Back to Journals » Drug Design, Development and Therapy » Volume 8
Diverse pathomechanisms leading to the breakdown of cellular estrogen surveillance and breast cancer development: new therapeutic strategies
Authors Suba Z
Received 3 July 2014
Accepted for publication 16 July 2014
Published 11 September 2014 Volume 2014:8 Pages 1381—1390
DOI https://doi.org/10.2147/DDDT.S70570
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Zsuzsanna Suba
National Institute of Oncology, Budapest, Hungary
Abstract: Recognition of the two main pathologic mechanisms equally leading to breast cancer development may provide explanations for the apparently controversial results obtained by sexual hormone measurements in breast cancer cases. Either insulin resistance or estrogen receptor (ER) defect is the initiator of pathologic processes and both of them may lead to breast cancer development. Primary insulin resistance induces hyperandrogenism and estrogen deficiency, but during these ongoing pathologic processes, ER defect also develops. Conversely, when estrogen resistance is the onset of hormonal and metabolic disturbances, initial counteraction is hyperestrogenism. Compensatory mechanisms improve the damaged reactivity of ERs; however, their failure leads to secondary insulin resistance. The final stage of both pathologic pathways is the breakdown of estrogen surveillance, leading to breast cancer development. Among premenopausal breast cancer cases, insulin resistance is the preponderant initiator of alterations with hyperandrogenism, which is reflected by the majority of studies suggesting a causal role of hyperandrogenism in breast cancer development. In the majority of postmenopausal cases, tumor development may also be initiated by insulin resistance, while hyperandrogenism is typically coupled with elevated estrogen levels within the low postmenopausal hormone range. This mild hyperestrogenism is the remnant of reactive estrogen synthesis against refractory ERs that were successfully counteracted at a younger age. When refractoriness of ERs is the initiator of pathologic processes, reactively increased estrogen levels may be found in both young and older breast cancer cases, while they may exhibit clinical symptoms of estrogen deficiency. Studies justifying a causal correlation between hyperestrogenism and tumor development compile such breast cancer cases. In conclusion, the quantitative evaluation of ER refractoriness in breast cancer cases has great importance, since the stronger the estrogen resistance, the higher the promising dose of estrogen therapy.
Keywords: sexual steroids, hyperandrogenism, hyperestrogenism, insulin resistance, estrogen receptors, estrogen resistance
Introduction
There is a prevailing concept suggesting a linear cause–effect relationship between serum levels of sexual steroids and the clinical manifestations of certain diseases, such as breast cancer, especially in postmenopausal women.1–3 Nevertheless, the associations between circulating concentrations of androgens and estrogens with breast cancer and related risk factors are not well understood until now.4 Results of studies on correlations between endogenous sexual steroid levels and breast cancer risk have yielded a wide range of controversies in both premenopausal and postmenopausal cases.
In premenopausal women, the majority of studies support that the increased serum concentrations of androgens are strongly associated with breast cancer risk.5–9 Moreover, in certain studies, both increased estrogen and androgen levels were found to be equally responsible for breast cancer development in young women.4,10 By contrast, a recent meta-analysis study found weak evidence of a positive association between premenopausal endogenous estradiol level and the risk of breast cancer.11
In postmenopausal women, the vast majority of publications suggest that within the range of low baseline serum sexual steroid levels, relatively higher estrogen and androgen concentrations are equally regarded to be dangerous for breast cancer development.2,3,12–20 In older women, a few studies justified that elevated androgen levels may be strongly associated with an increased risk of breast cancer,21,22 while another found an increased estradiol level to be dangerous for postmenopausal cases.23
Surprisingly, in certain studies, no convincing associations were found between serum sex hormone levels and breast cancer development among either premenopausal24 or postmenopausal cases.25–27
These highly controversial results, however, exhibited certain characteristic findings. Although high estrogen levels are regarded as causal factors for breast cancer, increased androgen levels, either with or without estrogen overproduction, were consistently associated with increased breast cancer risk. Elevated estrogen levels without androgen excess were a rare finding in both young and older breast cancer cases.
Proper estrogen receptor (ER) signaling is essential for the health of mammalian cells, as estrogens are the chief regulators of cellular metabolism, growth, differentiation, and proliferation.28–30 Difficulties in either the synthesis of estrogens or ER signal transduction mechanisms may increase the risk of clinically diagnosable diseases.31,32
There are strict crosstalks and mutual feedback mechanisms between estrogen synthesis and ER signaling. Critically low serum estrogen levels may induce alarming defense reactions in order to increase the transcriptional activity of ERs.33 Similarly, deficient ER signaling may be counteracted by reactively increased endogenous estrogen synthesis so as to save cellular estrogen surveillance.34,35 Pathologic states may develop when the interactions of these players cannot restore each other’s defect. Failures of estrogen synthesis or liganded ER signal transduction result in disturbances in cellular estrogen surveillance and glucose uptake, which is the fuel of all cellular functions.36 Appearances of the clinical biomarkers of these disorders may forecast the manifestation of serious diseases such as type 2 diabetes, cardiovascular diseases, cognitive disturbances, and malignancies.32,37
In conclusion, in order to evaluate the complex correlations between serum levels of sexual steroids and breast cancer development, one should be aware of all stimulatory and inhibitory players that have a role in the complex mechanisms of cellular estrogen surveillance. To clarify the background of puzzling correlations between serum sexual steroid levels and breast cancer risk, the following questions should be answered:
- Do uniform pathomechanisms lead to breast cancer development or are there diverse pathways leading to mammary malignancies?
- Is hyperestrogenism or hyperandrogenism the crucial factor causing metabolic disorders and their comorbidities, such as breast cancer?
- How can the lower postmenopausal range of endogenous estrogen levels be apparently in direct correlation with breast cancer risk, while this association cannot be observed at higher, premenopausal estrogen concentrations?
- Why cannot either the circulating levels of sex hormones or estrogen receptor gene (ESR) mutations in themselves predict the risk of breast cancer?
Decreased serum estrogen concentration and hyperandrogenism
The aromatase enzyme complex catalyzes the conversion of androgens to estrogens in a wide variety of tissues, including the breast, ovary, testis, placenta, brain, and adipose tissue.38,39 Estrogen synthesis may be altered or blocked by the mutation of human CYP19 gene encoding aromatase P450 enzyme.40
Aromatase activity and healthy estrogen synthesis may be endangered by various endogenous and exogenous factors as well, such as aging,41 insulin resistance,42–44 diabetes,45 low physical activity,41 and deficient natural light exposure associated with melatonin excess.46,47 In aromatase deficient cases, the excessive accumulation of androgen precursors is a typical laboratory finding, while estrogens exhibit extremely low serum levels.
Mutation in CYP19 in a sister and brother was published.38 The 28-year-old girl with XX chromosome presented the cardinal features of aromatase deficiency syndrome. At birth she had nonadrenal female pseudohermaphroditism. By the age of puberty, she developed progressive signs of virilization with no signs of estrogen action such as hypergonadotropic hypogonadism, polycystic ovaries, and tall stature. The basal concentrations of plasma androgens were highly elevated, whereas plasma estradiol was extremely low. Hormone replacement therapy led to breast development, menstrual cycles, resolution of ovarian cysts, and suppression of the elevated follicle-stimulating hormone and luteinizing hormone (LH) values.
The XY male sibling with aromatase coding gene (CYP19) mutation was examined at age 24 years. He was 204 cm tall with eunuchoid skeletal proportions.38 He was fully mature sexually and presented with macroorchidism. The plasma concentrations of androgens were elevated, while estradiol and estrone levels were <7 pg/mL. Plasma follicle-stimulating hormone and LH concentrations were more than three times the mean value. Hyperinsulinemia, increased serum total and low-density lipoprotein cholesterol, high triglyceride, and decreased high-density lipoprotein cholesterol levels were detected, suggesting insulin resistance.
Insulin resistance-associated compensatory hyperinsulinism is a double-edged sword against estrogen signaling. Excessive insulin synthesis inhibits aromatase activity, and the accumulation of precursor androgens results in hyperandrogenism.48 At the same time, decreased estrogen concentration hampers all phases of glucose metabolism from insulin synthesis to intracellular glucose transporter expression, provoking a further deepening of cellular insulin resistance.32 In women with type 2 diabetes, the ability of the ovaries to convert androgen to estrogen is reduced, possibly attributed to a reduction of the ovarian aromatase activity.45
Male-like central obesity in women is in close correlation with insulin resistance, hyperinsulinism, and excessive androgen synthesis, resulting in high morbidity.49 In centrally obese women, insulin resistance-associated sex hormone imbalance with defective estrogen synthesis is accompanied by increased risks of breast, endometrium, and ovarian cancers.43,44 In women with polycystic ovarian syndrome, anovulatory infertility, visceral obesity, and hirsutism are characteristic clinical symptoms associated with the laboratory findings of hyperinsulinism and androgen excess.50 In polycystic ovarian syndrome cases, dysmetabolism and ovarian failure are improved by either insulin-sensitizing metformin or oral contraceptive administration.51,52
In conclusion, insulin resistance, insufficient estrogen synthesis, and accumulation of androgen precursors all lead to further diverse morbidities, depending on the severity and duration of hormonal alterations.
Increased serum estrogen level in health and disease
Another end point of estrogen synthesis is excessive estrogen production. A high natural estrogen level is never harmful. Even sky high levels are well tolerable.
The physiologic example is pregnancy, in which state estrone, estriol, and estradiol levels are extremely high. Even a 200- to 300-fold serum estradiol concentration can be observed, as compared with the follicular phase of the menstrual cycle.53 This physiologically increased hormone level supervises the safety, explosion-like proliferation of fetal structures as chief regulator of abundant growth hormones, growth factors, and other important biologic mediators. At the same time, pregnancy-associated excessive estrogen concentration provides long-term deoxyribonucleic acid stabilization for parous women. Multiparity in women and risk for malignancies exhibit a strong inverse correlation, including breast cancer.54,55 Unique features of estrogens are their beneficial impacts on all the cellular mechanisms of privileged healthy cells,56 while estrogens are capable of recognizing mitotic failures and malignant cells and neutralize them by apoptosis or promotion of their differentiation.57,58
Pathologic, reactive hyperestrogenism may occur as a feedback mechanism against defective ER signaling.34,35 In these cases, excessive estrogen production is not a causal factor for diseases but serves as a breakthrough of blocked ER signaling mechanisms.
Reactive hyperestrogenism may be an embarrassing finding in certain young women with clinical symptoms of estrogen deficiency. In these cases, a feedback mechanism aims to break through the inherited or acquired defective ER signaling by increased estrogen synthesis.
Although BRCA gene mutation carriers usually exhibit clinical symptoms of defective estrogen signaling, the serum estrogen levels of these patients are consequently elevated. Median luteal phase titers for estradiol were 33% higher in BRCA1/2 mutation carrier women than in BRCA mutation-negative cases.59 Among breast cancer cases with BRCA2 gene mutation, the reactive increase in estradiol level is higher as compared with BRCA1 gene mutation carrier cases.60 The stronger estrogen synthesis seems to be more effective against refractory ERs and results in lower breast cancer risk in BRCA2 mutation carriers than in cases with BRCA1 mutation.61
In BRCA gene mutation carrier women, high risks for breast and ovarian cancers suggest low ER responsiveness to estrogen.61 Among these patients, low reactivity to fertility treatments62 and earlier age at natural menopause (<40 years)63,64 support the defect of estrogen surveillance. Among BRCA1/2 mutation carrier women with breast cancer, a high prevalence of type 2 diabetes and central obesity was observed,65 justifying the interplay between ER defect and insulin resistance in breast cancer development.
Elderly women with profound estrogen deficiency may have a higher risk of Alzheimer’s disease, as cognitive functions are in close correlation with sufficient estrogen supply.66 Nevertheless, the impact of increased endogenous estrogen level on the risk of dementia was established in a population-based prospective study including 5,644 postmenopausal women aged ≥65 years.67 Total estradiol values in both lower and upper quartiles were associated with an increased dementia risk (hazard ratio [HR] =2.2 and HR =2.4, respectively), showing a U-shaped relationship.
Taking into account that serum estrogen levels and cellular estrogen surveillance are not always in direct correlation, these remarkable results should be thoroughly re-evaluated. In postmenopausal women, an increase in years since menopause confers a negative influence on glucose tolerance.68 In elderly women, the lowest quartile of their decreased estradiol level means severely deficient estrogen synthesis associated with deepening insulin resistance, which endangers all highly estrogen-dependent organs, such as the brain.
In elderly women with estrogen levels in the upper quartile, the possibility emerges of a defective ER signaling and reactively increased but not sufficient estrogen synthesis. In postmenopausal women, higher estradiol levels were associated with Pvull polymorphism of the ESR1 gene and increased breast cancer risk, which was interpreted as harmful collaboration between defective ERs and increased estrogen levels.69 Nevertheless, a relatively increased estrogen level in elderly cases may be regarded as insufficient counteraction against the functional alterations of ERs instead of breast cancer initiator. In these cases, the majority of ER signaling defects was disguised by compensatory good estrogen synthesis at younger ages. With aging, however, the relatively higher but insufficient estradiol levels are not enough for the breakthrough of ER signal transduction defects.
Considering the complex mechanisms affecting correlations between serum estradiol levels and cellular estrogen surveillance, we can arrive at the comprehensive understanding that in elderly women either the lower or upper quartile of their low baseline serum estrogen concentrations may be associated with defective estrogen signaling.
Genetic defects of ER signaling
Rapid development of new methods for genetic investigations provided possibilities for the analysis of ERα gene (ESR1) polymorphism, which presumably may give a rational explanation for puzzling links among estrogen, metabolic disorders, and breast cancer risk. Inherited ESR gene mutation may be regarded as a ruined bridge between circulating endogenous hormones and the surveillance of cellular health causing complete or partial blockage of estrogen signaling.
A large number of naturally occurring splice variants of both ERα and ERβ have been identified in both normal epithelium and diseased tissues. By contrast, only a few point mutations have been identified to be pathogenic in human patient samples from a variety of diseases, including breast cancer, endometrial cancer, and psychiatric diseases.70 In young women, mild point mutations of ER regulator genes may be compensated by slightly elevated estrogen synthesis, and the patient remains clinically healthy until the maintenance of extra estrogen levels. With aging, dramatically decreasing serum estrogen levels may lead to the manifestation of earlier hidden point mutations of ER regulator genes resulting in signaling defects, especially in elderly women.32
The first mutation affecting ERs was reported 20 years ago in a 28-year-old man with knock-knees and serious signs of insulin resistance, obesity, and premature cardiovascular lesions.34 Examinations showed that his testosterone levels were normal, and although his estrogen hormone levels were extremely high, he had essentially no response to estrogen.
Recently, a case of an 18-year-old girl was published who experienced delayed puberty with the classic symptoms of too low estrogen level.35 Subsequent examinations revealed sky high levels of estrogens in her blood. In laboratory studies, 240 times the normal estrogen level was required to get a response out of the patient’s ERs. Without estrogen reactivity, insulin levels were also typically increased, and an unusual response to an oral glucose test indicated glucose intolerance problems.
Correlations between genetic polymorphisms in the ESR1 gene and breast cancer risk are highly inconclusive. In a Korean study, Pvull genotype distribution did not show any differences between breast cancer cases and controls, while the adjusted odds ratio (OR) for the Xbal allele containing genotypes was advantageously decreased (OR =0.4).71 Conversely, Pvull polymorphism was associated with a moderately increased risk for breast cancer, whereas Xbal polymorphism was related to nonsignificantly elevated risk confined to older postmenopausal women in Shanghai.72 Moreover, two single nucleotide polymorphisms in the ESR2 gene were convincingly associated with increased breast cancer risk in hormonally challenged postmenopausal women but not in premenopausal women.73
Correlations between hormone replacement therapy-associated mortality and the genetic variability of ERs suggested that some women are genetically more vulnerable to the effects of hormone replacement therapy in terms of their ER genotype.74 In genetically endangered older women, an alternative possibility emerges, according to which the lack of ER responsiveness would require a higher compensatory hormone dose as compared with the usual one, so as to achieve proper estrogen surveillance.
In conclusion, the controversial results of correlations between gene polymorphisms of ESRs and clinically manifested diseases suggest that genetic investigations in themselves are not enough to evaluate either the impairment of ER signaling or the activity of compensatory mechanisms. Results of gene polymorphism studies reveal that postmenopausal state and aging-associated estrogen deficiency may amplify the earlier hidden genetic defects of estrogen signaling, while, in young cases, sufficient or reactively high estrogen synthesis may disguise the malfunction of the ESR1 gene.32
Diverse molecular pathomechanisms leading to defective estrogen surveillance and breast cancer development
Sexual steroid level measurements among breast cancer cases were performed on randomly selected patients, disregarding their risk factors either for insufficient estrogen synthesis or defective ER signaling pathways. Analyzing the alteration patterns in the sexual steroid levels of breast cancer cases provides possibilities to estimate the frequency of the different pathologic mechanisms that may result in mammary malignancies.
Insulin resistance is the predominant, primary mechanism leading to breast cancer development in women (Figure 1). In the majority of young breast cancer cases, insulin resistance syndrome proved to be a typical finding associated with hyperandrogenism, menstrual cycle disorders, and anovulation, mirroring complex endocrine disturbances in the background of defective glucose uptake.44
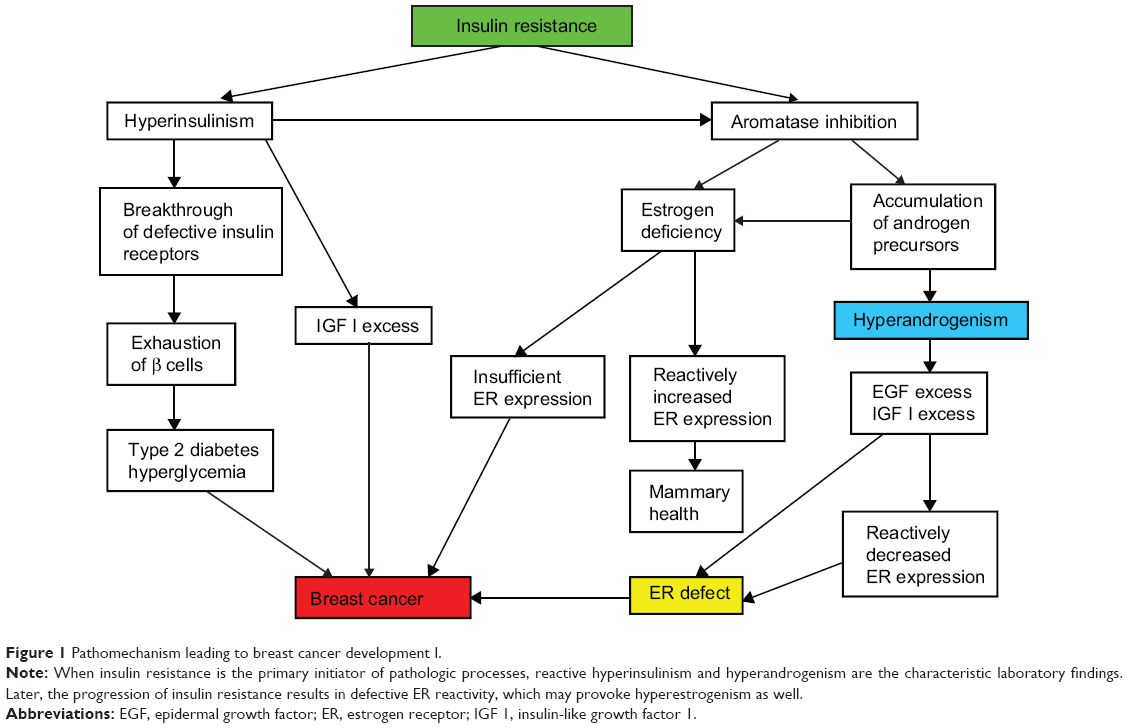 | Figure 1 Pathomechanism leading to breast cancer development I. |
Insulin resistance is an ongoing defect of insulin-mediated cellular glucose uptake, which induces a breakdown in the gene regulation of cellular metabolism, growth, differentiation, and mitotic activity.75 Progression of this disorder predisposes patients to a variety of diseases, such as metabolic syndrome, type 2 diabetes, cardiovascular lesions, and malignancies.76
In the first phase of insulin resistance, increased insulin synthesis results in reactive hyperinsulinism, which may transitorily bring about a breakthrough of refractory insulin receptors.77 Nevertheless, high insulin levels and the concomitant accumulation of insulin-like growth factors, such as IGF-1, have important roles in the alterations of cell proliferation and tumor growth.78,79
Insulin is a potent regulator of the balance of sexual steroid synthesis, and its overproduction contributes to further endocrine disturbances.80 Hyperinsulinism promotes a shift from estrogen to androgen synthesis by means of ovarian and adrenal aromatase inhibition.48 Moreover, a high insulin level may favor the LH secretion of the pituitary gland, which also stimulates androgen biosynthesis via activation of the adrenal gland and ovarian theca cells at the expense of reduced estrogen production.81,82 Accumulation of androgen precursors and estrogen deficiency are dangerous for all cellular mechanisms.
A low serum estrogen level is alarming considering that insufficient ER expression endangers the crucial cellular estrogen surveillance. In response to low estrogen exposure, such as after ovariectomy, a marked increase in the messenger ribonucleic acid levels of ERs may occur, ensuring a rise in the number of available receptors by feedback mechanism.83 This reactively increased ER expression may help to maintain mammary health, even in an estrogen-deficient milieu.
At the same time, hyperandrogenism provokes increased post-transcriptional gene expression of epidermal growth factor receptors,84 and a high androgen level may induce an excessive IGF-1 concentration.85,86 An excessive epidermal growth factor level induces reactively decreased ERα expression as well as suppression of ERα binding sites, ERα messenger ribonucleic acid level, and ERα gene transcription activity,87 which results in multifaceted ER defect. In emergency states of critically low estrogen levels, some growth factors via their own receptors may induce ER transcriptional activity, resulting in estrogen-like responses.33 Nevertheless, this physiologic coupling of growth factor and ER signaling pathways may work only in case of the availability of intact ERs.88 A decrease in both number and reactivity of ERs in an estrogen-deficient milieu completely disarms the self-defense of mammary cells, leading to high risk for cancer initiation.
In the meantime, the exhaustion of β cells in pancreatic Langerhans islands results in restrained insulin synthesis alongside the development of type 2 diabetes with hyperglycemia, which is a further pendulum swing against mammary health.89
Defective ER signaling is the second, less frequent initiator mechanism leading to breast cancer development (Figure 2). Missing or decreased ER reactivity provokes increased estrogen synthesis by feedback mechanism, resulting in hyperestrogenism;34,35 however, the consequence may be either a successful or an insufficient breakthrough of ER defect.
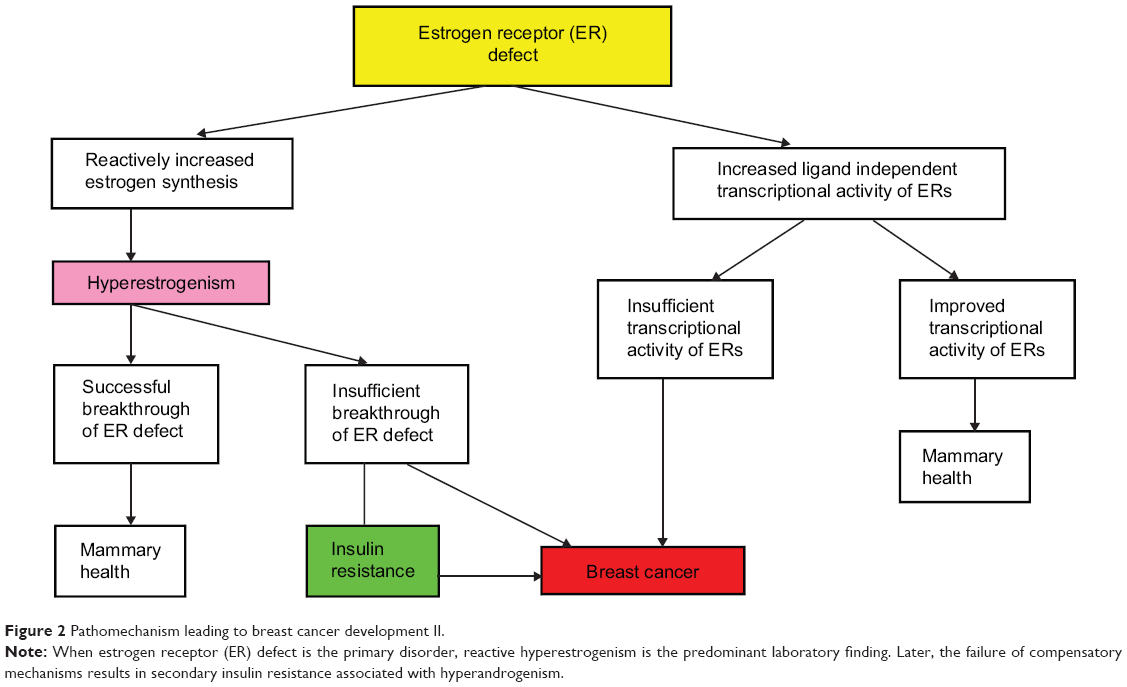 | Figure 2 Pathomechanism leading to breast cancer development II. |
In case of inherited severe ER mutation, the patient may exhibit the classic symptoms of extremely low estrogen levels, such as delayed puberty, whereas laboratory findings reveal sky high estrogen levels.35 By contrast, hyperestrogenism may also occur in apparently symptom-free cases, such as in certain women carrying BRCA gene mutations.59 In such cases, the compensatory estrogen production seems to be only transitorily defensive; these women have a lifelong increased risk for breast cancer. Considering these correlations, hyperestrogenism is not a causal factor of breast cancer but rather is a protective feedback mechanism trying to maintain the hormonal and metabolic equilibrium.
As physiologic ER signaling is crucial for all steps of cellular glucose uptake, even reactive hyperestrogenism may not always provide sufficient compensation for severe ER defect, and insulin resistance may develop.32 In BRCA mutation carrier women, breast cancer development is frequently associated with obesity and type 2 diabetes,65 justifying the close correlation between defective estrogen signaling and insulin resistance in the development of breast cancer.32,43
As an alternative possibility for the compensation of defective estradiol-mediated activation of ERα, an increased ligand-independent transcriptional activity of ERs may develop.90 In BRCA1-deficient cells, a reduction in estradiol-mediated activation of ERα was observed, while an increased estrogen-independent expression of estrogen response genes was monitored when compared with BRCA1-proficient cells.91 The improved transcriptional activity of ERs in a ligand-independent way may help to maintain mammary health. Conversely, in cases of insufficient transcriptional ER activity, the danger of breast cancer development is increased.
Conclusion
Recognition of the two main pathologic mechanisms equally leading to breast cancer development may provide explanations for the apparently chaotic results obtained by sexual hormone measurements in breast cancer cases.
Either insulin resistance or ER signaling defect may be the initiating pathologic process. They enter into a vicious circle. The final stage is the breakdown of estrogen surveillance with its severe comorbidities, such as breast cancer. In case of primary insulin resistance, hyperandrogenism is an early concomitant hormonal alteration, but during the ongoing pathologic changes the development of ER defect may induce hyperestrogenism as well. Conversely, when the ER signaling defect is the onset of hormonal and metabolic disturbances, the initial counteraction is a reactive hyperestrogenism, and further compensatory cellular mechanisms try to improve the damaged transcriptional activity of ERs. Finally, the insufficiency of defensive mechanisms leads to insulin resistance and the associated hyperandrogenism.
In the majority of young premenopausal women with breast cancer, insulin-resistant syndromes are the preponderant hormonal alterations with androgen excess. This prevailing pathomechanism is reflected in the majority of studies suggesting a presumed causal correlation between androgen excess and breast cancer risk. In these women, both insulin-sensitizing metformin and exogenous estrogen treatment may advantageously suppress the overwhelming excess of androgens.
In the second group of young breast cancer cases, hyperandrogenism may be coupled with reactive estrogen overproduction in a later stage of insulin resistance when growth factor abundance inhibits both the expression and transcriptional activity of ERs. Studies supporting the carcinogenic capacity of increased levels of both estrogens and androgens may preponderantly include these types of young breast cancer cases. In this group, a high dose of estrogen treatment is necessary to break through the ER resistance.
In the third, smaller group of premenopausal breast cancer cases, reactive but insufficient hyperestrogenism is associated with inherited, severe ER signaling defects. These types of patients are predominant in studies supporting the pivotal role of elevated levels of estrogens and their metabolites in malignant transformation without excessive androgen synthesis. In these women, a pregnancy mimicking high dose of estrogen may be a countermeasure against ER blockage.
In the vast majority of postmenopausal breast cancer cases, relatively increased circulating concentrations of both androgens and estrogens seem to be characteristic. Among these older breast cancer cases, tumor development was initiated by insulin resistance and the associated androgen excess. In such patients, slightly elevated estrogen concentration within the low postmenopausal hormone range may be regarded as an ineffective remnant of defensive estrogen synthesis against refractory ER signaling at a younger age. These cases require increased exogenous doses of estrogen, since usual hormone treatment may be ineffective.
In the second group of postmenopausal breast cancer cases, relatively high levels of circulating androgens define the hormonal imbalance without an increase in estrogens. ER signaling of these older women is not blocked either by progression of an insulin-resistant state or by ESR gene mutation. In this group of postmenopausal patients, even the usual dose of exogenous estrogen treatment may be effective.
In the third, smaller group of postmenopausal breast cancer cases, a reactively increased level of unconjugated estradiol without androgen excess may be strongly associated with refractory ERs. Patients included in this group exhibit estrogen resistance and need an extra high exogenous estrogen dose in order to get a response out of their defective ERs.
Recognition of the crucial role of cellular estrogen surveillance in mammary health may provide plausible answers to the emerging questions:
- At least two kinds of pathomechanisms (possibly more) may be involved in the breakdown of estrogen surveillance leading to breast cancer development. Either insulin resistance or estrogen resistance may be the initiator of pathologic processes.
- Neither hyperandrogenism nor hyperestrogenism is an etiologic factor of breast cancer. Both of them are links in the chain of pathologic processes. Nevertheless, there is a great difference between these two hormonal alterations. Androgen excess without counteractions leads to further breakdown of cellular mechanisms, while estrogen excess is beneficial, since only its insufficient overproduction leads to progressive pathologies.
- Endogenous estrogen level is not in direct correlation with breast cancer risk in either premenopausal or postmenopausal women. In young breast cancer cases, insulin resistance-associated androgen excess is the preponderant initiator of the breakdown of estrogen surveillance. In the majority of older breast cancer cases, insulin resistance induces increased androgen concentration, and the low but relatively elevated baseline estrogen level uncovers the earlier hidden refractoriness of ERs but is no longer defensive.
- Circulating sexual hormone levels and their receptors are in strict crosstalk and interplay with each other. Examined players randomly selected from the whole system cannot define the possible pathologic processes on their own.
Understanding the pathologic background of changes in sexual hormone levels enables us to plan the preventive and causal therapy of breast cancer. Paradoxically, the higher the increase in endogenous estrogen level, the higher the promising dose of estrogen therapy. Consequently, the quantitative evaluation of ER refractoriness in breast cancer patients will gain crucial importance in the therapeutic schedule. In the future, the development of estrogen sensitizer compounds will be a great challenge for the pharmaceutical industry instead of trying to produce an “ideal” selective estrogen receptor blocker.
Disclosure
The author reports no conflicts of interest in this work.
References
Dorgan JF, Longcope C, Stephenson HE Jr, et al. Relation of prediagnostic serum estrogen and androgen levels to breast cancer risk. Cancer Epidemiol Biomarkers Prev. 1996;5(7):533–539. | ||
Cauley JA, Lucas FL, Kuller LH, Stone K, Browner W, Cummings SR. Elevated serum estradiol and testosterone concentrations are associated with a high risk for breast cancer. Study of Osteoporotic Fractures Research Group. Ann Intern Med. 1999;130(4 Pt 1):270–277. | ||
The Endogenous Hormones and Breast Cancer Collaborative Group. Endogenous sex hormones and breast cancer in postmenopausal women: reanalysis of nine prospective studies. J Natl Cancer Inst. 2002;94(8):606–616. | ||
Endogenous Hormones and Breast Cancer Collaborative Group. Sex hormones and risk of breast cancer in premenopausal women: a collaborative reanalysis of individual participant data from seven prospective studies. Lancet Oncol. 2013;14(10):1009–1019. | ||
Kaaks R, Berrino F, Key T, et al. Serum sex steroids in premenopausal women and breast cancer risk within the European Prospective Investigation into Cancer and Nutrition (EPIC). J Natl Cancer Inst. 2005;97(10):755–765. | ||
Fortner RT, Eliassen AH, Spiegelman D, et al. Premenopausal endogenous steroid hormones and breast cancer risk: results from the Nurses’ Health Study II. Breast Cancer Res. 2013;15(2):R19. | ||
Zeleniuch-Jacquotte A, Afanasyeva Y, Kaaks R, et al. Premenopausal serum androgens and breast cancer risk: a nested case-control study. Breast Cancer Res. 2012;14(1):R32. | ||
Micheli A, Muti P, Secreto G, et al. Endogenous sex hormones and subsequent breast cancer in premenopausal women. Int J Cancer. 2004;112(2):312–318. | ||
Schernhammer ES, Sperati F, Razavi P, et al. Endogenous sex steroids in premenopausal women and risk of breast cancer: the ORDET cohort. Breast Cancer Res. 2013;15:R46. | ||
Eliassen AH, Missmer SA, Tworoger SS, et al. Endogenous steroid hormone concentrations and risk of breast cancer among premenopausal women. J Natl Cancer Inst. 2006;98(19):1406–1415. | ||
Walker K, Bratton DJ, Frost C. Premenopausal endogenous oestrogen levels and breast cancer risk: a meta-analysis. Br J Cancer. 2011;105(9):1451–1457. | ||
James RE, Lukanova A, Dossus L, et al. Postmenopausal serum sex steroids and risk of hormone receptor-positive and -negative breast cancer: a nested case-control study. Cancer Prev Res (Phila). 2011;4(10):1626–1635. | ||
Zhang X, Tworoger SS, Eliassen AH, Hankinson SE. Postmenopausal plasma sex hormone levels and breast cancer risk over 20 years of follow-up. Breast Cancer Res Treat. 2013;137(3):883–892. | ||
Farhat GN, Cummings SR, Chlebowski RT, et al. Sex hormone levels and risks of estrogen receptor-negative and estrogen receptor-positive breast cancers. J Natl Cancer Inst. 2011;103(7):562–570. | ||
Schoemaker MJ, Folkerd EJ, Jones ME, et al. Combined effects of endogenous sex hormone levels and mammographic density on postmenopausal breast cancer risk: results from the Breakthrough Generations Study. Br J Cancer. 2014;110:1898–1907. | ||
Zeleniuch-Jacquotte A, Shore RE, Koenig KL, et al. Postmenopausal levels of oestrogen, androgen, and SHBG and breast cancer: long-term results of a prospective study. Br J Cancer. 2004;90(1):153–159. | ||
Baglietto L, Severi G, English DR, et al. Circulating steroid hormone levels and risk of breast cancer for postmenopausal women. Cancer Epidemiol Biomarkers Prev. 2010;19(2):492–502. | ||
Manjer J, Johansson R, Berglund G, et al. Postmenopausal breast cancer risk in relation to sex steroid hormones, prolactin and SHBG (Sweden). Cancer Causes Control. 2003;14(7):599–607. | ||
Hankinson SE, Willett WC, Manson JE, et al. Plasma sex steroid hormone levels and risk of breast cancer in postmenopausal women. J Natl Cancer Inst. 1998;90(17):1292–1299. | ||
Missmer SA, Eliassen AH, Barbieri RL, Hankinson SE. Endogenous estrogen, androgen, and progesterone concentrations and breast cancer risk among postmenopausal women. J Natl Cancer Inst. 2004;96(24):1856–1865. | ||
Cummings SR, Lee JS, Lui LY, Stone K, Ljung BM, Cauley JA. Sex hormones, risk factors, and risk of estrogen receptor-positive breast cancer in older women: a long-term prospective study. Cancer Epidemiol Biomarkers Prev. 2005;14(5):1047–1051. | ||
Sieri S, Krogh V, Bolelli G, et al. Sex hormone levels, breast cancer risk, and cancer receptor status in postmenopausal women: the ORDET cohort. Cancer Epidemiol Biomarkers Prev. 2009;18(1):169–176. | ||
Fuhrman BJ, Schairer C, Gail MH, et al. Estrogen metabolism and risk of breast cancer in postmenopausal women. J Natl Cancer Inst. 2012;104(4):326–339. | ||
Sturgeon SR, Potischman N, Malone KE, et al. Serum levels of sex hormones and breast cancer risk in premenopausal women: a case-control study (USA). Cancer Causes Control. 2004;15(1):45–53. | ||
Endogenous Hormones and Breast Cancer Collaborative Group. Circulating sex hormones and breast cancer risk factors in postmenopausal women: reanalysis of 13 studies. Br J Cancer. 2011;105:709–722. | ||
Falk RT, Brinton LA, Dorgan JF, et al. Relationship of serum estrogens and estrogen metabolites to postmenopausal breast cancer risk: a nested case-control study. Breast Cancer Res. 2013;15(2):R34. | ||
Lamar CA, Dorgan JF, Longcope C, Stanczyk FZ, Falk RT, Stephenson HE Jr. Serum sex hormones and breast cancer risk factors in postmenopausal women. Cancer Epidemiol Biomarkers Prev. 2003;12(4):380–833. | ||
Björnström L, Sjöberg M. Mechanisms of estrogen receptor signaling: convergence of genomic and nongenomic actions on target genes. Mol Endocrinol. 2005;19(4):833–842. | ||
Ascenzi P, Bocedi A, Marino M. Structure-function relationship of estrogen receptor alpha and beta: impact on human health. Mol Aspects Med. 2006;27(4):299–402. | ||
Levin ER. Integration of the extranuclear and nuclear actions of estrogen. Mol Endocrinol. 2005;19(8):1951–1959. | ||
Suba Z. Common soil of smoking-associated and hormone-related cancers: estrogen deficiency. Oncol Rev. 2010;4(2):73–87. | ||
Suba Z. Low estrogen exposure and/or defective estrogen signaling induces disturbances in glucose uptake and energy expenditure. J Diabetes Metab. 2013;4:272. | ||
Smith CL. Cross-talk between peptide growth factor and estrogen receptor signaling pathways. Biol Reprod. 1998;58(3):627–632. | ||
Smith EP, Boyd J, Frank GR, et al. Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man. N Engl J Med. 1994;331:1056–1061. | ||
Quaynor SD, Stradtman EW, Kim HG, et al. Delayed puberty and estrogen resistance in a woman with estrogen receptor α variant. N Engl J Med. 2013;369(2):164–171. | ||
Barros RP, Machado UF, Gustafsson JA. Estrogen receptors: new players in diabetes mellitus. Trends Mol Med. 2006;12(9):425–431. | ||
Barros RP, Gustafsson JÅ. Estrogen receptors and the metabolic network. Cell Metab. 2011;14(3):289–299. | ||
Morishima A, Grumbach MM, Simpson ER, Fisher C, Qin K. Aromatase deficiency in male and female siblings caused by a novel mutation and the physiological role of estrogens. J Clin Endocrinol Metab. 1995;80(12):3689–3698. | ||
Simpson ER. Sources of estrogen and their importance. J Steroid Biochem Mol Biol. 2003;86(3–5):225–230. | ||
Maffei L, Rochira V, Zirilli L, et al. A novel compound heterozygous mutation of the aromatase gene in an adult man: reinforced evidence on the relationship between congenital oestrogen deficiency, adiposity and the metabolic syndrome. Clin Endocrinol (Oxf). 2007;67(2):218–224. | ||
Spangenburg EE, Wohlers LM, Valencia AP. Metabolic dysfunction under reduced estrogen levels: looking to exercise for prevention. Exerc Sport Sci Rev. 2012;40(4):195–203. | ||
Faulds MH, Zhao C, Dahlman-Wright K, Gustaffson J. The diversity of sex steroid action: regulation of metabolism by estrogen signaling. J Endocrinol. 2012;212:3–12. | ||
Suba Z. Interplay between insulin resistance and estrogen deficiency as co-activators in carcinogenesis. Pathol Oncol Res. 2012;18(2):123–133. | ||
Suba Z. Circulatory estrogen level protects against breast cancer in obese women. Recent Pat Anticancer Drug Discov. 2013;8(2):154–167. | ||
Stamataki KE, Spina J, Rangou DB, Chlouverakis CS, Piaditis GP. Ovarian function in women with non-insulin dependent diabetes mellitus. Clin Endocrinol (Oxf). 1996;45(5):615–621. | ||
Vriend J, Bertalanffy FD, Ralcewicz TA. The effects of melatonin and hypothyroidism on estradiol and gonadotropin levels in female Syrian hamsters. Biol Reprod. 1987;36:719–728. | ||
Suba Z. Light deficiency confers breast cancer risk by endocrine disorders. Recent Pat Anticancer Drug Discov. 2012;7(3):337–344. | ||
Nestler JE. Obesity, insulin, sex steroids and ovulation. Int J Obes Relat Metab Disord. 2000;24 Suppl 2: S71–S73. | ||
Björntorp P. The regulation of adipose tissue distribution in humans. Int J Obes Relat Metab Disord. 1996;20(4):291–302. | ||
Christakou CD, Diamanti-Kandarakis E. Role of androgen excess on metabolic aberrations and cardiovascular risk in women with polycystic ovary syndrome. Womens Health (Lond Engl). 2008;4(6):583–594. | ||
Attia GR, Rainey WE, Carr BR. Metformin directly inhibits androgen production in human thecal cells. Fertil Steril. 2001;76(3):517–524. | ||
Diamanti-Kandarakis E, Baillargeon JP, Iuorno MJ, Jakubowicz DJ, Nestler JE. A modern medical quandary: polycystic ovary syndrome, insulin resistance, and oral contraceptive pills. J Clin Endocrinol Metab. 2003;88(5):1927–1932. | ||
Lindberg BS, Johansson ED, Nilsson BA. Plasma levels of nonconjugated oestrone, oestradiol–17beta and oestriol during uncomplicated pregnancy. Acta Obstet Gynecol Scand Suppl. 1974;2:21–36. | ||
Key TJ. Hormones and cancer in humans. Mutat Res. 1995;333(1–2):59–67. | ||
Britt K, Ashworth A, Smalley M. Pregnancy and the risk of breast cancer. Endocr Relat Cancer. 2007;14(4):907–933. | ||
Maggi A. Liganded and unliganded activation of estrogen receptor and hormone replacement therapies. Biochim Biophys Acta. 2011;1812(8):1054–1060. | ||
Jordan VC, Ford LG. Paradoxical clinical effect of estrogen on breast cancer risk: a “new” biology of estrogen-induced apoptosis. Cancer Prev Res (Phila). 2011;4(5):633–637. | ||
Suba Z. Failures and controversies of the antiestrogen treatment of breast cancer. In: Suba Z, editor. Estrogen Prevention for Breast Cancer. New York, NY: Nova Science Publishers Inc.; 2013:105–125. | ||
Widschwendter M, Rosenthal AN, Philpott S, et al. The sex hormone system in carriers of BRCA1/2 mutations: a case-control study. Lancet Oncol. 2013;14(12):1226–1232. | ||
Kim J, Oktay K. Baseline E(2) levels are higher in BRCA2 mutation carriers: a potential target for prevention? Cancer Causes Control. 2013;24(3):421–426. | ||
Suba Z. Triple-negative breast cancer risk in women is defined by the defect of estrogen signaling: preventive and therapeutic implications. Onco Targets Ther. 2014;7:147–164. | ||
Oktay K, Kim JY, Barad D, Babayev SN. Association of BRCA1 mutations with occult primary ovarian insufficiency: a possible explanation for the link between infertility and breast/ovarian cancer risks. J Clin Oncol. 2010;28(2):240–244. | ||
Finch A, Valentini A, Greenblatt E, et al; Hereditary Breast Cancer Study Group. Frequency of premature menopause in women who carry a BRCA1 or BRCA2 mutation. Fertil Steril. 2013;99(6):1724–1728. | ||
Lin WT, Beattie M, Chen LM, et al. Comparison of age at natural menopause in BRCA 1/2 mutation carriers with a non-clinic-based sample of women in northern California. Cancer. 2013;119(9):1652–1659. | ||
Bordeleau L, Lipscombe L, Lubinski J, et al; Hereditary Breast Cancer Clinical Study Group. Diabetes and breast cancer among women with BRCA1 and BRCA2 mutations. Cancer. 2011;117(9):1812–1818. | ||
Wang KC, Woung LC, Tsai MT, Liu CC, Su YH, Li CY. risk of alzheimer’s disease in relation to diabetes: a population-based cohort study. Neuroepidemiology. 2012;38:237–244. | ||
Carcaillon L, Brailly-Tabard S, Ancelin ML, et al. High plasma estradiol interacts with diabetes on risk of dementia in older postmenopausal women. Neurology. 2014;82(6):504–511. | ||
Wu SI, Chou P, Tsai ST. The impact of years since menopause on the development of impaired glucose tolerance. J Clin Epidemiol. 2001;54(2):117–120. | ||
Onland-Moret NC, van Gils CH, Roest M, Grobbee DE, Peeters PH. The estrogen receptor alpha gene and breast cancer risk (the Netherlands). Cancer Causes Control. 2005;16(10):1195–1202. | ||
Herynk MH, Fuqua SA. Estrogen receptor mutations in human disease. Endocr Rev. 2004;25(6):869–898. | ||
Shin A, Kang D, Nishio H, et al. Estrogen receptor alpha gene polymorphisms and breast cancer risk. Breast Cancer Res Treat. 2003;80:127–131. | ||
Cai Q, Shu XO, Jin F, et al. Genetic polymorphisms in the estrogen receptor alpha gene and risk of breast cancer: results from the Shanghai Breast Cancer Study. Cancer Epidemiol Biomarkers Prev. 2003;12:853–859. | ||
Zheng SL, Zheng W, Chang BL, et al. Joint effect of estrogen receptor beta sequence variants and endogenous estrogen exposure on breast cancer risk in Chinese women. Cancer Res. 2003;63(22):7624–7629. | ||
Ryan J, Canonico M, Carcaillon L, et al. Hormone treatment, estrogen receptor polymorphisms and mortality: a prospective cohort study. PLoS One. 2012;7:e34112. | ||
Bloomgarden ZT. Second World Congress on the Insulin Resistance Syndrome: mediators, pediatric insulin resistance, the polycystic ovary syndrome, and malignancy. Diabetes Care. 2005;28(8):1821–1830. | ||
Suba Z, Ujpál M. [Correlations of insulin resistance and neoplasms]. Magy Onkol. 2006;50(2):127–135. Hungarian. | ||
Reaven GM. Banting lecture 1988: role of insulin resistance in human disease. Diabetes. 1988;37(12):1595–1607. | ||
Gupta K, Krishnaswamy G, Karnad A, Peiris A. Insulin: a novel factor in carcinogenesis. Am J Med Sci. 2002;323(3):140–145. | ||
Yu H, Rohan T. Role of insulin-like growth factor family in cancer development and progression. J Natl Cancer Inst. 2000;92(18):1472–1489. | ||
Sartor BM, Dickey RP. Polycystic ovarian syndrome and the metabolic syndrome. Am J Med Sci. 2005;330(6):336–342. | ||
Moghetti P, Castello R, Negri C, et al. Insulin infusion amplifies 17 alpha-hydroxycorticosteroid intermediates response to adrenocorticotropin in hyperandrogenic women: apparent relative impairment of 17, 20-lyase activity. J Clin Endocrinol Metab. 1996;81(3):881–886. | ||
Cara JF, Rosenfield RL. Insulin-like growth factor I and insulin potentiate luteinizing hormone-induced androgen synthesis by rat ovarian thecal-interstitial cells. Endocrinology. 1988;123(2):733–739. | ||
Shupnik MA, Gordon MS, Chin WW. Tissue-specific regulation of rat estrogen receptor mRNAs. Mol Endocrinol. 1989;3(4):660–665. | ||
Pascall JC. Post-transcriptional regulation of gene expression by androgens: recent observations from the epidermal growth factor gene. J Mol Endocrinol. 1997;18:177–180. | ||
Veldhuis JD, Frystyk J, Iranmanesh A, Ørskov H. Testosterone and estradiol regulate free insulin-like growth factor I (IGF-I), IGF binding protein 1 (IGFBP-1), and dimeric IGF-I/IGFBP-1 concentrations. J Clin Endocrinol Metab. 2005;90(5):2941–2947. | ||
Lukanova A, Lundin E, Zeleniuch-Jacquotte A, et al. Body mass index, circulating levels of sex-steroid hormones, IGF-I and IGF-binding protein-3: a cross-sectional study in healthy women. Eur J Endocrinol. 2004;150(2):161–171. | ||
Stoica A, Saceda M, Doraiswamy VL, Coleman C, Martin MB. Regulation of estrogen receptor-alpha gene expression by epidermal growth factor. J Endocrinol. 2000;165(2):371–378. | ||
Curtis SW, Washburn T, Sewall C, et al. Physiological coupling of growth factor and steroid receptor signaling pathways: estrogen receptor knockout mice lack estrogen-like response to epidermal growth factor. Proc Natl Acad Sci U S A. 1996;93(22):12626–12630. | ||
Larsson SC, Mantzoros CS, Wolk A. Diabetes mellitus and risk of breast cancer: a meta-analysis. Int J Cancer. 2007;121(4):856–862. | ||
Fan S, Ma YX, Wang C, et al. Role of direct interaction in BRCA1 inhibition of estrogen receptor activity. Oncogene. 2001;20(1):77–87. | ||
Zheng L, Annab LA, Afshari CA, Lee WH, Boyer TG. BRCA1 mediates ligand-independent transcriptional repression of the estrogen receptor. Proc Natl Acad Sci U S A. 2001;98(17):9587–9592. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.