")
Back to Journals » International Medical Case Reports Journal » Volume 7
Disseminated intravascular coagulopathy caused by Kikuchi–Fujimoto disease resulting in death: first case report in Turkey
Authors Uslu E, Gurbuz S, Erden A, Aykas F, Karagoz H, Karahan S, Karaman H, Cetinkaya A , Avci D
Received 8 December 2013
Accepted for publication 20 December 2013
Published 3 February 2014 Volume 2014:7 Pages 19—22
DOI https://doi.org/10.2147/IMCRJ.S58891
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Emine Uslu,1 Sibel Gurbuz,2 Abdulsamet Erden,1 Fatma Aykas,1 Hatice Karagoz,1 Samet Karahan,1 Hatice Karaman,3 Ali Cetinkaya,1 Deniz Avci1
1Internal Medicine Department, 2Infection Disease Department, 3Clinical Pathology Department, Kayseri Training and Research Hospital, Kayseri, Turkey
Abstract: Kikuchi disease, also called Kikuchi–Fujimoto disease or Kikuchi's histiocytic necrotizing lymphadenitis, is a rare, benign condition of unknown cause, usually characterized by cervical lymphadenopathy and fever. The diagnosis is based on histopathology. Our patient was a woman with bilateral cervical lymphadenopathy, fever, chest and abdominal pain, fatigue, maculopapular rash on her face, trunk, and upper and lower extremities. Immunological and rheumatological tests were negative. We took a cervical lymph node biopsy that showed a proliferative and necrotizing process centered in the paracortex characterized by patchy circumscribed or confluent areas of necrosis associated with karyorrhexis, and was remarkable by the absence of granulocytes and the paucity of plasma cells. These findings confirmed the diagnosis of Kikuchi's disease. The patient's hemoglobin values decreased, and the peripheral blood smear revealed schistocytes. Blood tests showed raised D-dimer, activated partial thromboplastin time, prothrombin time, and international normalized ratio with decreased fibrinogen. The patient's condition quickly worsened and disseminated intravascular coagulopathy eventually developed. Her initial management consisted of a corticosteroid and hydroxychloroquine.
Keywords: disseminated intravascular coagulopathy, Kikuchi–Fujimoto disease, necrotizing lymphadenitis
Introduction
Kikuchi–Fujimoto disease (KFD), or histiocytic necrotizing lymphadenitis, was originally described in Japan in 1972 by Kikuchi1 and Fujimoto et al.2 In the following years, several series of cases involving patients of different ages, races, and geographic origins were reported. This is a self-limiting process of unknown etiology, but in some reports, an immunologic or infectious event was speculated as being involved in the pathogenesis.3 However, it can rarely be fatal.4 KFD is more frequent among Asians, especially among Japanese individuals. It was thought to be much more common among women, with a female to male ratio of about 4:1, but in recent reports, the actual ratio is suggested to be closer to 1:1.5 Most of the patients are under the age of 30 years old.6 Regional tender lymphadenopathy (mostly cervical) and fever are the two most characteristic features of this disorder.7 Night sweats, weight loss, nausea, vomiting, and sore throat are the other findings that can be observed in this disease.7 Diagnosis is based on excisional biopsy of enlarged lymph nodes. The disease may be mistaken for and treated as lymphoma because it frequently mimics high-grade lymphoma.6 We report a case of disseminated intravascular coagulopathy caused by KFD.
Case report
A 32-year-old woman was admitted to hospital complaining of fever, fatigue, as well as chest and abdominal pain of 15 days’ duration, and with no personal and family history for tuberculosis and malignant diseases. Vital signs in the emergency department revealed a temperature of 39°C, a blood pressure level of 140/80 mmHg, a heart rate of 88 bpm, and a respiratory rate of 16 breaths/minute. On physical examination, she was lethargic and maculopapular rashes were patent on her face, trunk, and upper and lower extremities. She had bilateral enlarged cervical, axillary, and inguinal lymph nodes, which were freely mobile, painless on palpation, discrete and sized in the range of 0.5–3.7 cm. In Figure 1, computed tomography of the neck showed the presence of lymphadenomegalies of various sizes. Cardiovascular and respiratory examinations were unremarkable. Initial laboratory studies revealed hemoglobin levels of 10.2 g/dL, leukocytes of 17,800/mm3 with an abnormal differential count of eosinophils of 9.8%, neutrophils of 54.3%, monocytes of 14%, platelets of 205,000/mm3, lactate dehydrogenase of 578 U/L, C-reactive protein of 28.2 mg/L, and an erythrocyte sedimentation rate (ESR) of 77 mm/hour. Liver and renal tests were normal. Serologic tests for toxoplasma, Epstein–Barr virus, rubella, cytomegalovirus, herpes simplex type 1, hepatitis B, and hepatitis C were all negative. Blood, urine, and sputum cultures were negative. Sputum culture was also negative for acid-fast bacilli. Protein electrophoresis showed polyclonal gammopathy. Also, immunological and rheumatological tests, including antinuclear antibody, deoxyribonucleic acid antibody, perinuclear anti-neutrophil cytoplasmic antibodies, antineutrophil cytoplasmic antibodies, extractable nuclear antigens, and rheumatoid factor were all negative. Electrocardiogram and chest radiographs were normal.
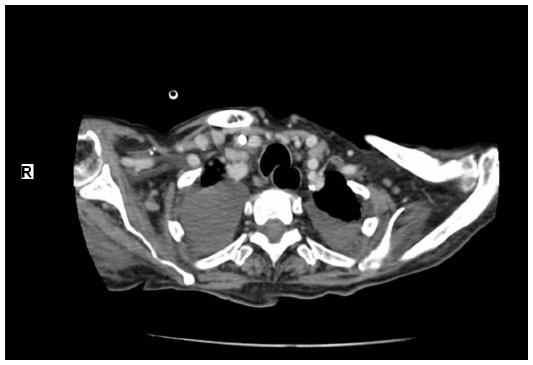 | Figure 1 Computed tomography of neck; lymphadenomegalies in various dimensions. |
Computed tomography demonstrated generalized lymphadenopathy in the axillae, cervical area, paraaortic areas, inguinal area, and mediastinum.
We took a cervical lymph node biopsy that showed karyorrhectic debris and the cellular infiltration of histiocytes and immunoblasts in the absence of polymorphonuclear leukocytes (Figure 2) and activated B-cells with histiocytes (Figure 3). An immunohistochemical study showed that the lymphoid cells adjacent to the necrotic foci were strongly positive for cluster of differentiation (CD)3, and the histiocytic cells at the periphery of the necrotic foci were strongly positive for CD68, so we thought that this histological pattern was suggestive of KFD.
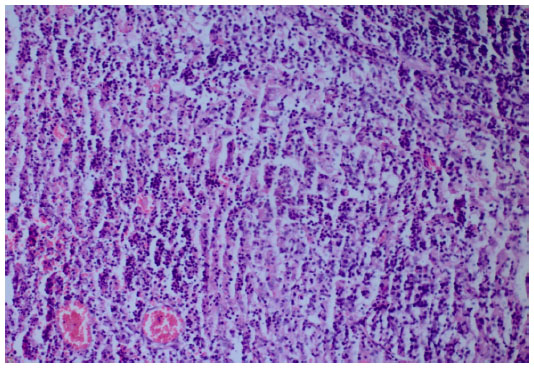 | Figure 2 Karyorrhectic debris and cellular infiltration of histiocytes and immunoblasts in the absence of polymorphonuclear leukocytes (hematoxylin and eosin stain ×200). |
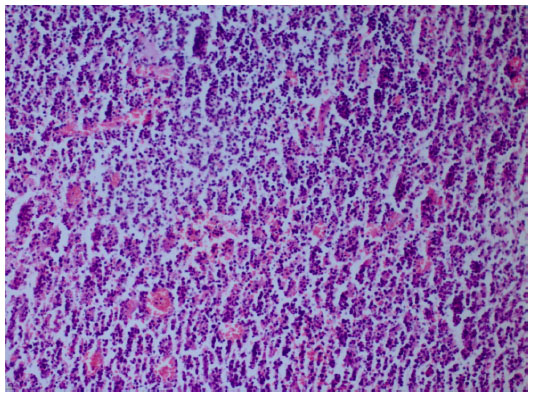 | Figure 3 Histiocytes and activated B-cells (hematoxylin and eosin stain ×200). |
After diagnosis, the patient received corticosteroid and hydroxychloroquine therapy. During treatment, the patient’s clinical and laboratory parameters worsened. The patient’s diagnosis was revised, but new findings were unremarkable for a particular disease. The patient’s hemoglobin values decreased, and the peripheral blood smear revealed schistocytes. Blood tests showed raised D-dimer, activated partial thromboplastin time, prothrombin time, and international normalized ratio with decreased fibrinogen. The patient’s condition quickly worsened and eventually disseminated intravascular coagulopathy (DIC) developed. She remained completely unresponsive to therapy and she subsequently died.
Discussion
KFD has a worldwide distribution, with higher prevalence noted among Japanese and other Asiatic people.5 Although a viral or autoimmune pathogenesis, notably with systemic lupus erythematosus, has been suggested, the etiology still remains unknown. The patients most often present with cervical lymphadenopathy, which may be painful and can be accompanied by fever and upper respiratory tract symptoms.7 However, the most common complaint is the unilateral involvement of the posterior cervical group. Other less common manifestations include fever, axillary and mesenteric lymphadenopathy, splenomegaly, parotid gland enlargement, cutaneous rash, arthralgias, myalgias, aseptic meningitis, bone marrow hemophagocytosis, and interstitial lung disease.7 Erythematous macules, papules, plaques, and nodules are the forms of cutaneous lesions that can be observed in this disease. Our patient had bilateral cervical lymphadenopathy, fever, fatigue, chest and abdominal pain, as well as a maculopapular rash on her face, trunk, and upper and lower extremities. Laboratory investigations are usually unremarkable, except for an elevated ESR, mild neutropenia, and lymphocytosis in some cases.8 Our patient had a high ESR and lymphocytosis. A pathologist, rather than a clinician, usually diagnoses KFD, and the diagnosis is generally based on an excisional biopsy of affected lymph nodes. Open biopsy seems to be the only reliable way to establish a diagnosis; however, some authors suggest that fine-needle biopsy may also be helpful.9 The characteristic histopathological features of the disease include geographic necrosis in the paracortical areas, karyorrhectic debris, and cellular infiltration of the histiocytes and immunoblasts in an absence of granulocytes. Mitoses are often seen. The accumulation of CD68-positive stained histiocytes in the necrotic areas of lymph nodes is helpful in the diagnosis. The ratio of misdiagnosis is defined as 40%, and it is reported that non-Hodgkin lymphoma is the most common disease that has a risk of interference with KFD.2 The frequent mitotic activity, especially in the areas of intensive immunoblasts and lymphoblasts, may result in a misdiagnosis of non-Hodgkin lymphoma.10 In our patient, we performed a lymph node biopsy for diagnosis. The lymph node biopsy showed karyorrhectic debris, and the cellular infiltration of histiocytes and immunoblasts in an absence of polymorphonuclear leukocytes, and so the diagnosis of KFD was confirmed.
Several non-neoplastic conditions such as systemic lupus erythematosus, toxoplasmic lymphadenitis, infectious mononucleosis and cat-scratch disease can mimic KFD; the main problem for the histopathologist is to distinguish this disease from non-Hodgkin’s lymphoma.8 In our patient, serological, immunological, and rheumatological tests, blood, urine and sputum cultures, and also sputum cultures for acid-fast bacilli were all negative.
Occasionally, the disease may progress to a mortal state, as in our case. Our patient rapidly developed DIC and she died. DIC, also called consumption coagulopathy and defibrination syndrome, is a systemic process characterized by both thrombosis and hemorrhage.11 During this process, endothelial damage, enhanced expression, and the release of granulocyte and macrophage procoagulant substances (such as tissue factor) activate the coagulation. Increased expression and release may be a direct action of endotoxin and other membrane lipopolysaccharides, or it may be an indirect action that results from mediation via the activation of cytokines, such as interleukin-6 and tumor necrosis factor alpha.12 In some studies, it has been reported that the serum concentrations of some inflammation mediators such as interferon-γ and interleukin-6 had increased during the acute phase of KFD, and they returned to normal during the convalescent phase; this suggests that these cytokines could play a role in the pathogenesis of this condition.13 The occurrence of DIC, which could be due to a massive cytokine release (mainly of tumor necrosis factor alpha, interleukin-1, and interleukin-6) by the activated mononuclear cells within the many lymph nodes affected by the disease is a remarkable feature for this case.14 In addition, massive release of interferon-γ probably played an important role as well. This cytokine release could explain the elevated lactate dehydrogenase levels in this patient.
No effective treatment has been established for KFD. It has a benign course and is self-limited. The patients usually require no specific treatment; the signs and symptoms usually resolve within 6 months from the time of diagnosis.8 Symptomatic treatment measures such as analgesics, antipyretics, rest, and so on, may be used to relieve patients’ complaints.7 Corticosteroids may be used for severe cases.8 There is also a report on treatment with chloroquine and hydroxychloroquine,15 and intravenous immunoglobulin was also used to effectively treat a patient with severe KFD.16 In our patient, DIC developed despite treatment with corticosteroids and hydroxychloroquine, and she subsequently died.
A clinician must consider DIC when a patient with KFD shows elevations in D-dimer, activated partial thromboplastin time, prothrombin time, and international normalized ratio, and decreases in fibrinogen and hemoglobin. It must be remembered that the patients with KFD who develop DIC may progress to the point where the disease is fatal.
Disclosure
The authors report no conflicts of interest in this work.
References
Kikuchi M. Lymphadenitis showing focal reticulum cell hyperplasia with nuclear debris and phagocytes: a clinicopathological study. Nippon Ketsueki Gakkai Zasshi. Acta Hematol Jpn. 1972;35:379–380. | |
Fujimoto Y, Kojima Y, Yamaguchi K. Cervical subacute necrotizing lymphadenitis. Naika. 1972;30:920–927. | |
Duskin-Bitan H, Kivity S, Olchovsky D, Schiby G, Ezra D, Mouallem M. Kikuchi-Fujimoto disease. Isr Med Assoc J. 2010;12(10):617–621. | |
Norris AH, Krasinskas AM, Salhany KE, Gluckman SJ. Kikuchi-Fujimoto disease: a benign cause of fever and lymphadenopathy. Am J Med. 1996;101(4):401–405. | |
Lin HC, Su CY, Huang CC, Hwang CF, Chien CY. Kikuchi’s disease: a review and analysis of 61 cases. Otolaryngol Head Neck Surg. 2003; 128(5):650–653. | |
Dorfman RF, Berry GJ. Kikuchi’s histiocytic necrotizing lymphadenitis: an analysis of 108 cases with emphasis on differential diagnosis. Semin Diagn Pathol. 1988;5(4):329–345. | |
Bosch X, Guilabert A, Miquel R, Campo E. Enigmatic Kikuchi-Fujimoto disease: a comprehensive review. Am J Clin Pathol. 2004; 122(1):141–152. | |
Kucukardali Y, Solmazgul E, Kunter E, Oncul O, Yildirim S, Kaplan M. Kikuchi-Fujimoto Disease: analysis of 244 cases. Clin Rheumatol. 2007;26(1):50–54. | |
Hsueh EJ, Ko WS, Hwang WS, Yam LT. Fine-needle aspiration of histiocytic necrotizing lymphadenitis (Kikuchi’s disease). Diagn Cytopathol. 1993;9(4):448–452. | |
Tong TR, Chan OW, Lee KC. Diagnosing Kikuchi disease on fine needle aspiration biopsy: a retrospective study of 44 cases diagnosed by cytology and 8 by histopathology. Acta Cytol. 2001;45(6):953–957. | |
Vincent JL, De Backer D. Does disseminated intravascular coagulation lead to multiple organ failure?. Crit Care Clin. Jul 2005;21(3):469–477. | |
Jansen PM, Pixley RA, Brouwer M, et al. Inhibition of factor XII in septic baboons attenuates the activation of complement and fibrinolytic systems and reduces the release of interleukin-6 and neutrophil elastase. Blood. 1996;87(6):2337–2344. | |
Kubota M, Tsukamoto R, Kurokawa K, Imai T, Furusho K. Elevated serum interferon gamma and interleukin-6 in patients with necrotizing lymphadenitis (Kikuchi’s disease). Br J Haematol. 1996;95(4):613–615. | |
Chan JK, Wong KC, Ng CS. A fatal case of multicentric Kikuchi’s histiocytic necrotizing lymphadenitis. Cancer. 1989;63(9):1856–1862. | |
Rezai K, Kuchipudi S, Chundi V, Ariga R, Loew J, Sha BE. Kikuchi-Fujimoto disease: hydroxychloroquine as a treatment. Clin Infect Dis. 2004;39(12):e124–e126. | |
Noursadeghi M, Agel N, Gibson P, Pasvol G. Successful treatment of severe Kikuchi’s disease with intravenous immunoglobulin. Rheumatology (Oxford). 2006;45(2):235–237. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.