Back to Journals » OncoTargets and Therapy » Volume 12
Dissecting the prevention of estrogen-dependent breast carcinogenesis through Nrf2-dependent and independent mechanisms
Authors Giudice A, Barbieri A ![]() , Bimonte S
, Bimonte S ![]() , Cascella M
, Cascella M ![]() , Cuomo A, Crispo A, D'Arena G
, Cuomo A, Crispo A, D'Arena G ![]() , Galdiero M, Della Pepa ME
, Galdiero M, Della Pepa ME ![]() , Botti G
, Botti G ![]() , Caraglia M
, Caraglia M ![]() , Capunzo M
, Capunzo M ![]() , Arra C, Montella M
, Arra C, Montella M ![]()
Received 8 August 2018
Accepted for publication 14 December 2018
Published 24 June 2019 Volume 2019:12 Pages 4937—4953
DOI https://doi.org/10.2147/OTT.S183192
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Tohru Yamada
Aldo Giudice,1 Antonio Barbieri,2 Sabrina Bimonte,3 Marco Cascella,3 Arturo Cuomo,3 Anna Crispo,1 Giovanni D’Arena,4 Massimiliano Galdiero,5 Maria Elena Della Pepa,5 Gerardo Botti,6 Michele Caraglia,7 Mario Capunzo,8 Claudio Arra,2 Maurizio Montella1
1Epidemiology Unit, Istituto Nazionale Tumori “Fondazione G. Pascale”, IRCCS, Naples, Italy; 2S.S.D Sperimentazione Animale, Istituto Nazionale Tumori “Fondazione G. Pascale”, IRCCS, Naples, Italy; 3Division of Anesthesia and Pain Medicine, Istituto Nazionale Tumori “Fondazione G. Pascale”, IRCCS, Naples, Italy; 4Hematology and Stem Cell Transplantation Unit, IRCCS Centro di Riferimento Oncologico della Basilicata, Rionero in Vulture, Italy; 5Department of Experimental Medicine, Università della Campania “Luigi Vanvitelli”, 80134 Naples, Italy; 6Scientific Direction, Istituto Nazionale Tumori-IRCCS “Fondazione G. Pascale”, Naples, Italy; 7Department of Biochemistry, Biophysics and General Pathology, University of Campania “Luigi Vanvitelli”, 80138 Naples, Italy; 8Department of Medicine, Surgery and Dentistry “Scuola Medica Salernitana”, University of Salerno, Baronissi, 84081 Salerno, Italy
Abstract: Breast cancer is the most common malignancy among women worldwide. Various studies indicate that prolonged exposure to elevated levels of estrogens is associated with development of breast cancer. Both estrogen receptor-dependent and independent mechanisms can contribute to the carcinogenic effects of estrogens. Among them, the oxidative metabolism of estrogens plays a key role in the initiation of estradiol-induced breast cancer by generation of reactive estrogen quinones as well as the associated formation of oxygen free radicals. These genotoxic metabolites can react with DNA to form unstable DNA adducts which generate mutations leading to the initiation of breast cancer. A variety of endogenous and exogenous factors can alter estrogen homeostasis and generate genotoxic metabolites. The use of specific phytochemicals and dietary supplements can inhibit the risk of breast cancer not only by the modulation of several estrogen-activating enzymes (CYP19, CYP1B1) but also through the induction of various cytoprotective enzymes (eg, SOD3, NQO1, glutathione S-transferases, OGG-1, catechol-O-methyltransferases, CYP1B1A, etc.) that reestablish the homeostatic balance of estrogen metabolism via nuclear factor erythroid 2-related factor 2 (Nrf2)-dependent and independent mechanisms.
Keywords: reactive estrogen quinones, breast carcinogenesis, depurinating estrogen–DNA adducts, dietary phytochemicals, nuclear factor erythroid 2-related factor 2
Introduction
An increasing body of evidence indicates that breast cancer is the most common malignancy among women, representing 23% of all diagnosed cancer cases, and the second leading cause of cancer deaths among women worldwide.1,2 Age is considered the most important risk factor. Unlike other malignancies that increase at the end of the fifth decade, breast cancer begins early in the third decade of life. This could be associated with the effects of estrogens and other ovarian hormones on breast tissue. Breast cancer is related to multiple risk factors. These are commonly subdivided into nonmodifiable, such as age, BRCA1, BRCA2 gene mutations, family history, reproductive history, and high-dose radiation to the chest, and modifiable, such as high endogenous estrogens, hormone therapy, and lifestyle including diet, physical activity, alcohol consumption, tobacco abuse, and other risky behaviors.3,4 Changing the Mediterranean diet to a westernized diet is recognized as a basic breast cancer risk factor.5,6 In addition, recent studies also suggest that the human papillomavirus infection may be considered a possible risk factor in the development of breast cancer among the female population.7 At the least, it is widely accepted that, among risk factors, sustained exposure to elevated levels of estrogens plays an important role in the initiation and development of breast cancer.8 In fact, studies in experimental animal models and cultured human cells strongly suggest that estradiol (E2), its interconvertible metabolite estrone (E1), and their estrogen quinones exert carcinogenic effects on breast tissue through several mechanisms.9,10 There are at least two major mechanisms involved in the development and progression of estrogen-induced breast cancer: 1) estrogen receptor (ER)-mediated stimulation of abnormal cell proliferation that generates random mutations; and 2) ER-independent mechanisms involving chemical (oxidative pathway) inflammatory, epigenetic, and cancer stem cell pathways.11–13 Among them, a major contribution has been attributed to unbalanced estrogen oxidative metabolism which generates genotoxic metabolites such as reactive estrogen quinones and oxygen free radicals that can react with DNA to form unstable estrogen–DNA adducts in critical genes leading to cancer initiation.14,15 Prevention of breast cancer can be achieved by inhibiting the formation of these DNA adducts which generate the mutations leading to the initiation, promotion, and progression of cancer.16 Various chemopreventive agents such as resveratrol (Res), sulforaphane, vitamin C, and N-acetylcysteine (NAC) as well as melatonin and lipoic acid have been reported in cell culture and in vivo animal models to inhibit oxidative metabolism of E2 and E1, and thus prevent DNA damage through nuclear factor-erythroid 2-related factor 2 (Nrf2)-dependent and independent mechanisms.17–25 Notably, Nrf2 is a major basic leucine zipper-containing transcription factor which controls gene expression of an elaborate network of cytoprotective proteins including antioxidant and detoxifying enzymes that defend cells from electrophiles and free radicals, playing a pivotal role in the prevention of human carcinogenesis.17 The purpose of this review is to shed light on the role of unbalanced oxidative estrogen metabolism on the initiation of breast cancer. Moreover, we will discuss the role of natural dietary phytochemicals in the prevention of estrogen-induced breast cancer by the modulation of several estrogen-activating enzymes (CYP19, CYP1B1) and through the induction of various cytoprotective enzymes (eg, SOD3, NQO1, glutathione S-transferases (GSTs), catechol-O-methyltransferases (COMTs), etc.) involved in the regulation of the homeostatic balance of estrogen metabolism through Nrf2-dependent and independent mechanisms.
The regulation of endogenous estrogen oxidative metabolism by cytochrome P450 enzymes and breast carcinogenesis
The importance of endogenous estrogens in the etiology of breast carcinogenesis has been widely recognized by the United States government since 2001. To date, several studies suggest that sustained exposure to endogenous estrogens is associated with the onset and progression of breast cancer.26 As mentioned above, there are different possible mechanisms by which estrogens can increase the risk of breast cancer.11,13 Among them, it has been suggested that the oxidative metabolism of estrogens plays a major role in the initiation of estrogen-induced breast cancer by the generation of reactive estrogen quinones as well as the associated formation of oxygen free radicals resulting from redox cycling of catechol estrogens and estrogen quinones.12,15,27 Notably, metabolic formation of estrogens mainly derives from the conversion of testosterone to E2 and androstenedione to E1 catalyzed by the enzyme aromatase (CYP19).15,27 Further, E1 can be reversibly conversed to E2 by the action of 17β-hydroxysteroid dehydrogenase enzyme.28 When an excess of estrogen is obtained, it can be stored as estrone sulfate.15,27 Briefly, once the estrogens are formed, they can also be metabolized through three competitive pathways catalyzed by the NADPH-dependent cytochrome P450 (CYP) enzymes such as CYP1A1, CYP1A2, CYP3A, and CYP1B1.29 E1 and E2 can be hydroxylated at positions C2, C4, and C16 and be converted to catechol estrogens (2-hydroxyestrone, 4-hydroxyestrone, 16α-hydroxyestrone, 2-hydroxyestradiol, and 4-hydroxyestradiol). Both 16α-hydroxyestrone and estradiol can also produce estriol by their hydroxylation. Specifically, CYP1A1 and CYP3A catalyze the hydroxylation preferentially at the 2 position, converting estrogens (E1/E2) to catechol estrogens 2-hydroxyestrone and 2-hydroxyestradiol. CYP1B1 instead catalyzes the hydroxylation almost exclusively at the 4 position, converting estrogens (E1/E2) to catechol estrogens 4-hydroxyestrone and 4-hydroxyestradiol.12,29 Experimental studies indicate that the 4-OH metabolites exert more carcinogenic effects than their 2-OH counterparts, 4-OH-E2 being a key step in inducing unstable DNA adducts.26,30 In fact, if the catechol estrogens are not conjugated by the COMT enzyme, further oxidation particularly of 4-OH-E1(E2) catechols by cytochrome P450 along with peroxidase enzymes can lead to formation of E2-3,4-semiquinones and E2-3,4-quinones that react with DNA to form unstable depurination adducts with adenine and guanine.14,31 These reactive quinones can also be reduced to semiquinones by CYP reductase through a redox cycle which produces ROS that cause oxidative DNA damage.12 Various phase 2 detoxification enzymes, such as COMT, GST, and quinone reductase (NOQ1, NQO2), play an important role in the inactivation of estrogen catechols, semiquinones, and quinones by limiting the formation of adducts and oxidative DNA damage.26,32 Specifically, catechol estrogens by the action of the COMT enzyme can be further metabolized (methylated) to nongenotoxic methoxyestrogens (2-methoxyestrone, 4-methoxyestrone, 2-methoxyestradiol, and 4-methoxyestradiol). Through this mechanism, COMT also plays an important role in preventing the transformation of catechol estrogens to quinone–DNA adducts and in the development of ROS capable of damaging cellular macromolecules such as DNA, proteins, and lipids.10,33,34 In addition to methylation, parent estrogens and catechol estrogens can be also excreted in the urine or feces by conjugation with glucuronic acid and sulfate by inducing hepatic phase 2 detoxification enzymes such as UDP-glucuronosyltransferases and sulfotransferases, respectively, through the Keap1–Nrf2–antioxidant responsive elements (ARE) pathway.35–38 As the COMT enzyme plays an essential role in the conversion of catechol estrogens to methoxyestrogens, insufficient activity of this enzyme due to polymorphic variations39,40 could increase the competitive catalytic oxidation of the catechol estrogens (4-OH-E1(E2)) to semiquinones and electrophilic catechol quinones. These products react with DNA by Michael addition to form adducts which detach from DNA, leaving behind apurinic sites.12,14,15,27,41,42 The apurinic sites can undergo error-prone DNA repair which results in point mutations that initiate the breast carcinogenesis process and other types of human cancer.12,14,15,27,41,42 In addition to electrophilic catechol quinones, oxidative stress produced by redox cycling between catechol estrogens and estrogen quinones also plays an important role in estrogen-induced breast cancer. Accordingly, it has been shown that the 8-hydroxydeoxyguanosine (8-OHdG) levels were higher in E2-exposed mammary tissue and in mammary tumors compared to age-matched controls.21,43
Taken together, these data suggest that the reactive estrogen quinones and ROS, by increasing DNA damage, may lead to specific mutations causative of the initiation of breast cancer and other malignancies.
The importance of unbalanced estrogen oxidative metabolism in breast carcinogenesis
The oxidative metabolism of estrogens is mediated by a balanced homeostatic set of activating (CYP19, CYP1B1) and deactivating/protective (eg, COMTs, SOD3, NQO1, GSTs, and CYP1B1A) enzymes.15,21,26,27,29,32,43 Homeostasis significantly reduces the metabolic oxidation of catechol estrogens to catechol quinones and their reaction with DNA.10,44 A variety of endogenous and exogenous factors (eg, diet, chronic inflammation, environment, lifestyle, aging, and genetic and epigenetic factors) can disrupt this homeostasis by shifting the equilibrium between these activating and deactivating enzymes toward ROS formation as well as estrogen quinones and depurinating estrogen–DNA adducts, which can lead to the initiation of breast cancer.10,44 The first important factor implicated in breast carcinogenesis process is the increased expression of activating enzyme CYP19 (aromatase) which converts androgens to estrogens in target tissues.45–47 A second factor that can disrupt homeostasis of the estrogen metabolism is the deregulation of sulfatase enzyme that converts excessive stored estrone sulfate into E1.48,49 A third critical factor involved in the disruption of estrogen homeostasis is the upregulation of CYP1B1 enzyme which promotes the production of high levels of 4-OH-E1(E2) from E1/E2.19,50–53 As mentioned above, higher levels of 4-OH-E1(E2) can give rise to higher levels of carcinogenic E1(E2)-3,4-quinones capable of generating unstable depurinating adducts with adenine and guanine, and subsequent point mutations that can initiate breast cancer and other prevalent types of cancer.12,42 A lack of or a low level of COMT activity due to polymorphic variations might produce a similar effect consisting of lower levels of methylation of 4-OH-E1(E2) catechols and increased competitive catalytic oxidation of 4-OH-E1(E2) catechols to E1(E2)-3,4-quinones.39,40 Once the catechol estrogen quinones (E1(E2)-3,4-quinones) are formed, they can be detoxified by GST through conjugation with glutathione (GSH) or by the reduction back to their catechol estrogens catalyzed by quinone reductase (NQO1, NQO2)15,54,55 which can be induced by numerous natural bioactive compounds via Nrf2-dependent mechanisms.17,18,43,56 Higher levels of reactive quinones available can be also produced by insufficient cellular levels of GSH, which reacts efficiently with the quinones. In addition, polymorphisms in NQO1 which reduce the conversion of quinones into catechols could be considered another important factor that produces higher levels of mutagenic reactive quinones.57 Imbalances in the estrogen metabolism have been also observed by comparing analyses of breast tissue from women with and without breast cancer.58 In fact, in nontumor breast tissue from women with breast carcinoma, the levels of 4-OH-E1(E2) are four times higher than the levels in breast tissue from women without breast cancer. Furthermore, evidence on the imbalance in estrogen homeostasis derives from a greater expression level of estrogen-protective enzymes, COMT and NQO1, in women without breast cancer and from a greater expression level of estrogen-activating enzymes, CYP19 and CYP1B1, in breast tissue of women with breast cancer.58 In addition, the imbalance in estrogen metabolism can also be induced by substances assumed by the mouth, skin, and nose. It has been reported that these environmental compounds (eg, dioxin) are able to modify the estrogen metabolism, leading to an increased formation of catechol estrogen quinones. For instance, dioxin induces expression of the activating enzyme CYPB1B1.19,53 These data indicate that the imbalances in the estrogen metabolism and the genetic polymorphisms can produce higher levels of mutagenic reactive quinones and ROS that are causative of breast cancer initiation and other malignancies (see Figure 1).
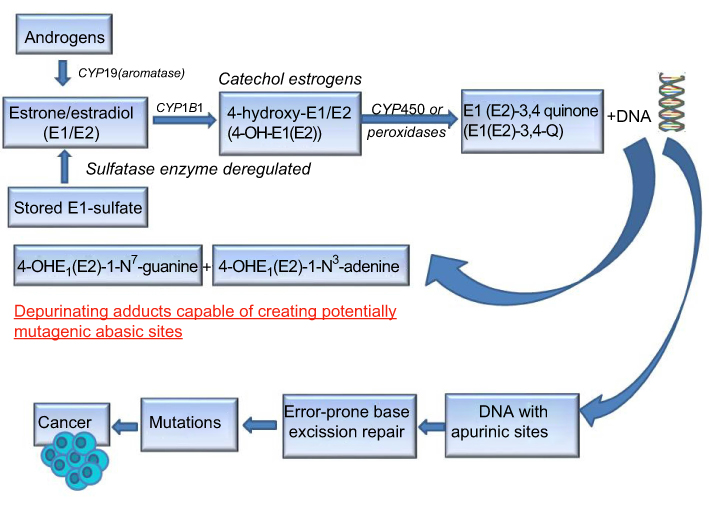 | Figure 1 Schematic representation of major metabolic pathways involved in breast cancer initiation by estrogens. Abbreviation: CYP, cytochrome P450. |
The role of chemopreventive agents in maintaining or reestablishing balanced estrogen metabolism by inducing cytoprotective enzymes through Nrf2-dependent and independent mechanisms
As mentioned above, E2 metabolism-induced electrophilic and oxidative stress plays an important role in breast carcinogenesis.15,27 To neutralize catechol-3,4-quinones and ROS, the major carcinogenic metabolites of estrogens, mammalian cells have evolved a hierarchy of sophisticated sensing and signaling mechanisms to turn on or off the endogenous cytoprotective response.38,59,60 One of the major strategies for inhibiting the overburden of oxidative stress and electrophilic catechol quinones involved in the estrogen-induced carcinogenic process15,27 is the use of specific phytochemicals (eg, curcumin, green tea catechins, genistein, Z-Ligustilide, Res, sulforaphane, vitamin C, tocopherols, etc.) and dietary supplements (NAC, melatonin, α-lipoic acid, etc.). These compounds are able to modulate several estrogen activating enzymes (CYP19, CYP1B1) and to induce the expression of cytoprotective enzymes (SOD3, NQO1, GST, COMT, CYP1A1, etc.) as indicated in Table 1 through Nrf2-dependent and independent mechanisms.17,18,21,22,24,25,43,61–66 Importantly, Nrf2 is a basic leucine zipper stress-sensitive transcription factor that maintains redox homeostasis by inducing the transcription of several cytoprotective genes through ARE/electrophile response elements in target gene promoters.59,60 In normal conditions, the activity of Nrf2 is suppressed in the cytosol by specific binding to the chaperone Keap1. However, upon stimulation by oxidants or electrophilic compounds having the ability to oxide or covalently modify thiol groups of critical cysteine residues in Keap1, Keap1-mediated degradation of Nrf2 is abrogated. Thus, Nrf2 is released and then translocated into the nucleus, thus potentiating the ARE response. This leads to the synthesis of a distinct set of defensive proteins and enzymes such as SOD3, NQO1, GSTs, UDP-glucuronosyltransferases, sulfotransferases, and 8-oxoguanine DNA glycosylase (OGG-1) that facilitate the excretion of carcinogens, enhance the removal of electrophiles and free radicals, increase cellular capacity to repair oxidatively damaged DNA and proteins, and, consequently, reduce the propensity of tissue and organisms to develop diseases or malignancies including E2-induced breast cancer21,38,43,59,60,67,68 (see Figure 2). Importantly, OGG-1 is the most important enzyme of the DNA base excision repair pathway induced through the Nrf2–ARE signaling-dependent mechanisms. Specifically, it is involved in the removal of 8-OHdG adducts from all regions of the genome.68 Interestingly, the Nrf2–ARE pathway can be mainly regulated through Keap1-dependent and independent mechanisms.69 Accordingly, Yang et al70 recently demonstrated that modified Xiaoyao powder (MXP) may play an important role in the prevention and treatment of breast cancer through modulating the Nrf2–ARE pathway. These authors sustain that MXP and its many active components including quercetin, kaempferol, and the sesquiterpene atractylenolide II exert chemopreventive, anti-inflammatory, and antioxidative stress effects by activating Nrf2–NQO1 signaling in MCF-7 cells. Specifically, these studies indicate that the flavonoids quercetin and kaempferol as well as atractylenolide II may play an essential role in E2-mediated breast cancer chemoprevention by acting as potent inducers of the Nrf2–NQO1 signaling pathway that catalyzes the reduction and detoxification of quinone substrates which are highly reactive molecules involved in cancers and other degenerative diseases. Other studies also demonstrated that cabbage juices and indoles such as indole-3-carbinol (I3C) and 3,3ʹ-diindolylmethane (DIM) can significantly influence E2-mediated breast cancer initiation and progression by modulating the expression of the estrogen metabolism key enzymes (CYP1A1, CYP1A2, CYP1B1) in breast epithelial cells, differing in ER status such as tumorigenic MCF7 and MDA-MB-231 cells and nontumorigenic MCF10A cells.71 Specifically, these authors suggest that both I3C and DIM can bind to and activate aryl hydrocarbon receptor (AhR) that induces expression of the CYP1 family genes or may also promote degradation of the ER and inhibit estrogen signaling. There is also evidence that these phytochemicals can modulate estrogen metabolism by preferentially inducing enzymes of the CYP1A1 family. In addition, these are also capable of binding to ER, competing with estrogen, and inhibiting the estrogen-dependent gene expression. Significant cross-talk between AhR, ER, and Nrf2 has also been identified in these cell lines. In fact, these studies reported that in estrogen-dependent MCF7 breast cancer cells, estrogen can also recruit ERα and class III histone deacetylase SIRT1 at the NQO1 promoter, leading to suppression of NQO1 gene transcription. By contrast, many of these phytochemicals present in cruciferous vegetables may reverse the inhibitory effect of estrogen on cytoprotective enzyme expression by significantly increasing transcription of the Nrf2 gene in all tested cell lines. This leads to enhanced expression of several detoxifying enzymes such as NQO1 and GSTP that play a key role in chemoprevention and therapy of breast cancer by reducing the level of estrogen quinones involved in toxicity and breast carcinogenicity. Recently, Peng et al72 also demonstrated that 4β-hydroxywithanolide E, an active component of the extract of Physalis peruviana, a member of the Solanaceae group, plays an important role in ROS production and subsequent ataxia-telangiectasia mutated protein-mediated apoptosis of breast cancer MCF-7 cells which is correlated with the expression of Nrf2-dependent antioxidative defense enzymes including SOD, CAT, and the glutathione system. Accordingly, pretreatment with antioxidants GSH and NAC was capable of reversing the 4β-hydroxywithanolide E-induced ROS accumulation and apoptotic cell death in MCF-7 cells. Role of the Nrf2-ARE pathway in inflammatory signaling involved in breast carcinogenesis
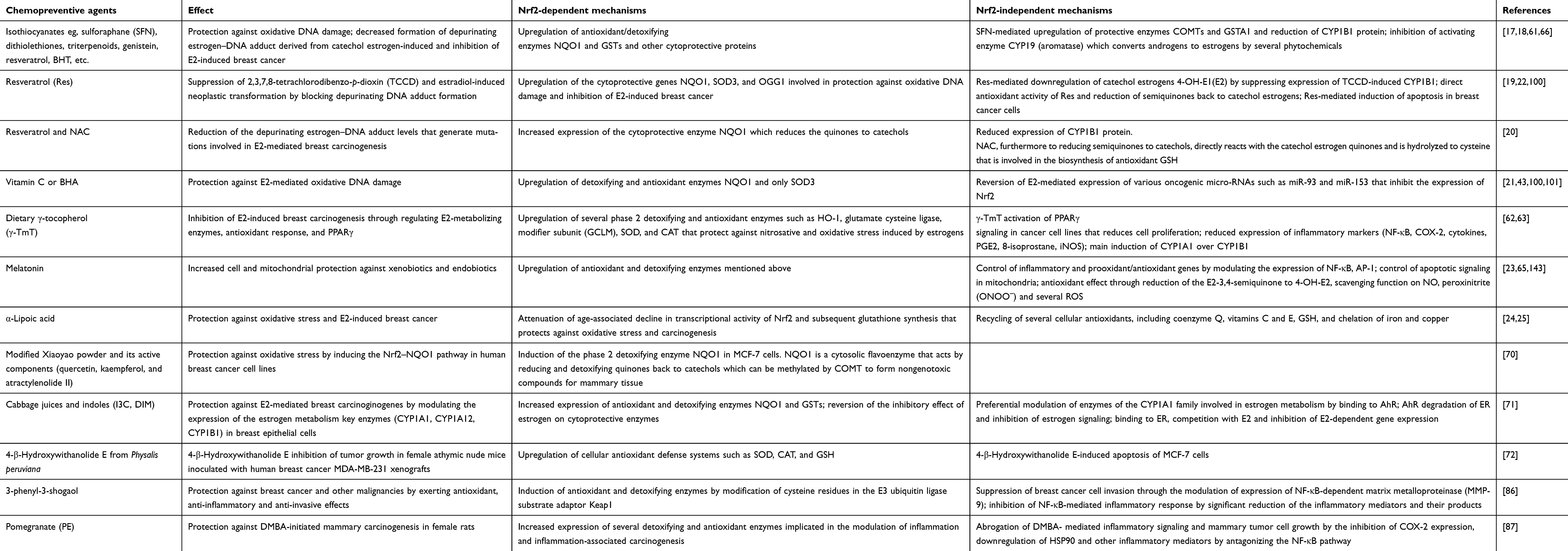 | Table 1 Chemopreventive agents capable of maintaining balanced estrogen metabolism through Nrf2-dependent and independent mechanisms |
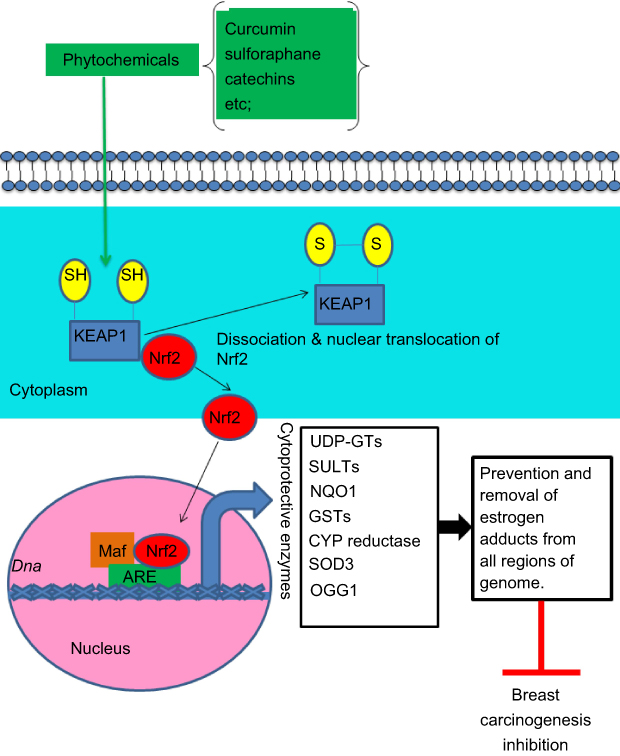 | Figure 2 Schematic representation of the Nrf2–ARE pathway activated by phytochemicals. Curcumin, sulforaphane, and green tea catechins are capable of inhibiting the formation of ROS and estrogen genotoxic metabolites such as catechol estrogens, semiquinones, and mutagenic/reactive estrogen quinones by inducing several cytoprotective enzymes such as UDP-GTs, SULTs, NQO1, GSTs, SOD3, and OGG1 through activation of the Nrf2–ARE pathway. OGG1 is the rate-limiting enzyme of the DNA base excision repair pathway implicated in the removal of 8-hydroxydeoxyguanosine adducts from DNA. Notably, the catechol-O-methyltransferase enzyme, which catalyzes the methylation of catechol estrogens to nongenotoxic methoxyestrogens, blocking their further oxidation to semiquinones and mutagenic quinones, is not represented because it is induced by phytochemicals through Nrf2–ARE-independent mechanisms.Abbreviations: ARE, antioxidant responsive elements; CYP, cytochrome P450; GST, glutathione S-transferase; Nrf2, nuclear factor-erythroid 2-related factor 2; NQO1, quinone reductase; OGG1, 8-oxoguanine DNA glycosylase; SULT, sulfotransferase; UDP-GT, UDP-glucuronosyltransferase. |
Role of the Nrf2–ARE pathway in inflammatory signaling involved in breast carcinogenesis
Besides its role in regulating carcinogen detoxification and cellular antioxidative defense, Nrf2 also appears to downregulate the expression of other genes encoding inflammatory molecules (eg, cytokines, prostaglandins, etc.)73,74 involved in the expression and activity of cytochrome P450 (CYP19) complex or aromatase which converts androgens to estrogens.75 There are many lines of evidence supporting cross-talk between Nrf2 and inflammatory signaling. For example, intracellular accumulation of ROS including superoxide anion (O2.–), hydrogen peroxide (H2O2), and peroxynitrite (ONOO–) plays an important role in proinflammatory responses by the activation of redox-sensitive transcription factors such as nuclear factor kappa B (NF-κB) and activator protein 1 (AP-1) and by the upmodulation of their kinases including mitogen-activated protein kinases (p38, ERK, and JNK) and phosphoinositide-3-kinase (PI3K).76 Notably, the transcription factor NF-κB modulates the de novo synthesis of a large array of proinflammatory mediators including different molecules such as chemokines, cytokines, immunoreceptors, cell-adhesion molecules, stress-response genes, regulators of apoptosis, growth factors, and transcription factors.77 Aberrant NF-κB activation is associated with a significant number of diseases ranging from inflammatory diseases to neurodegeneration, autoimmune disorders, and cancer.78,79 Since Nrf2–ARE-modulated cytoprotective genes confer protection against oxidative stresses and potentiate antioxidant defense capacity in cells, regulation of Nrf2–ARE signaling by various chemopreventive agents can exert significative effects on the redox-sensitive inflammation-modulating factors such as NF-κB and AP-1. For instance, many Nrf2-activating agents such as sulforaphane, curcumin, and avicins can inhibit NF-κB activation by acting directly on cysteine thiols present in Keap1, p50 (NF-κB), or IκB kinase.80,81 Similar to NF-κB inactivation, some Nrf2-activating dietary phytochemicals such as Res, sulforaphane, curcumin, and green tea catechins can also inhibit the AP-1 activation induced by different stimuli82-84 by directly modifying the DNA binding domain of AP-1.80,81 However, it should be noted that NF-κB also plays an important role in the negative regulation of the Nrf2–ARE pathway. Specifically, NF-κB p65 subunit can repress the Nrf2–ARE pathway at the transcriptional level when phosphorylated on S276 by either competitive interaction with the CH1-KIX domain of coactivator binding proteins or by recruiting histone deacetylase 3 (HDAC3), the corepressor, to the ARE, which can deacetylase histone H4 and MafK.85 Such a blockade may result in a cellular deficiency of antioxidant potential and subsequent increase of oxidative stress. A previous study proposed by Gan et al86 suggests that a synthetic novel shogaol analog, 3-phenyl-3-shogaol (3-Ph-3-SG), can suppress multiple steps in breast carcinogenesis through its in vitro anti-invasive, anti-inflammatory, and Nrf2-activating ability in several studies. Notably, inflammation can significantly contribute to the process of carcinogenesis not only through production of DNA-damaging free radicals but also via the increased production of proinflammatory mediators such as cytokines, chemokines, prostaglandins, and nitric oxide (NO) that exert their growth-promoting, invasive/metastatic, and angiogenic effects.73,74 These authors have demonstrated that shogaol compounds containing vanilloid and Michael acceptor moieties such as 3-Ph-3-SG may exert cytoprotective effects against oxidative and nitrosative stress and cancer-promoting inflammatory response by inducing several antioxidant and phase 2 detoxifying enzymes such as HO-1 and NQO1 through upregulation of the Nrf2–ARE pathway in the HEK 293 cell line, an immortalized human cell line of noncancerous origin. This cytoprotective activity by 3-Ph-3-SG was achieved at least partly by antagonizing the activity of Keap1 repressor by modifying its cysteine residues. Further, at nontoxic concentrations, 3-Ph-3-SG reduces the inflammatory response through inhibiting the expression of several inflammatory mediators such as NO, inducible nitric oxide synthase, cyclooxygenase-2, and prostaglandin-E2 in RAW 264.7 macrophage-like cells by suppressing the NF-κB signaling pathway. At similar concentrations, 3-Ph-3-SG also exerts anti-invasive effects on MDA-MB-231 and MCF7 breast cancer cell lines with invasive properties through inhibition of NF-κB-dependent matrix metalloproteinase-9 expression. Taken together, these results indicate that 3-Ph-3-SG, similarly to many abundant naturally-occurring shogaol compounds, blocks multiple steps of the carcinogenesis process; for this reason, it represents a promising candidate for use in chemotherapeutic and chemopreventive strategies against breast cancer and other malignancies. Another recent report by Mandal et al87 also suggests that pomegranate (PE), a unique fruit containing most bioactive phytochemicals, plays an important chemopreventive role against inflammation that contributes to dimethylbenz(a)anthracene (DMBA)-initiated mammary tumorigenesis in female rats through antiproliferative and apoptosis-inducing mechanisms. Notably, during DMBA-initiated rat mammary tumorigenesis, activation of NF-κB induces the expression of several inflammatory mediators such as cyclooxygenase-2, prostaglandin E2, and heat shock protein 90 implicated in proliferation, survival, inflammation, angiogenesis, invasion, and metastasis.73,74 These authors suggest that various phytochemicals present in PE can exert a synergistic effect in the modulation of inflammation and inflammation-associated breast carcinogenesis by involving anti-inflammatory mechanisms that require potential cross-talk between Nrf2 and NF-κB transcription factors. Accordingly, PE treatment not only may inhibit DMBA-induced degradation of IkBα protein in cytosol but may also be effective in increasing the expression and subsequent nuclear translocation of Nrf2 which in turn suppresses NF-κB-associated inflammatory activity.87 Collectively, these results indicate that PE treatment plays an important role in breast cancer development and prevention by suppressing NF-κB-mediated inflammatory pathways through Nrf2-dependent and independent mechanisms as indicated in Table 1. Numerous other phytochemicals present in plants and medical herbs can act with similar mechanisms against breast cancer.88–91 Therefore, the development of highly specific activators of Nrf2 can significantly improve the anti-inflammatory functions in oxidative stress-related diseases, especially in breast cancer.92 In addition, since 70% of breast cancers are estrogen dependent, the use of natural and synthetic aromatase inhibitors represents another important therapeutic strategy against estrogen-dependent breast cancer and other estrogen-dependent diseases such as endometriosis and endometrial cancer.75,93–98
Role of estrogen signaling in the repression of the Nrf2–ARE pathway
Notably, estrogens can also repress Nrf2-dependent transcription through ER-dependent and independent mechanisms.22,99,100 For instance, estrogens could increase breast cancer risk by enhancing the expression of several miRNAs with oncogenic activity (eg, miR-93, miR-153) and downregulating Nrf2 and its downstream cytoprotective genes.22,101 Additionally, they could also reduce the expression of other miRNAs with tumor-suppressor activity (eg, miR-663, miR-774, miR-141, and miR-200c) in mammary tissues and human breast cell lines.102,103 In these cases, it has been shown that both vitamin C and Res may exert chemopreventive effects against estrogen-induced breast cancer by decreasing the expression of various oncogenic miRNAs such as miR-93 and miR-153 and by inducing Nrf2 and its cytoprotective genes. Other studies by Chatterjee et al104 also suggested that cellular activity of the Nrf2–ARE pathway could be negatively modulated by Nrf3 protein induced by E2. Specifically, these authors reported that E2 treatment enhanced Nrf3 mRNA expression levels in a time-dependent manner in MCF-10A cells.105 In addition, treatment with vitamin C and Res can also reverse the estrogen-dependent increase in the expression of Nrf3 protein in tested cells.104 Another important function of Res is to reduce ROS accumulation and DNA damage in breast cancer type 1 susceptibility protein (BRCA1)-deficient mammary epithelial cells, MCF10A, by Nrf2 upregulation.106 In addition, during the carcinogenesis process, many oncogenes and tumor-suppressor genes including Nrf2 can be also silenced by their promoter hypermethylation and hypoacetylation.107–109 Many natural compounds mentioned above, in addition to reversing the inhibitory effects of estrogens on the Nrf2–ARE pathway,21,22,43,100,104 can also act as epigenetic modifiers because they are capable of demethylating the Nrf2 promoter and reactivating Nrf2 signaling in cancer cells, possibly through the inhibition of DNMT and HDAC expression and reduction of the binding of MeCP2, a methylation-dependent transcriptional repressor.61,110,111
Modulation of mammary stem cell properties by the transcription factor Nrf2
In addition to its roles in cellular detoxification and anti-inflammatory response, Nrf2 is also required to maintain homeostatic quiescence of stem cells.112–114 The dormancy is beneficial for stem cells to avert unnecessary DNA replication capable of generating DNA damage and mutations.112 In support of this notion, Zhang et al115 recently reported that Nrf2 can negatively modulate mammary stem cell expansion and protect against genotoxic effects of estrogen metabolites by cooperating with histone methyltransferase Enhancer of Zeste Homologue 2, a member of the Polycomb repressor complex, to silence long noncoding RNA (lncRNA) ROR expression. Accordingly, Nrf2 loss can lead to lncRNA ROR overexpression in mammary stem cells and subsequent mammary tumorigenesis. By contrast, ROR-overexpressing MCF10A cells treated with the chemopreventive compound Shikonin, a known activator of Nrf2, were protected against 4-OH-E2-induced DNA damage.116 Other studies also demonstrated that Nrf2 controls proliferation of stem cells and differentiation by regulating the cellular levels of ROS through the expression of several antioxidant/detoxification enzymes.17,21,43,113,117–119 In addition, estrogen can also regulate the breast cancer stem cell population through multiple pathways including Wnt/β-catenin, Hedgehog, Notch, and PI3K/mechanistic target of rapamycin (mTOR).120,121 Notably, estrogen induces expansion of breast cancer stem-like cell subpopulations and can lead to tumorigenic phenotypes in vivo through a paracrine FGF–FGFR–Tbx3 signaling pathway, which happens also to modulate normal embryonic breast stem cells, whereas cotreatment of cells with tamoxifen or a small molecule inhibitor of fibroblast growth factor receptor (FGFR) signaling was capable of preventing estrogen-induced expansion of cancer stem cells.122 The bioactive natural compounds mentioned above also have the ability to suppress breast cancer stem cells by modulation of self-renewal pathways such as Wnt/β-catenin, Hedgehog, Notch, and PI3K/mTOR.123–129 Therefore, it has been proposed that some of these bioactive natural compounds or their anologs may be utilized as cancer stem cell-eliminating agents to effectively prevent or treat cancer in the future, including breast cancer. Currently, available data provide only limited information on whether such phytochemicals are capable of suppressing estrogen-induced breast cancer stem cells. In addition, many studies on phytochemicals have been made in vitro and in animal models in vivo. Thus, the actual effectiveness of phytochemicals to inhibit the growth of cancer stem cells is extremely needed in patients through large-scale clinical studies.
Discussion and conclusions
Several clinical and experimental studies suggest that prolonged exposure to estrogens can significantly increase the risk of breast cancer and other malignancies mainly through two complementary pathways: 1) increased ER-mediated stimulation of breast cell proliferation with a concomitant enhanced rate of mutations; and 2) oxidative metabolism of estrogen to genotoxic metabolites that cause DNA damage and mutations by generation of unstable E2–DNA adducts as well as the associated formation of ROS resulting from redox cycling of 4-OH-E2 to the 3,4-estradiol quinone and back-conversion to 4-OH-E2.9 The oxidative metabolism of estrogens is mainly mediated by a balanced series of activating (CYP19 and CYP1B1) and deactivating (COMTs, SOD3, NQO1, and GSTs) enzymes. When homeostasis in the metabolism of estrogens is altered, excessive production of genotoxic metabolites such as ROS as well as estrogen quinones can generate mutations that initiate the breast carcinogenesis process and other types of human cancers.15 There is also evidence that estrogen metabolism plays an important role in the initiation and the development of breast cancer by reducing mRNA and protein expression levels not only of Nrf2 but also of several antioxidant/phase 2 detoxification enzymes.18,21,22,43 These enzymes can significantly contribute to cellular defensive mechanisms by facilitating the clearance of toxic metabolites and ROS and by enhancing cellular capacity to repair oxidatively damaged DNA and proteins.21,26,32,43 During the breast carcinogenesis process, many oncogenes and tumor-suppressor genes can frequently be inactivated by aberrant DNA methylation.108,109 The restoration of normal DNA methylation patterns on oncogenes and various tumor-suppressor genes can be achieved by many dietary phytochemicals.61,110,111 In addition, these natural bioactive compounds can also suppress the overburden of oxidative stress and reactive estrogen quinones in E2-induced breast cancer by inducing various cytoprotective enzymes mainly through the Nrf2-dependent and independent mechanisms.17,18,21,43 The main protective enzymes involved in the prevention of E2-induced breast cancer are antioxidant enzyme SOD3 as well as repair and phase 2 detoxification enzymes NQO1, COMTs, GSTs, and OGG-1.15,21,26,27,32,68 During oxidative metabolism of estrogens, redox cycling between catechol estrogens and their corresponding reactive estrogen quinones often produces potentially harmful superoxide anions. SOD3 induced by antioxidants vitamin C or butylated hydroxyanisole (BHA) through the Nrf2–ARE pathway may play a protective role against superoxide anion-mediated oxidative DNA damage by catalyzing their conversion to hydrogen peroxide.21 Subsequently, other antioxidant enzymes such as cellular catalase and peroxidases remove hydrogen peroxide.130 Other studies also demonstrated that vitamin C or BHA may exert chemopreventive effects against estrogen-induced breast cancer by reversing E2-mediated upregulation of various miRNAs with oncogenic activity such as miR-93 and miR-153, and inducing Nrf2 and its cytoprotective genes.100,101 In addition to the antioxidant SOD3 enzyme, the induction of repair and phase 2 detoxification enzymes NQO1, COMT, GSTs, and OGG-1 has also been reported to play an essential role in mammary carcinogenesis process chemoprevention.26,32,68 In fact, it was shown that the antioxidants vitamin C or BHA and E2-metabolic inhibitor α-naphthoflavone can reverse E2-mediated decrease in the protein expression levels of Nrf2, NQO1, and OGG-1 by increasing expression of these proteins.43,68 NQO1 is a key phase 2 detoxification enzyme induced by the Nrf2–ARE pathway that is capable of converting E2-quinones back to E2-catechol, and thus limiting oxidative DNA damage and the subsequent mammary carcinogenesis process.15,17,18,54–56 Another three cytoprotective enzymes against E2-mediated breast cancer are COMTs, GSTs, and OGG-1. These enzymes can be induced by various dietary phytochemicals through Nrf2-dependent and independent mechanisms.18,19,131 COMTs catalyze the methylation of catechol estrogens to nongenotoxic methoxyestrogens (4-OCH3E1/E2), impeding their further oxidation to semiquinones and carcinogenic quinones and the associated formation of ROS.10,33,34,53,132 whereas GSTs detoxify reactive estrogen quinones by conjugation with GSH.15 Instead, OGG-1 is the rate-limiting enzyme of the DNA base excision repair pathway induced by the Nrf2–ARE pathway which is involved in the removal of 8-OHdG adducts from DNA. Single or combined gene polymorphisms of COMT, GST, and NQO1 can significantly reduce the expression and activity of these enzymes, dramatically increasing the breast cancer risk by enhancing formation of the depurinating adducts and oxidative DNA damage.133,134 Other studies also demonstrated that dietary tocopherol mixture containing 58% γ-tocopherol (γ-TmT) can significantly reduce breast cancer development by inducing preferential gene expression of CYP1A1 over CYP1B1, in the rat mammary tissues. This effect may reduce the circulating levels of E2 in ACI rats which could contribute to its inhibitory effects on mammary tumorigenesis.135 Additionally, treatment with dietary γ-TmT can also upregulate Nrf2-dependent antioxidant enzymes as well as PPARγ and its downstream genes while significantly reducing oxidative stress and cell proliferation markers at all three stages of the breast carcinogenesis process in ACI rats.62,136 An important role in breast cancer prevention is also played by Res, melatonin, and lipoic acid. Res is a polyphenolic compound present in grapes and red wine137 that is capable of exerting chemopreventive effects against breast cancer by reducing expression of miR-93 with oncogenic activity and inducing Nrf2 and its cytoprotective enzymes.21,22,43,138 Additionally, it can also negatively modulate expression of estrogen-activating enzyme CYP1B1 implicated in the formation of the 4-OH-E1(E2) catechols from E1/E2 if it is overexpressed.19 Multiple chemopreventive and therapeutic effects against mammary carcinogenesis are also exerted by melatonin and lipoic acid.24,25,65 Melatonin is an indolic hormone produced mainly by the pineal gland only during the night, or more exactly in darkness.139 The nocturnal secretion of melatonin is very sensitive to the inhibitory effects of light. As a consequence, exposure to light at night, even if for a short time, can cause a reduction or suppression in melatonin secretion.140 In addition to acting as a direct free radical scavenger,141 melatonin can also stimulate the transcription of antioxidant and detoxification enzymes through the Nrf2–ARE pathways.23,65 Additionally, Kubatka et al142 suggest that melatonin exhibits prominent anticancer effects in chemically induced rat mammary carcinogenesis by significantly decreasing tumor volume by almost 70% vs control. Other studies also indicate that melatonin exerts an antioxidant effect by the reduction of the E2-3,4-semiquinone to 4-OH-E2, melatonin being incapable of reacting with E2-3,4-quinones.143 Therefore, factors which reduce pineal function such as exposure to light at night,140 surgical pinealectomy,144 and aging145 can significantly increase the risk of breast cancer. Many studies also suggest that α-lipoic acid and reduced lipoic acid (dihydro α-lipoic acid) can protect from a variety of human diseases and malignancies, including breast cancer, not only by inducing the transcription of several antioxidant and detoxifying enzymes through the Nrf2–ARE pathway but also by recycling many cellular antioxidants such as coenzyme Q, vitamins C and E, and chelating iron and copper.24,25 Anther interesting research area in the study of estrogen-induced breast carcinogenesis process is cancer stem cell biology. This population of cells has the capacity to self-renew, differentiate, and be capable of initiating and driving tumor growth.146,147 Additionally, cancer stem cells also possess the ability to promote cancer recurrence, standard chemotherapy resistance, and metastasis.148 Various reports also suggest that the Nrf2–ARE pathway is required not only to control homeostatic quiescence of stem cells,112–114 but also to modulate proliferation of stem cells and differentiation through regulating the cellular levels of ROS via the expression of antioxidant and detoxification enzymes.17,113,117 Further, recent studies also indicate that estrogens can regulate breast cancer stem cell expansion through multiple pathways including Wnt/β-catenin, Hedgehog, Notch, and PI3K/mTOR.120,121 Currently, there are only limited data available on whether certain natural bioactive compounds may effectively inhibit estrogen-induced breast cancer stem cells. Further studies using in vivo animal models and clinical trials are required to improve our knowledge on the mechanisms of action of dietary phytochemicals on the breast carcinogenesis process. At the same time, other studies are also needed to improve our understanding of the role of estrogen metabolites in breast cancer initiation or development. A better understanding of the mechanisms that drive the metabolism of estrogen to generate genotoxic metabolites and free radicals to induce E2-mediated breast cancer will be needed to optimize therapeutic strategies to effectively prevent or treat estrogen-induced breast cancer.
Acknowledgments
The authors thank Dr Alessandra Trocino, Librarian at the Library of NCI G. Pascale Foundation of Naples, Italy, for her excellent bibliographic service and assistance.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5–29. doi:10.3322/caac.21254
2. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi:10.3322/caac.20107
3. Hulka BS, Moorman PG. Breast cancer: hormones and other risk factors. Maturitas. 2001;38:
4. Howlader N, Noone AM, Krapcho M, editors. SEER Cancer Statistics Review, 1975-2010. Bethesda, MD: National Cancer Institute; 2012. Available from:
5. Henquin N, Trostler N, Horn Y. Nutritional risk factors and breast cancer in Jewish and Arab women. Cancer Nurs. 1994;17:326–333.
6. Ilic M, Vlajinac H, Marinkovic J. Cigarette smoking and breast cancer: a case-control study in Serbia. Asian Pac J Cancer Prev. 2014;14:6643–6647.
7. Fernandes A, Bianchi G, Feltri AP, Perez M, Correnti M. Presence of human papillomavirus in breast cancer and its association with prognostic factors. Ecancermedicalscience. 2015;9:548. doi:10.3332/ecancer.2015.548
8. Althuis MD, Fergenbaum JH, Garcia-Closas M, Brinton LA, Madigan MP, Sherman ME. Etiology of hormone receptor-defined breast cancer: a systematic review of the literature. Cancer Epidemiol Biomarkers Prev. 2004;13:1558–1568.
9. Yager JD. Mechanisms of estrogen carcinogenesis: the role of E2/E1-quinone metabolites suggests new approaches to preventive intervention–A review. Steroids. 2015;99:56–60. doi:10.1016/j.steroids.2014.08.006
10. Gaikwad NW, Yang L, Muti P, et al. The molecular etiology of breast cancer: evidence from biomarkers of risk. Int J Cancer. 2008;122:1949–1957. doi:10.1002/ijc.23329
11. Dietz BM, Hajirahimkhan A, Dunlap TL, Bolton JL. Botanicals and their bioactive phytochemicals for women’s health. Pharmacol Rev. 2016;68:1026–1073. doi:10.1124/pr.115.010843
12. Santen RJ, Yue W, Wang JP. Estrogen metabolites and breast cancer. Steroids. 2015;99:61–66. doi:10.1016/j.steroids.2014.08.003
13. Bak MJ, Das Gupta S, Wahler J, Suh N. Role of dietary bioactive natural products in estrogen receptor-positive breast cancer. Semin Cancer Biol. 2016;40–41:170–191. doi:10.1016/j.semcancer.2016.03.001
14. Cavalieri EL, Rogan EG. Depurinating estrogen-DNA adducts in the etiology and prevention of breast and other human cancers. Future Oncol. 2010;6:75–91. doi:10.2217/fon.09.137
15. Cavalieri EL, Rogan EG. Unbalanced metabolism of endogenous estrogens in the etiology and prevention of human cancer. J Steroid Biochem Mol Biol. 2011;125:169–180. doi:10.1016/j.jsbmb.2011.03.008
16. Hong WK, Sporn MB. Recent advances in chemoprevention of cancer. Science. 1997;278:1073–1077.
17. Kensler TW, Wakabayashi N. Nrf2: friend or foe for chemoprevention? Carcinogenesis. 2010;31:90–99. doi:10.1093/carcin/bgp231
18. Yang L, Zahid M, Liao Y, et al. Reduced formation of depurinating estrogen-DNA adducts by sulforaphane or KEAP1 disruption in human mammary epithelial MCF-10A cells. Carcinogenesis. 2013;34:2587–2592. doi:10.1093/carcin/bgt246
19. Lu F, Zahid M, Wang C, Saeed M, Cavalieri EL, Rogan EG. Resveratrol prevents estrogen-DNA adduct formation and neoplastic transformation in MCF-10F cells. Cancer Prev Res (Phila). 2008;1:135–145. doi:10.1158/1940-6207.CAPR-08-0037
20. Zahid M, Saeed M, Beseler C, Rogan EG, Cavalieri EL. Resveratrol and N-acetylcysteine block the cancer-initiating step in MCF-10F cells. Free Radic Biol Med. 2011;50:78–85. doi:10.1016/j.freeradbiomed.2010.10.662
21. Singh B, Bhat HK. Superoxide dismutase 3 is induced by antioxidants, inhibits oxidative DNA damage and is associated with inhibition of estrogen-induced breast cancer. Carcinogenesis. 2012;33:2601–2610. doi:10.1093/carcin/bgs300
22. Singh B, Shoulson R, Chatterjee A, et al. Resveratrol inhibits estrogen-induced breast carcinogenesis through induction of NRF2-mediated protective pathways. Carcinogenesis. 2014;35:1872–1880. doi:10.1093/carcin/bgu120
23. Vriend J, Reiter RJ. The Keap1-Nrf2-antioxidant response element pathway: a review of its regulation by melatonin and the proteasome. Mol Cell Endocrinol. 2015;401:213–220. doi:10.1016/j.mce.2014.12.013
24. Suh JH, Shenvi SV, Dixon BM, et al. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc Natl Acad Sci U S A. 2004;101:3381–3386. doi:10.1073/pnas.0400282101
25. Packer L, Roy S, Sen CK. Alpha-lipoic acid: a metabolic antioxidant and potential redox modulator of transcription. Adv Pharmacol. 1997;38:79–101.
26. Yager JD, Davidson NE. Estrogen carcinogenesis in breast cancer. N Engl J Med. 2006;354:270–282. doi:10.1056/NEJMra050776
27. Cavalieri EL, Rogan EG. Depurinating estrogen-DNA adducts, generators of cancer initiation: their minimization leads to cancer prevention. Clin Transl Med. 2016;5:12. doi:10.1186/s40169-016-0088-3
28. Ryan KJ. Biological aromatization of steroids. J Biol Chem. 1959;234:268–272.
29. Samavat H, Kurzer MS. Estrogen metabolism and breast cancer. Cancer Lett. 2015;356:231–243. doi:10.1016/j.canlet.2014.04.018
30. Cavalieri E, Chakravarti D, Guttenplan J, et al. Catechol estrogen quinones as initiators of breast and other human cancers: implications for biomarkers of susceptibility and cancer prevention. Biochim Biophys Acta. 2006;1766:63–78. doi:10.1016/j.bbcan.2006.03.001
31. Cavalieri EL, Stack DE, Devanesan PD, et al. Molecular origin of cancer: catechol estrogen-3,4-quinones as endogenous tumor initiators. Proc Natl Acad Sci U S A. 1997;94:10937–10942. doi:10.1073/pnas.94.20.10937
32. Yue W, Yager JD, Wang JP, Jupe ER, Santen RJ. Estrogen receptor-dependent and independent mechanisms of breast cancer carcinogenesis. Steroids. 2013;78:161–170. doi:10.1016/j.steroids.2012.11.001
33. Stack DE, Byun J, Gross ML, Rogan EG, Cavalieri EL. Molecular characteristics of catechol estrogen quinones in reactions with deoxyribonucleosides. Chem Res Toxicol. 1996;9:851–859. doi:10.1021/tx960002q
34. Circu ML, Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med. 2010;48:749–762. doi:10.1016/j.freeradbiomed.2009.12.022
35. Lakhani NJ, Venitz J, Figg WD, Sparreboom A. Pharmacogenetics of estrogen metabolism and transport in relation to cancer. Curr Drug Metab. 2003;4:505–513.
36. Raftogianis R, Creveling C, Weinshilboum R, Weisz J. Estrogen metabolism by conjugation. J Natl Cancer Inst Monogr. 2000;113–124. doi:10.1093/oxfordjournals.jncimonographs.a024234
37. Hobkirk R. Steroid sulfotransferases and steroid sulfate sulfatases: characteristics and biological roles. Can J Biochem Cell Biol. 1985;63:1127–1144.
38. Sumi D, Numasawa Y, Endo A, Iwamoto N, Kumagai Y. Catechol estrogens mediated activation of Nrf2 through covalent modification of its quinone metabolite to Keap1. J Toxicol Sci. 2009;34:627–635.
39. Yager JD. Catechol-O-methyltransferase: characteristics, polymorphisms and role in breast cancer. Drug Discov Today Dis Mech. 2012;9:e41–e46. doi:10.1016/j.ddmec.2012.10.002
40. Mitrunen K, Hirvonen A. Molecular epidemiology of sporadic breast cancer. The role of polymorphic genes involved in oestrogen biosynthesis and metabolism. Mutat Res. 2003;544:9–41.
41. Mailander PC, Meza JL, Higginbotham S, Chakravarti D. Induction of A.T to G.C mutations by erroneous repair of depurinated DNA following estrogen treatment of the mammary gland of ACI rats. J Steroid Biochem Mol Biol. 2006;101:204–215. doi:10.1016/j.jsbmb.2006.06.019
42. Cavalieri E, Rogan E. The molecular etiology and prevention of estrogen-initiated cancers: ockham’s Razor: pluralitas non est ponenda sine necessitate. Plurality should not be posited without necessity. Mol Aspects Med. 2014;36:1–55. doi:10.1016/j.mam.2013.08.002
43. Singh B, Bhat NK, Bhat HK. Induction of NAD(P)H-quinone oxidoreductase 1 by antioxidants in female ACI rats is associated with decrease in oxidative DNA damage and inhibition of estrogen-induced breast cancer. Carcinogenesis. 2012;33:156–163. doi:10.1093/carcin/bgr237
44. Gaikwad NW, Yang L, Pruthi S, et al. Urine biomarkers of risk in the molecular etiology of breast cancer. Breast Cancer (Auckl). 2009;3:1–8.
45. Miller WR, O’Neill J. The importance of local synthesis of estrogen within the breast. Steroids. 1987;50:537–548.
46. Simpson ER, Mahendroo MS, Means GD, et al. Aromatase cytochrome P450, the enzyme responsible for estrogen biosynthesis. Endocr Rev. 1994;15:342–355. doi:10.1210/edrv-15-3-342
47. Jefcoate CR, Liehr JG, Santen RJ, et al. Tissue-specific synthesis and oxidative metabolism of estrogens. In: Cavalieri E, Rogan E, editors. JNCI Monograph. PA: Oxford Press; 2000:95–112.
48. Santner SJ, Feil PD, Santen RJ. In situ estrogen production via the estrone sulfatase pathway in breast tumors: relative importance versus the aromatase pathway. J Clin Endocrinol Metab. 1984;59:29–33. doi:10.1210/jcem-59-1-29
49. Pasqualini JR, Chetrite G, Blacker C, et al. Concentrations of estrone, estradiol, and estrone sulfate and evaluation of sulfatase and aromatase activities in pre- and postmenopausal breast cancer patients. J Clin Endocrinol Metab. 1996;81:1460–1464. doi:10.1210/jcem.81.4.8636351
50. Spink DC, Hayes CL, Young NR, et al. The effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on estrogen metabolism in MCF-7 breast cancer cells: evidence for induction of a novel 17 beta-estradiol 4-hydroxylase. J Steroid Biochem Mol Biol. 1994;51:251–258.
51. Hayes CL, Spink DC, Spink BC, Cao JQ, Walker NJ, Sutter TR. 17 beta-estradiol hydroxylation catalyzed by human cytochrome P450 1B1. Proc Natl Acad Sci U S A. 1996;93:9776–9781. doi:10.1073/pnas.93.18.9776
52. Spink DC, Spink BC, Cao JQ, et al. Differential expression of CYP1A1 and CYP1B1 in human breast epithelial cells and breast tumor cells. Carcinogenesis. 1998;19:291–298.
53. Lu F, Zahid M, Saeed M, Cavalieri EL, Rogan EG. Estrogen metabolism and formation of estrogen-DNA adducts in estradiol-treated MCF-10F cells. The effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin induction and catechol-O-methyltransferase inhibition. J Steroid Biochem Mol Biol. 2007;105:150–158. doi:10.1016/j.jsbmb.2006.12.102
54. Gaikwad NW, Rogan EG, Cavalieri EL. Evidence from ESI-MS for NQO1-catalyzed reduction of estrogen ortho-quinones. Free Radic Biol Med. 2007;43:1289–1298. doi:10.1016/j.freeradbiomed.2007.07.021
55. Gaikwad NW, Yang L, Rogan EG, Cavalieri EL. Evidence for NQO2-mediated reduction of the carcinogenic estrogen ortho-quinones. Free Radic Biol Med. 2009;46:253–262. doi:10.1016/j.freeradbiomed.2008.10.029
56. Talalay P, Dinkova-Kostova AT, Holtzclaw WD. Importance of phase 2 gene regulation in protection against electrophile and reactive oxygen toxicity and carcinogenesis. Adv Enzyme Regul. 2003;43:121–134.
57. Singh S, Zahid M, Saeed M, et al. NAD(P)H:quinone oxidoreductase 1 Arg139Trp and Pro187Ser polymorphisms imbalance estrogen metabolism towards DNA adduct formation in human mammary epithelial cells. J Steroid Biochem Mol Biol. 2009;117:56–66. doi:10.1016/j.jsbmb.2009.07.003
58. Singh S, Chakravarti D, Edney JA, et al. Relative imbalances in the expression of estrogen-metabolizing enzymes in the breast tissue of women with breast carcinoma. Oncol Rep. 2005;14:1091–1096.
59. Giudice A, Arra C, Turco MC. Review of molecular mechanisms involved in the activation of the Nrf2-ARE signaling pathway by chemopreventive agents. Methods Mol Biol. 2010;647:37–74. doi:10.1007/978-1-60761-738-9_3
60. Giudice A, Montella M. Activation of the Nrf2-ARE signaling pathway: a promising strategy in cancer prevention. Bioessays. 2006;28:169–181. doi:10.1002/bies.20359
61. Qin S, Hou DX. Multiple regulations of Keap1/Nrf2 system by dietary phytochemicals. Mol Nutr Food Res. 2016;60:1731–1755. doi:10.1002/mnfr.201501017
62. Smolarek AK, So JY, Burgess B, et al. Dietary administration of delta- and gamma-tocopherol inhibits tumorigenesis in the animal model of estrogen receptor-positive, but not HER-2 breast cancer. Cancer Prev Res (Phila). 2012;5:1310–1320. doi:10.1158/1940-6207.CAPR-12-0263
63. Smolarek AK, So JY, Thomas PE, et al. Dietary tocopherols inhibit cell proliferation, regulate expression of ERalpha, PPARgamma, and Nrf2, and decrease serum inflammatory markers during the development of mammary hyperplasia. Mol Carcinog. 2013;52:514–525. doi:10.1002/mc.21886
64. Buchon N, Broderick NA, Chakrabarti S, Lemaitre B. Invasive and indigenous microbiota impact intestinal stem cell activity through multiple pathways in Drosophila. Genes Dev. 2009;23:2333–2344. doi:10.1101/gad.1827009
65. Luchetti F, Canonico B, Betti M, et al. Melatonin signaling and cell protection function. Faseb J. 2010;24:3603–3624. doi:10.1096/fj.10-154450
66. Nielsen AJ, McNulty J. Polyphenolic natural products and natural product-inspired steroidal mimics as aromatase inhibitors. Med Res Rev. 2018. doi:10.1002/med.21536
67. Itoh K, Chiba T, Takahashi S, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–322.
68. Singh B, Chatterjee A, Ronghe AM, Bhat NK, Bhat HK. Antioxidant-mediated up-regulation of OGG1 via NRF2 induction is associated with inhibition of oxidative DNA damage in estrogen-induced breast cancer. BMC Cancer. 2013;13:253. doi:10.1186/1471-2407-13-253
69. Tebay LE, Robertson H, Durant ST, et al. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic Biol Med. 2015;88:108–146. doi:10.1016/j.freeradbiomed.2015.06.021
70. Yang Y, Wang Y, Wang T, Jiang X, Wang L. Screening active components of modified Xiaoyao powder as NRF2 agonists. Cell Biochem Funct. 2017;35(8):518–526. doi:10.1002/cbf.3309
71. Szaefer H, Krajka-Kuźniak V, Licznerska B, Bartoszek A, Baer-Dubowska W. Cabbage juices and indoles modulate the expression profile of AhR, ERα, and Nrf2 in human breast cell lines. Nutr Cancer. 2015;67(8):1342–1354. doi:10.1080/01635581.2015.1082111
72. Peng CY, You BJ, Lee CL, et al. The roles of 4β-hydroxywithanolide e from physalis peruviana on the Nrf2-Anti-oxidant system and the cell cycle in breast cancer cells. Am J Chin Med. 2016;44(3):617–636. doi:10.1142/S0192415X16500348
73. Kobayashi EH, Suzuki T, Funayama R, et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat Commun. 2016;7:11624. doi:10.1038/ncomms11624
74. Kim J, Cha YN, Surh YJ. A protective role of nuclear factor-erythroid 2-related factor-2 (Nrf2) in inflammatory disorders. Mutat Res. 2010;690:12–23. doi:10.1016/j.mrfmmm.2009.09.007
75. Johnston SR, Dowsett M. Aromatase inhibitors for breast cancer: lessons from the laboratory. Nat Rev Cancer. 2003;3:821–831. doi:10.1038/nrc1211
76. Chen XL, Kunsch C. Induction of cytoprotective genes through Nrf2/antioxidant response element pathway: a new therapeutic approach for the treatment of inflammatory diseases. Curr Pharm Des. 2004;10:879–891.
77. Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1:a001651. doi:10.1101/cshperspect.a001651
78. Perkins ND. The Rel/NF-kappa B family: friend and foe. Trends Biochem Sci. 2000;25:434–440. doi:10.1016/S0968-0004(00)01617-0
79. Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat Immunol. 2011;12:715–723. doi:10.1038/ni.2060
80. Na HK, Surh YJ. Transcriptional regulation via cysteine thiol modification: a novel molecular strategy for chemoprevention and cytoprotection. Mol Carcinog. 2006;45:368–380. doi:10.1002/mc.20225
81. Surh YJ. Cancer chemoprevention with dietary phytochemicals. Nat Rev Cancer. 2003;3:768–780. doi:10.1038/nrc1189
82. Manna SK, Mukhopadhyay A, Aggarwal BB. Resveratrol suppresses TNF-induced activation of nuclear transcription factors NF-kappa B, activator protein-1, and apoptosis: potential role of reactive oxygen intermediates and lipid peroxidation. J Immunol. 2000;164:6509–6519.
83. Gopalakrishnan A, Tony Kong AN. Anticarcinogenesis by dietary phytochemicals: cytoprotection by Nrf2 in normal cells and cytotoxicity by modulation of transcription factors NF-kappa B and AP-1 in abnormal cancer cells. Food Chem Toxicol. 2008;46:1257–1270. doi:10.1016/j.fct.2007.09.082
84. Zhu M, Zhang Y, Cooper S, Sikorski E, Rohwer J, Bowden GT. Phase II enzyme inducer, sulforaphane, inhibits UVB-induced AP-1 activation in human keratinocytes by a novel mechanism. Mol Carcinog. 2004;41:179–186. doi:10.1002/mc.20052
85. Liu GH, Qu J, Shen X. NF-kappaB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim Biophys Acta. 2008;1783:713–727. doi:10.1016/j.bbamcr.2008.01.002
86. Gan FF, Ling H, Ang X, et al. A novel shogaol analog suppresses cancer cell invasion and inflammation,and displays cytoprotective effects through modulation of NF-κB and Nrf2-Keap1 signaling pathways. Toxicol Appl Pharmacol. 2013;272(3):852–862. doi:10.1016/j.taap.2013.07.011
87. Mandal A, Bhatia D, Bishayee A. Anti-inflammatory mechanism involved in pomegranate-mediated prevention of breast cancer: the role of NF-κB and Nrf2 signaling pathways. Nutrients. 2017;9(5). doi:10.3390/nu9050436
88. Mandal A, Bishayee A. Trianthema portulacastrum Linn. Displays anti-inflammatory responses during chemically induced rat mammary tumorigenesis through simultaneous and differential regulation of NF-κB and Nrf2 signaling pathways. Int J Mol Sci. 2015;16(2):2426–2445. doi:10.3390/ijms16022426
89. Jain A, Samykutty A, Jackson C, et al. Curcumin inhibits PhIP induced cytotoxicity in breast epithelial cells through multiple molecular targets. Cancer Lett. 2015;365(1):122–131. doi:10.1016/j.canlet.2015.05.017
90. Wu CF, Lee MG, El-Shazly M, et al. Isoaaptamine induces T-47D cells apoptosis and autophagy via oxidative stress. Mar Drugs. 2018;16(1):E18. doi:10.3390/md16010018
91. Wu JJ, Zhu YF, Guo ZZ, et al. Aconitum alkaloids, the major components of Aconitum species, affect expression of multidrug resistance-associated protein 2 and breast cancer resistance protein by activating the Nrf2-mediated signalling pathway. Phytomedicine. 2018;44:87–97.
92. Magesh S, Chen Y, Hu L. Small molecule modulators of Keap1-Nrf2-ARE pathway as potential preventive and therapeutic agents. Med Res Rev. 2012;32:687–726. doi:10.1002/med.21257
93. Brueggemeier RW, Hackett JC, Diaz-Cruz ES. Aromatase inhibitors in the treatment of breast cancer. Endocr Rev. 2005;26:331–345. doi:10.1210/er.2004-0015
94. Way TD, Lee HH, Kao MC, Lin JK. Black tea polyphenol theaflavins inhibit aromatase activity and attenuate tamoxifen resistance in HER2/neu-transfected human breast cancer cells through tyrosine kinase suppression. Eur J Cancer. 2004;40:2165–2174. doi:10.1016/j.ejca.2004.06.018
95. Li F, Wong TY, Lin SM, et al. Coadministrating luteolin minimizes the side effects of the aromatase inhibitor letrozole. J Pharmacol Exp Ther. 2014;351:270–277. doi:10.1124/jpet.114.216754
96. Zhang M, Huang J, Xie X, Holman CD. Dietary intakes of mushrooms and green tea combine to reduce the risk of breast cancer in Chinese women. Int J Cancer. 2009;124:1404–1408. doi:10.1002/ijc.24047
97. Liu H, Talalay P. Relevance of anti-inflammatory and antioxidant activities of exemestane and synergism with sulforaphane for disease prevention. Proc Natl Acad Sci U S A. 2013;110:19065–19070. doi:10.1073/pnas.1318247110
98. Wang Y, Lee KW, Chan FL, Chen S, Leung LK. The red wine polyphenol resveratrol displays bilevel inhibition on aromatase in breast cancer cells. Toxicol Sci. 2006;92:71–77. doi:10.1093/toxsci/kfj190
99. Ansell PJ, Lo SC, Newton LG, et al. Repression of cancer protective genes by 17beta-estradiol: ligand-dependent interaction between human Nrf2 and estrogen receptor alpha. Mol Cell Endocrinol. 2005;243:27–34. doi:10.1016/j.mce.2005.08.002
100. Singh B, Ronghe AM, Chatterjee A, Bhat NK, Bhat HK. MicroRNA-93 regulates NRF2 expression and is associated with breast carcinogenesis. Carcinogenesis. 2013;34:1165–1172. doi:10.1093/carcin/bgt026
101. Wang B, Teng Y, Liu Q. MicroRNA-153 regulates NRF2 expression and is associated with breast carcinogenesis. Clin Lab. 2016;62:39–47.
102. Vislovukh A, Kratassiouk G, Porto E, et al. Proto-oncogenic isoform A2 of eukaryotic translation elongation factor eEF1 is a target of miR-663 and miR-744. Br J Cancer. 2013;108:2304–2311. doi:10.1038/bjc.2013.243
103. Hagiwara K, Kosaka N, Yoshioka Y, Takahashi RU, Takeshita F, Ochiya T. Stilbene derivatives promote Ago2-dependent tumour-suppressive microRNA activity. Sci Rep. 2012;2:314. doi:10.1038/srep00386
104. Chatterjee A, Ronghe A, Singh B, Bhat NK, Chen J, Bhat HK. Natural antioxidants exhibit chemopreventive characteristics through the regulation of CNC b-Zip transcription factors in estrogen-induced breast carcinogenesis. J Biochem Mol Toxicol. 2014;28:529–538. doi:10.1002/jbt.21594
105. Sankaranarayanan K, Jaiswal AK. Nrf3 negatively regulates antioxidant-response element-mediated expression and antioxidant induction of NAD(P)H: quinoneoxidoreductase1 gene. J Biol Chem. 2004;279:50810–50817. doi:10.1074/jbc.M404984200
106. Kang HJ, Hong YB, Kim HJ, Wang A, Bae I. Bioactive food components prevent carcinogenic stress via Nrf2 activation in BRCA1 deficient breast epithelial cells. Toxicol Lett. 2012;209:154–160. doi:10.1016/j.toxlet.2011.12.002
107. Panahi Y, Beiraghdar F, Amirhamzeh A, Poursaleh Z, Saadat A, Sahebkar A. Environmental toxicant exposure and cancer: the role of epigenetic changes and protection by phytochemicals. Curr Pharm Des. 2016;22:130–140.
108. Guo Y, Yu S, Zhang C, Kong AN. Epigenetic regulation of Keap1-Nrf2 signaling. Free Radic Biol Med. 2015;88:337–349. doi:10.1016/j.freeradbiomed.2015.06.013
109. Wu TY, Khor TO, Su ZY, et al. Epigenetic modifications of Nrf2 by 3,3ʹ-diindolylmethane in vitro in TRAMP C1 cell line and in vivo TRAMP prostate tumors. Aaps J. 2013;15:864–874. doi:10.1208/s12248-013-9493-3
110. Khor TO, Huang Y, Wu TY, Shu L, Lee J, Kong AN. Pharmacodynamics of curcumin as DNA hypomethylation agent in restoring the expression of Nrf2 via promoter CpGs demethylation. Biochem Pharmacol. 2011;82:1073–1078. doi:10.1016/j.bcp.2011.07.065
111. Su ZY, Khor TO, Shu L, et al. Epigenetic reactivation of Nrf2 in murine prostate cancer TRAMP C1 cells by natural phytochemicals Z-ligustilide and radix angelica sinensis via promoter CpG demethylation. Chem Res Toxicol. 2013;26:477–485. doi:10.1021/tx300524p
112. Cheung TH, Rando TA. Molecular regulation of stem cell quiescence. Nat Rev Mol Cell Biol. 2013;14:329–340. doi:10.1038/nrm3591
113. Hochmuth CE, Biteau B, Bohmann D, Jasper H. Redox regulation by Keap1 and Nrf2 controls intestinal stem cell proliferation in Drosophila. Cell Stem Cell. 2011;8:188–199. doi:10.1016/j.stem.2010.12.006
114. Tsai JJ, Dudakov JA, Takahashi K, et al. Nrf2 regulates haematopoietic stem cell function. Nat Cell Biol. 2013;15:309–316. doi:10.1038/ncb2699
115. Zhang Y, Xia J, Li Q, et al. NRF2/long noncoding RNA ROR signaling regulates mammary stem cell expansion and protects against estrogen genotoxicity. J Biol Chem. 2014;289:31310–31318. doi:10.1074/jbc.M114.604868
116. Yao Y, Brodie AM, Davidson NE, Kensler TW, Zhou Q. Inhibition of estrogen signaling activates the NRF2 pathway in breast cancer. Breast Cancer Res Treat. 2010;124:585–591. doi:10.1007/s10549-010-1023-8
117. Ye ZW, Zhang J, Townsend DM, Tew KD. Oxidative stress, redox regulation and diseases of cellular differentiation. Biochim Biophys Acta. 2015;1850:1607–1621. doi:10.1016/j.bbagen.2014.11.010
118. Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi:10.1146/annurev.pharmtox.46.120604.141046
119. Reddy NM, Kleeberger SR, Yamamoto M, et al. Genetic dissection of the Nrf2-dependent redox signaling-regulated transcriptional programs of cell proliferation and cytoprotection. Physiol Genomics. 2007;32:74–81. doi:10.1152/physiolgenomics.'00126.2007
120. Valkenburg KC, Graveel CR, Zylstra-Diegel CR, Zhong Z, Williams BO. Wnt/beta-catenin Signaling in Normal and Cancer Stem Cells. Cancers (Basel). 2011;3:2050–2079. doi:10.3390/cancers3022050
121. Velasco-Velazquez MA, Homsi N, De La Fuente M, Pestell RG. Breast cancer stem cells. Int J Biochem Cell Biol. 2012;44:573–577. doi:10.1016/j.biocel.2011.12.020
122. Fillmore CM, Gupta PB, Rudnick JA, et al. Estrogen expands breast cancer stem-like cells through paracrine FGF/Tbx3 signaling. Proc Natl Acad Sci U S A. 2010;107:21737–21742. doi:10.1073/pnas.1007863107
123. Charpentier MS, Whipple RA, Vitolo MI, et al. Curcumin targets breast cancer stem-like cells with microtentacles that persist in mammospheres and promote reattachment. Cancer Res. 2014;74:1250–1260. doi:10.1158/0008-5472.CAN-13-1778
124. Li Y, Zhang T, Korkaya H, et al. Sulforaphane, a dietary component of broccoli/broccoli sprouts, inhibits breast cancer stem cells. Clin Cancer Res. 2010;16:2580–2590. doi:10.1158/1078-0432.CCR-09-2937
125. Montales MT, Rahal OM, Kang J, et al. Repression of mammosphere formation of human breast cancer cells by soy isoflavone genistein and blueberry polyphenolic acids suggests diet-mediated targeting of cancer stem-like/progenitor cells. Carcinogenesis. 2012;33:652–660. doi:10.1093/carcin/bgr317
126. Mineva ND, Paulson KE, Naber SP, Yee AS, Sonenshein GE. Epigallocatechin-3-gallate inhibits stem-like inflammatory breast cancer cells. PLoS One. 2013;8:e73464. doi:10.1371/journal.pone.0073464
127. Fu Y, Chang H, Peng X, et al. Resveratrol inhibits breast cancer stem-like cells and induces autophagy via suppressing Wnt/beta-catenin signaling pathway. PLoS One. 2014;9(7):e102535. doi:10.1371/journal.pone.0102535
128. Kubatka P, Kello M, Kajo K, et al. Oregano demonstrates distinct tumour-suppressive effects in the breast carcinoma model. Eur J Nutr. 2017;56(3):1303–1316. doi:10.1007/s00394-016-1181-5
129. Kubatka P, Uramova S, Kello M, et al. Antineoplastic effects of clove buds (Syzygium aromaticum L.) in the model of breast carcinoma. J Cell Mol Med. 2017;21(11):2837–2851. doi:10.1111/jcmm.13197
130. Zelko IN, Mariani TJ, Folz RJ. Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic Biol Med. 2002;33:337–349.
131. Agyeman AS, Chaerkady R, Shaw PG, et al. Transcriptomic and proteomic profiling of KEAP1 disrupted and sulforaphane-treated human breast epithelial cells reveals common expression profiles. Breast Cancer Res Treat. 2012;132:175–187. doi:10.1007/s10549-011-1536-9
132. Zahid M, Saeed M, Lu F, Gaikwad N, Rogan E, Cavalieri E. Inhibition of catechol-O-methyltransferase increases estrogen-DNA adduct formation. Free Radic Biol Med. 2007;43:1534–1540. doi:10.1016/j.freeradbiomed.2007.08.005
133. Park SK, Yim DS, Yoon KS, et al. Combined effect of GSTM1, GSTT1, and COMT genotypes in individual breast cancer risk. Breast Cancer Res Treat. 2004;88:55–62. doi:10.1007/s10549-004-0745-x
134. Peng Q, Lu Y, Lao X, et al. The NQO1 Pro187 Ser polymorphism and breast cancer susceptibility: evidence from an updated meta-analysis. Diagn Pathol. 2014;9:100. doi:10.1186/1746-1596-9-100
135. Das Gupta S, Sae-Tan S, Wahler J, et al. Dietary gamma-tocopherol-rich mixture inhibits estrogen-induced mammary tumorigenesis by modulating estrogen metabolism, antioxidant response, And PPARgamma. Cancer Prev Res (Phila). 2015;8:807–816. doi:10.1158/1940-6207.CAPR-15-0154
136. Das Gupta S, So JY, Wall B, et al. Tocopherols inhibit oxidative and nitrosative stress in estrogen-induced early mammary hyperplasia in ACI rats. Mol Carcinog. 2015;54:916–925. doi:10.1002/mc.22164
137. Catalgol B, Batirel S, Taga Y, Ozer NK. Resveratrol: french paradox revisited. Front Pharmacol. 2012;3:141. doi:10.3389/fphar.2012.00141
138. Singh B, Bhat NK, Bhat HK. Partial inhibition of estrogen-induced mammary carcinogenesis in rats by tamoxifen: balance between oxidant stress and estrogen responsiveness. PLoS One. 2011;6:e25125. doi:10.1371/journal.pone.0025125
139. Brainard GC, Rollag MD, Hanifin JP. Photic regulation of melatonin in humans: ocular and neural signal transduction. J Biol Rhythms. 1997;12:537–546. doi:10.1177/074873049701200608
140. Grant SG, Melan MA, Latimer JJ, Witt-Enderby PA. Melatonin and breast cancer: cellular mechanisms, clinical studies and future perspectives. Expert Rev Mol Med. 2009;11:e5. doi:10.1017/S1462399409000982
141. Bonnefont-Rousselot D, Collin F. Melatonin: action as antioxidant and potential applications in human disease and aging. Toxicology. 2010;278:55–67. doi:10.1016/j.tox.2010.04.008
142. Kubatka P, Bojková B, M ciková-Kalická K, et al. Effects of tamoxifen and melatonin on mammary gland cancer induced by N-methyl-N-nitrosourea and by 7,12-dimethylbenz(a)anthracene, respectively, in female. Sprague-Dawley Rats Folia Biol (Praha). 2001;47(1):5–10.
143. Zahid M, Gaikwad NW, Rogan EG, Cavalieri EL. Inhibition of depurinating estrogen-DNA adduct formation by natural compounds. Chem Res Toxicol. 2007;20:1947–1953. doi:10.1021/tx700269s
144. Cohen M, Lippman M, Chabner B. Role of pineal gland in aetiology and treatment of breast cancer. Lancet. 1978;2:814–816.
145. Hill SM, Cheng C, Yuan L, et al. Age-related decline in melatonin and its MT1 receptor are associated with decreased sensitivity to melatonin and enhanced mammary tumor growth. Curr Aging Sci. 2013;6:125–133.
146. Liu S, Dontu G, Wicha MS. Mammary stem cells, self-renewal pathways, and carcinogenesis. Breast Cancer Res. 2005;7:86–95. doi:10.1186/bcr1021
147. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi:10.1038/35102167
148. Dandawate PR, Subramaniam D, Jensen RA, Anant S. Targeting cancer stem cells and signaling pathways by phytochemicals: novel approach for breast cancer therapy. Semin Cancer Biol. 2016;40–41:192–208. doi:10.1016/j.semcancer.2016.09.001
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.