Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 15
Disrupting Interleukin 12 Improves Microvascular Endothelial Function in Type 2 Diabetes Through ER Stress CHOP and Oxidative Stress Mechanisms
Authors Radwan E, Belmadani S, Matrougui K
Received 6 April 2022
Accepted for publication 8 July 2022
Published 30 August 2022 Volume 2022:15 Pages 2633—2642
DOI https://doi.org/10.2147/DMSO.S369488
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Antonio Brunetti
Eman Radwan,1,2 Souad Belmadani,1 Khalid Matrougui1
1Department of Physiological Sciences, EVMS, Norfolk, VA, 23501, USA; 2Department of Medical Biochemistry, Faculty of Medicine, Assiut University, Asyut, Egypt
Correspondence: Khalid Matrougui, Department of Physiological Sciences, EVMS, Norfolk, VA, 23501, USA, Tel +1 757-446-5278, Email [email protected]
Purpose: Vascular endothelial dysfunction is well established in type 2 diabetes. Interleukin-12 (IL-12) and endoplasmic reticulum (ER) stress are up-regulated in type 2 diabetic patients and animal models of type 2 diabetes. However, the role and underlying mechanisms of IL-12 and the ER stress CHOP in endothelial dysfunction are not fully understood.
Methods: We generated double knockout mice between db−/db− and p40IL-12−/− mice (db−/db−p40-IL− 12-/-) and endoplasmic (ER) stress-CHOP−/− mice (db−/db−CHOP-/-). We performed a glucose tolerance test (GTT) to determine the effect of IL-12 and ER stress CHOP on glucose metabolism. We assessed the endothelial function and determined the phosphorylation level of eNOS, Akt, AMPK, and the expression of ER stress (CHOP, BIP), and oxidative stress (Nox2 and Nox4 and NADPH oxidase activity).
Results: The results showed that GTT was improved in db-/db−p40-IL− 12-/- and db−/db−CHOP-/- suggesting IL-12 and CHOP as parts of a mechanism involved in the development of type 2 diabetes. The microvascular endothelial dysfunction in db−/db− mouse is associated with decreased phosphorylated eNOS, Akt, AMPK, and increased CHOP, BIP, Nox2, and Nox4 expressions. Interestingly, disrupting IL-12 and ER stress CHOP in db−/db− mice significantly improved endothelial function, increased survival markers expression and decreased ER and oxidative stress.
Conclusion: Using a genetic approach, these findings provide evidence that IL-12 and ER stress CHOP play a significant role in microvascular endothelial dysfunction in type 2 diabetes.
Keywords: type 2 diabetes, interleukin-12, ER stress, microvascular endothelial dysfunction, oxidative stress
Introduction
Diabetes has affected lives for thousands of years. Type 2 diabetes is a complex metabolic disease that fundamentally arises from the loss of functional pancreatic beta cell mass associated with a significant damage to the cardiovascular system. Microvascular endothelial cell dysfunction is a clinically significant problem in patients with type 2 diabetes as it causes myocardial infarction, stroke, peripheral artery disease, retinopathy, nephropathy, cardiomyopathy and delaying wound healing.1–9 Moreover, complications of the microcirculation often lead to hypertension, which substantially aggravates the health of diabetic patients. As a result, the concomitant cardiovascular complications are mainly responsible for the high morbidity and mortality in patients with type 2 diabetes. Despite decades of extensive biomedical research in this area, current therapies neither halt nor reverse microvascular complications that occur in type 2 diabetes. Thus, there is a critical need for the identification of mechanism-based, treatable targets to improve the microvascular cell function and limit the progression of this disease. Inflammation is a key component in the pathogenesis of type 2 diabetes and cardiovascular diseases (CVD).10 Type 2 diabetes is associated with an increase of cytokines and adipokines secretion11–17 capable of affecting microvascular cell function and structure.4–7 Excessive microvascular inflammation evokes endothelial dysfunction seen in patients and mimicked in animal models with type 2 diabetes.5–9,18–21 IL-12 is a class-I helical cytokine produced mostly by macrophages, neutrophils, and dendritic cells.22–24 It consists of a light chain (p35 subunit) and a heavy chain (p40 subunit) that both are covalently linked forming the p70 unit.25,26 IL-12 helps in the differentiation of T helper 1 immune cells and induces the secretion of pro-inflammatory cytokines.25 Specifically, the level of the pro-inflammatory cytokine interleukin-12 (IL-12) is increased in adolescent and adult T2D patients and could be involved in the development of insulin resistance and vascular complications such as atherosclerosis.26–28 However, it is unknown whether the inhibition of IL-12 protects microvascular function in type 2 diabetes. Furthermore, the mechanism by which increased IL-12 might cause vascular endothelial dysfunction is still unknown. We previously reported that endoplasmic reticulum (ER) stress is an important factor associated with cardiovascular complications in hypertension and diabetes.6,29,30 It has been reported that ER stress signaling increases oxidative stress causing apoptosis induction associated with vascular endothelial dysfunction.31 ER stress-CHOP was proven to increase protein synthesis leading to protein misfolding and induction of oxidative stress contributing to cell damage.32 In the present study, we determined whether the genetic deletion of IL-12 and ER stress CHOP in type 2 diabetic mice could rescue the microvascular endothelial function.
Materials and Methods
Animals and Research Design
All procedures involving animals followed the NIH Guide for the Care and Use of Laboratory Animals and were approved by the institutional animal care and use committee (IACUC) at EVMS, VA, USA. Eight weeks old male C57BL/6J, db−/db−, IL-12−/− and CHOP−/− mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA). We generated double knockout mice between db−/db− and p40IL- 12−/− mice (db−/db−p40-IL−12-/-) and ER Stress-CHOP−/− mice (db−/db−CHOP-/-). Mice were housed under pathogen-free conditions at a temperature of 23°C with 12 hr light/dark cycles with unrestricted access to food and water. Body weights were measured weekly throughout the experiment. After 1 week of acclimatization, mice were divided into the following groups: Group 1: Wild type controls (C57/Bl6), Group 2: Mice with type 2 diabetes (db−/db−), Group 3: IL-12 knockout mice (IL-12−/−), Group 4: IL-12 double knockout mice (db−/db−IL−12-/-), Group 5: ER stress CHOP knockout mice (CHOP−/−) and Group 6: CHOP double knockout mice (db−/db−CHOP-/-). Mice were sacrificed, and the mesenteric resistant arteries (MRAs) were immediately harvested. The MRAs were divided for vascular reactivity assessment, which was performed immediately, Western blot analysis, and NADPH oxidase assay.
Glucose Tolerance Test
After overnight fasting, blood samples were obtained by lancing the mouse-tail vein, and the blood glucose level was measured using an Ultra Touch glucometer (True test; Trividia Health Inc, Fort Lauderdale, FL, USA). Mice then received an IP injection of 2 g/kg glucose. Blood glucose levels were measured at 10-, 20-, 30-, 60-, 90-, and 120-minutes following glucose injection.
Microvascular Reactivity
Microvascular reactivity was performed as reported previously33 in MRA isolated from male C57BL/6J, db−/db−, IL-12−/−, db−/db−IL−12-/-, CHOP−/−, and db−/db−CHOP-/- mice. The MRAs were immediately placed in PSS solution, pH=7.4 (NaCl 118 mM; KCl 4.7 mM; KH2PO4 1.2 mM; CaCl2 2.5 mM; NaHCO3 25 mM; MgSO4x7H2O 1.2 mM and glucose 11 mM). MRAs were cleaned from fat and connective tissue and cut into 2 mm rings. Then rings were mounted in a small vessel dual chamber myograph for isometric tension measurements. After a 30 min calibration period in PSS solution bubbled with carbogen at 37°C, arteries were stretched to their optimal lumen diameter for active tension development. After 60 min incubation, cumulative concentration response curves to phenylephrine (PE, 10−8-10−4 M) were obtained. Rings were washed, then pre-constricted with PE (10−5 M) and when a steady maximal contraction was reached, cumulative concentration responses were obtained for acetylcholine (ACh, 10−9-10−5 M) and sodium nitroprusside (SNP, 10−9-10−5 M). All chemicals were purchased from Sigma-Aldrich, MO, USA.
Western Blot Analysis
Western Blot was used to detect specific proteins in lysates of MRA tissue as previously described.34 Mice were sacrificed then MRA were immediately harvested and frozen in liquid nitrogen and stored at −80°C. Tissue lysates were prepared by homogenizing in ice cooled Tissue protein Extraction Reagent (Prod# 78510, Thermo Scientific, MA, USA), sonicated for 5 seconds and centrifuged for 15 min at 13,000 rpm. Protein quantification was performed according to Pierce™ BCA Protein Assay Kit (Product No. 23225, Thermo scientific, MA, USA). Specific antibodies against Anti-Phospho-Akt (ser473, Cat #9271, Cell Signaling, MA, USA), Anti-total-Akt (Cell Signaling, #9272), Anti-Bip (C50B12, Cell Signaling, #3177), Anti-CHOP (Cell Signaling, #2895), Anti-Phospho-AMPKa (Thr172, Cell Signaling, #2351), Anti-AMPKα (Cell Signaling, #2352), Anti-Phospho-eNOS (Ser1177, Cell Signaling, #9571), Anti-NOX2/gp91phox (Cat #ab80508, Abcam, MA, USA), Anti-NADPH oxidase 4 (#ab133303), Anti-eNOS/NOS Type III (Cat #610296, BD biosciences, CA, USA) and β-actin (Santa Cruz Biotechnology, TX, USA) were purchased and used. All dilutions were prepared according to manufacturer recommendations. Membranes were developed using odyssey-imaging system (LICOR, NE, USA), and band quantification was performed using image J software. Data are expressed after normalization to β-actin as % compared to control.
NADPH Oxidase Activity Assay
Superoxide anion levels generated by NADPH oxidase were measured in lysates of mice MRA using lucigenin chemiluminescence as previously described.29,34 MRA were homogenized in iced sucrose buffer containing KH2PO4 50 mM; EGTA 1 mM, sucrose 150 mM; pH=7.0 and the “Complete-C mini” protease inhibitor cocktail (Roche Diagnostics, IN, USA). The aliquots of the homogenates were used immediately. About 100 μL of each lysate was added to PBS buffer preheated at 37°C, containing lucigenin (5 μM) and NADPH (100 μM) to a total volume of 1 mL. Blank samples were prepared using 100 μL of sucrose buffer. Lucigenin activity was measured every 30 seconds till enzymatic activity reached a plateau in a luminometer (Turner Biosystems 20/20, single tube luminometer, Turner BioSystems, Sunnyvale, CA, USA). The NADPH oxidase activity was performed in the presence or absence of Apocynin and L-NAME inhibitors (Sigma-Aldrich, USA). Data were normalized to protein content and are expressed as % compared to control.
Statistical Analysis
Data are expressed as mean ± SEM. Concentration–response curves were analyzed using the GraphPad Prism 8.0 software (GraphPad, USA). Statistical calculations for significant differences were performed using one-way followed by Post-Hoc test or two-way ANOVA as appropriate. Comparisons are considered significant when p <0.05.
Results
Body weights of C57/Bl6, IL-12−/− and CHOP−/− mice are identical. The deletion of IL-12 and ER stress CHOP in db−/db− mice did not affect the body weight, suggesting that IL-12 and the ER stress CHOP are not involved in obesity development in type 2 diabetes (Figure 1A). The disruption of IL-12 and ER stress CHOP in db−/db− mice significantly improved the glucose test tolerance (GTT) indicating that IL-12 and ER stress CHOP are playing a significant role in the glucose metabolism disorder in type 2 diabetes (Figure 1B).
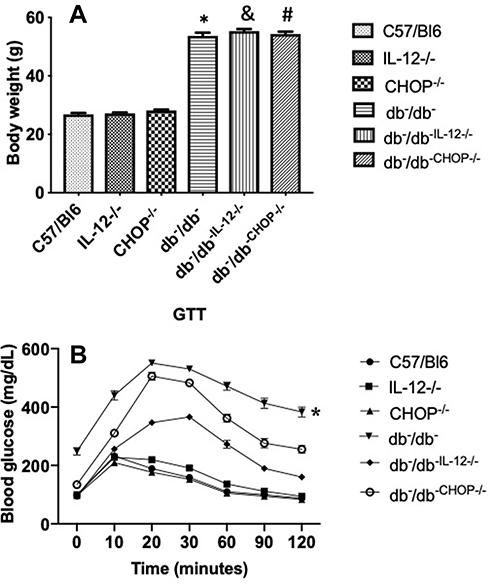 |
Figure 1 (A) Body weights measured at the end of experiment for C57/Bl6, IL-12−/−, CHOP−/−, db−/db−, db−/db−IL−12-/-, and db−/db-chop-/- mice. Data are expressed as mean ± S.E.M. One way ANOVA was used for comparison between the groups followed by Tukey’s post-hoc test. *p<0.05 for db−/db− vs C57/Bl6, IL-12−/−, and CHOP−/−. #p<0.05 for db−/db−chop-/- vs C57/Bl6, IL-12−/−, and CHOP−/−. &p<0.05 for db−/db−IL12-/- vs C57/Bl6, IL-12−/−, and CHOP−/− mice. (B) Glucose tolerance test (GTT) of C57/Bl6, IL-12−/−, CHOP−/−, db−/db−, db−/db−IL−12-/-, and db−/db-chop-/- mice. Data are expressed as mean ± SEM. Two-way ANOVA was used for comparison between the groups followed by Tukey’s post-hoc test. *p<0.05 for db−/db− vs C57/Bl6, IL-12−/−, CHOP−/−, db−/db−IL−12-/-, and db−/db-chop-/. |
We previously reported that ER stress induction in db−/db− mice is associated with microvascular endothelial dysfunction.35,36 The relaxation of MRA in response to exogenous nitric oxide donor (sodium nitroprusside: SNP) is similar in all groups (Figure 2A) indicating that the endothelial dysfunction is mainly related to the lack of endothelial cell to release enough vaso-relaxing factors. Figure 2B shows endothelial dysfunction in db−/db− compared to C57/Bl6, IL-12−/−, and ER stress CHOP−/− mice. Interestingly, the disruption of IL-12 and the ER stress CHOP in db−/db− mice significantly improved MRA endothelial function (Figure 2B) suggesting that IL-12 and ER stress CHOP are parts of mechanisms involved in endothelial dysfunction in type 2 diabetes. Mesenteric resistance artery (MRA) constriction induced by phenylephrine was similar between all groups (C57/Bl6, db−/db−, IL-12−/−, CHOP−/−, db−/db−IL−12-/-, db−/db−CHOP-/-) (Figure 2C). The endothelial dysfunction, assessed by a reduction in MRA relaxation in response to acetylcholine, is an important event involved in cardiovascular diseases.
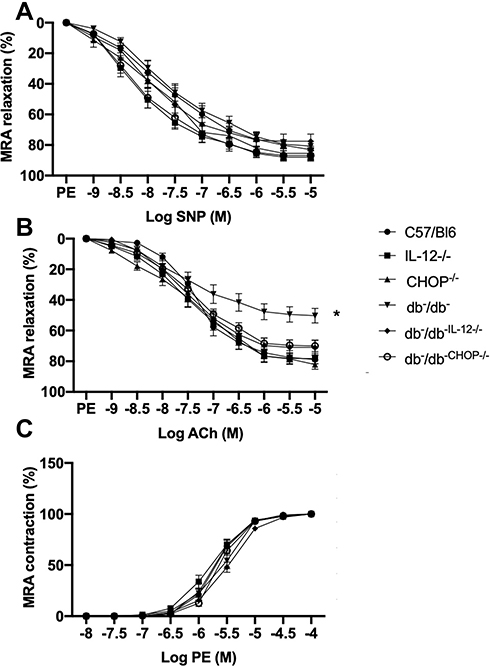 |
Figure 2 Mesenteric resistance arteries reactivity showing (A) endothelium-independent relaxation in response to sodium nitroprusside (SNP). (B) endothelium-dependent relaxation in response to acetylcholine (ACh). (C) contractility in response to sympathetic stimulation by phenylephrine (PE) in C57/Bl6, IL-12−/−, CHOP−/−, db−/db−, db−/db−IL−12-/-, and db−/db-chop-/- mice. Data are expressed as mean ± SEM. Two-way ANOVA was used for comparison between the groups followed by Tukey’s post-hoc test. *p<0.05 for db−/db− vs C57/Bl6, IL-12−/−, CHOP−/−, db−/db−IL−12-/-, and db−/db-chop-/- mice. |
These functional studies are further supported by the assessment of signaling pathways (phosphorylated (P) AMPK, eNOS, and Akt) involved in the endothelial function (Figure 3). Western blot analysis of MRA tissue shows a decrease in phosphorylated-AMPK, eNOS, and Akt in db−/db− compared to C57/Bl6, IL-12−/−, CHOP−/−, db−/db−IL−12-/-, and db−/db−CHOP-/- mice (Figure 3A-C). These data indicate that IL-12 and ER stress CHOP are drivers of microvascular endothelial dysfunction in type 2 diabetes likely through eNOS, Akt, and AMPK signaling.
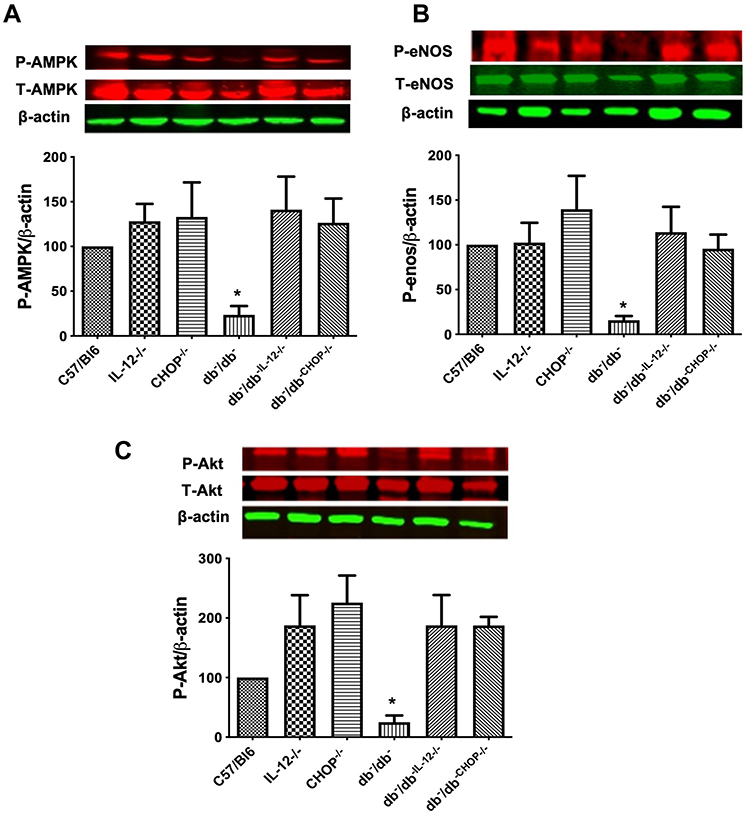 |
Figure 3 Western blot analysis of survival markers (A) Phosphorylated and total AMPK. (B) Phosphorylated and total eNOS. (C) Phosphorylated and total Akt. β-actin was used as a loading control. Data are expressed as % compared to control. One way ANOVA was used for comparison between the groups followed by Tukey’s post-hoc test. *p<0.05 for db−/db− vs C57/Bl6, IL-12−/−, CHOP−/−, db−/db−IL−12-/-, and db−/db-chop-/- mice. |
In addition, Western blot data illustrate an increase in ER stress CHOP and BIP expression in db−/db− compared to C57/Bl6, IL-12−/−, CHOP−/−, db−/db−IL−12-/-, and db−/db−CHOP-/- mice (Figure 4). The data also showed that IL-12 disruption in db−/db− mice significantly reduced ER stress CHOP and BIP induction, suggesting that IL-12 is an upstream mechanism for the induction of the ER stress in type 2 diabetes (Figure 4).
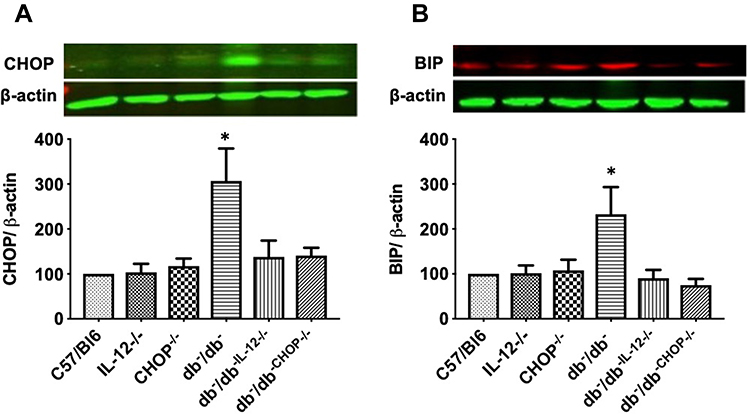 |
Figure 4 Western blot analysis of ER markers (A) CHOP. (B) BIP. β-actin was used as a loading control. Data are expressed as % compared to control. One way ANOVA was used for comparison between the groups followed by Tukey’s post-hoc test. *p<0.05 for db−/db− vs C57/Bl6, IL-12−/−, CHOP−/−, db−/db−IL−12-/-, and db−/db-chop-/- mice. |
Furthermore, NADPH oxidase activity was significantly increased in db−/db− compared to C57/Bl6, IL-12−/−, CHOP−/−, db−/db−IL−12-/-, and db−/db−CHOP-/- mice (Figure 5A). To study the involvement of eNOS coupling in vascular function, NADPH oxidase activity was performed in the presence of a direct inhibitor of nitric oxide synthase (L-NAME) or an inhibitor of NADPH oxidase (Apocynin). Our data showed that the addition of L-NAME or Apocynin inhibited NADPH oxidase activity, indicating the involvement of both uncoupled eNOS and NADPH in the production of ROS. Furthermore, Western blot analysis showed an increase in Nox4 and Nox 2 expression in db−/db− compared to C57/Bl6, IL-12−/−, CHOP−/−, db−/db−IL−12-/-, and db−/db−CHOP-/- mice (Figure 5B and C) supporting the augmented NADPH oxidase activity in db−/db− mice.
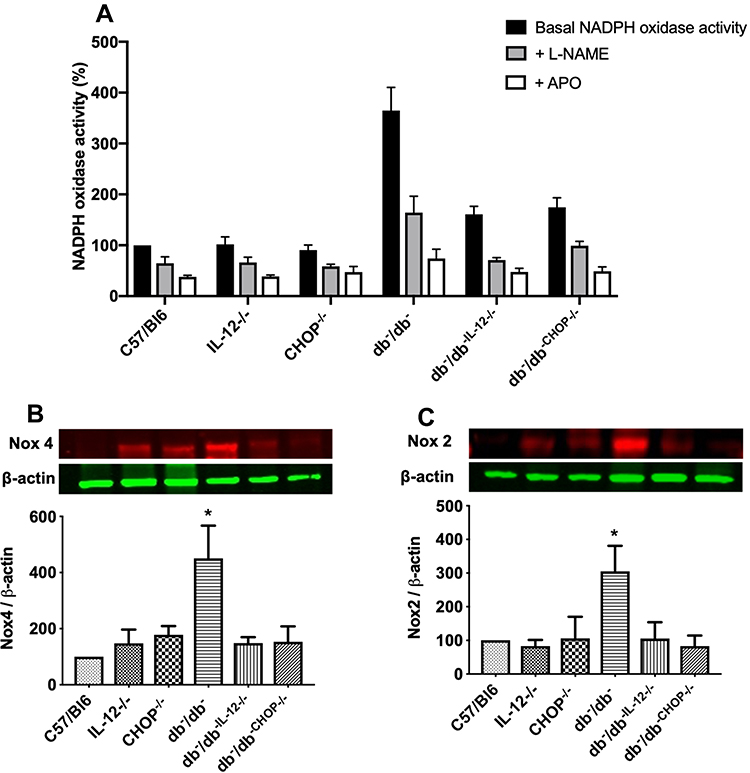 |
Figure 5 (A) NADPH oxidase activity with and without incubation with L-NAME and Apocynin (APO) in mesenteric resistance arteries from C57/Bl6, IL-12−/−, CHOP−/−, db−/db−, db−/db−IL−12-/-, and db−/db-chop-/- mice. Data were normalized to protein content and are expressed as % compared to control. One way ANOVA was used for comparison between the groups followed by Tukey’s post-hoc test. *p<0.05 for db−/db− vs C57/Bl6, IL-12−/−, CHOP−/−, db−/db−IL−12-/-, and db−/db-chop-/- mice. Western blot analysis of oxidative stress markers (B) Nox 4. (C) Nox 2. β-actin was used as a loading control. Data are expressed as % compared to control. One way ANOVA was used for comparison between the groups followed by Tukey’s post-hoc test. *p<0.05 for db−/db− vs C57/Bl6, IL-12−/−, CHOP−/−, db−/db−IL−12-/-, and db−/db-chop-/- mice. |
Discussion
Type 2 diabetes is associated with chronic inflammation where a variety of cytokines, adipokines and stress pathways are increased11–17,37,38 and could affect vascular function and structure.5–8 Significantly, IL-12 is increased in adolescent and adult patients with type 2 diabetes,28,39 in addition to animal models of type 2 diabetes.40–42 Moreover, IL-12 administration to non-obese mice leads to diabetes,43 liver fibrosis,44 kidney damage,45 and atherosclerosis46 supporting a detrimental role of IL-12 in the etiology of diabetes and abnormalities of multiple tissues and organs. Furthermore, a previous study showed a correlation between elevated levels of serum IL-12 and vascular endothelial dysfunction in type 2 diabetes.27 These studies suggest that IL-12 is a key player involved in type 2 diabetes-induced cardiovascular complications. However, the mechanism by which increased IL-12 in type 2 diabetes causes microvascular endothelial dysfunction is not completely understood. It is well established that type 2 diabetes is associated with endoplasmic reticulum (ER) stress induction and increase in oxidative stress.47 We and other laboratories previously reported endothelial dysfunction associated with ER stress induction in type 2 diabetic mice.48,49 We also elucidated the involvement of the ER stress and oxidative stress in vascular endothelial dysfunction in type 2 diabetic mice.48
The present study aimed to determine whether IL-12 and ER stress CHOP play a key role in obesity, glucose metabolism, and microvascular endothelial dysfunction. Our results showed that disruption of IL-12 and ER stress CHOP in db/db mice did not affect the body weight, however significantly reduced glucose test tolerance, suggesting that IL-12 and ER stress CHOP in this genetic type 2 diabetic (db/db) model play a significant role in controlling glucose homeostasis. Previous study reported that mice lacking IL-12 and fed a high fat diet for 16 weeks showed no improvement in glucose tolerance test.50 This difference in results could be related to the genetic deletion of IL-12 in db/db in addition to the severe hyperglycemia in db/db mice (550 mg/dL of blood glucose) compared to moderate hyperglycemia (300 mg/dL of blood glucose) induced by high fat diet.
Interestingly, our data showed that the microvascular endothelial function was significantly improved in db−/db−IL−12-/- compared to db−/db− mice. These data provide evidence that increases in IL-12 in type 2 diabetes causes microvascular endothelial dysfunction. At this stage, we do not know whether the effect of IL-12 on microvascular endothelial dysfunction is a direct effect on endothelial cells or through immune cells infiltrated into microvessels wall that release factors and compromise endothelial cell function. It is well known that immune cells, such as macrophages and dendritic cells release IL-12 and respond to IL-12 by releasing a variety of factors.51 Moreover, type 2 diabetes is associated with activated macrophages and dendritic cells.52 Therefore, it is possible that in type 2 diabetes, macrophages and dendritic cells increase the production and release of IL-12, which in turn acts on macrophages and dendritic cells leading to vascular endothelial dysfunction. Further studies are required to confirm this hypothesis.
In the present study, we examined the mechanism that triggers ER stress and oxidative stress in type 2 diabetes. Data showed that increased IL-12 augmented ER stress CHOP and BIP in microvessels, together with elevating oxidative stress markers. Importantly, disruption of IL-12 protects against the induction of ER and oxidative stress, suggesting that IL-12 is an upstream mechanism that dictates the regulation of the ER stress and oxidative stress in type 2 diabetes. Our data are supported by the fact that disrupting the ER stress CHOP in db/db mice significantly improved microvascular endothelial function. Our findings are supported by previous studies showing that deletion of ER stress CHOP alleviates ER stress, corrects hepatic steatosis, beta cell viability, and participates in islet amyloid polypeptide-induced beta cell apoptosis.53–55 Prospectively, future research should determine how IL-12 is increased in type 2 diabetes and what are the cellular and molecular targets that compromise the cardiovascular system in type 2 diabetes.
Limitation
The limitation of this study is the sole use of the db−/db− genetic model mouse. This model has a mutation in the gene encoding the leptin receptor, causing obesity, insulin resistance, and T2D. However, human diabetes is more complicated with several interplaying factors, including obesity, bad dietary habits, and sedentary lifestyle. Therefore, further evaluation of the role of IL-12 signaling in other animal models as diet-induced diabetes is of relevance.
Conclusion
In the present study, we determined that the increase in IL-12 is a significant mechanism responsible for microvascular endothelial dysfunction in type 2 diabetes. We also delineated that the ER stress and oxidative stress are mechanisms by which IL-12 causes vascular endothelial dysfunction in type 2 diabetes. The disruption of IL-12 in db−/db− mice significantly reduced ER stress and oxidative stress, which improved vascular endothelial function. Furthermore, the disruption of the ER stress CHOP in db−/db− mice also improved vascular endothelial function supporting the role of the ER stress CHOP induction in endothelial dysfunction. Our genetic approach (disrupting IL-12 and the ER stress CHOP in db−/db− mice) provides clear evidence that the IL-12 in type 2 diabetes causes microvascular dysfunction through the ER stress CHOP and oxidative stress mechanisms. Overall, the present study uncovered the importance of IL-12 cytokine in regulating microvascular endothelial function through a mechanism involving the ER stress CHOP and oxidative stress. Understanding the mechanism by which IL-12 causes microvascular endothelial dysfunction and determining a cause-and-effect relationship will help in the design of additional new therapeutic tools to protect from type 2 diabetes and its microvascular complications.
Acknowledgments
The abstract of this paper was presented at the American Heart Association’s 2019 Scientific Sessions meeting as a poster presentation with interim findings. The poster’s abstract was published in “Poster Abstracts” in 2019 Journal name Circulation.
Funding
The Study is supported by Funding from the NIH HL150014 (KM) and NIH HL151616 (KM).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Simpson HC, Mann JI, Chakrabarti R, et al. Effect of high-fibre diet on haemostatic variables in diabetes. Br Med J. 1982;284(6329):1608.
2. Everhart JE, Pettitt DJ, Knowler WC, Rose FA, Bennett PH. Medial arterial calcification and its association with mortality and complications of diabetes. Diabetologia. 1988;31(1):16–23.
3. Nelson RG, Gohdes DM, Everhart JE, et al. Lower-extremity amputations in NIDDM. 12-yr follow-up study in Pima Indians. Diabetes Care. 1988;11(1):8–16.
4. Rogers MA, Aikawa E. Modifying vascular calcification in diabetes mellitus: contribution of O-GlcNAcylation. Circ Res. 2014;114(7):1074–1076.
5. Belmadani S, Palen DI, Gonzalez-Villalobos RA, Boulares HA, Matrougui K. Elevated epidermal growth factor receptor phosphorylation induces resistance artery dysfunction in diabetic db/db mice. Diabetes. 2008;57(6):1629–1637.
6. Galan M, Kassan M, Choi SK, et al. A novel role for epidermal growth factor receptor tyrosine kinase and its downstream endoplasmic reticulum stress in cardiac damage and microvascular dysfunction in type 1 diabetes mellitus. Hypertension. 2012;60(1):71–80.
7. Choi SK, Galan M, Kassan M, Partyka M, Trebak M, Matrougui K. Poly(ADP-ribose) polymerase 1 inhibition improves coronary arteriole function in type 2 diabetes mellitus. Hypertension. 2012;59(5):1060–1068.
8. Choi SK, Galan M, Partyka M, Trebak M, Belmadani S, Matrougui K. Chronic inhibition of epidermal growth factor receptor tyrosine kinase and extracellular signal-regulated kinases 1 and 2 (ERK1/2) augments vascular response to limb ischemia in type 2 diabetic mice. Am J Pathol. 2012;180(1):410–418.
9. Kassan M, Choi SK, Galan M, et al. Enhanced NF-kappaB Activity Impairs Vascular Function Through PARP-1-, SP-1-, and COX-2-Dependent Mechanisms in Type 2 Diabetes. Diabetes. 2013;62(6):2078–2087.
10. Chen L, Deng H, Cui H, et al. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget. 2017;9(6):7204–7218.
11. Gao X, Belmadani S, Picchi A, et al. Tumor necrosis factor-alpha induces endothelial dysfunction in Lepr(db) mice. Circulation. 2007;115(2):245–254.
12. Garcia C, Feve B, Ferre P, et al. Diabetes and inflammation: fundamental aspects and clinical implications. Diabetes Metab. 2010;36(5):327–338.
13. Kaul K, Hodgkinson A, Tarr JM, Kohner EM, Chibber R. Is inflammation a common retinal-renal-nerve pathogenic link in diabetes? Curr Diabetes Rev. 2010;6(5):294–303.
14. Al-Aly Z. Arterial calcification: a tumor necrosis factor-alpha mediated vascular Wnt-opathy. Transl Res. 2008;151(5):233–239.
15. Das UN. Acetylcholinesterase and butyrylcholinesterase as possible markers of low-grade systemic inflammation. Med Sci Monit. 2007;13(12):RA214–21.
16. Savoia C, Schiffrin EL. Vascular inflammation in hypertension and diabetes: molecular mechanisms and therapeutic interventions. Clin Sci (Lond). 2007;112(7):375–384.
17. Diamanti-Kandarakis E, Paterakis T, Kandarakis HA. Indices of low-grade inflammation in polycystic ovary syndrome. Ann N Y Acad Sci. 2006;1092:175–186.
18. Domingueti CP, Dusse LM, Carvalho M, de Sousa LP, Gomes KB, Fernandes AP. Diabetes mellitus: the linkage between oxidative stress, inflammation, hypercoagulability and vascular complications. J Diabetes Complications. 2016;30(4):738–745.
19. Seferovic PM, Paulus WJ. Clinical diabetic cardiomyopathy: a two-faced disease with restrictive and dilated phenotypes. Eur Heart J. 2015;36(27):
20. Lu Q, Lu L, Chen W, Chen H, Xu X, Zheng Z. RhoA/mDia-1/profilin-1 signaling targets microvascular endothelial dysfunction in diabetic retinopathy. Graefes Arch Clin Exp Ophthalmol. 2015;253(5):669–680.
21. Huang R, Abdelmoneim SS, Nhola LF, Basu R, Basu A, Mulvagh SL. Relationship between glycosylated hemoglobin A1c and coronary flow reserve in patients with Type 2 diabetes mellitus. Expert Rev Cardiovasc Ther. 2015;13(4):445–453.
22. Schoenhaut DS, Chua AO, Wolitzky AG, et al. Cloning and expression of murine IL-12. J Immunol. 1992;148(11):3433.
23. Cooper AM, Khader SA. IL-12p40: an inherently agonistic cytokine. Trends Immunol. 2007;28(1):33–38.
24. Khader SA, Partida-Sanchez S, Bell G, et al. Interleukin 12p40 is required for dendritic cell migration and T cell priming after Mycobacterium tuberculosis infection. J Exp Med. 2006;203(7):1805.
25. Strissel KJ, DeFuria J, Shaul ME, Bennett G, Greenberg AS, Obin MS. T-cell recruitment and Th1 polarization in adipose tissue during diet-induced obesity in C57BL/6 mice. Obesity. 2010;18(10):1918–1925.
26. Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133.
27. Mishra M, Kumar H, Bajpai S, Singh RK, Tripathi K. Level of serum IL-12 and its correlation with endothelial dysfunction, insulin resistance, proinflammatory cytokines and lipid profile in newly diagnosed type 2 diabetes. Diabetes Res Clin Pract. 2011;94(2):255–261.
28. Wegner M, Winiarska H, Bobkiewicz-Kozlowska T, Dworacka M. IL-12 serum levels in patients with type 2 diabetes treated with sulphonylureas. Cytokine. 2008;42(3):312–316.
29. Kassan M, Galan M, Partyka M, et al. Endoplasmic reticulum stress is involved in cardiac damage and vascular endothelial dysfunction in hypertensive mice. Arterioscler Thromb Vasc Biol. 2012;32(7):1652–1661.
30. Amin A, Choi SK, Galan M, et al. Chronic inhibition of endoplasmic reticulum stress and inflammation prevents ischaemia-induced vascular pathology in type II diabetic mice. J Pathol. 2012;227(2):165–174.
31. Zhang Y, Ren J. Thapsigargin triggers cardiac contractile dysfunction via NADPH oxidase-mediated mitochondrial dysfunction: role of Akt dephosphorylation. Free Radic Biol Med. 2011;51(12):2172–2184.
32. Han J, Back SH, Hur J, et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol. 2013;15(5):481–490.
33. Radwan E, Mali V, Haddox S, et al. Treg cells depletion is a mechanism that drives microvascular dysfunction in mice with established hypertension. Biochimica et biophysica acta Mol dis. 2019;1865(2):403–412.
34. Kassan M, Galan M, Partyka M, Trebak M, Matrougui K. Interleukin-10 released by CD4(+)CD25(+) natural regulatory T cells improves microvascular endothelial function through inhibition of NADPH oxidase activity in hypertensive mice. Arterioscler Thromb Vasc Biol. 2011;31(11):2534–2542.
35. Galán M, Kassan M, Kadowitz PJ, Trebak M, Belmadani S, Matrougui K. Mechanism of endoplasmic reticulum stress-induced vascular endothelial dysfunction. Biochim Biophys Acta. 2014;1843(6):1063–1075.
36. Kassan M, Galán M, Partyka M, et al. Endoplasmic Reticulum Stress Is Involved in Cardiac Damage and Vascular Endothelial Dysfunction in Hypertensive Mice. Arterioscler Thromb Vasc Biol. 2012;32(7):1652–1661.
37. Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. 2005;115(5):1111–1119.
38. Arcidiacono B, Chiefari E, Foryst-Ludwig A, et al. Obesity-related hypoxia via miR-128 decreases insulin-receptor expression in human and mouse adipose tissue promoting systemic insulin resistance. EBioMedicine. 2020;59:102912.
39. Lichtenauer M, Franz M, Fritzenwanger M, Figulla HR, Gerdes N, Jung C. Elevated plasma levels of interleukin-12p40 and interleukin-16 in overweight adolescents. Biomed Res Int. 2015;2015:940910.
40. Wen Y, Gu J, Li S-L, Reddy MA, Natarajan R, Nadler JL. Elevated glucose and diabetes promote interleukin-12 cytokine gene expression in mouse macrophages. Endocrinology. 2006;147(5):2518–2525.
41. Strissel KJ, DeFuria J, Shaul ME, Bennett G, Greenberg AS, Obin MS. T‐cell recruitment and Th1 polarization in adipose tissue during diet‐induced obesity in C57BL/6 mice. Obesity. 2010;18(10):1918–1925.
42. Ali M, Mali V, Haddox S, et al. Essential role of IL-12 in angiogenesis in type 2 diabetes. Am J Pathol. 2017;187(11):2590–2601.
43. Trembleau S, Penna G, Gregori S, Giarratana N, Adorini L. IL-12 administration accelerates autoimmune diabetes in both wild-type and IFN-gamma-deficient nonobese diabetic mice, revealing pathogenic and protective effects of IL-12-induced IFN-gamma. J Immunol. 2003;170(11):5491–5501.
44. Dibra D, Cutrera J, Xia X, Kallakury B, Mishra L, Li S. Interleukin-30: a novel antiinflammatory cytokine candidate for prevention and treatment of inflammatory cytokine-induced liver injury. Hepatology. 2012;55(4):1204–1214.
45. Schwarting A, Tesch G, Kinoshita K, Maron R, Weiner HL, Kelley VR. IL-12 drives IFN-gamma-dependent autoimmune kidney disease in MRL-Fas(lpr) mice. J Immunol. 1999;163(12):6884–6891.
46. Lee TS, Yen HC, Pan CC, Chau LY. The role of interleukin 12 in the development of atherosclerosis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 1999;19(3):734–742.
47. Burgos-Morón E, Abad-Jiménez Z, Marañón A, et al. Relationship Between Oxidative Stress, ER Stress, and Inflammation in Type 2 Diabetes: the Battle Continues. J Clin Med. 2019;8(9):1385.
48. Kassan M, Choi S-K, Galán M, Lee Y-H, Trebak M, Matrougui K. Enhanced p22phox expression impairs vascular function through p38 and ERK1/2 MAP kinase-dependent mechanisms in type 2 diabetic mice. Am J Physiol Heart Circ Physiol. 2014;306(7):H972–H80.
49. Molnar J, Yu S, Mzhavia N, Pau C, Chereshnev I, Dansky Hayes M. Diabetes Induces Endothelial Dysfunction but Does Not Increase Neointimal Formation in High-Fat Diet Fed C57BL/6J Mice. Circ Res. 2005;96(11):1178–1184.
50. Ali M, Mali V, Haddox S, et al. Essential Role of IL-12 in Angiogenesis in Type 2 Diabetes. Am J Pathol. 2017;187(11):2590–2601.
51. Liu J, Cao S, Kim S, et al. Interleukin-12: an update on its immunological activities, signaling and regulation of gene expression. Curr Immunol Rev. 2005;1(2):119–137.
52. Kraakman MJ, Murphy AJ, Jandeleit-Dahm K, Kammoun HL. Macrophage polarization in obesity and type 2 diabetes: weighing down our understanding of macrophage function? Front Immunol. 2014;5:470.
53. Yong J, Parekh VS, Reilly SM, et al. Chop/Ddit3 depletion in beta cells alleviates ER stress and corrects hepatic steatosis in mice. Sci Transl Med. 2021;13:604.
54. McKimpson WM, Zheng M, Chua SC, Pessin JE, Kitsis RN. ARC is essential for maintaining pancreatic islet structure and beta-cell viability during type 2 diabetes. Sci Rep. 2017;7(1):7019.
55. Song B, Scheuner D, Ron D, Pennathur S, Kaufman RJ. Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. J Clin Invest. 2008;118(10):3378–3389.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.