Back to Journals » Neuropsychiatric Disease and Treatment » Volume 15
Disturbances of mitochondrial parameters to distinguish patients with depressive episode of bipolar disorder and major depressive disorder
Authors Zvěřová M ![]() , Hroudová J
, Hroudová J ![]() , Fišar Z
, Fišar Z ![]() , Hansíková H
, Hansíková H ![]() , Kališová L
, Kališová L ![]() , Kitzlerová E, Lambertová A, Raboch J
, Kitzlerová E, Lambertová A, Raboch J ![]()
Received 3 October 2018
Accepted for publication 13 December 2018
Published 14 January 2019 Volume 2019:15 Pages 233—240
DOI https://doi.org/10.2147/NDT.S188964
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Martina Zvéřová,1 Jana Hroudová,1,2 Zdeněk Fišar,1 Hana Hansíková,3 Lucie Kališová,1 Eva Kitzlerová,1 Alena Lambertová,1 Jiří Raboch1
1Department of Psychiatry, First Faculty of Medicine, Charles University and General University Hospital in Prague, 120 00 Prague 2, Czech Republic; 2Institute of Pharmacology, First Faculty of Medicine, Charles University and General University Hospital in Prague, 128 00 Prague 2, Czech Republic; 3Department of Pediatrics and Adolescent Medicine, First Faculty of Medicine, Charles University and General University Hospital in Prague, 120 00 Prague 2, Czech Republic
Background: Mitochondrial dysfunctions are implicated in the pathophysiology of mood disorders. We measured and examined the following selected mitochondrial parameters: citrate synthase (CS) activity, electron transport system (ETS) complex (complexes I, II, and IV) activities, and mitochondrial respiration in blood platelets.
Patients and methods: The analyses were performed for 24 patients suffering from a depressive episode of bipolar affective disorder (BD), compared to 68 patients with MDD and 104 healthy controls. BD and unipolar depression were clinically evaluated using well-established diagnostic scales and questionnaires.
Results: The CS, complex II, and complex IV activities were decreased in the depressive episode of BD patients; complex I and complex I/CS ratio were significantly increased compared to healthy controls. We observed significantly decreased complex II and CS activities in patients suffering from MDD compared to controls. Decreased respiration after complex I inhibition and increased residual respiration were found in depressive BD patients compared to controls. Physiological respiration and capacity of the ETS were decreased, and respiration after complex I inhibition was increased in MDD patients, compared to controls. Increased complex I activity can be a compensatory mechanism for decreased CS and complex II and IV activities.
Conclusion: We can conclude that complex I and its abnormal activity contribute to the defects in cellular energy metabolism during a depressive episode of BD. The observed parameters could be used in a panel of biomarkers that could selectively distinguish BD depression from MDD and can be easily examined from blood elements.
Keywords: affective disorder, biomarker, oxidative phosphorylation, mitochondrial enzyme, platelet
Introduction
Bipolar affective disorder (BD) is a major mental disorder characterized by fluctuations in mood state and energy with intermittent episodes of depressive episodes and hypomania or mania; rapid cycling, mixed states and psychotic symptoms can occur in some cases.1 BD is a common, severe, recurrent chronic, and often life-threatening illness with high prevalence of psychiatric and medical comorbidities. Life expectancy in BD is reduced by 10 years or more.2 It affects >1% of the world’s population irrespective of ethnic origin, nationality, or socioeconomic status.
The depressive phase contributes to the majority of morbidity and mortality (an increased risk of suicide) in BD. Depression also leads to impairment in cognitive and social functioning, which is often as severe as that in other chronic medical illnesses.3 In typical mild, moderate, or severe depressive episodes, the patient suffers from lower mood, decrease in activity, decrease in pleasure (anhedonia), and reduction of energy. Sleep is usually disturbed, and appetite is diminished. Some ideas of worthlessness or guilt, even in the mild form, are often present. The worst consequence of depression could be suicidality. Depressive symptoms may be similarly expressed in unipolar depression (MDD) and BD disorder, but changes in specific brain networks can be very distinct.4
Psychiatric nosology since the Diagnostic and Statistical Manual of Mental Disorders, Third Edition (DSM-III) has classified MDD separately from BD. BD and MDD have been categorized as separate branches on the mood disorder diagnostic tree with different pharmacological approaches. MDD has been seen as less endogenous than BD depression.5
BD is commonly misdiagnosed as other mental disorders, including depression, anxiety disorders, obsessive–compulsive disorders, schizophrenia, and personality disorders, resulting in the mistreatment of clinical symptoms and an increase in recurrent episodes.6 Compared to MDD, BD depression appears to be associated with greater short-term mood variability, more frequent episodes, and a younger age of onset. No consistent differences have been found between episode lengths, although some studies suggest that unipolar depression episodes are longer than bipolar depression episodes. The severity of depression appears comparable in patients with MDD and bipolar disorders.7 Distinguishing BD depression and MDD as early as possible is fundamental for correct clinical choices and, consequently, better short- and long-term outcomes. Only 20% of the BD patients receive the correct diagnosis within the first year, and >60% of them are misdiagnosed as MDD patients.8
Because MDD and BD depressions are different disorders, observable differences in symptomatology, course, biology, or psychosocial factors would be expected. A stronger genetic contribution to BD depression than MDD has been confirmed.2
The idea of the “same disorder” model could be drawn from the absence of replicable differences in symptomatology, course, biology, or psychosocial cofactors of BD depression and MDD.5
Although there is evidence for altered functional and structural brain connectivity and changes in mitochondrial function, dopamine, markers of oxidative stress, inflammation, and circadian rhythms, it remains difficult to separate primary causative changes from those that are secondary to the disorder and/or its treatment.9 There are currently no valid biomarkers for BD or MDD. For example, metabolic changes have been observed with opposite variations of uric acid in BD and MDD.10,11 Patients with MDD had lower levels of uric acid,11 whereas BD patients in mania and augmented treatment with allopurinol showed decrease in manic symptoms.10 It was suggested that uric acid could stimulate ATP production in mania of BD patients.12
The association of mitochondrial dysfunctions with psychiatric illnesses has been observed both in vitro and in vivo.13–15 Mitochondria strongly affect all intracellular processes coupled to signal transduction, neuronal survival, and plasticity. In addition to their crucial role in the generation of ATP, mitochondria are involved in other important intracellular processes, including the regulation of ROS, calcium signaling, and apoptosis. Mitochondrial dysfunctions and defects in oxidative metabolism are characteristics of many complex illnesses that are not necessarily categorized as mitochondrial diseases.1 The hypothesis of mitochondrial dysfunctions in BD is based on the findings from mitochondrial respiration, activities of mitochondrial enzyme complexes, ROS production and antioxidant activity, and magnetic resonance spectroscopy.16,17
Mitochondrial dysfunctions in BD have not been observed consistently.1 Studies are often limited by a small number of recruited BD patients, patients’ noncompliance, different pharmacotherapies, and differences in the BD episodes, when patients were recruited. No differences in the CS, succinate dehydrogenase, and malate dehydrogenase activities were found in leukocytes from drug-naïve BD patients during depressive episodes compared to healthy controls.18 However, the study was limited by a small sample size (18 subjects); the young ages of the subjects, who were minimally exposed to medication; and the recent onset of BD. No differences in complex I activity were found postmortem in prefrontal cortex mitochondria from BD and controls.19 The activity of other complexes or mitochondrial respiration has not been determined. High complex I activity was found in patients without medication; decreased complex I activity was found in patients with detectable psychotropic medications.19
The hypothesis was tested that the activities of respiratory chain complexes and functioning of the OXPHOS system differ in BD and MDD. The recent studies showed decreased mitochondrial respiration, decreased ATP and high-energy phosphates, and decreased pH in the brain of BD patients.20 A bioenergetic model of BD is based on disturbed energy availability, as a biphasic disorder, and increased ATP levels in mania have been discussed.12 Therefore, we hypothesized that depressive patients in BD could have decreased mitochondrial energy metabolism and in this matter observed parameters could serve as distinguishing markers of BD and MDD. We do not expect disturbed activity of respiratory chain complexes in MDD and controls.
The aim of this study is to compare mitochondrial dysfunctions in peripheral blood of patients with MDD, patients with depressive episodes of BD, and controls. The activities of CS and ETS complexes and changes in mitochondrial respiration were studied in blood platelets from BD and MDD patients as potential biological markers of these mood disorders that could be easily examined using peripheral blood samples.
Patients and methods
Study design and participants
Patients in a depressive episode of BD and patients with MDD were recruited for this study. The control group consisted of healthy volunteers. The baseline characteristics of participants are summarized in Table 1. The severity of depression was evaluated by the Hamilton Depressive Rating Scale, 21-item (HDRS-21) and by Clinical Global Impression - severity scale (CGI-01). The patients met the following inclusion criteria: age 18–65 years, no psychoactive substance abuse, no cognitive impairment, and organic brain disease. The patients did not have a serious somatic disease or chronic somatic pharmacotherapy. The patients did not take mitochondria-targeting compounds, such as L-carnitine, coenzyme Q10, vitamin E, antibiotics (chloramphenicol, doxycycline, and quinolones), monoclonal antibodies, or immunosuppressive drugs (azathioprine, cyclosporine, everolimus, and tacrolimus).
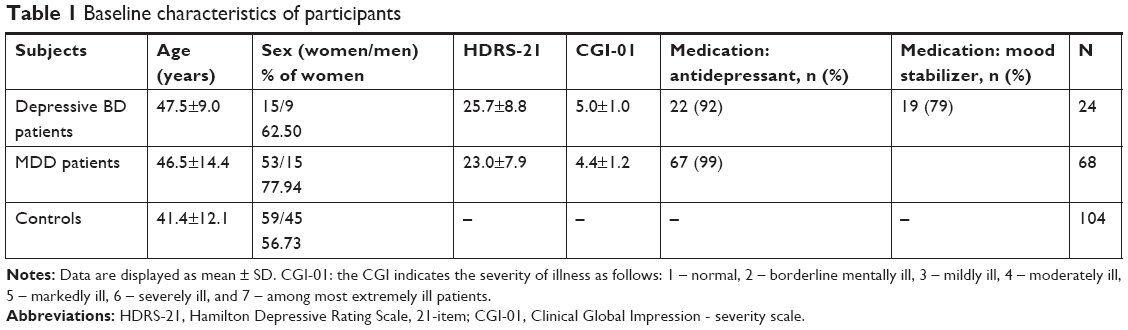 | Table 1 Baseline characteristics of participants |
The study was conducted according to the principles expressed in the Declaration of Helsinki, and the study protocol was approved by the ethical review board of the First Faculty of Medicine and General University Hospital in Prague, Czech Republic. Written informed consent was obtained from all participants.
Chemicals and solutions
The following buffers were used for platelet isolation: buffer A (120 mM NaCl, 13 mM sodium citrate, and 30 mM dextrose, pH 7.0); buffer B (154 mM NaCl, 10 mM Tris/HCl, and 1 mM EDTA, pH 7.4); and buffer C (138 mM NaCl, 2.9 mM KCl, 12 mM NaHCO3, 0.36 mM NaH2PO4, 5.5 mM glucose, 1.8 mM CaCl2, and 0.4 mM MgCl2, pH 7.4). Intact platelets in the platelet-rich plasma were diluted in Krebs–Henseleit (KH) medium without Ca2+, which consisted of 118 mM NaCl, 4.7 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4·7H2O, 25 mM NaHCO3, and 11.1 mM glucose (pH 7.4); the KH medium was saturated with oxygen prior to use by percolation with a 95% O2 and 5% CO2 gas mixture.
The following stock solutions were used: 4 mg/mL of oligomycin, 1 mM and 10 mM carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP), 1 mM rotenone, and 0.5 mg/mL of antimycin A.
All chemicals were purchased from Sigma-Aldrich Co. (St Louis, MO, USA).
Platelet preparation
Blood samples were taken from the antecubital vein of each fasting participant (refrained from coffee and cigarettes) before administration of the morning medication. Peripheral blood was drawn into BD Vacutainer® blood collection tubes with an anticoagulant. Buffered sodium citrate was used as the anticoagulant for the CS and complex I, II, and IV activity measurements, and K2EDTA was used as the anticoagulant for the platelet respiratory rate measurement. Platelet-rich plasma was separated by centrifugation at 150 × g for 20 minutes at 25°C. Subsequently, the platelets were counted by microscopy using a counting chamber and used for the mitochondrial respiratory rate measurements.
For measurement of the activities of complexes I, II, and IV and CS, platelets were isolated from 9 mL blood by the modified method according to Fox et al.21 The plasma with platelets was centrifuged at 730 × g for 20 minutes. The pelleted platelets were resuspended in 5 mL of buffer A, and the suspension was centrifuged at 730 × g for 10 minutes. Pellet was resuspended in 5 mL of buffer B and centrifuged in the same conditions. The final pellet of platelets was resuspended in 500 μL of buffer C. Fresh cells were used for the measurement of complexes I and II; frozen aliquots of the cells stored at −80°C were used for the determination of complex IV and CS activities.
Activities of respiratory chain complexes and CS
Activities of complex I (NADH:ubiquinone reductase; EC 1.6.5.3), complex II (succinate dehydrogenase [quinone]; EC 1.3.5.1), complex IV (cytochrome c oxidase; EC 1.9.3.1), and CS (EC 2.3.3.1) were measured spectrophotometrically, as described previously.22–25
High-resolution respirometry
The mitochondrial respiratory rate was measured at 37°C using a high-resolution oxygraph (Oxygraph-2k; Oroboros Instruments, Innsbruck, Austria). The sample was placed in the incubation chamber and stirred continuously; the oxygen concentration was monitored over time, and the rate of oxygen consumption (O2 flow per cell, respiratory rate) was calculated. Assessments of cellular respiration were performed immediately after blood collection to avoid a decline in ATP synthase and complex IV activities during storage.26
To distinguish between specific mitochondrial defects and respiratory alterations related to the cellular status, the respiratory rates were normalized to 1) the platelet concentration and 2) the maximal mitochondrial oxygen consumption rate.27,28
Platelet-rich plasma was diluted twofold in the KH medium. We measured mitochondrial parameters in the intact platelets using the modified method of Sjövall et al.29 Mitochondrial respiration in intact platelets was determined in the following parameters: PR – physiological respiration of intact platelets without any additions, measured in diluted platelet-rich plasma; LEAK – nonphosphorylating respiration, respiration independent of ADP phosphorylation, measured after addition of oligomycin (state 4o); ETSC – maximal capacity of ETS measured as noncoupled respiration by titration with uncoupler (FCCP); Rot – respiration after complex I inhibition, this state was measured after the addition of rotenone (complex I inhibitor); and ROX – state after addition of antimycin A (complex III inhibitor); ROX was subtracted from all other respiratory rates.
Data analysis
Data were collected and analyzed using the DatLab 4.3 software (Oroboros Instruments) by displaying the real-time oxygen concentration and oxygen flux, which was the negative time derivative of the oxygen concentration. The oxygen solubility factor relative to distilled water was set at 0.905 for plasma diluted 1:1 with KH buffer.29 The respiratory rates (oxygen fluxes) were expressed as pmol O2 consumed per second relative to 106 platelets.
The flux control ratios were calculated to evaluate the mitochondrial parameters independent of the mitochondrial contents and sizes. These calculations were used to determine the relative contributions of the different respiratory states. When residual respiration (ROX) was subtracted, the flux control ratios in the intact platelets were calculated as PR/ETSC, LEAK/ETSC, NetPR = (PR – LEAK)/ETSC and Rot/ETSC.
All data are presented as mean ± SD. The statistical analysis was performed in R statistical package version 3.4.2, R Core Team (2017).30 A nonparametric Kruskal–Wallis test was applied to compare the BD depression, MDD depression, and control groups. In the case of comparing only two groups (BD depression and MDD), a nonparametric Mann–Whitney–Wilcoxon test was used. For post hoc comparisons, a set of Mann–Whitney–Wilcoxon tests was adopted with P-values adjusted for multiple comparisons by a Holm’s method.
Results
Twenty-four patients in a depressive episode of BD, 68 patients in depressive episode of MDD, and 104 age-matched healthy controls were recruited for this study. Patients were consecutively recruited, and the number of selected subjects was arbitrarily chosen. Healthy controls were selected mostly from the staff of Department of Psychiatry. The Kruskal–Wallis test was applied for the statistical evaluation of results.
Clinicians provided the clinical evaluation of both groups of depressive patients; the severity of depression was evaluated by HDRS-21 and CGI-01. The psychopathology level was high – corresponding to at least medium severity depression, and all the included patients were in a marked depressive state; the baseline characteristics of participants are summarized in Table 1. The Mini-Mental State Examination was used to measure the cognitive impairment. The results could be transposed only for population from 18 to 65 years; the activity of complexes is decreasing with the age.22
Sixteen MDD patients were drug naïve, and other patients were on pharmacotherapy (some of them malcompliant or noncompliant). Details are mentioned in Table 1.
Activities of respiratory chain complexes and CS
Complex I activity was significantly increased in BD patients compared to the controls (BD vs controls: P=0.016) and MDD patients (BD vs MDD: P=0.004). Complex II (BD vs controls: P=0.009, MDD vs controls: P=0.006) was decreased in patients suffering from depression; no significant difference was found between BD and MDD depression. Complex IV was significantly decreased in BD patients compared to controls and MDD patients (BD vs controls: P=0.001, BD vs MDD: P=0.007). No significant difference was found in complex I and complex IV activities between MDD patients and controls (Figure 1A).
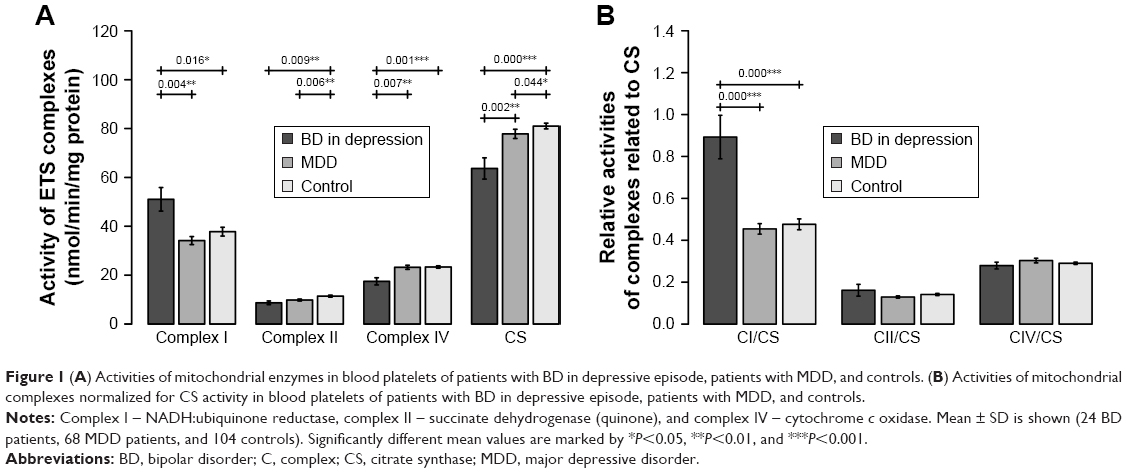 | Figure 1 (A) Activities of mitochondrial enzymes in blood platelets of patients with BD in depressive episode, patients with MDD, and controls. (B) Activities of mitochondrial complexes normalized for CS activity in blood platelets of patients with BD in depressive episode, patients with MDD, and controls. |
CS activity was significantly decreased in BD patients compared to both controls and MDD patients (BD vs controls: P<0.001, BD vs MDD: P=0.002) and in MDD vs controls (P=0.044) (Figure 1A).
An increased complex I/CS ratio was found in BD patients compared to both controls and MDD patients (BD vs controls <0.001, BD vs MDD: <0.001) (Figure 1B).
Respirometry in blood platelets
Mitochondrial respiration was measured in intact platelets, in diluted plasma without addition of substrates. PR was significantly decreased in MDD patients compared to both BD patients (BD vs MDD: P<0.001) and controls (MDD vs controls: P<0.001). PR of BD patients was not significantly different from PR in controls. Interestingly, respiration after complex I inhibition (Rot) was increased in MDD compared to both BD patients (MDD vs BD: P<0.001) and controls (MDD vs controls: P=0.004). Furthermore, Rot was significantly decreased in BD patients compared to both MDD and controls (BD vs MDD: P<0.001, BD vs controls: P<0.001) (Figure 2A).
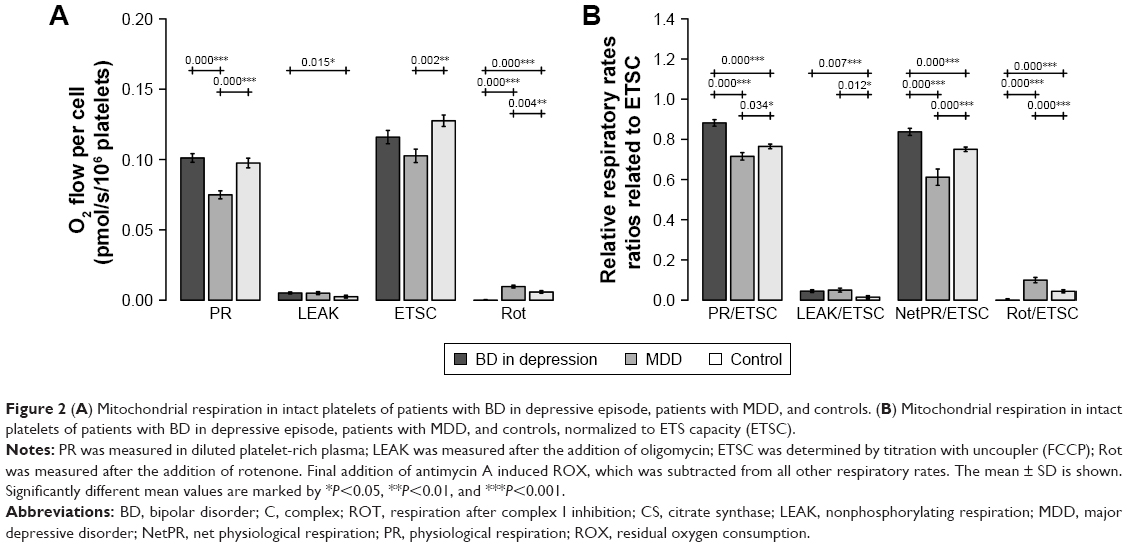 | Figure 2 (A) Mitochondrial respiration in intact platelets of patients with BD in depressive episode, patients with MDD, and controls. (B) Mitochondrial respiration in intact platelets of patients with BD in depressive episode, patients with MDD, and controls, normalized to ETS capacity (ETSC). |
Parameter LEAK was found significantly increased in BD patients compared to controls (BD vs controls: P=0.015). Parameter ETSC was decreased in MDD patients compared to controls (MDD vs controls: P=0.002).
When mitochondrial respiration was normalized to the ETS capacity (ETSC), which represents an internal functional mitochondrial marker, PR/ETSC (BD vs controls: P<0.001, BD vs MDD: P<0.001) and NetPR/ETSC (BD vs controls: P<0.001, BD vs MDD: P<0.001) ratios were significantly increased in BD patients compared to controls and MDD patients. The Rot/ETSC (BD vs controls: P<0.001, BD vs MDD: P<0.001) ratio was significantly decreased in blood platelets from the BD patients compared to controls and MDD patients. Interestingly, PR/ETSC and NetPR/ETSC ratios in MDD patients were decreased compared to those in controls (MDD vs controls: P=0.034, P<0.001, respectively). The LEAK/ETSC ratio was increased in both groups of depressive patients (BD vs controls: P=0.007, MDD vs controls: P=0.012). All significant differences are depicted in Figure 2B.
Discussion
Summary of results
We examined and compared mitochondrial respiration in blood platelets of BD patients in depressive episode, MDD patients in depressive episode, before treatment with antidepressant, and controls. Platelet mitochondria enable both in situ and in vitro measurements; they are used as a suitable model for studying the activities of both ETS complexes and mitochondrial respiration.29,31 Collection of blood platelets is only a mildly invasive method with minimal risk to study participants; low platelet sensitivity toward the mechanisms of cellular compensation enables the study of physiological and pathogenic mechanisms of diseases with mitochondrial dysfunctions.31,32
The severity of depression was high and comparable between the group of BD and MDD patients.
Interpretation and generalizability of findings
When we compared BD patients with MDD patients and controls, complex I was significantly increased, and complexes IV and CS were significantly decreased in blood platelets of BD patients. These mitochondrial parameters could be used in a panel of biomarkers to distinguish BD depression from MDD and can be easily examined from blood elements.
The CS was used to normalize the activity of the complexes and as a reference value for the number of mitochondria; decreased CS activity was observed in both BD patients and MDD patients. Only the complex I/CS ratio was increased in BD patients compared to MDD patients and controls. Increased complex I activity and an increased complex I/CS ratio were also found in blood platelets of Alzheimer’s disease patients;22 these data indicate that increased complex I activity is not a specific marker of BD and may be related to a common pathophysiology of BD and neurodegenerative disorders. We speculate that increased complex I activity can be a compensatory mechanism in response to disturbances in the OXPHOS system, eg, decreased complex II and complex IV activities.33
In intact platelets, PR was increased, and respiration after complex I inhibition was decreased in BD patients compared to MDD patients. Rotenone-induced decreases could be caused not only by the inhibition of complex I but also by an impaired complex II activity. Rot approached zero, which might suggest insufficient complex II-linked respiration in BD patients.
Increased LEAK, a nonphosphorylating state and respiration independent of ADP, supports the hypothesis that there are changes in the permeability of mitochondrial membranes in mood disorders. The parameters PR and ETS capacity were comparable to those in controls. From these results, we can assume that ETS components are not lacking; the activity of transfer molecules and coenzymes entering into OXPHOS is sufficient. More likely, the function of the complexes is disturbed and can be compensated for by increased complex I activity.
Study limitations
Some limitations of the study involve the relatively small number of BD patients, undefined lifestyle, and current pharmacotherapy. Most depressive patients were treated by antidepressants, primarily by selective serotonin reuptake inhibitors. Some patients received antidepressants with pharmacologically different mechanisms of action (eg, mirtazapine, trazodone, venlafaxine, and agomelatine). BD patients in depression were treated mostly with a combination of antidepressant and mood stabilizer: ten patients were medicated with valproate, five patients received lithium, and four patients received lamotrigine. Furthermore, the therapy could be augmented with antipsychotics (mostly atypical), benzodiazepines and hypnotics. Although higher antidepressant concentrations have been found to inhibit mitochondrial respiration in vitro,34 we were not able to observe significant differences in the mean values of the measured mitochondrial parameters in vivo. Only a part of patients were drug-naïve, most of the patients were treated with antidepressants, but we could not distinguish low compliant or noncompliant patients. Previously, mitochondrial dysfunction was associated with vulnerability to psychopathology, and antidepressant medication did not affect mitochondrial biochemical analyses.35 Therefore, we suppose only a small in vivo effect of antidepressants on the observed mitochondrial parameters. Mitochondrial respiration was measured in blood platelets, not in brain cells. Generally, the relationship between parameters measured in peripheral blood and brain has been controversial and continuously discussed. We assume that mitochondrial dysfunctions accompanying neuropsychiatric disorders are either systematic or transmitted from the brain to the periphery.
The strength of this study is the well-established clinical evaluation of patients and experimental design. This study is the first to compare mitochondrial respiration in blood platelets among BD patients in depressive episodes, MDD patients in depressive episodes, and controls.
Conclusion
We showed that changes in the activity of mitochondrial complexes and oxygen consumption may be involved in the pathophysiology of depression. Complex I, complex IV, and CS were significantly changed in the blood platelets of BD patients compared to both MDD patients and healthy controls. Therefore, these parameters can be used in a panel of biomarkers that could differentiate MDD from BD depression. Increased complex I activity can be a compensatory mechanism for decreased complex II, complex IV, and CS activities and ROS production. Underlying molecular mechanisms elucidating whether mitochondrial dysfunctions in depressive disorders are causal or consequential events should be further examined.
Supposing that the disturbances of mitochondrial functions in the brain are mirrored in platelets, our results support the hypothesis that changes in the rate of mitochondrial oxygen consumption and the activity of ETS complexes are involved in the pathophysiology of BD. The observed parameters could be further evaluated in different episodes of BD with the aim of determining whether they are state or trait markers of the disease. Distinguishing BD depression and MDD as early as possible is fundamental for correct clinical choices and, consequently, better short- and long-term outcomes.
Data sharing statement
Data are available upon request, and for further information contact the corresponding author.
Abbreviations
BD, bipolar affective disorder; CS, citrate synthase; CGI-01, Clinical Global Impression – severity scale; ETS, electron transport system; ETSC, maximal capacity of the electron transfer system; HDRS-21, Hamilton Depressive Rating Scale, 21 item; LEAK, nonphosphorylating state of respiration; MDD, major depressive disorder; OXPHOS, oxidative phosphorylation; PR, physiological respiration; Rot, respiration after complex I inhibition by rotenone; ROX, residual oxygen consumption.
Acknowledgments
The authors thank Blanka Hrubá, Marie Balíková, Eva Štolová, Suzana Knopová, Daniela Sedláčková, and Zdeněk Hanuš for their assistance. This work was supported by a grant AZV 15-28616A from the Ministry of Health, Czech Republic.
Disclosure
The authors report no conflicts of interest in this work.
References
Cikánková T, Sigitova E, Zverova M, Fisar Z, Raboch J, Hroudova J. Mitochondrial dysfunctions in bipolar disorder: Effect of the disease and pharmacotherapy. CNS Neurol Disord Drug Targets. 2017;16(2):176–186. | ||
Harrison PJ, Geddes JR, Tunbridge EM. The emerging neurobiology of bipolar disorder. Trends Neurosci. 2018;41(1):18–30. | ||
Grande I, Berk M, Birmaher B, Vieta E. Bipolar disorder. Lancet. 2016;387(10027):1561–1572. | ||
Phillips ML, Kupfer DJ. Bipolar disorder diagnosis: challenges and future directions. Lancet. 2013;381(9878):1663–1671. | ||
Cuellar AK, Johnson SL, Winters R. Distinctions between bipolar and unipolar depression. Clin Psychol Rev. 2005;25(3):307–339. | ||
Hirschfeld RM, Lewis L, Vornik LA. Perceptions and impact of bipolar disorder: how far have we really come? Results of the national depressive and manic-depressive association 2000 survey of individuals with bipolar disorder. J Clin Psychiatry. 2003;64(2):161–174. | ||
Shao L, Martin MV, Watson SJ, et al. Mitochondrial involvement in psychiatric disorders. Ann Med. 2008;40(4):281–295. | ||
Shen H, Zhang L, Xu C, Zhu J, Chen M, Fang Y. Analysis of misdiagnosis of bipolar disorder in an outpatient setting. Shanghai Arch Psychiatry. 2018;30(2):93–101. | ||
Parker G, McCraw S, Hadzi-Pavlovic D, Fletcher K. Costs of the principal mood disorders: a study of comparative direct and indirect costs incurred by those with bipolar I, bipolar II and unipolar disorders. J Affect Disord. 2013;149(1–3):46–55. | ||
Bartoli F, Crocamo C, Clerici M, Carrà G. Allopurinol as add-on treatment for mania symptoms in bipolar disorder: systematic review and meta-analysis of randomised controlled trials. Br J Psychiatry. 2017;210(1):10–15. | ||
Bartoli F, Trotta G, Crocamo C, Malerba MR, Clerici M, Carrà G. Antioxidant uric acid in treated and untreated subjects with major depressive disorder: a meta-analysis and meta-regression. Eur Arch Psychiatry Clin Neurosci. 2018;268(2):119–127. | ||
Morris G, Walder K, McGee SL, et al. A model of the mitochondrial basis of bipolar disorder. Neurosci Biobehav Rev. 2017;74(Pt A):1–20. | ||
Rezin GT, Amboni G, Zugno AI, Quevedo J, Streck EL. Mitochondrial dysfunction and psychiatric disorders. Neurochem Res. 2009;34(6):1021–1029. | ||
Jou SH, Chiu NY, Liu CS. Mitochondrial dysfunction and psychiatric disorders. Chang Gung Med J. 2009;32(4):370–379. | ||
Schapira AH, Diseases M. Mitochondrial diseases. Lancet. 2012;379(9828):1825–1834. | ||
Stork C, Renshaw PF. Mitochondrial dysfunction in bipolar disorder: evidence from magnetic resonance spectroscopy research. Mol Psychiatry. 2005;10(10):900–919. | ||
Yildiz-Yesiloglu A, Ankerst DP. Neurochemical alterations of the brain in bipolar disorder and their implications for pathophysiology: a systematic review of the in vivo proton magnetic resonance spectroscopy findings. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30(6):969–995. | ||
de Sousa RT, Streck EL, Forlenza OV, et al. Regulation of leukocyte tricarboxylic acid cycle in drug-naïve bipolar disorder. Neurosci Lett. 2015;605:65–68. | ||
Rollins BL, Morgan L, Hjelm BE, et al. Mitochondrial complex I deficiency in schizophrenia and bipolar disorder and medication influence. Mol Neuropsychiatry. 2018;3(3):157–169. | ||
Clay HB, Sillivan S, Konradi C. Mitochondrial dysfunction and pathology in bipolar disorder and schizophrenia. Int J Dev Neurosci. 2011;29(3):311–324. | ||
Fox JE, Reynolds CC, Boyles JK. Studying the platelet cytoskeleton in Triton X-100 lysates. Methods Enzymol. 1992;215:42–58. | ||
Fišar Z, Hroudová J, Hansíková H, et al. Mitochondrial respiration in the platelets of patients with Alzheimer’s disease. Curr Alzheimer Res. 2016;13(8):930–941. | ||
Böhm M, Papezova H, Hansikova H, Wenchich L, Zeman J. Activities of respiratory chain complexes in isolated platelets in females with anorexia nervosa. Int J Eat Disord. 2007;40(7):659–663. | ||
Rustin P, Chretien D, Bourgeron T, et al. Biochemical and molecular investigations in respiratory chain deficiencies. Clin Chim Acta. 1994;228(1):35–51. | ||
Hroudova J, Fisar Z. Activities of respiratory chain complexes and citrate synthase influenced by pharmacologically different antidepressants and mood stabilizers. Neuroendocrinology Letters. 2010;31(3):336–342. | ||
Diab YA, Thomas A, Luban NL, Wong EC, Wagner SJ, Levy RJ. Acquired cytochrome C oxidase impairment in apheresis platelets during storage: a possible mechanism for depletion of metabolic adenosine triphosphate. Transfusion. 2012;52(5):1024–1030. | ||
Renner K, Amberger A, Konwalinka G, Kofler R, Gnaiger E. Changes of mitochondrial respiration, mitochondrial content and cell size after induction of apoptosis in leukemia cells. Biochim Biophys Acta. 2003;1642(1–2):115–123. | ||
Stadlmann S, Renner K, Pollheimer J, et al. Preserved coupling of oxidative phosphorylation but decreased mitochondrial respiratory capacity in IL-1beta-treated human peritoneal mesothelial cells. Cell Biochem Biophys. 2006;44(2):179–186. | ||
Sjövall F, Morota S, Hansson MJ, Friberg H, Gnaiger E, Elmér E. Temporal increase of platelet mitochondrial respiration is negatively associated with clinical outcome in patients with sepsis. Crit Care. 2010;14(6):R214. | ||
R Core Team. A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2017. | ||
Sjövall F, Ehinger JK, Marelsson SE, et al. Mitochondrial respiration in human viable platelets – methodology and influence of gender, age and storage. Mitochondrion. 2013;13(1):7–14. | ||
Wang L, Wu Q, Fan Z, Xie R, Wang Z, Lu Y. Platelet mitochondrial dysfunction and the correlation with human diseases. Biochem Soc Trans. 2017;45(6):1213–1223. | ||
Lim SC, Hroudová J, van Bergen NJ, Lopez Sanchez MI, Trounce IA, McKenzie M. Loss of mitochondrial DNA-encoded protein ND1 results in disruption of complex I biogenesis during early stages of assembly. FASEB J. 2016;30(6):2236–2248. | ||
Hroudová J, Fišar Z. In vitro inhibition of mitochondrial respiratory rate by antidepressants. Toxicol Lett. 2012;213(3):345–352. | ||
Gardner A, Johansson A, Wibom R, et al. Alterations of mitochondrial function and correlations with personality traits in selected major depressive disorder patients. J Affect Disord. 2003;76(1–3):55–68. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.