")
Back to Journals » Drug Design, Development and Therapy » Volume 9
DNA stabilization by the upregulation of estrogen signaling in BRCA gene mutation carriers
Authors Suba Z
Received 11 March 2015
Accepted for publication 1 April 2015
Published 15 May 2015 Volume 2015:9 Pages 2663—2675
DOI https://doi.org/10.2147/DDDT.S84437
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Shu-Feng Zhou
Zsuzsanna Suba
Surgical and Molecular Tumor Pathology Centre, National Institute of Oncology, Budapest, Hungary
Abstract: Currently available scientific evidence erroneously suggests that mutagenic weakness or loss of the BRCA1/2 genes may liberate the proliferative effects of estrogen signaling, which provokes DNA damage and genomic instability. Conversely, BRCA mutation seems to be an imbalanced defect, crudely inhibiting the upregulation of estrogen receptor expression and liganded transcriptional activity, whereas estrogen receptor-repressor functions become predominant. In BRCA-proficient cases, estrogen signaling orchestrates the activity of cell proliferation and differentiation with high safety, while upregulating the expression and DNA-stabilizing impact of BRCA genes. In turn, BRCA proteins promote estrogen signaling by proper estrogen synthesis via CYP19 gene regulation and by induction of the appropriate expression and transcriptional activity of estrogen receptors. In this exquisitely organized regulatory system, the dysfunction of each player may jeopardize genome stability and lead to severe chronic diseases, such as cancer development. Female organs, such as breast, endometrium, and ovary, exhibiting regular cyclic proliferative activity are particularly vulnerable in case of disturbances in either estrogen signaling or BRCA-mediated DNA repair. BRCA mutation carrier women may apparently be healthy or exhibit clinical signs of deficient estrogen signaling in spite of hyperestrogenism. Even women who enjoy sufficient compensatory DNA-defending activities are at risk of tumor development because many endogenous and environmental factors may jeopardize the mechanisms of extreme compensatory processes. Natural estrogens have numerous benefits in tumor prevention and therapy even in BRCA mutation carriers. There are no toxic effects even in sky-high doses and all physiologic cellular functions are strongly upregulated, while malignant tumor cells are recognized and killed in a Janus-faced manner.
Keywords: BRCA1/2 mutations, breast cancer, estrogen receptors, ER signaling, genome stabilization, cancer prevention
Introduction
A more holistic therapeutic approach that works with the body and is mindful of biological principles of evolution and adaptation is certainly worthy of consideration.[Seymour CB and Mothersill C]1
The identification of the BRCA1 gene was announced in 1994, the first gene known to strongly predispose subjects to breast cancer when becoming mutated.2 Identification of the BRCA2 gene was revealed the next year, in 1995.3 BRCA1 and BRCA2 genes have been implicated in a number of important cellular functions and may be regarded as safeguards of the genome, having DNA-stabilizing and tumor-suppressor capacities. Their ubiquitously expressed BRCA protein products control DNA replication, including transcriptional processes and recombination, as well as the repair of DNA damages.4
Mutation or loss of BRCA genes leads to disruption of BRCA proteins in mutation carriers and results in failures in the homologous recombination of defective DNA double-strand breaks. Decreased genome stability induces tumor development with conspicuous specificity in highly estrogen-dependent female organs, such as the breast and ovary, despite the crucial role of functional BRCA in the health of all cell types.5,6
Discovery of BRCA genes proved to be a milestone in medical sciences, and this great possibility inspired very hard work to find the holy grail of cancer research. Currently available scientific evidence strongly suggests that mutagenic weakness or loss of the BRCA1/2 genes may liberate the proliferative effects of estrogens and estrogen receptors (ERs). This extreme estrogen signaling is erroneously regarded as the initiator of DNA damage and genomic instability, being involved in the malignant transformation of hormone-sensitive epithelial cells.5–7 In spite of enormous efforts worldwide, there are numerous embarrassing controversies concerning the presumed causal correlations between unrestrained estrogen signaling and BRCA mutation-linked tumors, suggesting that the well-known traditional concept should be revisited.
All studies analyzing the associations among estrogens, ERs, and BRCA genes are important pieces leading to the solution of the enormous puzzle of carcinogenesis; however, the prevention and treatment of breast cancer seem to be a great challenge even today. The aims of the present study are to assemble the most relevant results published so far and to provide a line of logical arguments from evidence to conclusion in an attempt to achieve a better approach to gene-stabilizing processes.
Triangular partnership among estrogens, ERs, and BRCA genes
When examining the strong interplay and feedback mechanisms among BRCA genes, ERs, and estrogens, it appears that all players together provide the exquisite safeguard of genome integrity. Possible failures of either estrogen signaling or the BRCA function induce strong counteractions that aim to restore the surveillance of DNA replication, but the insufficiency of defensive mechanisms may lead to cancer initiation.
Upregulation of BRCA genes by estrogens
In the circulation, the most potent and abundant estrogen is estradiol (E2), whereas estrone (E1) and estriol (E3) also have estrogenic activities. Estrogen binding activates ERs, which act as ligand-activated transcription factor proteins in the promoter region of the target genes by means of classic genomic mechanisms. ERs can also regulate gene expression without direct binding to DNA via interaction with transcription factor proteins in the nucleus. Moreover, estrogen action also has nongenomic signaling cascades through cell membrane-associated ERs. Finally, genomic and nongenomic pathways of ER signaling converge on the target genes.8
ER isoforms, ER-alpha and ER-beta, exhibit strong crosstalk and interplay, by which cellular health may be ensured in both resting and proliferative biologic structures. ER-beta is responsible mainly for cell growth, while the role of ER-alpha is crucial in the course of cell proliferation.9
BRCA1 gene upregulation is required for the controlled proliferation and differentiation of breast cells as functional BRCA1 protein is necessary to prevent malignant transformation.10 By contrast, BRCA1 mutation results in impaired differentiation but increased poorly controlled proliferation in human mammary epithelial cell line.5
During puberty and pregnancy, when estrogen levels exhibit a dramatic increase and an excessive development of mammary gland takes place, wild-type BRCA1 gene expression is strongly enhanced.11 In pregnancy, the explosion-like cell proliferation in the female organs and fetal structures is associated with extreme increases in estrogen levels, which strongly stimulate the expression of DNA-stabilizing BRCA genes.12
In animal models, before the onset of puberty, administration of 17β-estradiol reduced the later risk of breast cancer by the increased expression of BRCA1 gene.13 In the mammary gland of ovariectomized mice, estradiol substitution increased the level of BRCA1 expression.11 In mice, high expression of BRCA1 was associated with the terminal differentiation of both ectodermally and mesodermally derived tissues.14
In human ER-positive breast cancer cell lines, depletion of estrogens significantly reduced BRCA1 mRNA expression, while the expression was increased again by estradiol treatment.15 Upregulation of BRCA1 mRNA expression by estradiol administration in tumor cells suggests that proper estrogen signaling has a pivotal role in DNA repair even in malignancies.
In conclusion, clinical and experimental data support that estradiol-induced mammary cell proliferation and differentiation are strongly associated with DNA-stabilizing effect by means of BRCA1 gene upregulation.
Mutual upregulation between ERs and BRCA proteins
ER-alpha expression has been studied extensively in BRCA1/2 mutation-associated cancers, while there are limited data on ER-beta and its isoforms. A significantly high proportion of ER-alpha-negative familial breast cancers were positive for both ER-beta isoforms: nuclear ER-beta1 and cytoplasmic ER-beta2.16 In the case of BRCA1-associated tumors, the expression of ER-beta was significantly higher than the weak expression of ER-alpha.17 These results suggest that BRCA1 mutation is predominantly in correlation with the defect of ER-alpha expression, while the significance of the different ER-beta isoform expressions and their intracellular locations may need further investigations.
There is strong regulatory protein–protein interplay between BRCA1 and ER-alpha expressions during cell proliferation (Figure 1). These interactions are fairly complex; therefore, the majority of studies mistakenly support the concept that both functional BRCA1 gene and BRCA1 protein suppress ESR1 gene expression and ER-alpha-mediated transactivation of its downstream target genes so as to keep the “dangerous” estrogen signaling in check.
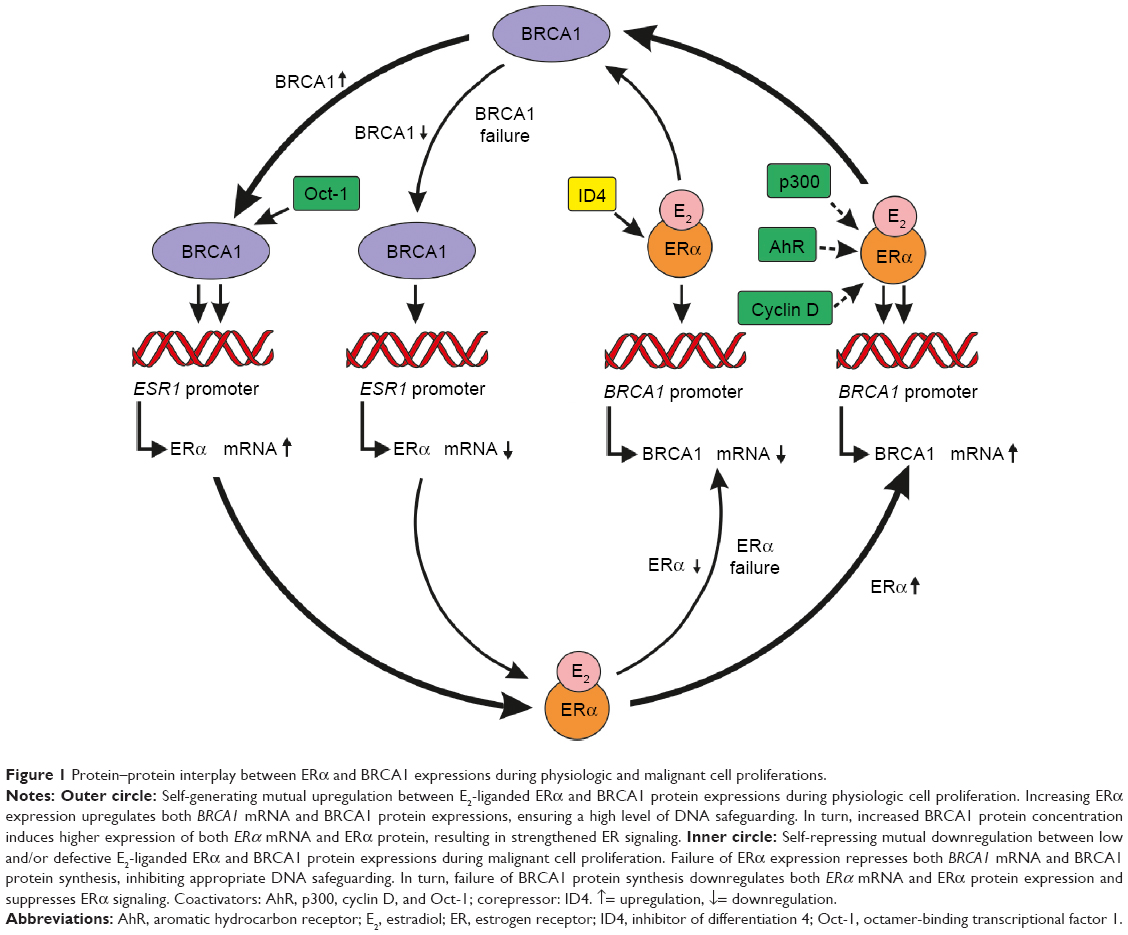 | Figure 1 Protein–protein interplay between ERα and BRCA1 expressions during physiologic and malignant cell proliferations. |
ER-alpha signaling upregulates BRCA1 expression
ER-alpha regulates BRCA1 protein mainly through its increased expression as BRCA1 gene is ER responsive. In MCF-7 tumor cells, a delayed response of BRCA1 protein expression was achieved by estrogen treatment, arriving at the peak value after 24 hours, as new protein synthesis takes a long time.18
Coregulators along with the other ER partner proteins all significantly contribute to estradiol signaling and mammary cell development.19 Coactivator protein functions define the genomic targets (cistromes) of ligand-activated ERs. Selective activation of a subset of the ER-alpha cistrome in MCF-7 breast cancer cells provided high specificity to the estradiol response.20 An upregulation of the expression levels of both ER isoforms and their coactivators was observed during the development of intraductal carcinomas from normal mammary glands, and a decrease was observed in their expression levels during the progression of breast cancer.21 These observations suggest that in the initial phase of cancer development, the upregulation of ERs provides stronger, defensive estrogen surveillance, while decreased transcriptional activity of ERs is associated with aggressive cancer spread.
E2 regulation of BRCA mRNA expression seems to be a highly complex process involving a variety of ER-alpha cofactors that may compete for ER-alpha binding sites, influencing BRCA1 promoter occupancy. BRCA1 upregulation after E2 stimulation was illuminated by showing that ER-alpha and its cofactor p300 are recruited to an activator protein site on the BRCA1 promoter region.22 ER stimulation of BRCA1 mRNA expression was also possible via the complex of ligand-bound ER-alpha and unliganded aromatic hydrocarbon receptor, which was capable of occupying the BRCA1 promoter region.23 Cyclin D binding also increased ER-alpha transactivation, presumably allowed by BRCA1.24 Finally, sequences resembling estrogen-responsive elements on the BRCA1 promoter may also be directly responsive to E2 stimulation.5
Estrogen and ER signaling may primarily define the levels of cell proliferation, differentiation, and apoptosis while exquisitely regulating the DNA-stabilizing surveillance of BRCA1 as well.
BRCA1 upregulates ER-alpha expression and transcriptional activity
BRCA1 was shown to be involved in ER-alpha-mediated transactivation after estrogen stimulation, but the results are apparently controversial.
Wild-type BRCA1 was demonstrated to inhibit the ability of ER-alpha to transactivate reporter constructs under the control of estrogen-responsive elements.25 It was also reported that BRCA1 could abrogate the induction of >90% of known E2-inducible genes.26 In BRCA1-deficient human ovarian cancer cells, ER-alpha exhibited ligand-independent transcriptional activity that was not observed in BRCA1-proficient cells.27 It was erroneously proposed that functional BRCA1 defensively blocks not only the liganded but also the ligand-independent transcriptional activity of ER-alpha. In addition, BRCA1 was able to inhibit p300-mediated ER acetylation,28 which is essential for the transactivation function of ERs.29 These observations apparently supported the preconception of the fundamental suppressor impact of BRCA1 on ER-mediated functions.
Considering the increased ligand-independent transcriptional activity of ERs in BRCA-mutant cells, it may be regarded as a defensive alarm reaction in answer to the danger of defective liganded ER transcription and low estrogen signaling. Moreover, the justified inhibitory impacts of wild-type BRCA1 on ER functions are randomly selected from the complex regulatory system.
By contrast, it was also shown that BRCA1 may induce an increased transcriptional activity of ER-alpha by upregulation of p300 expression, a coactivator of ER-alpha. It was theoretically supposed that BRCA1 and p300 coactivator are most likely in competition for the same binding site on ER-alpha and the winner may define the repressive or upregulative impact.30 Similarly, cyclin D binding was also capable of inducing ER-alpha transactivation.24
These findings reveal the complexity of regulatory processes, which include both activation and repression. In conclusion, estrogen-liganded ER-alpha may choose the momentarily most suitable cofactors, promoter regions, and transcriptional pathways in harmony with optimal BRCA1 expression and activity (Figure 1).
BRCA promotes expression of ESR1
BRCA is able to promote expression of ESR1, the coding gene for ER-alpha, mediated by octamer-binding transcription factor 1 (Oct-1) in breast cancer cells.31 According to an additional interesting finding, BRCA1 could also transcriptionally induce ER-alpha mRNA expression.31 Because functional BRCA1 upregulates ER-alpha mRNA expression, the predominance of ER-alpha-deficient cancers among BRCA1 loss-linked tumors may be at least partially illuminated.
Nevertheless, upregulation of ER-alpha signaling by BRCA1 also is incongruent with the novel finding that BRCA1 mutation-linked ER-negative breast cancer cells may be derived from either ER-positive or ER-negative luminal progenitor cells.6 This observation suggests that the neoplastic transformation of epithelial breast cells seems to be irrelevant to the ER status of progenitor cells, and BRCA mutation-linked breast tumors may not be initiated by excessive estrogen signaling.
BRCA1 may not act as a simple brake on ER-alpha-driven “unrestrained” cell proliferation. Liganded ER-alpha induces the appropriate transcriptional activities that orchestrate the proliferation, differentiation, and apoptosis of all cell types, while strongly interacting with BRCA1 to guarantee the exquisite regulation of DNA repair in the phases of both proliferation and involution.
Crosstalk between estrogens and ERs
Alterations in either estrogen concentrations or the expression of ERs may provoke quick counteractions so as to restore physiologic cellular estrogen surveillance.12
ER expression is adapted to estrogen levels in a U-shaped manner. Low estrogen level increases the expression of ERs by a positive feedback mechanism so as to strengthen the utilization of hormone supply. Physiologic high estrogen levels, as in the case of pregnancy, also promote overexpression of ERs, inducing a self-generating upregulation of ER signaling and protective genome-stabilizing effects of BRCA proteins for the enormously proliferating maternal and fetal structures. At the same time, high estrogen levels also have autoregulative increasing effects on estrogen synthesis (Figure 2).
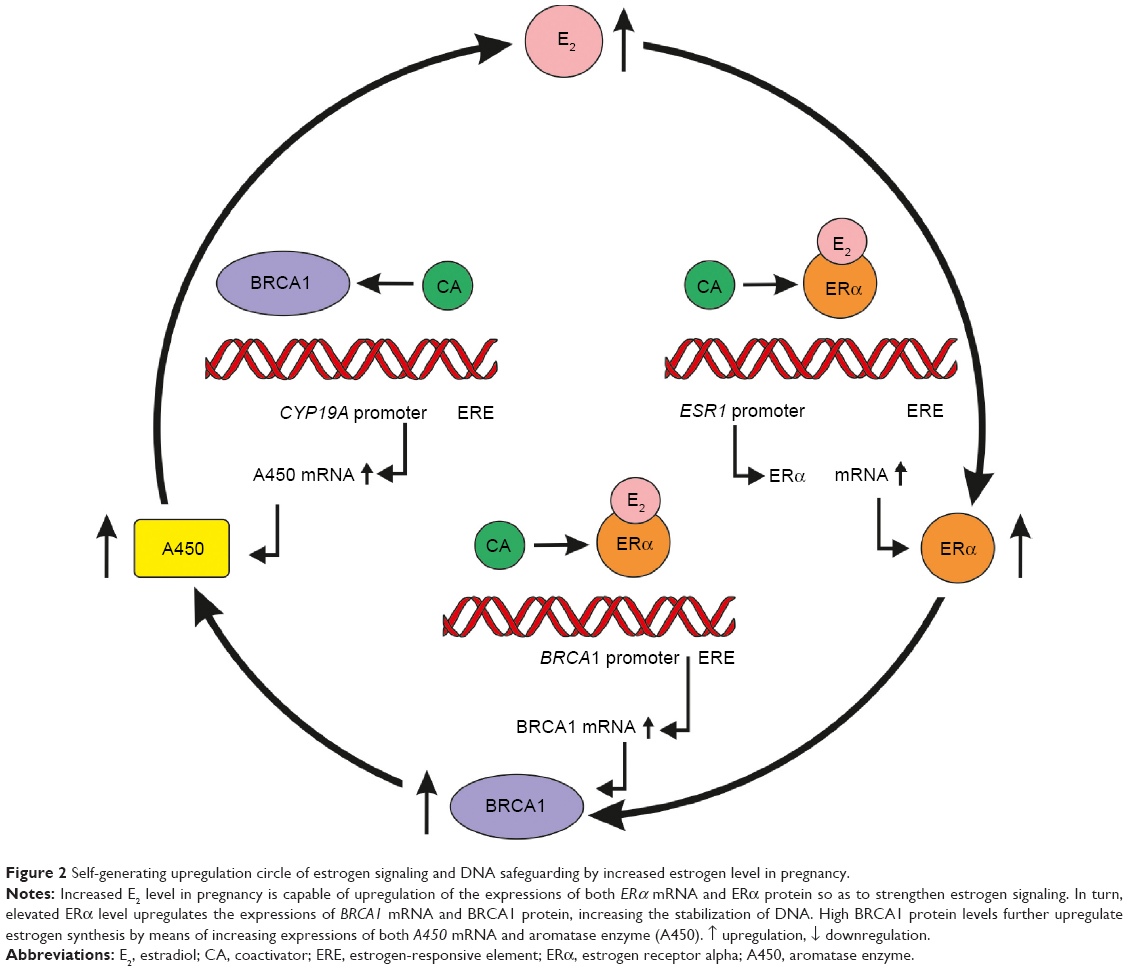 | Figure 2 Self-generating upregulation circle of estrogen signaling and DNA safeguarding by increased estrogen level in pregnancy. |
Decreased estrogen levels induce increased ER expression
Low estrogen concentrations promote increased ER expression by a positive feedback mechanism. In female rats, ovariectomy significantly increased the levels of ER-alpha expression in the uterus, kidney, and cerebral cortex.32
In benign proliferative breast lesions with known outcome, the expressions of ER-alpha and Ki-67 were studied.33 Patients who later experienced breast cancer development showed significantly higher ER-alpha and Ki-67 expressions in their previously benign lesion when compared with controls remaining cancer free. The authors suggested that higher ER-alpha expression in benign breast lesions was presumably associated with stronger pathogenic hormonal signaling. Alternatively, it can be concluded that in benign lesions later dedifferentiating into breast cancer, the higher ER-alpha expression reflected a strong but insufficient alarm reaction against dangerously defective estrogen signaling.
In hormonally challenged women, both anovulatory infertility and nulliparity are associated with increased breast cancer risk.34 On the other hand, multiparity, associated with strong estrogen signaling, is regarded as being protective against mammary cancer development.35 In the resting breast of parous women, significantly lower ER-alpha and 2-fold higher ER-beta expressions were detectable when compared with nulliparous subjects, which were regarded as intriguing paradigms between the effects of parity and breast cancer risk.36
Nevertheless, in parous women, safe estrogen exposure is plausibly linked with lower ER-alpha/ER-beta ratio. Conversely, in hormonally challenged nulliparous women, the higher ER-alpha/ER-beta ratio of mammary tissue cells calls attention to the danger of inadequate estrogen supply and risk for malignant transformation.12 In a breast cancer cell line, long-term estradiol deprivation threw light on the mechanism of estrogen hypersensitivity mediated by highly increased expression of ER-alpha. Hypersensitivity was characterized by the ability of tumor cells to respond to levels of estrogens at concentrations two-to-three logs lower than that required to stimulate wild-type cells.37 This positive feedback mechanism between estrogen depletion and the increased expression of ERs illuminates that acquired estrogen hypersensitivity may help the tumor killer activity of estrogen even in estrogen-deficient milieu.
Antiestrogen treatment of ER-alpha-positive tumors involves a chemical block of available ERs. Treatment with either raloxifene or tamoxifen strongly stimulated antiestrogen-resistant MCF-7/Ral tumor growth. When a 9-week raloxifene or tamoxifen treatment of tumors was followed by a 5-week estradiol treatment, estradiol statistically significantly reduced the size of tumors earlier stimulated by raloxifene or tamoxifen pretreatment.38 In clinical practice, physiologic estrogen-induced apoptosis is successfully applied for breast cancer prevention and treatment following estrogen deprivation.39
These observations justify that the antiestrogen blockade of estrogen signaling may result in defensive ER overexpression (estrogen hypersensitivity) even in tumor cells, which seems to be an unexpected kamikaze action. Transitional anticancer effect of antiestrogens in nearly 30% of breast cancer cases is a misleading experience because tumor regression may be attributed to the alarmed overexpression of ERs counteracting estrogen withdrawal or receptor block. Later, the exhaustion of the abundant, defensive ER expression results in tumor spread, being termed “secondary antiestrogen resistance.”
Estrogen-induced regression of even antiestrogen-resistant tumors strongly supports that estrogen is capable of the restoration of ER expression, reactivating the tumor killer capacity of estrogen signaling.
Increased estrogen levels augment ER expression and estrogen signaling
During pregnancy, highly increased levels of circulating estrogens are essential for the upregulation of ERs, which provide the appropriate hormone sensitivity and ensure the estrogen-mediated transcriptional activity of target genes in proliferating structures.
In pregnancy, extreme increase in the estradiol level was shown to promote uterine blood flow, rapid myometrial growth, and breast growth at term, mediated by the increased expression of myometrial and mammary ERs.40 Estrogen-mediated upregulation of ER expression in pregnancy may explain why defensive estrogen effects are prolonged and powerful in multiparous women.
Estradiol is able to increase both nongenomic and genomic ER signaling even in tumor cells. In ER-alpha-positive breast cancer cell line, estradiol binding can rapidly increase the expression and transcriptional activity of membrane-associated ER-alpha via the phosphatidylinositol 3-kinase (PI3K)/Akt system through the nongenomic pathway. Moreover, estradiol activation of Akt may interact with nuclear ER-alpha as well, promoting its expression and activity.41 These observations justify that estradiol-induced upregulation of ER signaling is capable of tumor killer activity even without antiestrogen pretreatment or estrogen withdrawal.
Increased estrogen level counteracts ESR1mutation-associated ER-alpha defects
Inherited ESR1 gene mutation may cause complete or partial blockage of estrogen signaling.42 Inherited, severe mutation of the ESR1 gene was published in the case of an 18-year-old girl who exhibited delayed puberty.43 The serum estrogen levels were reactively sky high but her ERs had essentially no response to estrogen.
Correlations between milder genetic defects in the ESR1 gene and breast cancer risk are highly inconclusive. Certain gene polymorphism studies revealed that the postmenopausal state may amplify the earlier hidden genetic defects of estrogen signaling.44,45 The results suggest that aging-associated decline of estrogen levels increases the breast cancer risk in women with ESR1 gene mutation, while in young women, reactively elevated estrogen synthesis may disguise the mild malfunction of the ESR1 gene.12,42
There is no direct link between estrogen supply and ER expression in breast cancers
In breast cancer cases, the controversial results of sexual hormone measurements may be explained by the diverse pathomechanisms leading to the breakdown of cellular estrogen surveillance.12,42 Failure of ER expression or transcriptional activity (estrogen resistance) leads to reactive hyperestrogenism, while estrogen deficiency is a consequence of defective aromatase activity.42,45 Both alterations lead to defective estrogen signaling and the breakdown of cellular estrogen surveillance. Female organs, such as the breast, exhibit cyclic proliferative activity and require strong estrogen surveillance, which can explain why mammary tissue is highly endangered by either estrogen deficiency or estrogen resistance.
In BRCA mutation-carrier women, defective estrogen signaling may be coupled with high but occasionally insufficient estrogen levels. Their high risk for ER-negative tumors may be attributed to the challenged estrogen signaling and insufficient compensatory mechanisms, instead of increased estrogen levels.12
It is a well-known fact that of all immunohistochemical markers, the ER negativity of breast cancers is the crucial factor in defining the biologic aggressivity and fatal outcome of the disease.46 The upregulation of ER expression and transcriptional activity by exogenous estrogen administration provides a plausible possibility for tumor therapy. High estrogen supply is suppressive against all subtypes of breast cancer, although the lower the ER expression of tumors, the higher is the promising dose of estrogens.12
In malignancies, a dynamic inverse relationship was revealed between the expressions of growth factor receptors (GFRs) and membrane-associated ERs. Excessive epidermal growth factor administration in the human breast cancer cell line MCF-7 resulted in persistent decreases in ER-alpha protein concentration, estradiol binding sites, the amount of ER-alpha mRNA, and ER-alpha gene transcription.47 Alternatively, high estrogen doses could inhibit lung carcinogenesis by reducing the level of insulin-like growth factor-I, which is a potent mitogenic agent for several malignancies.48
In apparently ER-negative breast cancer cells, inhibition of growth factor signaling provided the possibility for restoration of ER expression.49 Conversely, the inverse relationship between GFRs and ERs in tumors explains why a successful tamoxifen blockage of ERs may liberate overwhelming GFR predominance, leading to a rapidly fatal outcome of the disease.12
BRCA1/2 gene mutation-associated defective estrogen signaling
There is a strong interplay among mutation-free BRCA genes, CYP19-aromatase gene, and the ESR genes of ERs, providing the harmony among estrogen synthesis, estrogen signaling, and the safe DNA repair of proliferating cellular systems (Figure 2). Conversely, BRCA gene mutation is associated with defects in both estrogen signaling and the safeguarding of DNA.
Clinical manifestations of defective estrogen signaling in BRCA mutation carrier women
BRCA gene mutation carrier women frequently exhibit the clinical symptoms of estrogen deficiency.50,51 In certain premenopausal cases with BRCA gene mutation, the milder defects of ER signaling are clinically masked by reactively increased estrogen synthesis and/or other compensatory mechanisms. With aging, however, the relatively elevated but decreasing estradiol levels are not enough for the breakthrough of ER signal transduction defects, increasing the risk for breast cancer.42
Both germ line mutations in BRCA genes and anovulatory infertility are associated with high susceptibility for breast and ovarian cancers. In BRCA1 mutation-positive women, the low response rate of ovaries to fertility treatment was significantly increased (33.3%) when compared with BRCA1 mutation-negative patients (3.3%).50 These results support that BRCA mutations are associated with defective estrogen signaling.
Young women with BRCA1/2 mutations experienced natural menopause at earlier age significantly more frequently than mutation-free cases (P<0.001).51 The high risk of premature ovarian failure among BRCA1/2 mutation carriers reflects the fact that disorders of estrogen signaling may confer the risk of tumors.
Decreased ER-alpha expression in cells with BRCA gene mutation
In BRCA gene mutation carrier women, difficulties in estrogen signaling may be associated with the fairly decreased cellular expression and/or transcriptional activity of ERs.
In the breast of BRCA1 mutation carrier women, the persistent presence of least-differentiated type-1 lobules is associated with low ER-alpha expression,52 suggesting that functional BRCA1 is necessary for proper estrogen signaling and normal mammary differentiation. Selective inactivation of BRCA1 gene in mammary epithelial cells resulted in blunted ductal development, breast hyperplasia, and tumor formation.53 In BRCA1-mutant tumors, ESR1 gene expression was found to be 5.4-fold lower than that in BRCA1 gene-proficient ones.31
These findings support that among BRCA1 mutation carriers, increased tumor risk and the predominance of poorly differentiated ER-negative breast cancers may be explained by the low ER-alpha expression in both developing mammary glands and breast cancers.
Decreased estradiol-liganded transcriptional activity of ERs in BRCA gene-deficient cells
Interestingly, the inhibition of ER-alpha activity by BRCA1 protein was demonstrated in cell lines with either wild-type or defective BRCA1 gene.54 It can be proposed that the interaction between functional BRCA1 protein and the transcriptional activity of ERs is a complex regulatory process, including both activation and repression.
The amino terminal region of the BRCA1 protein was capable of binding with ER-alpha by an estrogen-independent interaction. This direct binding with the amino terminus of BRCA1 protein was able to upregulate the transcriptional activity of ER-alpha. At the same time, the carboxyl terminus of BRCA1 functioned as a simple transcriptional repression domain.29 A further study established that the BRCA1 gene is upregulated in response to estradiol administration in mammary epithelial cells, and BRCA1 in turn positively regulates the transcriptional activity of ERs.5 This implies the predominantly inhibitory effect of BRCA gene mutation on the transcriptional activity of ERs.
BRCA1 mutation seems to be an imbalanced defect, inhibiting more strongly the upregulation of ER expression and liganded transcriptional activity, whereas the ER-repressor functions become predominant. In BRCA1 mutation carriers, the security of cellular estrogen surveillance is endangered, resulting in accumulated mutations in both mammary tissue and breast cancer cells, liberating the initiation and progression of malignancies.
Compensatory mechanisms against BRCA gene mutations
The vast majority of BRCA gene mutations lead to a functionally defective BRCA protein synthesis.55 Although BRCA gene mutation carrier women have an increased lifelong risk for cancer development, there are certain defensive counteractions, which may help to improve the safeguarding of cell proliferation.42
Increased aromatase activity associated with BRCA gene mutation
BRCA1 gene transcriptionally regulates aromatase CYP19A1 gene, which defines the expression of aromatase enzyme that catalyzes the conversion of C19 steroids into bioactive estrogens.56 Gene silencing of BRCA1 seemed to be associated with liberation of aromatase gene expression in human stromal adipose cells, resulting in increased enzyme activity.57 BRCA1 gene mutation may upregulate estrogen synthesis, which is erroneously regarded as the causal factor for the development of cancers of breast and ovary.
Defective BRCA1 protein correlated to higher aromatase levels in 85% of BRCA1 mutation carriers, either in patients who had experienced breast cancer or in women who had prophylactic removal of their breast tissues because of the high risk for breast cancer.58 In BRCA1 mutation carriers, increases in aromatase and its proximal promoter I.3/II transcripts justify that decreased BRCA1 function upregulates aromatase transcription, leading to increased estrogen synthesis.
Aromatase activity in the different quadrants of mastectomy specimens from patients with breast cancer was always higher in quadrants associated with tumor when compared with that in noninvolved quadrants.59 These results were mistakenly estimated as the causal role of local estrogen synthesis within the breast in terms of both the natural history and behavior of breast cancers. Considering the tumor killer capacity of estrogens, their increased synthesis in the neighborhood of cancers may be regarded as a defensive local mechanism instead of as pathogenic agents.
Recent data support that local aromatase activity has close correlation with breast cancer regression. In young breast cancer cases, locoregional control after breast-conserving surgery highlighted that the absence of CYP19-aromatase activity in removed tumor samples carried a strongly significant risk for locoregional tumor recurrence and rapid tumor spread.60 Lack of intratumoral estrogen synthesis seems to be strongly associated with low differentiation of breast cancers and, at the same time, prophesy poor prognosis.
Hyperestrogenism as compensatory mechanism in BRCA gene mutation carriers
Elevated estrogen synthesis in BRCA1 mutation carr-iers counteracts decreased ER-alpha expression and transcriptional activity so as to preserve cellular estrogen surveillance.
Higher serum titers of estradiol were observed in BRCA1/BRCA2 mutation carriers when compared with women who were free of mutations.61 Stronger hyperestrogenism (71.7 pg/mL) was observed in BRCA2 mutation carrier patients when compared with the modestly increased estrogen levels of women with BRCA1 mutations (45.5 pg/mL) and the normal levels in cases without BRCA mutation (38.5 pg/mL).62
Higher estrogen production in BRCA2 mutation carriers seems to be an effective counteraction against defective estrogen signaling, mediating their markedly lower cancer risk, while the moderate estrogen overproduction in BRCA1 mutation carriers is not enough for tumor prevention.
Estradiol is beneficial even in sky-high doses and its physiologic overproduction in pregnancy may ensure long-lasting DNA-stabilizing and tumor-preventive effect.12,42 After in vitro fertilization-assisted childbirth, a significantly decreased overall cancer risk was reported in anovulatory women, mainly due to a lower-than-expected incidence of breast cancer.63
Use of oral contraceptives (OCs) was found to highly reduce the risk of ovarian cancers in women with both BRCA1 (odds ratio [OR]: 0.56) and BRCA2 mutations (OR: 0.39).64 The OC-associated risk of breast cancer in BRCA mutation carriers seemed to be controversial.12 Nevertheless, the use of OCs for at least 12 months was associated with strongly decreased breast cancer risk for BRCA1 mutation carriers (OR: 0.22).65
Increased ligand-independent transcriptional activity in cells with BRCA1 mutation
A relative decrease in estradiol-activated transcription of ERs was observed in BRCA1 gene-deficient human ovarian cancer cells.25 Conversely, ER-alpha showed an unexpectedly increased ligand-independent transcriptional activity in cells with BRCA1 mutation, which was not observed in BRCA1-proficient cells.27 This ligand-independent ER activation could partially be attributed to the increase in PI3K/Akt signaling. The authors mistakenly concluded that BRCA1 mutation liberates harmful estrogen signaling via ligand-independent pathways, while functional BRCA1 gene suppresses the transcription of unliganded ERs.
Increased estrogen-independent stimulation of ERs in BRCA1-deficient tumor cells may be a compensatory action against the defect of estrogen-activated ER transcription so as to restore estrogen signaling in emergency situations. Cancer may develop and progress when the defect of ligand-activated ER signal is not supplemented by compensatory unliganded ER transcription.42
Insulin sensitivity is defined by appropriate estrogen signaling
Complex correlations between estrogen signaling and several diseases have led to the erroneous concept that either low or high estrogen levels may cause disorders in glucose metabolism and chronic morbidity, the so-called estrogenic diseases.66 In women with irregular menstrual cycles, as well as in gestational diabetes, hyperestrogenism is mistakenly presumed to be a contributor to insulin resistance. Conversely, high estrogen level is an insufficient counteraction to break through the defect of ERs, which have a crucial role in insulin sensitivity.42,45
Glucose is the crucial fuel for mammalian cells and its uptake is fundamental for cellular metabolism, differentiation, survival, and proliferation.66 Insulin signaling regulates glucose uptake and whole-body metabolic homeostasis via insulin receptors (IRs). Dysregulation of insulin secretion or alterations of IR signal leads to progressive insulin resistance, in thorough interplay with defective estrogen synthesis.34 Insulin resistance and the concomitant hormonal and metabolic alterations predispose patients to a variety of life-threatening diseases, such as metabolic syndrome, type 2 diabetes, cardiovascular and neurodegenerative diseases, as well as malignancies.45
Experimental and clinical data justifying the significance of estrogen signaling in glucose homeostasis
Mice lacking ER-alpha exhibit insulin resistance, impaired glucose tolerance, and adiposity affecting both males and females. By contrast, in ER-beta knockout mice, the diabetogenic impact of ER-alpha loss may be reversed.67 These data suggest that healthy cellular glucose uptake requires a balanced equilibrium between functional ER isoforms, and the pharmaceutical manipulation of ER signal transduction pathways leads to crude metabolic anomalies.45 Aromatase knockout mice, with the inactivation of estrogen synthesis, exhibit insulin resistance, increased adiposity, and hyperinsulinism in both sexes, while estradiol administration is able to reverse insulin resistance even in male mice.68
Congenital estrogen deficiency or estrogen resistance has been of fundamental importance in understanding the close correlation between lack of estrogen signaling and insulin resistance.
Mutation of the human CYP19 gene was reported in a sister and brother.69 The 28-year-old girl presented progressive signs of virilization and polycystic ovaries with strikingly high androgen concentration in the fluid of cysts. Plasma androgen concentrations were highly elevated, whereas plasma estradiol was extremely low. Estrogen replacement therapy led to breast development, menstrual cycles, and resolution of the ovarian cysts. This case illustrates that polycystic ovarian syndrome (PCOS), the most frequent hormonal disorder in young women, may be derived from defective estrogen signaling.
The XY male sibling with CYP19 mutation was examined at 24 years of age.69 He was 204 cm tall, presenting with macroorchidism. The plasma concentrations of androgens were elevated, while estrogen levels were very low. Hyperinsulinemia, increased levels of serum total and low-density lipoprotein cholesterol, and high triglyceride levels were detected, suggesting insulin resistance.
Mutation affecting ERs was reported in a 28-year-old man with serious signs of insulin resistance.70 He exhibited diabetes, obesity, and premature cardiovascular disease. His testosterone levels were normal and, although his estrogen levels were extremely high, he had essentially no response to estrogen.
Hereditary estrogen resistance was published in the case of an 18-year-old girl experiencing delayed puberty.43 Laboratory examinations revealed sky-high serum levels of estrogens, and in vitro, 240 times the normal estrogen level was required to get a response from the patient’s ERs. Defective estrogen reactivity resulted in hyperinsulinism and glucose intolerance.
These findings justify that suitable estrogen signaling is necessary for appropriate glucose metabolism in both women and men.
Mechanisms of estrogen signaling in the regulation of glucose homeostasis
Estrogen signaling has crucial impact on glucose homeostasis by several pathways.45 In the pancreatic islets, ER-alpha activation promotes beta-cell mass proliferation and insulin biosynthesis even in cases with diabetes and obesity, whereas ER-beta stimulation improves glucose-stimulated insulin secretion.71
ER signaling regulates the activation of liganded IR protein via tyrosine phosphorylation and promotes the glucose transporter (GLUT4) accumulation in the cell membrane to facilitate glucose uptake.72 In conclusion, ER signaling has essential roles in all phases of insulin-stimulated cellular glucose uptake (Figure 3).
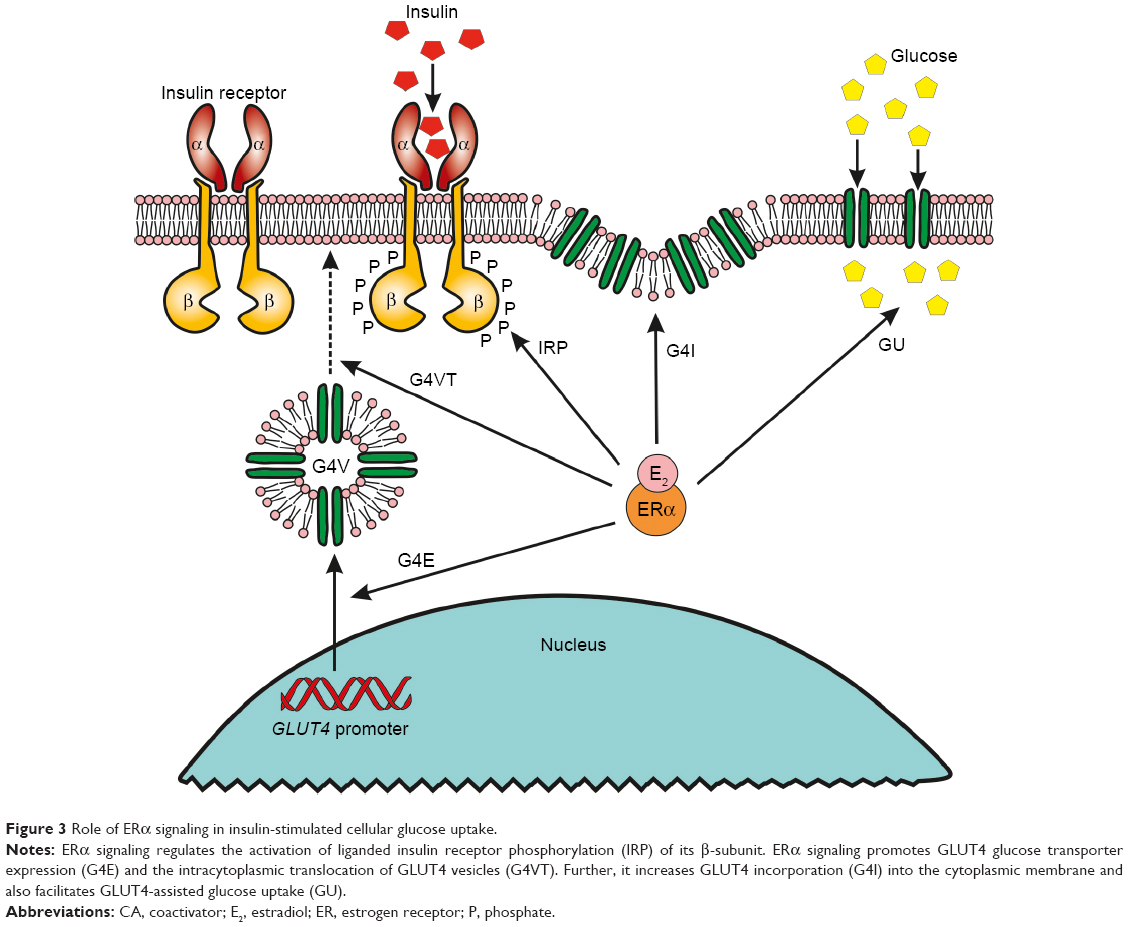 | Figure 3 Role of ERα signaling in insulin-stimulated cellular glucose uptake. |
Disorders associated with insulin resistance, such as PCOS, metabolic syndrome, type 2 diabetes, and central obesity, are in close correlation with increased breast cancer risk, while they exhibit advantageous improvements by proper estrogen administration.12,42,45
The BRCA1 protein seems to play a key role in insulin response, utilization of fatty acids, and mitochondrial function in both mouse and human skeletal muscle cells. Loss of BRCA1 function results in increased lipid storage, decreased insulin signaling, and increased oxidative stress in muscle cells.73 Functional BRCA1 may be protective against insulin resistance by ER-alpha upregulation.
BRCA1 mutation may be a risk factor for metabolic diseases conferred by defective ER-alpha signaling. In BRCA mutation carrier women, breast cancer development is frequently associated with insulin-resistant states, such as central obesity and type 2 diabetes.74,75 These observations are supportive of the concept that in BRCA mutation carriers, uncompensated defective estrogen signaling and the associated insulin resistance are strong risk factors for breast malignancies.12
Conclusion
Comprehensive analysis of experimental and clinical–epidemiologic results suggests that ER signaling is the chief safeguard of genome stability in the strong interplay with DNA-controlling and -repairing systems, such as BRCA genes and their protein products. Loss or mutation of BRCA genes results in imbalanced defect, crudely inhibiting the upregulation of ER expression and its liganded transcriptional activity, while ER-repressor functions become predominant. At the same time, even mutant BRCA genes are capable of upregulating the expression of aromatase enzyme and the increased compensatory estrogen synthesis may counteract defective estrogen signaling.
BRCA mutation carrier women may apparently be healthy or exhibit clinical signs of deficient estrogen signaling, such as amenorrhea, irregular cycles, anovulatory infertility, PCOS, or early menopause, in spite of increased estrogen levels. Nevertheless, tumor development endangers even those women enjoying temporarily sufficient DNA-defending activities because many endogenous and environmental factors may jeopardize the mechanisms of extreme compensatory processes.
In BRCA mutation carriers, breast cancer risk may not be predictable either by the compensatory level of estrogens or by the defect of ER expression and/or transcriptional activity. Complex evaluation of all players of estrogen signaling may help to plan tumor-preventive and -curative measures in both BRCA-mutation-positive and -negative cases.
Use of natural estradiol is appropriate for upregulation of defective BRCA genes and ERs while also having autoregulative increasing effects on estrogen synthesis. In the context of tumor therapy, estradiol is suppressive against all subtypes of breast cancers, although the lower the ER expression of tumors, the higher is the promising therapeutic dose even in BRCA mutation carriers with hyperestrogenism.
Natural estrogens have numerous benefits in tumor prevention and therapy. There are no toxic effects even at sky-high doses and all physiologic cellular functions are quite strongly upregulated, while malignant tumor cells are recognized and killed in a Janus-faced manner. Should we search for a better medicament?
Disclosure
The author reports no conflicts of interest in this work.
References
Seymour CB, Mothersill C. Breast cancer causes and treatment: where are we going wrong? (Breast Cancer (Dove Med Press)). 2013;5: 111–119. | ||
Miki Y, Swensen J, Shattuck-Eidens D, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994; 266(5182):66–71. | ||
Wooster R, Bignell G, Lancaster J, et al. Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995;378(6559): 789–792. | ||
Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell. 2002;108:171–182. | ||
Gorski JJ, Kennedy RD, Hosey AM, Harkin DP. The complex relationship between BRCA1 and ERalpha in hereditary breast cancer. Clin Cancer Res. 2009;15(5):1514–1518. | ||
Wang L, Di LJ. BRCA1 and estrogen/estrogen receptor in breast cancer: where they interact? Int J Biol Sci. 2014;10(5):566–575. | ||
Savage KI, Matchett KB, Barros EM, et al. BRCA1 deficiency exacerbates estrogen-induced DNA damage and genomic instability. Cancer Res. 2014;74(10):2773–2784. | ||
Maggi A. Liganded and unliganded activation of estrogen receptor and hormone replacement therapies. Biochim Biophys Acta. 2011;1812(8): 1054–1060. | ||
Helguero LA, Faulds MH, Gustafsson JA, Haldosén LA. Estrogen receptors alfa (ERalpha) and beta (ERbeta) differentially regulate proliferation and apoptosis of the normal murine mammary epithelial cell line HC11. Oncogene. 2005;24(44):6605–6616. | ||
Mueller CR, Roskelley CD. Regulation of BRCA1 expression and its relationship to sporadic breast cancer. Breast Cancer Res. 2003;5: 45–52. | ||
Marquis ST, Rajan JV, Wynshaw-Boris A, et al. The developmental pattern of BRCA1 expression implies a role in differentiation of the breast and other tissues. Nat Genet. 1995;11:17–26. | ||
Suba Z. Triple-negative breast cancer risk in women is defined by the defect of estrogen signaling: preventive and therapeutic implications. Onco Targets Ther. 2014;7:147–164. | ||
Cabanes A, Wang M, Olivo S, et al. Prepubertal estradiol and genistein exposures up-regulate BRCA1 mRNA and reduce mammary tumorigenesis. Carcinogenesis. 2004;25(5):741–748. | ||
Lane TF, Deng C, Elson A, Lyu MS, Kozak CA, Leder P. Expression of Brca1 is associated with terminal differentiation of ectodermally and mesodermally derived tissues in mice. Genes Dev. 1995;9:2712–2722. | ||
Spillman M, Bowcock A. BRCA1 and BRCA2 mRNA levels are coordinately elevated in human breast cancer cells in response to estrogen. Oncogene. 1996;13:1639–1645. | ||
Yan M, Rayoo M, Takano EA, kConFab Investigators, Fox SB. Nuclear and cytoplasmic expressions of ERβ1 and ERβ2 are predictive of response to therapy and alters prognosis in familial breast cancers. Breast Cancer Res Treat. 2011;126(2):395–405. | ||
Litwiniuk MM, Roznowski K, Filas V, et al. Expression of estrogen receptor beta in the breast carcinoma of BRCA1 mutation carriers. BMC Cancer. 2008;8:100. | ||
Kininis M, Chen BS, Diehl AG, et al. Genomic analyses of transcription factor binding, histone acetylation, and gene expression. Mol Cell Biol. 2007;27:5090–5104. | ||
Manavathi B, Samanthapudi VS, Gajulapalli VN. Estrogen receptor coregulators and pioneer factors: the orchestrators of mammary gland cell fate and development. Front Cell Dev Biol. 2014;2:34. | ||
Lupien M, Eeckhoute J, Meyer CA, et al. Coactivator function defines the active estrogen receptor alpha cistrome. Mol Cell Biol. 2009;29(12):3413–3423. | ||
Kurebayashi J, Otsuki T, Kunisue H, Tanaka K, Yamamoto S, Sonoo H. Expression levels of estrogen receptor-alpha, estrogen receptor-beta, coactivators, and corepressors in breast cancer. Clin Cancer Res. 2000;6(2):512–518. | ||
Jeffy BD, Hockings JK, Kemp MQ, et al. An estrogen receptor-alpha/p300 complex activates the BRCA-1 promoter at an AP-1 site that binds Jun/Fos transcription factors: repressive effects of p53 on BRCA-1 transcription. Neoplasia. 2005;7:873–882. | ||
Hockings JK, Thorne PA, Kemp MQ, Morgan SS, Selmin O, Romagnolo DF. The ligand status of the aromatic hydrocarbon receptor modulates transcriptional activation of BRCA-1 promoter by estrogen. Cancer Res. 2006;66:2224–2232. | ||
Wang C, Fan S, Li Z, et al. Cyclin D1 antagonizes BRCA1 repression of estrogen receptor alpha activity. Cancer Res. 2005;65:6557–6567. | ||
Fan S, Wang J, Yuan R, et al. BRCA1 inhibition of estrogen receptor signaling in transfected cells. Science. 1999;284:1354–1356. | ||
Xu J, Fan S, Rosen EM. Regulation of the estrogen-inducible gene expression profile by the breast cancer susceptibility gene BRCA1. Endocrinology. 2005;146:2031–2047. | ||
Zheng L, Annab LA, Afshari CA, Lee WH, Boyer TG. BRCA1 mediates ligand-independent transcriptional repression of the estrogen receptor. Proc Natl Acad Sci U S A. 2001;98:9587–9592. | ||
Ma Y, Fan S, Hu C, et al. BRCA1 regulates acetylation and ubiquitination of estrogen receptor-alpha. Mol Endocrinol. 2010;24:76–90. | ||
Kim MY, Woo EM, Chong YT, Homenko DR, Kraus WL. Acetylation of estrogen receptor alpha by p300 at lysines 266 and 268 enhances the deoxyribonucleic acid binding and transactivation activities of the receptor. Mol Endocrinol. 2006;20:1479–1493. | ||
Fan S, Ma YX, Wang C, et al. p300 modulates the BRCA1 inhibition of estrogen receptor activity. Cancer Res. 2002;62:141–151. | ||
Hosey AM, Gorski JJ, Murray MM, et al. Molecular basis for estrogen receptor alpha deficiency in BRCA1-linked breast cancer. J Natl Cancer Inst. 2007;99:1683–1694. | ||
Mohamed MK, Abdel-Rahman AA. Effect of long-term ovariectomy and estrogen replacement on the expression of estrogen receptor gene in female rats. Eur J Endocrinol. 2000;142(3):307–314. | ||
Shaaban AM, Sloane JP, West CR, Foster CS. Breast cancer risk in usual ductal hyperplasia is defined by estrogen receptor-alpha and Ki-67 expression. Am J Pathol. 2002;160(2):597–604. | ||
Suba Z. Circulatory estrogen level protects against breast cancer in obese women. Recent Pat Anticancer Drug Discov. 2013;8(2):154–167. | ||
Britt K, Ashworth A, Smalley M. Pregnancy and the risk of breast cancer. Endocr Relat Cancer. 2007;14(4):907–933. | ||
Asztalos S, Gann PH, Hayes MK, et al. Gene expression patterns in the human breast after pregnancy. Cancer Prev Res (Phila). 2010;3(3):301–311. | ||
Santen RJ, Song RX, Zhang Z, et al. Long-term estradiol deprivation in breast cancer cells up-regulates growth factor signaling and enhances estrogen sensitivity. Endocr Relat Cancer. 2005;12:S61–S73. | ||
Liu H, Lee ES, Gajdos C, et al. Apoptotic action of 17beta-estradiol in raloxifene-resistant MCF-7 cells in vitro and in vivo. J Natl Cancer Inst. 2003;95(21):1586–1597. | ||
Jordan VC. The new biology of estrogen-induced apoptosis applied to treat and prevent breast cancer. Endocr Relat Cancer. 2015;22(1): R1–R31. | ||
Schumacher A, Costa SD, Zenclussen AC. Endocrine factors modulating immune responses in pregnancy. Front Immunol. 2014;5:196. | ||
Stoica GE, Franke TF, Moroni M, et al. Effect of estradiol on estrogen receptor-alpha gene expression and activity can be modulated by the ErbB2/PI3-K/Akt pathway. Oncogene. 2003;22(39):7998–8011. | ||
Suba Z. Diverse pathomechanisms leading to the breakdown of cellular estrogen surveillance and breast cancer development: new therapeutic strategies. Drug Des Devel Ther. 2014;8:1381–1390. | ||
Quaynor SD, Stradtman EW, Kim HG, et al. Delayed puberty and estrogen resistance in a woman with estrogen receptor α variant. N Engl J Med.2013;369(2):164–171. | ||
Shin A, Kang D, Nishio H, et al. Estrogen receptor alpha gene polymorphisms and breast cancer risk. Breast Cancer Res Treat. 2003;80:127–131. | ||
Suba Z. Low estrogen exposure and/or defective estrogen signaling induces disturbances in glucose uptake and energy expenditure. J Diabet Metab. 2013;4:272–281. | ||
Hartley MC, McKinley BP, Rogers EA, et al. Differential expression of prognostic factors and effect on survival in young (< or =40) breast cancer patients: a case-control study. Am Surg. 2006;72(12):1189–1194. | ||
Stoica A, Saceda M, Doraiswamy VL, Coleman C, Martin MB. Regulation of estrogen receptor-alpha gene expression by epidermal growth factor. J Endocrinol. 2000;165(2):371–378. | ||
Marquez-Garban DC, Chen HW, Fishbein MC, Goodglick L, Pietras RJ. Estrogen receptor signaling pathways in human non-small cell lung cancer. Steroids. 2007;72:135–143. | ||
Massarweh S, Schiff R. Resistance to endocrine therapy in breast cancer: exploiting estrogen receptor/growth factor signaling crosstalk. Endocr Relat Cancer. 2006;13(suppl 1):S15–S24. | ||
Oktay K, Kim JY, Barad D, Babayev SN. Association of BRCA1 mutations with occult primary ovarian insufficiency: a possible explanation for the link between infertility and breast/ovarian cancer risks. J Clin Oncol. 2010;28(2):240–244. | ||
Lin WT, Beattie M, Chen LM, et al. Comparison of age at natural menopause in BRCA1/2 mutation carriers with a non-clinic-based sample of women in northern California. Cancer. 2013;119(9):1652–1659. | ||
Russo J, Russo IH. Toward a unified concept of mammary carcinogenesis. In: Aldaz MC, Gould MN, McLachlan J, Slaga TJ, editors. Progress in Clinical and Biological Research. New York: Wiley-Liss; 1997:1–16. | ||
Xu X, Wagner KU, Larson D, et al. Conditional mutation of Brca1 in mammary epithelial cells results in blunted ductal morphogenesis and tumour formation. Nat Genet. 1999;22:37–43. | ||
Fan S, Ma YX, Wang C, et al. Role of direct interaction in BRCA1 inhibition of estrogen receptor activity. Oncogene. 2001;20(1):77–87. | ||
Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434: 917–921. | ||
Simpson ER, Dowsett M. Aromatase and its inhibitors: significance for breast cancer therapy. Recent Prog Horm Res. 2002;57:317–338. | ||
Ghosh S, Lu Y, Katz A, Hu Y, Li R. Tumor suppressor BRCA1 inhibits a breast cancer-associated promoter of the aromatase gene (CYP19) in human adipose stromal cells. Am J Physiol Endocrinol Metab. 2007; 292:246–252. | ||
Chand AL, kConFab, Simpson ER, Clyne CD. Aromatase expression is increased in BRCA1 mutation carriers. BMC Cancer. 2009;9:148. | ||
Simpson ER. Sources of estrogen and their importance. J Steroid Biochem Mol Biol. 2003;86(3–5):225–230. | ||
Bollet MA, Savignoni A, De Koning L, et al. Tumor aromatase expression as a prognostic factor for local control in young breast cancer patients after breast-conserving treatment. Breast Cancer Res. 2009; 11(4):R54. | ||
Widschwendter M, Rosenthal AN, Philpott S, et al. The sex hormone system in carriers of BRCA1/2 mutations: a case-control study. Lancet Oncol. 2013;14(12):1226–1232. | ||
Kim J, Oktay K. Baseline E(2) levels are higher in BRCA2 mutation carriers: a potential target for prevention? Cancer Causes Control. 2013;24(3):421–426. | ||
Källén B, Finnström O, Lindam A, Nilsson E, Nygren KG, Olausson PO. Malignancies among women who gave birth after in vitro fertilization. Hum Reprod. 2011;26(1):253–258. | ||
McLaughlin JR, Risch HA, Lubinski J, et al; Hereditary Ovarian Cancer Clinical Study Group. Reproductive risk factors for ovarian cancer in carriers of BRCA1 or BRCA2 mutations: a case-control study. Lancet Oncol. 2007;8(1):26–34. | ||
Milne RL, Knight JA, John EM, et al. Oral contraceptive use and risk of early-onset breast cancer in carriers and noncarriers of BRCA1 and BRCA2 mutations. Cancer Epidemiol Biomarkers Prev. 2005;14(2): 350–356. | ||
Barros RP, Gustafsson JÅ. Estrogen receptors and the metabolic network. Cell Metab. 2011;14:289–299. | ||
Heine PA, Taylor JA, Iwamoto GA, Lubahn DB, Cooke PS. Increased adipose tissue in male and female estrogen receptor-alpha knockout mice. Proc Natl Acad Sci U S A. 2000;97(23):12729–12734. | ||
Jones ME, Thorburn AW, Britt KL, et al. Aromatase-deficient (ArKO) mice have a phenotype of increased adiposity. Proc Natl Acad Sci U S A. 2000;97:12735–12740. | ||
Morishima A, Grumbach MM, Simpson ER, Fisher C, Qin K. Aromatase deficiency in male and female siblings caused by a novel mutation and the physiological role of estrogens. J Clin Endocrinol Metab. 1995;80(12):3689–3698. | ||
Smith EP, Boyd J, Frank GR, et al. Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man. N Engl J Med. 1994;331:1056–1061. | ||
Tiano JP, Mauvais-Jarvis F. Importance of oestrogen receptors to preserve functional β-cell mass in diabetes. Nat Rev Endocrinol. 2012;8:342–351. | ||
Barros RPA, Machadon UF, Gustafsson JA. Estrogen receptors: new players in diabetes mellitus. Trends Mol Med. 2006;9:425–431. | ||
Jackson KC, Gidlund EK, Norrbom J, et al. BRCA1 is a novel regulator of metabolic function in skeletal muscle. J Lipid Res. 2014;55(4):668–680. | ||
Bordeleau L, Lipscombe L, Lubinski J, et al; Hereditary Breast Cancer Clinical Study Group. Diabetes and breast cancer among women with BRCA1 and BRCA2 mutations. Cancer. 2011;117(9):1812–1818. | ||
Kotsopoulos J, Olopado OI, Ghadirian P, et al. Changes in body weight and the risk of breast cancer in BRCA1 and BRCA2 mutation carriers. Breast Cancer Res. 2005;7(5):R833–R843. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.