Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 20
DNA Methylation–Mediated Downregulation of SDK1 Promotes COPD Progression: A Multi-Omics Mendelian Randomization Study
Authors Xue Z ![]() , Xue Q, Deng X, Li S, Liang P, Chen H, Xue H, Fang G
, Xue Q, Deng X, Li S, Liang P, Chen H, Xue H, Fang G
Received 14 April 2025
Accepted for publication 5 October 2025
Published 15 October 2025 Volume 2025:20 Pages 3387—3397
DOI https://doi.org/10.2147/COPD.S534335
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Zhichun Xue,1,2,* Qing Xue,1,2,* Xinyu Deng,1,2 Shuqi Li,1,2 Peng Liang,1,2 Huiling Chen,3 Hong Xue,4– 6 Guiju Fang1,2
1Department of Respiratory and Critical Care Medicine, Ningde Clinical Medical College of Fujian Medical University, Ningde, Fujian, People’s Republic of China; 2Department of Respiratory and Critical Care Medicine, Ningde Municipal Hospital of Ningde Normal University, Ningde, Fujian, People’s Republic of China; 3College of Marine Sciences, Ningde Normal University, Ningde, Fujian, People’s Republic of China; 4Department of Respiratory and Critical Care Medicine, Shengli Clinical Medical College of Fujian Medical University, Fuzhou, Fujian, People’s Republic of China; 5Fuzhou University Affiliated Provincial Hospital, School of Medicine, Fuzhou University, Fuzhou, Fujian, People’s Republic of China; 6Chest Medical Center, Fujian Provincial Hospital; Fujian Provincial Research Laboratory of Respiratory Diseases, Fuzhou, Fujian, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Guiju Fang, Department of Respiratory and Critical Care Medicine, Ningde Clinical Medical College of Fujian Medical University, Ningde, Fujian, People’s Republic of China, Email [email protected] Hong Xue, Department of Respiratory and Critical Care Medicine, Shengli Clinical Medical College of Fujian Medical University, Fuzhou, Fujian, 350001, People’s Republic of China, Email [email protected]
Purpose: To systematically explore the potential causal relationships among gene expression, DNA methylation, and chronic obstructive pulmonary disease (COPD) susceptibility using a multi-omics Mendelian randomization (MR) framework, and to further investigate key regulatory genes and methylation sites potentially involved in COPD pathogenesis.
Patients and Methods: We integrated genome-wide association study (GWAS) data from 635,145 individuals, expression quantitative trait loci (eQTL) data (N=15,695) from the eQTLGen Consortium, and methylation quantitative trait loci (mQTL) data from the Genetics of DNA Methylation Consortium (GoDMC). Two-sample Mendelian randomization was performed using genome-wide significant, linkage disequilibrium (LD)-independent (P < 5× 10−⁸, r² < 0.1) instruments filtered by Steiger analysis. Sensitivity analyses included inverse-variance weighted (IVW), MR-Egger, weighted median, and leave-one-out approaches. Colocalization analysis (posterior probability H₄ ≥ 0.75) and summary data-based Mendelian randomization (SMR) with heterogeneity in dependent instruments (HEIDI) test (P > 0.05) were used to validate shared causal variants. A three-step Mendelian randomization assessed mediation through methylation, gene expression, and COPD risk.
Results: We identified eight putative causal genes for COPD based on Mendelian randomization and colocalization analyses. SDK1 demonstrated consistent statistical significance across all subsequent steps. Increased SDK1 expression was significantly associated with a reduced risk of COPD (β = – 0.124, P = 0.002). Methylation at the intronic CpG site cg07526904 within SDK1 was associated with lower SDK1 expression (β = – 0.148, P = 0.002) and elevated COPD susceptibility (β = 0.036, P = 0.038). Mediation analysis indicated that SDK1 expression mediated approximately 51.9% of the total effect of cg07526904 on COPD risk (β = 0.018, P = 0.038), supporting a potential epigenetic pathway.
Conclusion: This analysis suggests that SDK1 methylation may affect COPD risk by regulating gene expression, highlighting a potential epigenetic mechanism. These findings offer preliminary insights into COPD pathogenesis and may help identify targets for future biomarker-based interventions.
Keywords: chronic obstructive pulmonary disease, COPD, SDK1, DNA methylation, gene expression, Mendelian randomization
Introduction
Chronic obstructive pulmonary disease (COPD), a progressive respiratory disorder characterized by persistent airflow limitation and chronic inflammation, imposes a substantial global health and economic burden owing to its escalating morbidity and mortality.1–3 Although environmental risk factors, including cigarette smoking and air pollution, are well documented, the genetic and epigenetic mechanisms driving COPD heterogeneity remain incompletely understood.4–6 Although genome-wide association studies (GWAS) have identified multiple genetic loci associated with COPD susceptibility, these findings still lack mechanistic insights and causal validation.7–9 Epigenetic regulation, particularly DNA methylation, has emerged as a critical modulator of gene expression and disease development.10,11 However, its specific role in COPD remains poorly characterized, and existing studies are often limited by observational design and inconsistent results. Mendelian Randomization (MR) leverages genetic variants as instrumental variables, enabling causal inference by reducing confounding and reverse causation.12,13 Integrating GWAS findings with expression quantitative trait loci (eQTL) and methylation quantitative trait loci (mQTL) data provides a powerful framework for systematically elucidating how gene expression and DNA methylation contribute to disease risk.14–16 However, to our knowledge, such approaches are still underexplored in COPD.
To address these knowledge gaps, we implemented a multi-omics Mendelian randomization framework that integrates large-scale GWAS, blood eQTL, and mQTL datasets. We hypothesized that this integrative approach would uncover causal regulatory genes and methylation sites underlying COPD susceptibility, including potential roles for SDK1 and its methylation-mediated expression regulation. Our study aims to provide novel mechanistic insights into the genetic and epigenetic architecture of COPD, potentially guiding future biomarker discovery and targeted interventions.
Material and Methods
Study Design and Data Sources
We conducted a two-sample, multi-omics MR analysis by integrating three major types of large-scale, publicly available summary statistics: GWAS, eQTL, and mQTL. The GWAS summary statistics for COPD were obtained from a meta-analysis including 13,530 European ancestry cases, 454,945 European controls, 4,017 East Asian cases, and 162,653 East Asian controls.17 The blood cis-eQTL data were derived from the eQTLGen consortium, covering 31,684 individuals and 15,695 genes,18 while the blood mQTL data were obtained from the GoDMC database, comprising approximately 32,000 individuals.19 Detailed information on data sources, sample size, ancestry, references, URLs, and download dates is provided in Table 1. All datasets were accessed in October 2024 and had received the appropriate ethical approvals. An overview of the study design is presented in Figure 1.
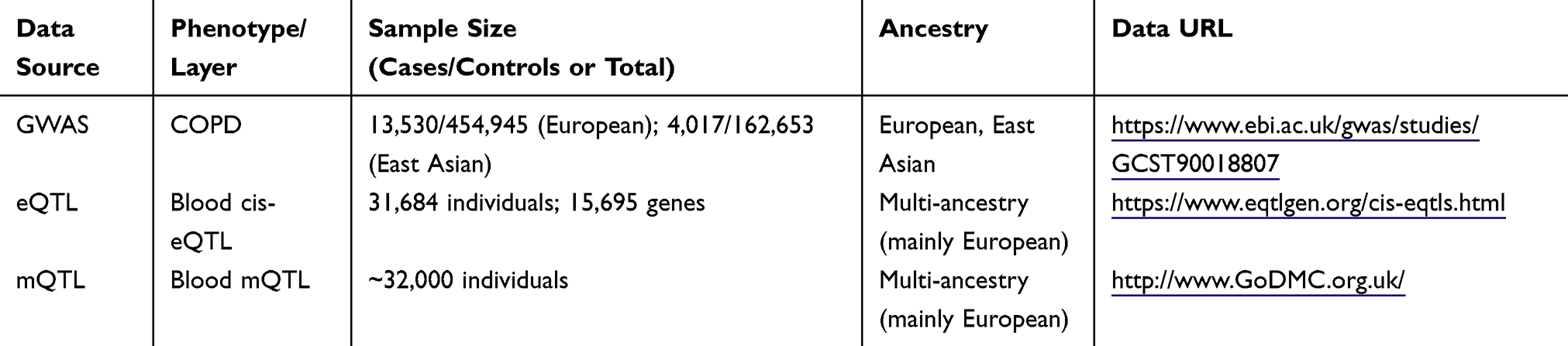 |
Table 1 Summary of Primary Data Sources Used in This Study |
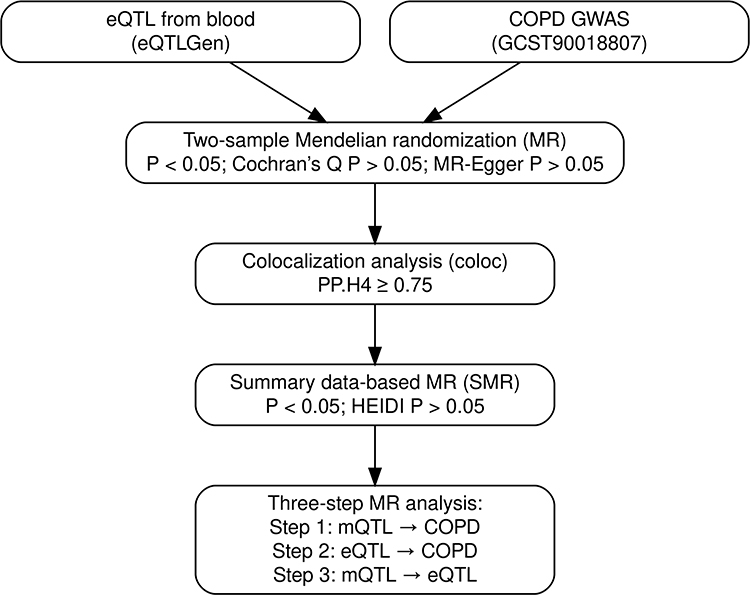 |
Figure 1 Multi-omics Mendelian randomization (MR) framework. Schematic overview of the analytical pipeline. Blood-based expression quantitative trait loci (eQTL) and COPD genome-wide association study (GWAS) summary statistics were integrated using two-sample MR. Colocalization analysis (coloc) was performed to assess whether the same causal variants underlie both gene expression and COPD risk (PP.H4 ≥ 0.75). The summary data-based MR (SMR) approach with the HEIDI test was used to further validate single-variant causality. Finally, a three-step MR analysis evaluated the directional relationships among methylation QTLs (mQTL), gene expression (eQTL), and COPD susceptibility. |
Instrument Selection and Quality Control
Across all datasets, candidate SNPs were selected using stringent criteria: strong associations with the exposure traits (P < 5 × 10−⁸) and low linkage disequilibrium (LD; r² < 0.1 within a 10,000 kb window), with clumping performed using the default parameters in the TwoSampleMR package. SNPs with inconsistent causal directionality were removed using Steiger filtering (P < 0.05). Instrument strength was assessed using the F statistic (F = β²_exposure/SE²_exposure), and only SNPs with F > 10 were retained to minimize instrument bias.20
Mendelian Randomization Analysis and Sensitivity Analyses
MR analyses were conducted using the TwoSampleMR package (v0.5.6) in R. For single SNP analyses, the Wald ratio method was used, whereas multiple SNP analyses primarily employed the inverse variance weighted (IVW) method to estimate causal effects. Sensitivity analyses, including MR-Egger regression, weighted median, simple mode, and weighted mode, were performed to evaluate the robustness of the causal estimates and detect potential horizontal pleiotropy. Additional diagnostic tests included the MR-Egger intercept test, with a P-value > 0.05, indicating no evidence of horizontal pleiotropy, and leave-one-out analysis to assess the influence of individual SNPs. Genetic heterogeneity was assessed using Cochran’s Q test, where a P-value > 0.05 was interpreted as no significant heterogeneity among instrumental variables, suggesting consistent causal estimates. To account for multiple hypothesis testing, we applied the Benjamini-Hochberg false discovery rate (FDR) correction. For each set of association analyses, raw P-values were adjusted using the p.adjust() function in R (method = “fdr”), controlling the expected proportion of false discoveries.21
Colocalization Analysis
To assess whether gene expression and COPD risk associations, identified through GWAS and eQTL datasets, shared the same causal variant, we performed colocalization analysis using the coloc package in R. Posterior probabilities (PP) were calculated for five mutually exclusive hypotheses: (1) H₀, no association; (2) H₁, association only with gene expression; (3) H₂, association only with COPD risk; (4) H₃, associations with both traits driven by distinct causal variants; and (5) H₄, associations with both traits driven by the same causal variant. Strong evidence supporting colocalization was defined as a posterior probability (PP) of H ≥ 0.75.22,23
Summary-Data-Based MR (SMR) Analysis and HEIDI Test
We further conducted SMR analysis coupled with the heterogeneity in dependent instruments (HEIDI) test to validate whether the associations identified between gene expression (eQTL) and COPD risk were likely driven by a single genetic variant, thereby minimizing genetic heterogeneity. A HEIDI test P-value > 0.05 indicated that associations were likely attributable to a single variant, supporting robust causal inference.24
Three-Step Mendelian Randomization Analysis
To systematically explore potential multi-omics causal pathways among genetic variation, DNA methylation, gene expression, and COPD susceptibility, we adopted a three-step MR approach: (1) DNA methylation → COPD risk and evaluated the causal effect of DNA methylation (mQTL) on COPD susceptibility. (2) Gene expression → COPD risk: We assessed the causal association between gene expression levels (eQTL) and COPD susceptibility. (3) DNA methylation → gene expression: We examined whether DNA methylation regulates COPD risk by modulating gene expression, thereby elucidating the possible epigenetic pathways.25
External Validation of Candidate Gene Expression Using Single-Cell RNA-Sequencing Data
To further validate our multi-omics Mendelian randomization findings, we performed an external expression analysis using the COPD Cell Atlas (http://www.copdcellatlas.com/), which contains single-cell RNA-sequencing profiles from lung tissues of COPD patients and healthy controls. We assessed the expression of SDK1 and other candidate genes across major lung cell types, comparing between disease and control groups.
Code and Data Availability
All custom scripts and code used in this study have been deposited in a public GitHub repository (https://github.com/YINJUNXUE/Analysis-code-for-COPD-Mendelian-randomization-study).
Results
MR Analysis of Gene Expression and COPD Risk
We performed two-sample MR analysis by integrating eQTL data with COPD GWAS summary statistics to investigate the potential causal effects of gene expression on COPD risk. A total of 1,781 genes showed suggestive evidence of association (P < 0.05), among which SDK1 exhibited a significant inverse association with COPD susceptibility (β = − 0.124, p = 0.002) (Figure 2). Sensitivity analyses revealed no evidence of genetic heterogeneity or horizontal pleiotropy (all P > 0.05) (Table S1).
 |
Figure 2 Mendelian randomization estimates of gene expression effects on COPD risk. Each dot represents a gene, with the x-axis showing the estimated causal effect (beta coefficient) of gene expression on chronic obstructive pulmonary disease (COPD) risk, and the y-axis representing the significance level as –log₁₀(P-value). Red dots indicate genes with significantly positive associations (upregulated), while blue dots represent significantly negative associations (downregulated). Gray dots denote genes with no statistically significant association. |
Colocalization Analysis of Gene Expression and COPD Risk
To determine whether the observed associations between gene expression and COPD susceptibility were attributable to shared causal variants, we performed colocalization analysis. Colocalization analysis identified 29 genes with strong evidence of a shared causal variant for both gene expression and COPD risk (posterior probability for hypothesis H₄ ≥ 0.75) Notably, SDK1 exhibited a posterior probability of 0.755, indicating a likely shared genetic basis for its expression and susceptibility to COPD (Figure 3 and Table S2).
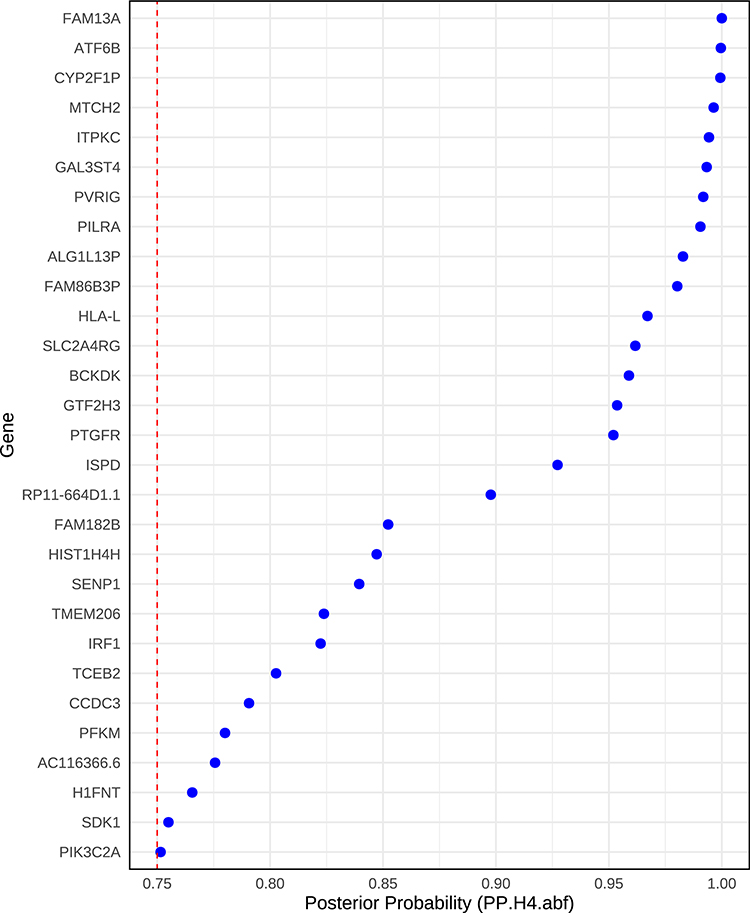 |
Figure 3 Colocalization of gene expression and COPD-associated loci. Each point represents a gene, with the x-axis indicating the posterior probability (PP.H4.abf) that the same genetic variant is responsible for both gene expression and COPD association signals. The y-axis lists candidate genes prioritized by Mendelian randomization analysis. The vertical red dashed line marks the significance threshold (PP.H4.abf = 0.75) for strong colocalization evidence. |
Summary-Data-Based MR (SMR) Analysis and HEIDI Test
To further assess whether the observed associations were attributable to a single causal variant, we performed SMR analysis in conjunction with the HEIDI test. Eight genes demonstrated suggestive evidence of a potential causal association with COPD risk (P < 0.05) and passed the HEIDI test P-value > 0.05 (Figure 4 and Table S3). Among them, SDK1 showed a significant negative association with COPD (β = − 0.385, p = 0.001) with no evidence of heterogeneity (HEIDI P > 0.05), further supporting its potential causal role (Figure 4 and Table S3).
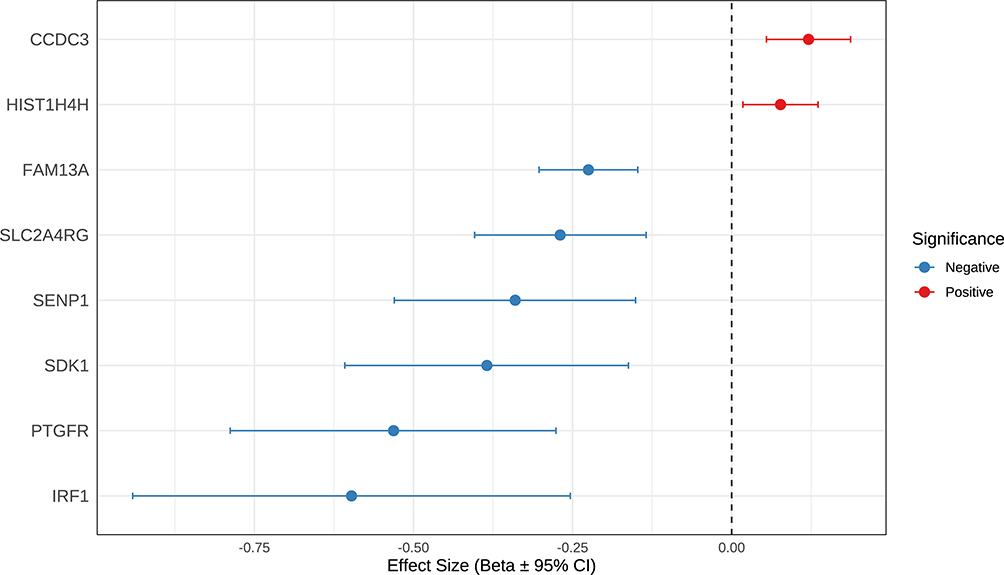 |
Figure 4 SMR-based associations between gene expression and COPD risk. This forest plot presents the summary data-based Mendelian randomization (SMR) results for candidate genes, showing the estimated effect sizes (Beta) and their 95% confidence intervals (CIs) for the association between gene expression and COPD risk. Blue dots indicate genes where higher expression is associated with reduced COPD risk (negative effect), while red dots represent genes where higher expression is linked to increased COPD risk (positive effect). Genes included in the plot passed both SMR and HEIDI filtering criteria, indicating that the associations are likely driven by single, shared genetic variants rather than confounded by linkage disequilibrium. |
Mendelian Randomization Analysis of DNA Methylation and COPD Risk
To investigate whether DNA methylation at loci corresponding to the candidate genes identified in the previous SMR analysis may contribute to COPD risk, we extracted CpG sites annotated to these eight genes based on the UCSC_RefGene_Name field from the Illumina 450k annotation file and performed Mendelian randomization (MR) analysis using mQTL and GWAS summary data. A total of 43 CpG sites were significantly associated with COPD risk (SMR, P < 0.05; HEIDI, P > 0.05; Table S4), including 15 sites associated with increased susceptibility and 28 sites with reduced susceptibility. Notably, cg07526904, an intronic CpG site within the SDK1 locus, showed a significant positive association with COPD (β = 0.036, p = 0.038), suggesting a potential epigenetic mechanism underlying its effect (Table S4).
Three-Step Mendelian Randomization Analysis
Mediation analysis revealed that DNA methylation at cg07526904 significantly downregulated SDK1 expression (β = –0.148, p = 0.002), whereas higher SDK1 expression was associated with a reduced risk of COPD (β = –0.124, p = 0.002). In addition, cg07526904 methylation was directly and positively associated with COPD susceptibility (Î ² = 0.036, p = 0.038). The indirect effect mediated by SDK1 expression was estimated at β = 0.018 (p = 0.038), accounting for 51.9% of the total effect. These results suggest that hypermethylation of cg07526904 may contribute to increased COPD risk by suppressing SDK1 expression (Figure 5).
 |
Figure 5 Hypothetical model of SDK1 regulation via DNA methylation. Schematic representation of the causal pathway linking DNA methylation at cg07526904, SDK1 gene expression, and COPD risk. Statistical effects and mediation proportion are shown along the arrows. β: effect size; p: p-value. |
External Validation of Candidate Gene Expression Using Single-Cell RNA-Sequencing Data
To further assess the cellular expression context of the identified candidate genes, we analyzed their single-cell transcriptional profiles in lung tissue using data from the COPD Cell Atlas. Among the candidate genes, only SDK1 demonstrated appreciable expression across multiple epithelial cell subtypes, particularly in PNEC, goblet, and ciliated cells. In contrast, CCDC3 and IRF1 showed low to negligible expression in these populations. Notably, there were no significant differences in SDK1 expression between COPD and control samples within each epithelial cell type. The expression profiles of CCDC3, IRF1, and the other candidate genes showed little to no difference between COPD and control groups (Figure 6).
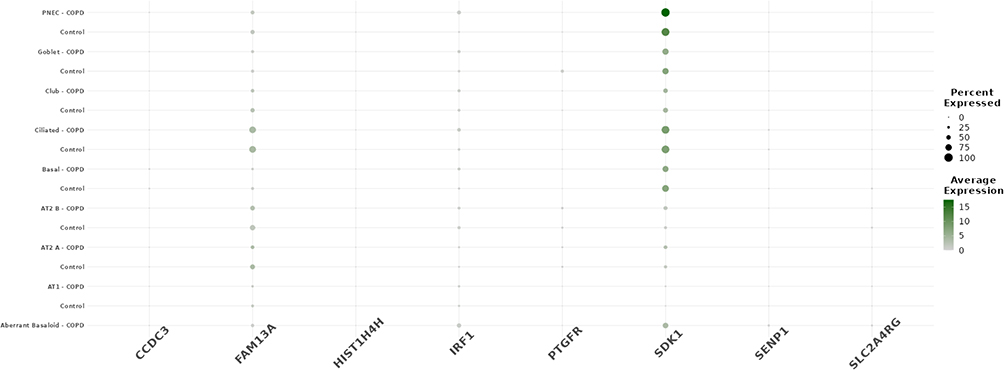 |
Figure 6 Single-cell RNA-seq expression landscape of candidate genes in lung epithelial cell subtypes from COPD and control samples. Dot plot showing single-cell RNA-seq expression of eight candidate genes (CCDC3, FAM13A, HIST1H4H, IRF1, PTGFR, SDK1, SENP1, and SLC24ARG) across major airway epithelial cell subtypes using data from the COPD Cell Atlas. Each dot represents the percentage of cells expressing the gene (dot size) and the average expression level. |
Discussion
Our study pioneered the application of a multi-omics Mendelian randomization (MOMR) framework to deconvolute the causal triad of DNA methylation, gene expression, and COPD susceptibility. Through systematic integration of GWAS and blood eQTL data, we identified SDK1 as the sole gene surviving rigorous colocalization, SMR-HEIDI filtering, and methylation-mediated pathway validation. Notably, intronic methylation at cg07526904 exhibited bidirectional causality, positively influencing COPD risk (β=0.036, P=0.038) while suppressing SDK1 expression (β=−0.148, P=0.002), and SDK1-mediated effects accounted for 51.9% of the methylation-disease association (β_indirect=0.018, P=0.038). To our knowledge, this is the first study to systematically explore the potential epigenetic regulation of SDK1 in COPD using a Mendelian randomization framework, providing preliminary evidence of an epigenetic mechanism linking DNA methylation, gene expression, and COPD susceptibility.
SDK1 is a transmembrane protein belonging to the immunoglobulin superfamily and is characterized by six immunoglobulin-like domains and 13 fibronectin type III domains.26 These structural features facilitate homophilic interactions, which are critical for maintaining cell adhesion and tissue integrity.27 Tan et al demonstrated that oxidative stress and inflammatory cytokines can impair airway epithelial integrity.28 Although SDK1 was not specifically mentioned in their study, its well-established role in mediating homophilic cell adhesion suggests that it may be functionally relevant in such pathological processes. Previous studies have reported that SDK1 expression is significantly downregulated in the small airway epithelial cells of COPD patients compared to healthy individuals.29 This downregulation has been hypothesized to compromise epithelial adhesion, increase airway permeability, and promote chronic inflammation.30 However, the upstream regulatory mechanisms responsible for SDK1 suppression in COPD remain poorly understood and few studies have systematically explored its genetic or epigenetic regulation.
Emerging evidence suggests that SDK1 expression is influenced by DNA methylation.31,32 An epigenome-wide study in individuals with HIV-associated COPD has implicated SDK1 methylation as a potential factor influencing lung function trajectories. However, specific CpG sites directly linked to COPD risk remain poorly characterized.33 Additionally, although GWAS have implicated the SDK1 locus in immune-related diseases, including atopic dermatitis, its specific epigenetic regulation in COPD remains unclear.34 Given the critical role of SDK1 in maintaining epithelial integrity and its potential involvement in COPD pathogenesis, elucidating the regulatory effects of specific methylation sites on SDK1 expression is essential to understand COPD susceptibility and progression.
Our results demonstrated that SDK1 expression was inversely associated with COPD risk, suggesting a potentially protective role. Notably, cg07526904 is an intronic mQTL located within the SDK1. This CpG site is significantly associated with decreased SDK1 expression and increased susceptibility to COPD. Although prior studies have not established a direct mQTL-eQTL-COPD linkage for SDK1, our findings highlight epigenetic regulation at this CpG site as a plausible intermediary connecting SDK1 expression to COPD pathogenesis.
Compared to previous studies based primarily on preclinical or observational designs,35–37 our study is, to our knowledge, the first to apply Mendelian randomization at the population level to explore SDK1 methylation in relation to COPD. This provides a robust analytical framework for inferring causality. Although the clinical utility of cg07526904 methylation requires further validation, our findings suggest that it may serve as a potential biomarker for early diagnosis or risk stratification, and offer a promising starting point for exploring SDK1-related precision medicine strategies.
Importantly, while SDK1 stood out as a significant candidate in our analysis, none of the other seven initially prioritized genes (SENP1, SLC2A4RG, PTGFR, HIST1H4H, IRF1, FAM13A, and CCDC3) demonstrated a similarly robust methylation-mediated causal pathway in the subsequent analyses. Specifically, SENP1 and PTGFR lacked annotated CpG sites, precluding further methylation analysis. Although FAM13A, HIST1H4H, and SLC2A4RG had annotated CpG sites, none showed significant associations with COPD risk in methylation-based MR analyses, indicating that methylation-driven regulation of these genes may not be detected in the current reference dataset. While CCDC3 and IRF1 initially exhibited several significant methylation–COPD associations in preliminary MR screening, neither demonstrated significant mediation via gene expression in the comprehensive three-step MR framework, suggesting that their methylation signals may not translate into meaningful transcriptional dysregulation relevant to COPD.
Several factors may account for these negative findings. First, tissue and cell-type specificity may limit the detection of regulatory effects when using mQTL and eQTL data derived from peripheral blood.38 Additionally, single-cell RNA sequencing data from the COPD Cell Atlas indicate that, with the exception of SDK1, these genes display minimal or negligible expression in key airway epithelial cell populations. This cell-type–specific expression profile is consistent with our MR findings: if a gene is scarcely expressed in the critical cell types of the lung, it is unlikely to influence COPD pathogenesis through gene expression or epigenetic modifications.38 Previous studies further support this inference, showing that the functional effects of many COPD susceptibility genes are highly tissue- and cell-type specific and become apparent only in disease-relevant cellular contexts.39
Furthermore, the general absence of significant expression changes in lung tissue for these candidate genes suggests that their direct pathogenic roles in COPD may be limited. For example, although FAM13A is one of the earliest identified COPD risk genes and is broadly expressed in human lung epithelial cells and macrophages,29,40 its mRNA levels do not differ significantly between patients with moderate COPD and controls. Only in very severe cases of COPD is increased FAM13A protein observed in lung tissue, and FAM13A-deficient mice show marked resistance to cigarette smoke–induced emphysema.41 This suggests that the functional effects of certain susceptibility genes, such as FAM13A, may require specific disease environments or a high threshold of severity to manifest, and may not be detectable at baseline expression levels. For the other genes identified in our screen, it is likely that they do not play a major role in COPD pathogenesis, and that the initial association signals may have resulted from non–lung-specific regulatory effects or limited statistical power.
It should also be noted that our multi-omics analyses relied primarily on eQTL and mQTL reference datasets from peripheral blood or bulk tissue, which may not fully capture the regulatory variation present in lung-specific cell types. For certain genes with inherently low expression, the strength of available genetic instruments may also be limited, further reducing the power of causal inference. Even after rigorous multiple testing correction, some true but subtle effects may have been missed.42
This study has several limitations that should be considered when interpreting the findings. First, the GWAS datasets analyzed were predominantly derived from individuals of European and East Asian ancestry, which may limit the generalizability of our findings to other ethnic populations. Although we performed extensive sensitivity analyses to account for genetic heterogeneity and horizontal pleiotropy, the possibility of residual confounding and undetected bias cannot be completely ruled out. Importantly, the causal inferences in this study were entirely based on statistical evidence; thus, functional validation in cellular or animal models is essential to substantiate our findings.
Conclusion
This study employed a multi-omics Mendelian randomization framework to identify a potential epigenetic pathway involving SDK1 in COPD pathogenesis. Specifically, methylation of cg07526904 may contribute to increased COPD risk by downregulating SDK1 expression. These findings advance our understanding of the molecular mechanisms underlying COPD and may support the development of biomarker-based strategies for early detection and personalized interventions.
Data Sharing Statement
All analyses in this study were based on data obtained from publicly available databases and from enrolled patients. The complete set of custom scripts used for data cleaning, statistical analysis, and visualization has been deposited in a public GitHub repository (https://github.com/YINJUNXUE/Analysis-code-for-COPD-Mendelian-randomization-study). The raw datasets analyzed during the current study are available from the corresponding author upon reasonable request, in accordance with relevant ethical and privacy regulations.
Ethics Approval and Consent to Participate
The study protocol and all procedures were reviewed and approved by the Ethics Committee of Ningde Municipal Hospital of Ningde Normal University (Approval Number: NSYKLL-2025-115).
Acknowledgments
The authors thank all those who participated in this study.
Funding
This work was supported by the Research Projects of Ningde Normal University (2023ZX708 and 2023ZX218), the Fujian Provincial Key Specialty Construction Project, the Natural Science Foundation of Fujian Province (2024J01998), the Joint Funds for the Innovation of Science and Technology of Fujian Province (2024Y9013), and the Fujian Provincial Health Technology Project (2021QNA004). Additional funding was provided by the Fujian Provincial Key Clinical Specialty Construction Project in 2022 (Fujian Medical Policy Letter [2022] No. 884).
Disclosure
The authors declare that they have no known competing financial interests or personal relationships that could have influenced the work reported in this study.
References
1. Mei F, Dalmartello M, Bonifazi M, et al. Chronic obstructive pulmonary disease (COPD) mortality trends worldwide: an update to 2019. Respirology. 2022;27(11):941–950. doi:10.1111/resp.14328
2. Yin P, Wu J, Wang L, et al. The burden of COPD in China and its provinces: findings from the Global Burden of Disease Study 2019. Front Public Health. 2022;10:859499. doi:10.3389/fpubh.2022.859499
3. Erhabor GE, Adeniyi B, Arawomo AO, et al. Acute Exacerbation of COPD: clinical Perspectives and Literature Review. West Afr J Med. 2021;38(11):1129–1142. doi:10.55891/wajm.v38i11.25. PMID: 34922414.Raherison C, Gi
4. Huo X, Jin S, Wang Y, Ma L. DNA methylation in chronic obstructive pulmonary disease. Epigenomics. 2021;13(14):1145–1155. doi:10.2217/epi-2021-0111
5. Schamberger AC, Mise N, Meiners S, Eickelberg O. Epigenetic mechanisms in COPD: implications for pathogenesis and drug discovery. Expert Opin Drug Discov. 2014;9(6):609–628. doi:10.1517/17460441.2014.913020
6. Ragusa R, Bufano P, Tognetti A, Laurino M, Caselli C. Recent evidences of epigenetic alterations in chronic obstructive pulmonary disease (COPD): a systematic review. Int J Mol Sci. 2025;26(6):62571. doi:10.3390/ijms26062571
7. Silverman EK. Genetics of COPD. Annu Rev Physiol. 2020;82(1):413–431. doi:10.1146/annurev-physiol-021317-121224. PMID: 31730394; PMCID: PMC7193187.
8. Lin WD, Liao WL, Chen WC, Liu TY, Chen YC, Tsai FJ. Genome-wide association study identifies novel susceptible loci and evaluation of polygenic risk score for chronic obstructive pulmonary disease in a Taiwanese population. BMC Genomics. 2024;25(1):607. doi:10.1186/s12864-024-10526-5
9. Werder RB, Zhou X, Cho MH, Wilson AA. Breathing new life into the study of COPD with genes identified from genome-wide association studies. Eur Respir Rev. 2024;33(172):230019. doi:10.1183/16000617.0019-2024
10. Rotondo DG, Lanzolla G, Comi S, et al. Insights into the role of DNA methylation and gene expression in Graves orbitopathy. J Clin Endocrinol Metab. 2023;108(5):e160–e168. doi:10.1210/clinem/dgac645
11. Yoo S, Takikawa S, Geraghty P, et al. Integrative analysis of DNA methylation and gene expression data identifies EPAS1 as a key regulator of COPD. PLoS Genet. 2015;11(1):e1004898. doi:10.1371/journal.pgen.1004898
12. Carter AR, Sanderson E, Hammerton G, et al. Mendelian randomisation for mediation analysis: current methods and challenges for implementation. Eur J Epidemiol. 2021;36(5):465–478. doi:10.1007/s10654-021-00757-1
13. Larsson SC, Butterworth AS, Burgess S. Mendelian randomization for cardiovascular diseases: principles and applications. Eur Heart J. 2023;44(47):4913–4924. doi:10.1093/eurheartj/ehad736
14. Wang Z, Li S, Cai G, et al. Mendelian randomization analysis identifies druggable genes and drugs repurposing for chronic obstructive pulmonary disease. Front Cell Infect Microbiol. 2024;14:1386506. doi:10.3389/fcimb.2024.1386506
15. Liu Y, Li B, Ma Y, Huang Y, Ouyang F, Liu Q. Mendelian randomization integrating GWAS, eQTL, and mQTL data identified genes pleiotropically associated with atrial fibrillation. Front Cardiovasc Med. 2021;8:745757. doi:10.3389/fcvm.2021.745757
16. Grau-Perez M, Agha G, Pang Y, Bermudez JD, Tellez-Plaza M. Mendelian randomization and the environmental epigenetics of health: a systematic review. Curr Environ Health Rep. 2019;6(1):38–51. doi:10.1007/s40572-019-0226-3
17. Sakaue S, Kanai M, Tanigawa Y, et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat Genet. 2021;53(10):1415–1424. doi:10.1038/s41588-021-00931-x
18. Võsa U, Claringbould A, Westra HJ, et al. Unraveling the polygenic architecture of complex traits using blood eQTL meta-analysis. bioRxiv. 2018:447367. doi:10.1101/447367.
19. Min JL, Hemani G, Hannon E, et al. Genomic and phenotypic insights from an atlas of genetic effects on DNA methylation. Nat Genet. 2021;53(9):1311–1321. doi:10.1038/s41588-021-00923-0
20. Yan K, Li C, Chen B, Tao Y, Zhang D, Zhang P. Identification of potential pathogenic genes for urolithiasis through multi-omics Mendelian randomization analysis. Urolithiasis. 2024;53(1):6. doi:10.1007/s00240-024-01675-z
21. Zhang Z, Fang T, Chen L, Ji F, Chen J. Multi-omics Mendelian randomization integrating GWAS, eQTL, and mQTL data identified genes associated with breast cancer. Am J Cancer Res. 2024;14(3):1433–1445. doi:10.62347/BCZW1355
22. Sheng D, Wang S, Xiao Z, Liu W, Xiao B, Zhou L. Unraveling the association between retinal thickness and Alzheimer’s disease, and circulating total-tau levels: insights from genetic evidence. Research Square Preprint. 2025;1:1
23. Teng M, Wang J, Su X, et al. Associations between immune cells signatures and osteoarthritis: an integrated analysis of bidirectional Mendelian randomization and Bayesian colocalization. Cytokine. 2024;179:156633. doi:10.1016/j.cyto.2024.156633
24. Jacobs BM, Taylor T, Awad A, et al. Summary-data-based Mendelian randomization prioritizes potential druggable targets for multiple sclerosis. Brain Commun. 2020;2(2):fcaa119. doi:10.1093/braincomms/fcaa119
25. Cao Z, Wu T, Fang Y, et al. Dissecting causal relationships between immune cells, plasma metabolites, and COPD: a mediating Mendelian randomization study. Front Immunol. 2024;15:1406234. doi:10.3389/fimmu.2024.1406234
26. Raredon MSB, Adams TS, Suhail Y, et al. Single-cell connectomic analysis of adult mammalian lungs. Sci Adv. 2019;5(12):eaaw3851. doi:10.1126/sciadv.aaw3851
27. Tang H, Chang H, Dong Y, et al. Architecture of cell-cell adhesion mediated by sidekicks. Proc Natl Acad Sci U S A. 2018;115(37):9246–9251. doi:10.1073/pnas.1801810115
28. Tan CL, Chan Y, Candasamy M, et al. Unravelling the molecular mechanisms underlying chronic respiratory diseases for the development of novel therapeutics via in vitro experimental models. Eur J Pharmacol. 2022;919:174821. doi:10.1016/j.ejphar.2022.174821
29. Chang WA, Tsai MJ, Jian SF, Sheu CC, Kuo PL. Systematic analysis of transcriptomic profiles of COPD airway epithelium using next-generation sequencing and bioinformatics. Int J Chron Obstruct Pulmon Dis. 2018;13:2387–2398. doi:10.2147/COPD.S173206
30. Carlier FM, de Fays C, Pilette C. Epithelial barrier dysfunction in chronic respiratory diseases. Front Physiol. 2021;12:691227. doi:10.3389/fphys.2021.691227
31. Nada S, Kahaleh B, Altorok N. Genome-wide DNA methylation pattern in systemic sclerosis microvascular endothelial cells: identification of epigenetically affected key genes and pathways. J Scleroderma Relat Disord. 2022;7(1):71–81. doi:10.1177/23971983211033772
32. Bueno AC, Da SR, Stecchini MF, et al. DNA methylation is a comprehensive marker for pediatric adrenocortical tumors. Endocr Relat Cancer. 2022;29(11):599–613. doi:10.1530/ERC-22-0145
33. Hernández CA, Yang CX, Yang J, et al. Airway aging and methylation disruptions in HIV-associated chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2022;206(2):150–160. doi:10.1164/rccm.202106-1440OC
34. Johansson Å, Rask-Andersen M, Karlsson T, Ek WE. Genome-wide association analysis of 350,000 Caucasians from the UK Biobank identifies novel loci for asthma, hay fever and eczema. Hum Mol Genet. 2019;28(23):4022–4041. doi:10.1093/hmg/ddz175
35. De Smet EG, Van Eeckhoutte HP, Avila Cobos F, et al. The role of miR-155 in cigarette smoke-induced pulmonary inflammation and COPD. Mucosal Immunol. 2020;13(3):423–436. doi:10.1038/s41385-019-0235-0
36. Qiu W, Baccarelli A, Carey VJ, et al. Variable DNA methylation is associated with chronic obstructive pulmonary disease and lung function. Am J Respir Crit Care Med. 2012;185(4):373–381. doi:10.1164/rccm.201108-1382OC
37. Faner R, Agusti A, Network C. Multilevel omics in COPD: integrating gene expression, methylation, and clinical data. Chest. 2016;149(1):32–43. doi:10.1378/chest.15-0974
38. Battle A, Brown CD, Engelhardt BE, et al. Genetic effects on gene expression across human tissues. Nature. 2017;550(7675):204–213. doi:10.1038/nature24277
39. Lamontagne M, Bérubé JC, Obeidat M, et al. Leveraging lung tissue transcriptome to uncover candidate causal genes in COPD genetic associations. Hum Mol Genet. 2018;27(10):1819–1829. doi:10.1093/hmg/ddy091
40. Cho MH, Boutaoui N, Klanderman BJ, et al. Variants in FAM13A are associated with chronic obstructive pulmonary disease. Nat Genet. 2010;42(3):200–202. doi:10.1038/ng.535
41. Zhu J, Wang F, Feng X, Li B, Ma L, Zhang J. Family with sequence similarity 13 member A mediates TGF-β1-induced EMT in small airway epithelium of patients with chronic obstructive pulmonary disease. Respir Res. 2021;22(1):192. doi:10.1186/s12931-021-01783-z
42. Zhu Z, Zhang F, Hu H, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet. 2016;48(5):481–487. doi:10.1038/ng.3538
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.