Back to Journals » Cancer Management and Research » Volume 11
DNA methylation-mediated silencing of miR-204 is a potential prognostic marker for papillary thyroid carcinoma
Authors Xia F, Wang W ![]() , Jiang B, Chen Y, Li X
, Jiang B, Chen Y, Li X ![]()
Received 20 August 2018
Accepted for publication 8 January 2019
Published 8 February 2019 Volume 2019:11 Pages 1249—1262
DOI https://doi.org/10.2147/CMAR.S184566
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Chien-Feng Li
Fada Xia, Wenlong Wang, Bo Jiang, Yong Chen, Xinying Li
Department of General Surgery, Xiangya Hospital, Central South University, Changsha 410008, People’s Republic of China
Background: Papillary thyroid carcinoma (PTC) is the most common endocrine malignancy and its incidence has increased over the last few decades. The molecular mechanisms underlying PTC tumorigenesis and progression are still unclear.
Patients and methods: The microRNA (miRNA) expression patterns of PTC were revealed by miRNA microarray analysis and validated with The Cancer Genome Atlas (TCGA) data. Promoter DNA methylation rates of miR-204 were analyzed by Agena Methylation MassARRAY analysis and validated with TCGA data. The underlying molecular mechanisms of miR-204 involved in PTC were studied by bioinformatics analyses.
Results: A total of 181 differentially expressed miRNAs (89 downregulated and 92 upregulated miRNAs) between PTC and normal tissues were detected in this study. We identified miR-204 as one of the most significantly downregulated miRNAs in PTC. Downregulation of miR-204 was related to PTC extrathyroidal extension, high T-stage, lymph metastasis, BRAF V600E mutation, and aggressive tall cell variant. The Agena MassARRAY results indicated that 12 CpG sites located at the promoter of miR-204 were hypermethylated in PTC tissues compared to normal tissues. The promoter methylation rates of miR-204 in PTC were negatively correlated with the expression levels of miR-204 and its host gene TRPM3. Downregulated miR-204 expression was related to several important pathways and mechanisms involved in tumorigenesis and progression.
Conclusion: Promoter DNA methylation-silenced miR-204 could serve as a potential diagnostic biomarker of PTC. Downregulation of miR-204 may play an important role in PTC via its involvement in many tumor-related pathways. Novel target genes and putative mechanisms of miR-204 in PTC need to be further validated.
Keywords: papillary thyroid carcinoma, miRNA, promoter DNA methylation, bioinformatics
Introduction
Papillary thyroid carcinoma (PTC), a differentiated thyroid cancer, is the most common endocrine malignancy, and its incidence has increased over the last few decades. The prognosis of PTC is excellent. If treatment based on the current guidelines is given, cases of recurrence or persistence are rare. Surgery and radioiodine therapy followed by levothyroxine substitution remain the established therapeutic treatment.1 However, in patients with metastatic and/or radioiodine refractory tumors, additional treatment options are urgently needed. Over the last two decades, our understanding of the genetic mechanisms of the thyroid cancer has dramatically expanded. BRAF mutation is the first single gene mutational marker used for the diagnosis of PTC. Currently, several more advanced molecular tests are available for clinical use. These tests are based on multiple molecular markers and have an increasing impact on the clinical management of patients with thyroid nodules.2 The molecular mechanisms underlying PTC tumorigenesis and progression are still unclear. Therefore, a better understanding of these mechanisms will facilitate the development of novel diagnostic and even therapeutic targets for PTC.
MicroRNAs (miRNAs) are noncoding RNAs that range in size from 19 to 25 nucleotides. They are important molecules with a vital role in several cellular functions, such as cell differentiation, cell cycle regulation, apoptosis, and angiogenesis in human diseases.3,4 Typically, miRNAs basepair to target the 3′ UTR of the mRNAs within the RNA-induced silencing complex and block translation and stimulate mRNA transcript degradation.5 Numerous studies have reaffirmed the existence of mature miRNAs in the nucleus, and the nucleus-cytoplasm transport mechanism has also been illustrated. Moreover, active regulatory functions of nuclear miRNAs, including posttranscriptional gene silencing, transcriptional gene silencing, and transcriptional gene activation (TGA), in which miRNAs bind nascent RNA transcripts, gene promoter regions, or enhancer regions and exert further effects via epigenetic pathways, have been identified.6 MiRNAs have emerged as important regulators of human carcinogenesis by affecting the expression of key oncogenes and tumor suppressor genes.7 A new strategy of miRNA therapy has been developed in the last 5 years. There are two miRNA-based treatment approaches for cancer: miRNA reduction or inhibition therapy and miRNA replacement or restoration therapy.3 Accumulating evidence has revealed that miRNAs can serve as diagnostic and therapeutic targets for human cancers.
Research on miRNA expression in thyroid tumors has provided new insights into the development of novel biomarkers that can be used to diagnose thyroid cancer and optimize its management.8 In the present study, we analyzed the expression patterns of miRNAs in five pairs of PTC vs normal tissues and compared our findings with the results derived from data deposited in The Cancer Genome Atlas (TCGA). We identified miR-204 as one of the most significantly downregulated miRNAs in PTC. The relationship between miR-204 and the clinicopathologic characteristics of PTC and the putative mechanism underlying the downregulation of miR-204 were explored. Promoter DNA methylation rates of miR-204 were analyzed by Agena Methylation MassARRAY analysis and validated with TCGA data. Furthermore, we studied the underlying molecular mechanism of miR-204 involved in PTC by bioinformatics analyses. This study revealed that methylation-silenced miR-204 acts as an important biomarker in PTC and miR-204 may be involved in various pathway abnormalities in the tumorigenesis and progression of PTC.
Patients and methods
Patient samples
Fifteen pairs of human PTC and matched adjacent normal tissues were collected from the Department of General Surgery of Xiangya Hospital of Central South University, People’s Republic of China. All tissues were immediately snap-frozen in liquid nitrogen. The cases selected were patients who had not received previous chemotherapy or radiotherapy before surgery and were based on clear pathological diagnosis. Among the 15 pairs of tissues used in our study, ten pairs of tissues were used for methylation array and quantitative real-time PCR (qRT-PCR) analyses, whereas others were used for miRNA expression analysis. This study was approved by the Institutional Ethics Committee of Xiangya Hospital. According to the Declaration of Helsinki, written informed consent permitting the use of their samples and publication of relevant data for the purpose of the present study was obtained from all patients.
RNA extraction and miRNA microarrays
Total RNA was extracted with TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA) and purified using a mirVana™ miRNA Isolation Kit (AM1561). RNA dephosphorylation reactions were performed at 37°C for 30 minutes using a calf intestinal phosphatase master mix. Dephosphorylated RNA was labeled using the T4 RNA ligase labeling method with 4.5 µL ligation master mix (including 1.0 µL 10× T4 RNA ligase buffer, 3.0 µL Cyanine 3-pCP, and 0.5 µL T4 RNA ligase). The labeling reaction was performed at 16°C for 2 hours. The hybridization mixture was composed of 17.0 µL labeled miRNA sample, 1.0 µL Hyb Spike-In, 4.5 µL 10× gene expression (GE) (Agilent, Santa Clara, CA, USA) blocking agent, and 22.5 µL 2× Hi-RPM hybridization buffer. Hybridization was performed on a microarray chip (Agilent-070156 human miRNA V21.0 microarray), which contained the latest 2,549 miRNA probes spotted in triplicates. Hybridization reactions were performed at 20 rpm for 16 hours in an Agilent hybridization oven. The hybridized five pairs of chips were scanned with an Agilent G2565CA laser confocal scanner and visualized with Agilent feature extraction (v 10.7) software. Deregulated miRNAs were characterized by a fold change (FC) >2 and P-value <0.05. The microarray data have been deposited in the Gene Expression Omnibus (GEO) database (accession code GSE113629).
Cell culture and qRT-PCR
Human PTC cell lines BCPAP and TPC-1 and the normal cell line Nthy-ori 3-1 were used in this study. Cells were cultured in RPMI 1640 supplemented with FBS (10%). Isolated total RNA from tissues and cells was reversed transcribed using the miRNA Reverse Transcription Kit (Thermo Fisher Scientific, Inc) according to the manufacturer’s protocol. cDNA was mixed with TaqMan MicroRNA Assay Kit and amplified using Applied Biosystems Instruments (ABI) 7900HT system. The primer sequences9 were as follows: U6 forward, 5′-GCTTCGGCAGCACATATACTAAAAT-3′, reverse 5′-CGCTTCACGAATTTGCGTGTCAT-3′; miR-204 forward, 5′-CTGTCACTCGAGCTGCTGGAATG-3′, and reverse, 5′-ACCGTGTCGTGGAGTCGGCAATT-3′.
Identification of the miR-204 promoter and validation of promoter DNA methylation with TCGA data
As intronic miRNAs are transcribed as a result of their host genes, the putative promoter region of miR-204 was predicted from the genomic sequence of the miR-204 host gene TRPM3 for the 2,000 upstream and 1,000 downstream sequences around its first exon based on the validated National Center for Biotechnology Information (NCBI) nucleotide sequences (Figure S1). The promoter CpG islands were predicted using designing primers for methylation PCRs at Chinese Academy of Medical Sciences (MethPrimer, http://www.urogene.org/cgi-bin/methprimer/methprimer.cgi). DNA methylation data of TCGA samples were downloaded and processed with the database of DNA methylation and gene expression in human cancer (MethHC, http://MethHC.mbc.nctu.edu.tw). The methylation levels of the identified CpG sites in tumor and normal tissues were analyzed on the basis of the gene chip data (methylation 450K) downloaded from the University of California Santa Cruz Xena Cancer Browser (UCSC) (http://xena.ucsc.edu/).
DNA extraction and methylation analyses
Genomic DNA was extracted from fresh tissues using a QIAamp DNA Mini Kit (Qiagen NV, Venlo, the Netherlands) and modified by sulfite with an EZ DNA Methylation Kit (ZYMO, Irvine, CA, USA) according to the manufacturer’s protocols. Modified DNA was used for PCR in a total reaction volume of 10 µL as follows: ten cycles of melting (45 seconds at 94°C), annealing (48 seconds at 62°C), and extension (60 seconds at 72°C) and 35 cycles of melting (45 seconds at 94°C), annealing (48 seconds at 57°C), and extension (60 seconds at 72°C). PCR primers were designed using the online tools of Agena Bioscience, Inc. (San Diego, CA, USA) (https://agenacx.com/online-tools/) to amplify miR-204 promoter regions with CpG islands. Two promoter regions (regions 9 and 15 that contain 50 and 18 CpG sites, respectively) were selected for the next amplification. The following primers were used: region 9, 5′-GTGGGTTTTGTATTTTTTAGAGAAG-3′ and 3′-AACCCCTACTTAAAACTTAACCTTTTCC-5′ and region 15, 5′-GTTTTTTTAAGGGTGAGAGTTAGGG-3′ and 3′-CAAACACCTAAAATATTCTTACTATTCTC-5′. PCR products (2 µL) modified by alkaline phosphatase were used as a template for in vitro transcription. RNase A cleavage (MassCleave Kit; Agena Bioscience, Inc.) was used for the reverse. The purified samples were spotted on a 384-pad SPectroCHIP (Agena Bioscience, Inc.) using a MassARRAY Nanodispenser RS 1000 (Agena Bioscience, Inc.). The spotted chips were placed in the MassARRAY Compact System (Agena Bioscience, Inc.) for detection, followed by spectral acquisition on a MALDI-TOF analyzer (Agena Bioscience, Inc.). The test data were analyzed and quantified by EpiTYPER software (Agena Bioscience, Inc.). To validate the promoter methylation regulation of miR-204, PTC cell lines were treated with the DNA methylation inhibitor 5-aza-2′-deoxycytidine (5-Aza). Cells were seeded in six-well plates, allowed to attach for 96 hours, and treat with different dose of 5-Aza. miR-204 expression levels were then detected by qRT-PCR.
TCGA RNA sequencing and clinical data collection
The TCGA thyroid carcinoma dataset (TCGA-THCA) consisted of 507 cases of PTC and 59 normal tissue samples. Merged level 3 mRNA and miRNA sequencing data and clinical information were downloaded from cBioPortal for Cancer Genomics (http://www.cbioportal.org/index.do) and Firebrowse (http://firebrowse.org). PTC cases and normal samples with both complete mRNA and miRNA sequencing and clinical data were identified. The RNAseq by expectation-maximization and reads per kilobase per million (RPKM) miRNA mapped values were used to quantify mRNA and miRNA expression levels, respectively.
Screening differentially expressed miRNAs (DEMis) and genes
The Limma package (version 3.32.5) of R (version 1.1.143) was used to identify DEMis between PTC and normal tissues from TCGA. According to the median expression values of miR-204, patients from TCGA were divided into the miR-204 high expression group and low expression group. The Limma package of R was used to identify differentially expressed genes (DEGs) between these two groups. Hierarchical analyses of DEMis identified from the miRNA microarray and TCGA data were performed by Cluster 3.0 and then visualized via Java TreeView 1.16r4 (http://www.treeview.net).
Gene functional eanalysis
Bioinformatics methods were utilized to identify the pathways that are significantly associated with the downregulation of miR-204. We performed functional annotation clustering using the Database for Annotation, Visualization and Integrated Discovery ([DAVID], https://david.ncifcrf.gov/) online system. Application of ggplot2 (version 2.2.1) in R studio was used to visualize the gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses. To further investigate the associations between downregulation of miR-204 and cancer-related pathways, we conducted gene set enrichment analysis (GSEA) using GSEA JAVA version 2.1 (http://www.broadinstitute.org/gsea). GSEA calculates a gene set enrichment score that is estimated from predefined gene sets. The expression level of miR-204 was served as a phenotype, and KEGG pathway enrichment maps were used for visualization of the GSEA results.
Statistical analysis
All statistical analyses were performed using the SPSS 21.0 package and R studio. Application of Limma and ggplot2 in R studio was used as described above. Continuous variables are presented as the mean ± SD and were analyzed using the Student’s t-test or one-way ANOVA. Survival analysis was performed using Kaplan–Meier and log-rank tests. A P-value of <0.05 was considered statistically significant.
Results
Downregulation of miR-204 in PTC is associated with aggressive clinicopathologic characteristics
Five pairs of PTC tumor and para-tumor tissues were analyzed for expression of miRNAs using the most updated miRNA array panel containing 2,549 human mature miRNAs (release 21.0 from the miRBase database). With a fold change >2 and P-value <0.05 as the cutoff point, 181 DEMis (89 downregulated and 92 upregulated miRNAs; Figure 1) were detected in this study. Among these DEMis, miR-146b, miR-551b, miR-375, miR-31, and miR-221 were the most significantly upregulated miRNAs, whereas miR-5703, miR-630, miR-718, miR-4646, and miR-6794 were the most significantly downregulated miRNAs (ranked by fold change values, for more details, see GSE113629). Then, we screened DEMis with TCGA data. Differential analysis revealed 86 DEMis between PTC and normal tissues, of which 66 were downregulated and 20 were upregulated. We confirmed only six downregulated miRNAs in this independent and large TCGA cohort (miR-139, miR-486, miR-3652, miR-1275, miR-144, and miR-204). Consistent with the result from TCGA-THCA, qRT-PCR results indicated that miR-204 expression was markedly downregulated in thyroid cancer tissues (Figure 2A). Notably, miR-204 was also lowly expressed in PTC cell lines compared to its expression level in normal cell line (Figure 2B). Dysregulated miR-204 has been observed in various types of cancers, including thyroid cancer.10,11 However, the underlying mechanisms of miR-204 in PTC remain to be further elucidated. To address the clinicopathological significance of the decreased expression of miR-204 in PTC, we used clinical data from TCGA PTC cohort. The data revealed that downregulation of miR-204 was frequently found in PTC tissues characterized by extrathyroidal extension, high T stage, lymph metastasis, BRAF V600E mutation, and aggressive tall cell variant (Table 1). By contrast, no significant differences were found based on patient gender, age, tumor multifocality, M stage, and TNM stage. Furthermore, we divided patients into two groups depending on the median expression value of miR-204 (miR-204 low expression group and high expression group). However, Kaplan–Meier survival analysis demonstrated that patients with lower miR-204 expression showed no differences in disease-free and overall survival rates compared to patients with higher miR-204 expression.
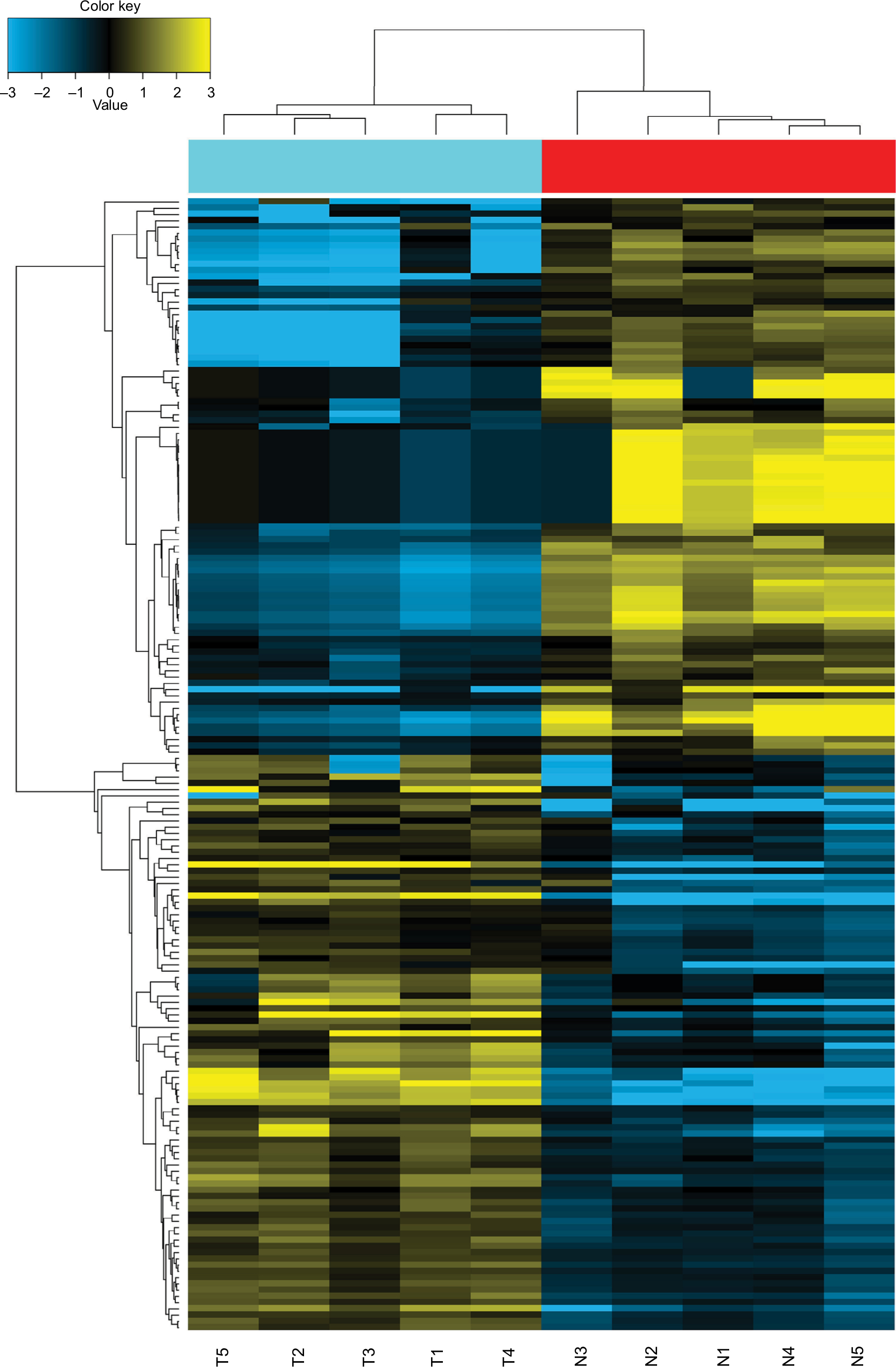 | Figure 1 Heatmap of the 181 dysregulated miRNAs. Notes: Hierarchical analysis of the 181 dysregulated miRNAs based on the comparison of their expression values in PTC and normal tissues. Data have been deposited in GEO (accession code GSE113629). Abbreviations: GEO, Gene Expression Omnibus; miRNAs, microRNAs; PTC, papillary thyroid carcinoma; T, tumor tissues; N, normal tissues. |
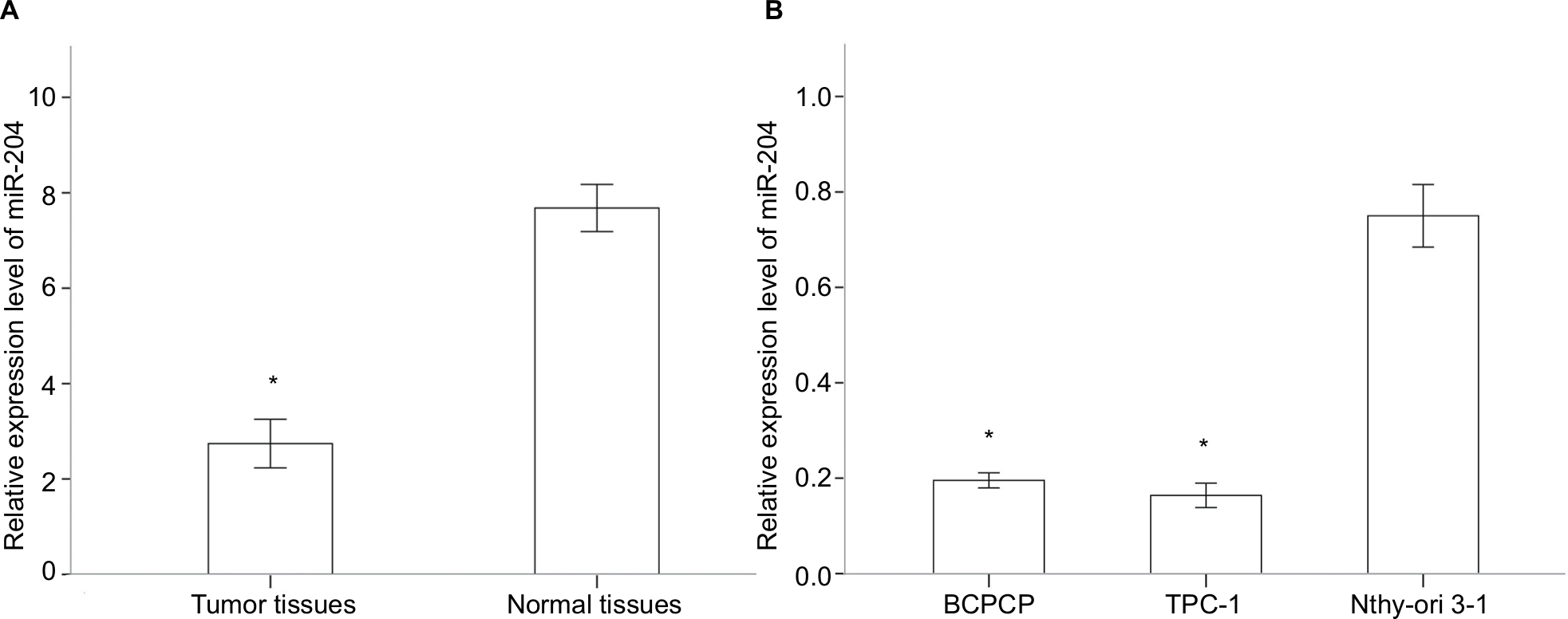 | Figure 2 Downregulated miR-204 expression in PTC tissues and cell lines. Notes: (A) miR-204 expression levels are downregulated in human PTC tissues compared to those in paired adjacent normal tissues. (B) miR-204 expression levels are downregulated in human PTC cell lines compare to that in normal cell line. *P<0.05. Abbreviation: PTC, papillary thyroid carcinoma. |
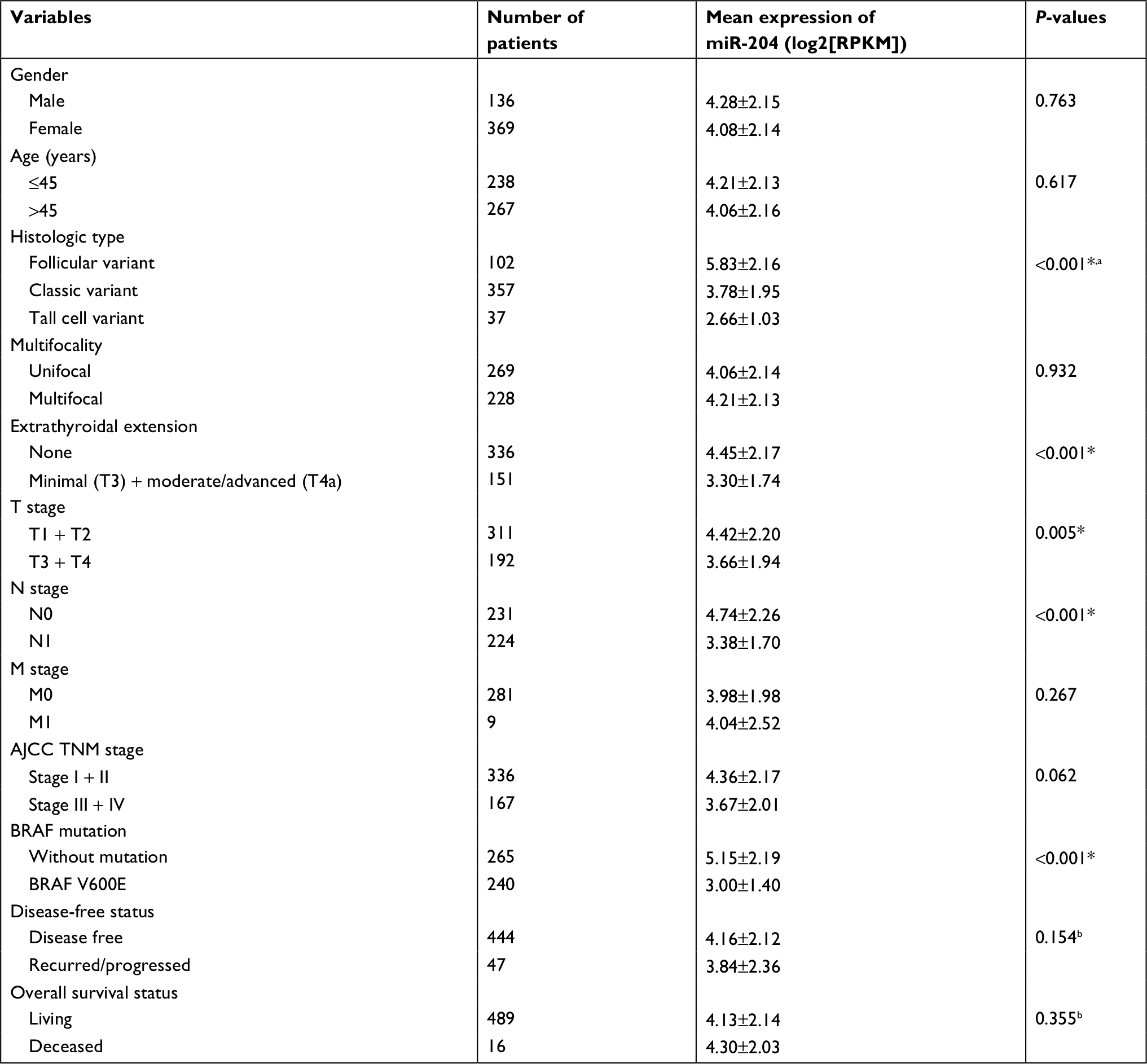 | Table 1 The relationship between miR-204 expression and clinicopathological features in PTC patients from TCGA data set Notes: Mean expression of miR-204 is represented as the mean value ± SD. Patients with missed data were excluded. *P<0.05. aP-value by one-way ANOVA. bP-value by log-rank test. Abbreviations: AJCC, American Joint Committee on Cancer; PTC, papillary thyroid carcinoma; RPKM, per kilobase per million; TCGA, the Cancer Genome Atlas. |
Downregulated expression of miR-204 in PTC is correlated with promoter DNA methylation of its host gene TRPM3
Little is known about the genetic and regulatory factors contributing to the altered expression of miR-204 in PTC. Approximately, half of all miRNA genes are encoded in the introns of protein-encoding genes, and these miRNA genes are likely to be susceptible to transcriptional repression by aberrant DNA methylation of CpG islands located in the host genes.12 The transcriptional regulation of miR-204 may be controlled by the regulatory mechanism of its host gene. We found that miR-204 is located within the introns of TRPM3 (located at 9q21, Figure 3A) using the UCSC genome browser. The chromatin patterns, genomic structure of the miR-204 and TRPM3 gene, and the location of neighboring genes are shown in Figure S1. TCGA data indicated a coordinated downregulation of miR-204 expression with that of host gene TRPM3 (with log2FCs =−9.99 and −3.41 respectively, data not shown). Two CpG islands around the miR-204 promoter were identified (Figure 3B). Therefore, we assessed the methylation status of promoter CpG islands of miR-204/TRPM3. Agena MassARRAY results indicated that 12 CpG sites located at the promoter of miR-204 were hypermethylated in primary tumor tissues compared to the matched normal tissues (Table 2). Given the limited sample size of our study, data from TCGA database were used to confirm the DNA methylation status of the promoter of miR-204. Four CpG sites (cg00840310, cg02654358, cg14223966, and cg23553442) in the promoter region of TRPM3 were identified, which are also parts of our assessed CpG islands. Data from MethHC showed a significant negative correlation between miR-204 expression and methylation levels in its promoter region (Figure 4A). Furthermore, three of the four CpG sites were strongly hypermethylated in PTC tissues compared to normal tissues (Figure 4B). As shown in Figure 4C and D, pretreatment with 5-Aza in PTC cells for 48 hours or 96 hours led to remarkable miR-204 expression, indicating a significantly negative correlation between niR-204 expression and methylation levels. Importantly, we observed higher methylation rates of the miR-204 promoter in tumor tissues than in normal tissues in 13 cancers of the total 18 cancers with the data deposited in MethHC (Figure 5). Among these 13 types of cancers, downregulated miR-204 expression was also observed in nine cancers (data not shown). Thus, we revealed that promoter DNA methylation is a common mechanism of miR-204 downregulation in cancers, including PTC.
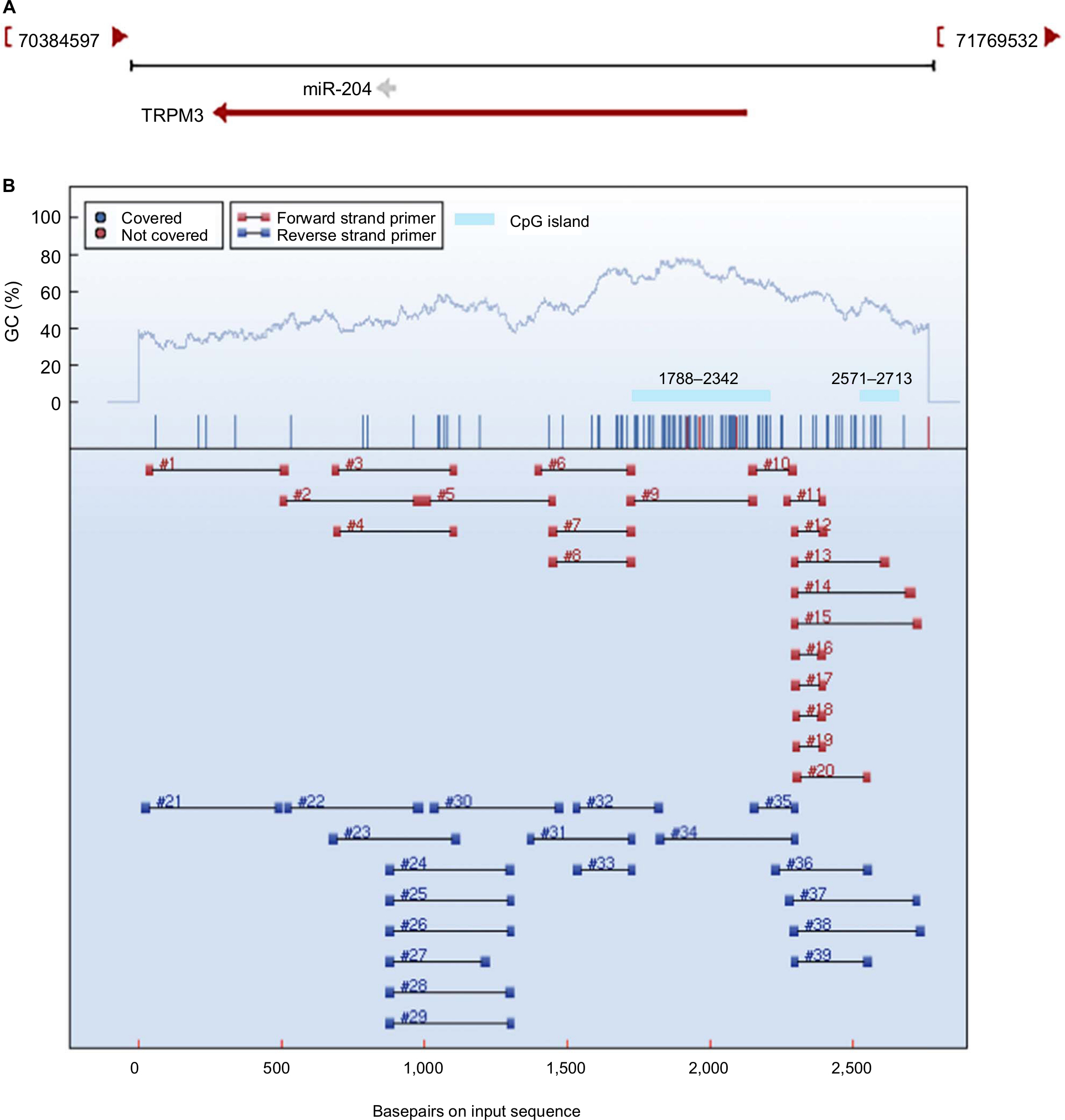 | Figure 3 Promoter DNA methylation analysis of miR-204. Notes: (A) Schematic representation of the location of miR-204 in chromosome 9. miR-204 is located within the introns of TRPM3 (located at 9q21). (B) CpG island prediction and primer design of the miR-204 promoter region. Two CpG islands around the miR-204 promoter were identified using MethPrimer. Regions 9 and 15 that completely cover the two CpG islands were selected for the amplification. Each numbered region represents a region that can be amplified. |
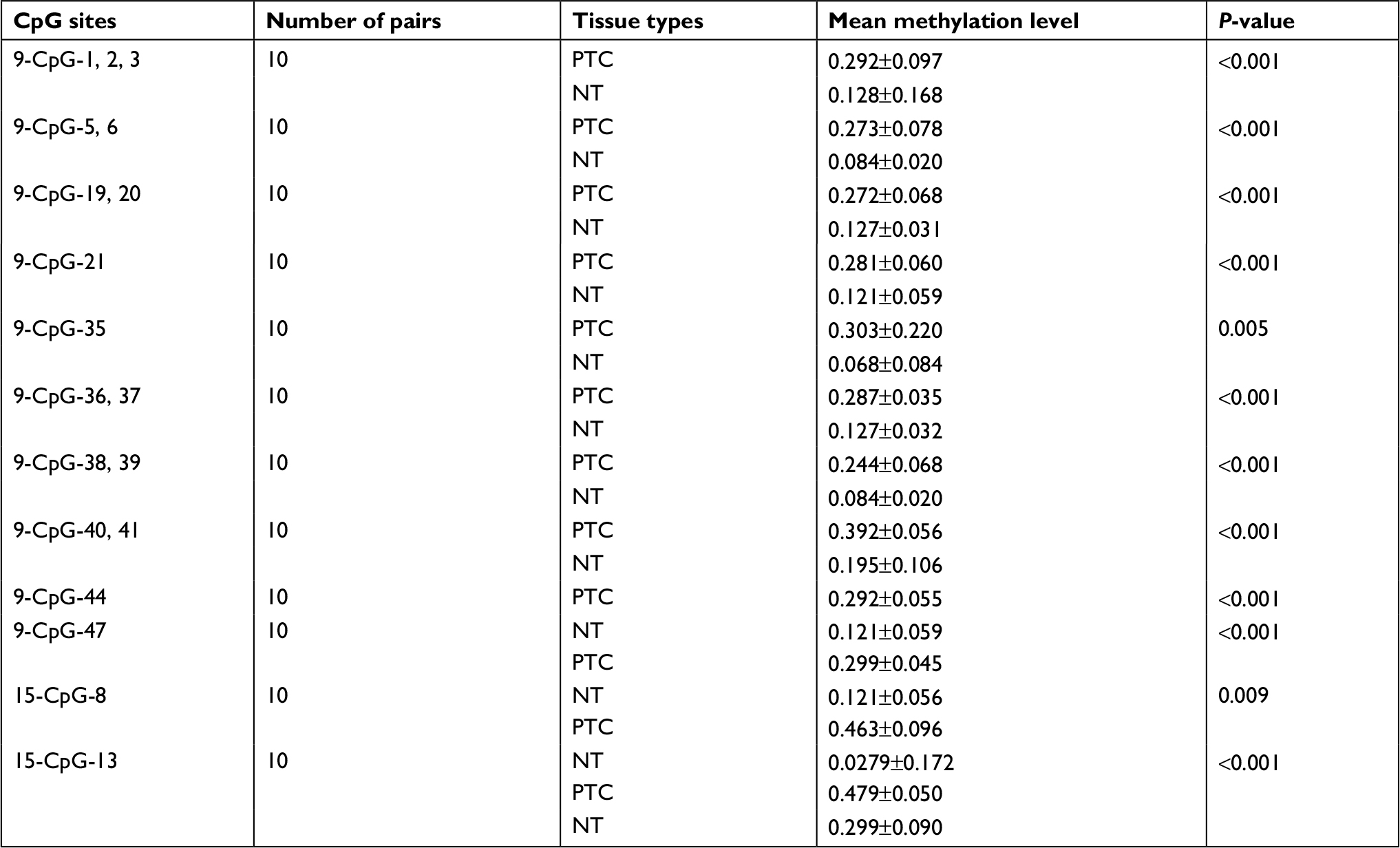 | Table 2 Comparison of methylation levels in CpG sites between PTC and adjacent normal tissues Notes: CpG sites represent region number-CpG site numbers. Mean methylation level is represented as the mean value ± SD. Abbreviations: NT, normal tissue; PTC, papillary thyroid carcinoma. |
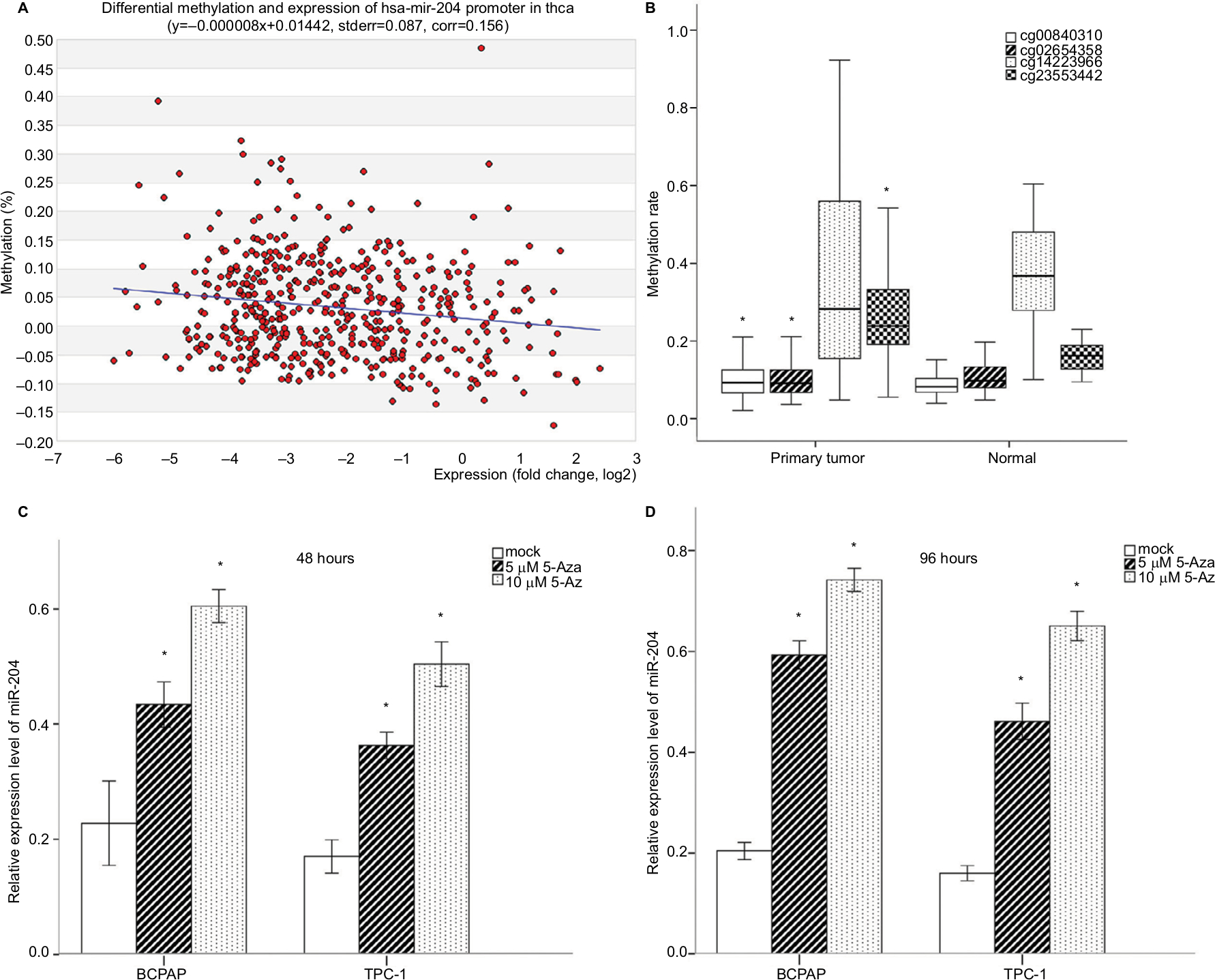 | Figure 4 Promoter DNA methylation analysis of miR-204 with TCGA data. Notes: (A) Correlation analysis between miR-204 expression levels and promoter methylation levels. The expression levels of miR-204 in tumor tissues were inversely correlated with the promoter DNA methylation rates. (B) Methylation levels of CpG sites from TCGA data. Three of the four CpG sites were hypermethylated in tumor tissues compared to normal tissues. One CpG site was hypomethylated (cg14223966). (C and D) qRT-PCR demonstrated increased expression of miR-204 in PTC cells after treatment with 5-Aza-dye for 48 and 98 hours compared to mock-treated cells. *P<0.05. Abbreviations: 5-Aza, 5-aza-2′-deoxycytidine; PTC, papillary thyroid carcinoma; qRT-PCR, quantitative real-time PCR. |
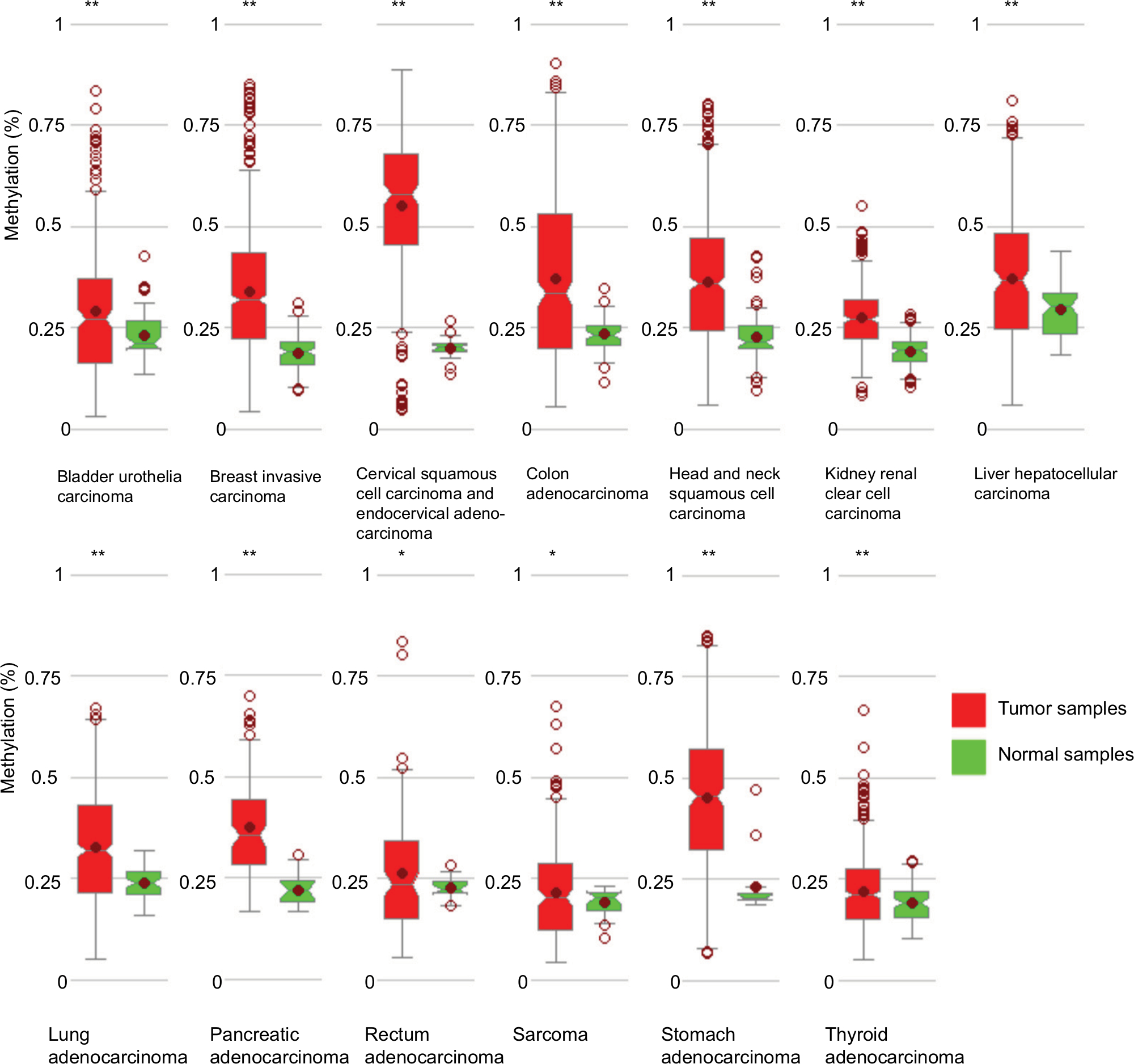 | Figure 5 miR-204 promoter methylation rates in multiple cancers. Notes: In 13 types of cancers, higher methylation rates were observed in cancer tissues than in normal tissues. *P<0.05, **P<0.005. |
Identification of related pathways involved in the downregulation of miR-204
Differential analysis revealed 1,273 DEGs (615 upregulated genes and 658 downregulated genes) between the miR-204 low expression group and high expression group. To gain a better understanding of gene function and signaling pathways related to the downregulation of miR-204 in PTC, we used the DAVID online tool to classify these DEGs using the GO and KEGG pathway references. For miR-204-related DEGs, the top enriched GO terms for biological process were cell adhesion, extracellular matrix organization, collagen catabolic process, chemical synaptic transmission, and signal transduction. The top enriched GO terms for cellular component were extracellular space, extracellular region, plasma membrane, integral component of plasma membrane, and proteinaceous extracellular matrix. For molecular function, heparin binding, calcium ion binding, potassium channel activity, serine-type endopeptidase activity, and receptor binding were considerably enriched (Figure 6A). Furthermore, the top significantly enriched pathways with the largest gene numbers of these DEGs included pathway in cancer, PI3K-Akt signaling pathway, cytokine–cytokine receptor interaction, focal adhesion, and cell adhesion molecules (Figure 6B). GSEA is based on the overall expression trend of a group of genes to determine the differences between two biological states. We also carried out GSEA to identify the cancer-related pathways associated with the downregulated miR-204 expression. The top significantly enriched pathways related to the downregulated miR-204 expression included pathogenic Escherichia coli infection, DNA replication, adherens junction, tight junction, nucleotide excision repair, P53 signaling pathway, endocytosis, apoptosis, and chronic myeloid leukemia (ranked by P-value, Figure 6C). These results suggested that downregulated miR-204 expression in PTC was related to a series of important pathways and mechanisms involved in tumorigenesis and progression.
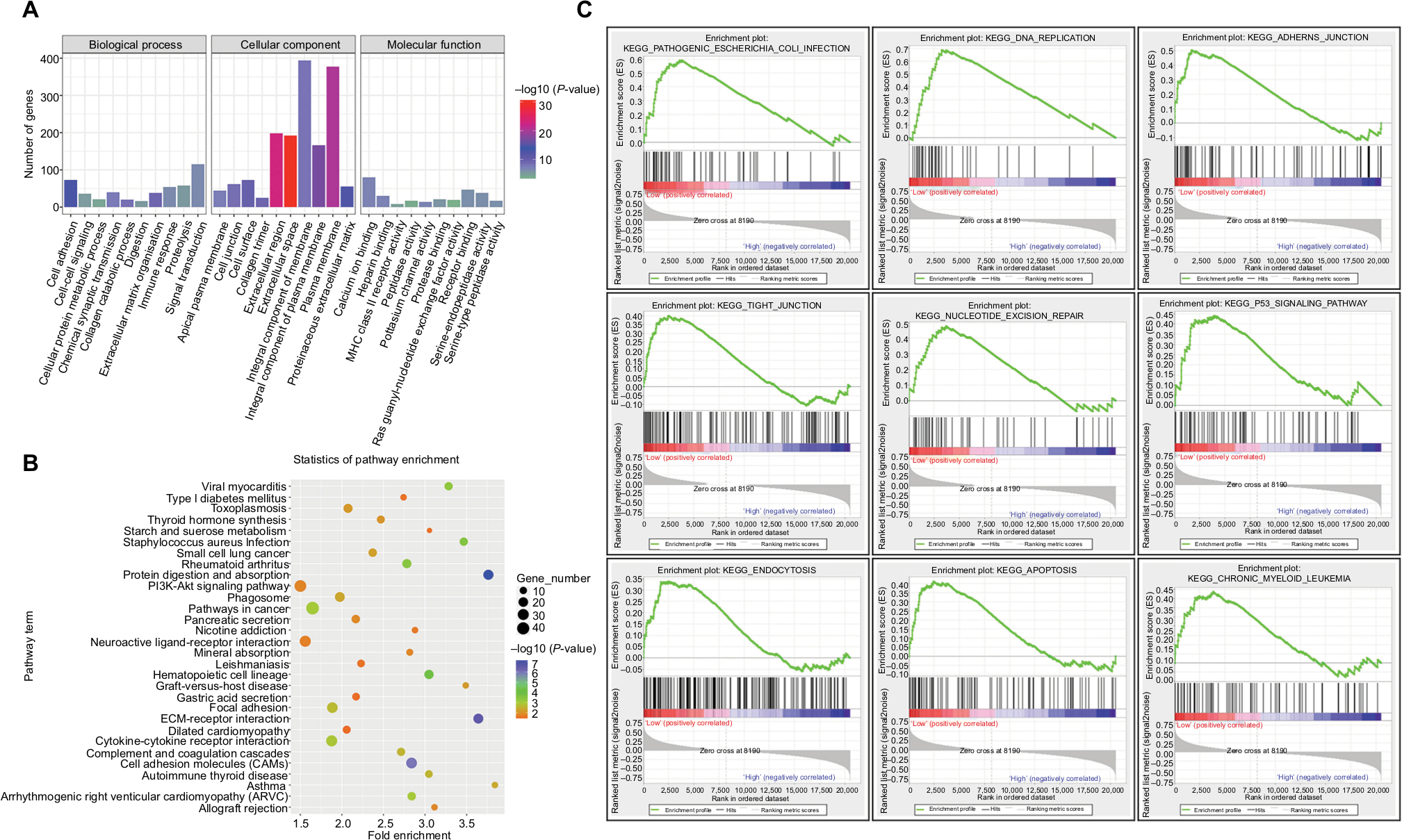 | Figure 6 GO and KEGG enrichment analyses of miR-204-related DEGs. Notes: (A) Significantly enriched GO terms of DEGs in PTC based on their functions. GO term classifications include biological process, cellular component, and molecular function. (B) Enriched KEGG pathways of miR-204-related DEGs. KEGG terms, gene numbers, P-values, and fold enrichment scores are presented in the pathway enrichment analysis. (C) Downregulation of miR-204-related pathway enrichment analysis by GSEA. The top enriched pathways showed significant enrichment in PTC tissues with lower miR-204 expression levels compared to tissues with higher miR-204 expression levels. Abbreviations: DEG, differentially expressed gene; GO, gene ontology; GSEA, gene set enrichment analysis; KEGG, Kyoto Encyclopedia of Genes and Genomes; PTC, papillary thyroid carcinoma. |
Discussion
In the present study, we evaluated the expression patterns of miRNAs in patients with PTC using miRNA microarrays and demonstrated that miR-204 was significantly downregulated in PTC tissues. Downregulated miR-204 expression was associated with aggressive clinicopathologic characteristics of PTC. For the first time, we revealed that downregulation of miR-204 was partly due to the promoter DNA methylation status in PTC tissues. Furthermore, bioinformatics analyses suggest that downregulation of miR-204 was involved in multiple biological processes and cancer-related pathways.
Several studies have reported the miRNA expression patterns in PTC. For example, Yang et al used miRNA expression arrays to identify a subset of DEMis between aggressive and nonaggressive PTCs. The results revealed 14 upregulated miRNAs and ten downregulated miRNAs.13 In this study, we used miRNA expression array analysis as an initial screening. Furthermore, we screened DEMis in patients with PTC from TCGA data set. We found that a number of miRNAs are differentially regulated in PTC tissues compared to normal tissues. Consistent with the findings of other studies, some of the most commonly deregulated miRNAs in PTC, including miR-146b,14 miR-221, miR-222,15 miR-144,16 and miR-7,17 were also identified in our study. Our findings indicated that miR-204 could serve as a potential biomarker in PTC diagnosis. Downregulation of miR-204 was frequently found in PTC tissues with aggressive clinicopathologic characteristics. However, the preoperative diagnostic and recurrence predictive values of miR-204 in PTC should be validated in a large cohort of patients. The cutoff value of miR-204 used for predictive analyses need to be evaluated.
Several causes may be associated with a defect in miRNA production in cancers, such as upstream oncogenic/tumor-suppressor defects, alterations in the miRNA processing machinery, and epigenetic inactivation.18 DNA methylation involves the addition of a methyl group at the 5-carbon position of the cytosine ring at CpG dinucleotides within a promoter and/or enhancer and is a common reversible epigenetic alteration that has been reported in many cancers.19 The transcriptional repression of miRNAs by DNA hypermethylation of their corresponding promoter loci seems to be a common feature of all human tumors.18 DNA methylation was shown to contribute to the downregulated expression of miRNAs such as miR-224,20 miR-145,21 and miR-29c.22 The 9q21.12 chromosomal region containing miR-204 is frequently lost in multiple types of cancers, suggesting that loss of genomic loci containing miR-204 is directly linked with the downregulation of miR-204.10 Another mechanism is that the activated signal transducer and activator of transcription 3 binds the TRPM3 promoter and eventually suppresses expression of miR-204 in cancer cells.10,23 Compared to normal tissues, the primary PTC tissues showed dense methylation of promoter CpG sites. The methylation rates were consistent with the expression levels of miR-204 and its host gene TRPM3. This study is the first to identify promoter DNA hypermethylation as a potential factor in the downregulation of miR-204 and its host gene TRMP3 in PTC. Importantly, analyses of data from TCGA suggested that the promoter DNA hypermethylation might be a common mechanism contributing to the downregulation of miR-204 in multiple cancers. These results are consistent with the findings in non-small-cell lung carcinoma,24 glioma,25 and cutaneous squamous cell carcinoma.26 Not all tumors with promoter DNA hypermethylation showed miR-204 down-expression. This observed discordance indicates that other causes may be associated with a defect in miR-204 production in cancers.
Pathological functions of miR-204 have been observed in various types of cancers. It is believed that miR-204 acts as a tumor suppressor via promoting apoptosis, conferring the resistance of cancer stem cells and epithelia to mesenchymal transition.10 It has been reported that miR-204 suppressed thyroid cancer cell proliferation and induced cell cycle arrest and apoptosis by directly targeting insulin-like growth factor-binding protein 5 and high mobility group AT-hook 2 mRNA.11,27 However, the involvement of miR-204 in relevant pathway abnormalities has not been comprehensively studied. The most commonly deregulated pathways in PTC include the MAPK signaling pathway, PI3K-Akt signaling pathway, NF-κB signaling pathway, Wnt-β-catenin signaling pathway, and hypoxia inducible factor-1 α (HIF-1α) signaling pathway.28 In this study, bioinformatics analysis results suggested that downregulated miR-204 expression was related to a series of important pathways and mechanisms involved in the tumorigenesis and progression. Downregulation of miR-204 may play an important role in PTC via interaction with many target genes and involvement in many tumor-related pathways, including classical pathways, such as the PI3K-Akt signaling pathway identified in this study. Novel target genes and putative mechanisms of miR-204 in PTC need to be further validated.
Acknowledgment
This work was funded by the National Natural Science Foundation of China (81672885 and 81372860) and Innovative Foundation for graduate students of Central South University (2017zzts232).
Disclosure
The authors report no conflicts of interest in this work.
References
Schmidbauer B, Menhart K, Hellwig D, Grosse J. Differentiated thyroid cancer—treatment: state of the art. Int J Mol Sci. 2017;18(6):E1292. | ||
Nikiforov YE. Role of molecular markers in thyroid nodule management: then and now. Endocr Pract. 2017;23(8):979–988. | ||
Chakraborty C, Sharma AR, Sharma G, Sarkar BK, Lee SS. The novel strategies for next-generation cancer treatment: miRNA combined with chemotherapeutic agents for the treatment of cancer. Oncotarget. 2018;9(11):10164–10174. | ||
Wang Y, Wang L, Chen C, Chu X. New insights into the regulatory role of microRNA in tumor angiogenesis and clinical implications. Mol Cancer. 2018;17(1):22. | ||
Klinge CM. Non-coding RNAs: long non-coding RNAs and microRNAs in endocrine-related cancers. Endocr Relat Cancer. 2018;25(4):R259–R282. | ||
Liu H, Lei C, He Q, Pan Z, Xiao D, Tao Y. Nuclear functions of mammalian microRNAs in gene regulation, immunity and cancer. Mol Cancer. 2018;17(1):64. | ||
Vannini I, Fanini F, Fabbri M. Emerging roles of microRNAs in cancer. Curr Opin Genet Dev. 2018;48:128–133. | ||
Celano M, Rosignolo F, Maggisano V, et al. MicroRNAs as biomarkers in thyroid carcinoma. Int J Genomics. 2017;2017(4):1–11. | ||
Yang YN, Zhang XH, Wang YM, Zhang X, Gu Z. miR-204 reverses temozolomide resistance and inhibits cancer initiating cells phenotypes by degrading FAP-α in glioblastoma. Oncol Lett. 2018;15(5):7563–7570. | ||
Li T, Pan H, Li R. The dual regulatory role of miR-204 in cancer. Tumor Biol. 2016;37(9):11667–11677. | ||
Wu ZY, Wang SM, Chen ZH, et al. MiR-204 regulates HMGA2 expression and inhibits cell proliferation in human thyroid cancer. Cancer Biomark. 2015;15(5):535–542. | ||
Dohi O, Yasui K, Gen Y, et al. Epigenetic silencing of miR-335 and its host gene mest in hepatocellular carcinoma. Int J Oncol. 2013;42(2):411–418. | ||
Yang Z, Yuan Z, Fan Y, Deng X, Zheng QI. Integrated analyses of microRNA and mRNA expression profiles in aggressive papillary thyroid carcinoma. Mol Med Rep. 2013;8(5):1353–1358. | ||
Ramírez-Moya J, Wert-Lamas L, Santisteban P. MicroRNA-146b promotes PI3K/Akt pathway hyperactivation and thyroid cancer progression by targeting PTEN. Oncogene. 2018;37(25):3369–3383. | ||
Dai L, Wang Y, Chen L, Zheng J, Li J, Wu X. MiR-221, a potential prognostic biomarker for recurrence in papillary thyroid cancer. World J Surg Oncol. 2017;15(1):11. | ||
Guan H, Liang W, Xie Z, et al. Down-regulation of miR-144 promotes thyroid cancer cell invasion by targeting ZEB1 and ZEB2. Endocrine. 2015;48(2):566–574. | ||
Hua K, Jin J, Zhang H, et al. MicroRNA-7 inhibits proliferation, migration and invasion of thyroid papillary cancer cells via targeting CKS2. Int J Oncol. 2016;49(4):1531–1540. | ||
Lopez-Serra P, Esteller M. DNA methylation-associated silencing of tumor-suppressor microRNAs in cancer. Oncogene. 2012;31(13):1609–1622. | ||
Shenoy N, Vallumsetla N, Zou Y, et al. Role of DNA methylation in renal cell carcinoma. J Hematol Oncol. 2015;8:88. | ||
Kristensen H, Haldrup C, Strand S, et al. Hypermethylation of the GABRE~miR-452~miR-224 promoter in prostate cancer predicts biochemical recurrence after radical prostatectomy. Clin Cancer Res. 2014;20(8):2169–2181. | ||
Xia W, Chen Q, Wang J, et al. DNA methylation mediated silencing of microRNA-145 is a potential prognostic marker in patients with lung adenocarcinoma. Sci Rep. 2015;5:16901. | ||
Poli E, Zhang J, Nwachukwu C, et al. Molecular subtype-specific expression of microRNA-29c in breast cancer is associated with CpG dinucleotide methylation of the promoter. PLoS One. 2015;10(11):e0142224. | ||
Ma L, Deng X, Wu M, Zhang G, Huang J. Down-regulation of miRNA-204 by LMP-1 enhances Cdc42 activity and facilitates invasion of EBV-associated nasopharyngeal carcinoma cells. FEBS Lett. 2014;588(9):1562–1570. | ||
Shi L, Zhang B, Sun X, et al. MiR-204 inhibits human NSCLC metastasis through suppression of NUAK1. Br J Cancer. 2014;111(12):2316–2327. | ||
Ying Z, Li Y, Wu J, et al. Loss of miR-204 expression enhances glioma migration and stem cell-like phenotype. Cancer Res. 2013;73(2):990–999. | ||
Toll A, Salgado R, Espinet B, et al. MiR-204 silencing in intraepithelial to invasive cutaneous squamous cell carcinoma progression. Mol Cancer. 2016;15(1):53. | ||
Liu L, Wang J, Li X, et al. MiR-204-5p suppresses cell proliferation by inhibiting IGFBP5 in papillary thyroid carcinoma. Biochem Biophys Res Commun. 2015;457(4):621–626. | ||
Xing M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat Rev Cancer. 2013;13(3):184–199. |
Supplementary material
 | Figure S1 (A) Schematic representation of the chromatin patterns of miR-204. miR-204 is located within the introns of TRPM3 at 9q21. The genomic structure of the miR-204 and TRPM3 gene (currently known transcripts), the location of neighboring genes, and CpG islands are presented in each line. All data are obtained from the UCSC genome browser. (B) The genomic sequence of TRPM3 DNA promoter region. The promoter region was predicted from the genomic sequence of TRPM3 for the 2,000 upstream and 1,000 downstream sequences around its first exon (transcriptional start site). All of the CpG sites have been highlighted. CpG sites in red color are identified CpG sites of the gene chip data from TCGA. Abbreviations: TCGA, the Cancer Genome Atlas; UCSC, University of California Santa Cruz. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.