Ma Q. Pharmacokinetics and disposition of various drug loaded biodegradable poly(lactide-co-glycolide) (PLGA) nanoparticles. Curr Drug Metab. 2010;11(10):859–869. | ||
Rafiei P, Michel D, Haddadi A. Application of a rapid ESI–MS/MS method for quantitative analysis of docetaxel in polymeric matrices of PLGA and PLGA–PEG nanoparticles through direct injection to mass spectrometer. Am J Anal Chem. 2015;6:164–175. | ||
Moghimi SM, Hunter AC, Andresen TL. Factors controlling nanoparticle pharmacokinetics: an integrated analysis and perspective. Annu Rev Pharmacol Toxicol. 2012;52:481–503. | ||
Toy R, Hayden E, Shoup C, Baskaran H, Karathanasis E. The effects of particle size, density and shape on margination of nanoparticles in microcirculation. Nanotechnology. 2011;22(11):115101. | ||
Rao J. Shedding light on tumors using nanoparticles. ACS Nano. 2008;2(10):1984–1986. | ||
Kircher MF, Mahmood U, King RS, Weissleder R, Josephson L. A multimodal nanoparticle for preoperative magnetic resonance imaging and intraoperative optical brain tumor delineation. Cancer Res. 2003;63(23):8122–8125. | ||
Owens Iii DE, Peppas NA. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm. 2006;307(1):93–102. | ||
Yoo JW, Chambers E, Mitragotri S. Factors that control the circulation time of nanoparticles in blood: challenges, solutions and future prospects. Curr Pharm Des. 2010;16(21):2298–2307. | ||
Wang J, Sui M, Fan W. Nanoparticles for tumor targeted therapies and their pharmacokinetics. Curr Drug Metab. 2010;11(2):129–141. | ||
Zhang JS, Liu F, Huang L. Implications of pharmacokinetic behavior of lipoplex for its inflammatory toxicity. Adv Drug Deliv Rev. 2005;57(5):689–698. | ||
Cedervall T, Lynch I, Foy M, et al. Detailed identification of plasma proteins adsorbed on copolymer nanoparticles. Angew Chem Int Ed Engl. 2007;46(30):5754–5756. | ||
Gessner A, Waicz R, Lieske A, Paulke B, Mader K, Muller RH. Nanoparticles with decreasing surface hydrophobicities: influence on plasma protein adsorption. Int J Pharm. 2000;196(2):245–249. | ||
Nagayama S, Ogawara K, Fukuoka Y, Higaki K, Kimura T. Time-dependent changes in opsonin amount associated on nanoparticles alter their hepatic uptake characteristics. Int J Pharm. 2007;342(1–2):215–221. | ||
Patel B, Gupta N, Ahsan F. Particle engineering to enhance or lessen particle uptake by alveolar macrophages and to influence the therapeutic outcome. Eur J Pharm Biopharm. 2015;89:163–174. | ||
Hamidi M, Azadi A, Rafiei P. Pharmacokinetic consequences of PEGylation. Drug Deliv. 2006;13(6):399–409. | ||
Avgoustakis K, Beletsi A, Panagi Z, et al. Effect of copolymer composition on the physicochemical characteristics, in vitro stability, and biodistribution of PLGA–mPEG nanoparticles. Int J Pharm. 2003;259(1–2):115–127. | ||
Mosqueira VC, Legrand P, Morgat JL, et al. Biodistribution of long-circulating PEG-grafted nanocapsules in mice: effects of PEG chain length and density. Pharm Res. 2001;18(10):1411–1419. | ||
Keum CG, Noh YW, Baek JS, et al. Practical preparation procedures for docetaxel-loaded nanoparticles using polylactic acid-co-glycolic acid. Int J Nanomedicine. 2011;6:2225–2234. | ||
Scholes PD, Coombes AG, Illum L, et al. Detection and determination of surface levels of poloxamer and PVA surfactant on biodegradable nanospheres using SSIMS and XPS. J Control Release. 1999;59(3):261–278. | ||
Esmaeili F, Atyabi F, Dinarvand R. Preparation and characterization of estradiol-loaded PLGA nanoparticles using homogenization-solvent diffusion method. DARU. 2008;16:196–202. | ||
Zhang H, Cui W, Bei J, Wang S. Preparation of poly(lactide-co-glycolide-co-caprolactone) nanoparticles and their degradation behaviour in aqueous solution. Polym Degrad Stab. 2006;91(9):1929–1936. | ||
Astete CE, Sabliov CM. Synthesis and characterization of PLGA nanoparticles. J Biomater Sci Polym Ed. 2006;17(3):247–289. | ||
Koopaei MN, Dinarvand R, Amini M, et al. Docetaxel immunonanocarriers as targeted delivery systems for HER 2-positive tumor cells: preparation, characterization, and cytotoxicity studies. Int J Nanomedicine. 2011;6:1903–1912. | ||
Senthilkumar M, Mishra P, Jain NK. Long circulating PEGylated poly(D,L-lactide-co-glycolide) nanoparticulate delivery of docetaxel to solid tumors. J Drug Target. 2008;16(5):424–435. | ||
Esmaeili F, Dinarvand R, Ghahremani MH, Ostad SN, Esmaily H, Atyabi F. Cellular cytotoxicity and in-vivo biodistribution of docetaxel poly(lactide-co-glycolide) nanoparticles. Anticancer Drugs. 2010;21(1):43–52. | ||
Musumeci T, Ventura CA, Giannone I, et al. PLA/PLGA nanoparticles for sustained release of docetaxel. Int J Pharm. 2006;325(1–2):172–179. | ||
Dhankar R, Rathee P, Jain AK, et al. HER-2 targeted immunonanoparticles for breast cancer chemotherapy. J Appl Pharm Sci. 2011;1(3):132–139. | ||
Vaishampayan U, Parchment RE, Jasti BR, Hussain M. Taxanes: an overview of the pharmacokinetics and pharmacodynamics. Urology. 1999;54(6A suppl):22–29. | ||
Baker SD, Sparreboom A, Verweij J. Clinical pharmacokinetics of docetaxel: recent developments. Clin Pharmacokinet. 2006;45(3):235–252. | ||
Clarke SJ, Rivory LP. Clinical pharmacokinetics of docetaxel. Clin Pharmacokinet. 1999;36(2):99–114. | ||
Sparreboom A, van Tellingen O, Nooijen WJ, Beijnen JH. Preclinical pharmacokinetics of paclitaxel and docetaxel. Anticancer Drugs. 1998;9(1):1–17. | ||
Loos WJ, Baker SD, Verweij J, Boonstra JG, Sparreboom A. Clinical pharmacokinetics of unbound docetaxel: role of polysorbate 80 and serum proteins. Clin Pharmacol Ther. 2003;74(4):364–371. | ||
Yu Y, Tan S, Zhao S, et al. Antitumor activity of docetaxel-loaded polymeric nanoparticles fabricated by Shirasu porous glass membrane-emulsification technique. Int J Nanomedicine. 2013;8:2641–2652. | ||
Chu KS, Hasan W, Rawal S, et al. Plasma, tumor and tissue pharmacokinetics of docetaxel delivered via nanoparticles of different sizes and shapes in mice bearing SKOV-3 human ovarian carcinoma xenograft. Nanomedicine. 2013;9(5):686–693. | ||
Gupta M, Chashoo G, Sharma PR, et al. Dual targeted polymeric nanoparticles based on tumor endothelium and tumor cells for enhanced antitumor drug delivery. Mol Pharm. 2014;11(3):697–715. | ||
Jain S, Spandana G, Agrawal AK, Kushwah V, Thanki K. Enhanced antitumor efficacy and reduced toxicity of docetaxel loaded estradiol functionalized stealth polymeric nanoparticles. Mol Pharm. 2015;12(11):3871–3884. | ||
Zhang YN, Poon W, Tavares AJ, McGilvray ID, Chan WC. Nanoparticle-liver interactions: cellular uptake and hepatobiliary elimination. J Control Release. 2016;240:332–348. | ||
Park JK, Utsumi T, Seo YE, et al. Cellular distribution of injected PLGA-nanoparticles in the liver. Nanomedicine. 2016;12(5):1365–1374. | ||
Sarin H. Physiologic upper limits of pore size of different blood capillary types and another perspective on the dual pore theory of microvascular permeability. J Angiogenes Res. 2010;2:14. | ||
Rensen PC, Sliedregt LA, Ferns M, et al. Determination of the upper size limit for uptake and processing of ligands by the asialoglycoprotein receptor on hepatocytes in vitro and in vivo. J Biol Chem. 2001;276(40):37577–37584. | ||
Kumari A, Singla R, Guliani A, Yadav SK. Biodegradable nanoparticles and their in vivo fate. In: Yadav KS, editor. Nanoscale Materials in Targeted Drug Delivery, Theragnosis and Tissue Regeneration. Singapore: Springer Singapore; 2016:21–39. | ||
Ernsting MJ, Murakami M, Roy A, Li SD. Factors controlling the pharmacokinetics, biodistribution and intratumoral penetration of nanoparticles. J Control Release. 2013;172(3):782–794. | ||
Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5(4):505–515. | ||
Gref R, Domb A, Quellec P, et al. Long-circulating Drug Delivery Systems. The controlled intravenous delivery of drugs using PEG-coated sterically stabilized nanospheres. Adv Drug Deliv Rev. 1995;16(2):215–233. | ||
Hoyt RF Jr, Hawkins JV, St Clair MB, Kennett JM. Chapter 2 – mouse physiology A2. In: Fox JG, Davisson MT, Quimby FW, Barthold SW, Newcomer CE, Smith AL, editors. The Mouse in Biomedical Research (Second Edition). Burlington: Academic Press; 2007:23–XVI. | ||
White E. Mechanical modulation of cardiac microtubules. Pflugers Arch. 2011;462(1):177–184. | ||
Aquila-Pastir LA, DiPaola NR, Matteo RG, Smedira NG, McCarthy PM, Moravec CS. Quantitation and distribution of β-tubulin in human cardiac myocytes. J Mol Cell Cardiol. 2002;34(11):1513–1523. | ||
Jin C, Bai L, Wu H, Song W, Guo G, Dou K. Cytotoxicity of paclitaxel incorporated in PLGA nanoparticles on hypoxic human tumor cells. Pharm Res. 2009;26(7):1776–1784. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.