Back to Archived Journals » Oncolytic Virotherapy » Volume 8
Directed evolution as a tool for the selection of oncolytic RNA viruses with desired phenotypes
Authors Zainutdinov SS ![]() , Kochneva GV, Netesov SV
, Kochneva GV, Netesov SV ![]() , Chumakov PM, Matveeva OV
, Chumakov PM, Matveeva OV ![]()
Received 3 March 2019
Accepted for publication 7 June 2019
Published 12 July 2019 Volume 2019:8 Pages 9—26
DOI https://doi.org/10.2147/OV.S176523
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tommy Alain
Sergei S Zainutdinov,1 Galina V Kochneva,1 Sergei V Netesov,2 Peter M Chumakov3,4, Olga V Matveeva5
1State Research Center of Virology and Biotechnology “Vector”, Koltsovo 630559, Russia; 2Department of Natural Sciences, Novosibirsk State University, Novosibirsk 630090, Russia; 3Engelhardt Institute of Molecular Biology, Moscow 119991, Russia; 4Chumakov Federal Scientific Center for Research and Development of Immune-and-Biological Products, Moscow 108819, Russia; 5SATOR Therapeutics LLC, Cleveland, OH 44106, USA
Abstract: Viruses have some characteristics in common with cell-based life. They can evolve and adapt to environmental conditions. Directed evolution can be used by researchers to produce viral strains with desirable phenotypes. Through bioselection, improved strains of oncolytic viruses can be obtained that have better safety profiles, increased specificity for malignant cells, and more efficient spread among tumor cells. It is also possible to select strains capable of killing a broader spectrum of cancer cell variants, so as to achieve a higher frequency of therapeutic responses. This review describes and analyses virus adaptation studies performed with members of four RNA virus families that are used for viral oncolysis: reoviruses, paramyxoviruses, enteroviruses, and rhabdoviruses.
Keywords: oncolytic viruses, virus selection, virus adaptation, directed viral evolution
Introduction
Tumor regression after naturally acquired viral infections or vaccination was observed more than 100 years ago.1–3 Following these earlier random observations, the number of studies in which a virus was used as an anticancer therapeutic increased dramatically during the 20th century.3,4 As a result, the term oncolytic virus (OV) was introduced. OV has tumor-selective replication abilities and can kill tumor cells directly through infection and lysis of cancer cells, as well as indirectly through initiation of systemic antitumor immune responses.3–5 The viruses that have been investigated as oncolytic agents belong to different families and represent a diverse group of wild-type and genetically engineered viral strains. OV research has intensified in the 1990s leading in 2015 to the first US Food and Drug Administration-approved OV strain (Talimogene laherparepvec (T-VEC or Imlygic)) for the treatment of advanced melanoma.6 This first approved oncolytic viral agent is based on genetically engineered herpes simplex virus (HSV-1). While T-VEC showed encouraging durability of responses, its overall efficacy remains limited.7 The field of oncolytic viral therapy faces multiple challenges, including optimizing viral delivery and viral spread inside tumors, as well as overcoming tumor resistance and antiviral immunity. So far no known biomarkers can reliably identify cancer patients, who are likely to respond to viral therapy. However, extensive preclinical and clinical studies are gradually addressing these challenges.
This review describes and analyzes studies that employed directed evolution via multiple rounds of virus selection for improving, testing, or studying OV properties. In most selection experiments, an OV is passaged in a target cell line for achieving genetic changes that produce desirable characteristics (Figure 1). Alternative routes of viral selection include selective passaging in embryonated chicken eggs or tumor tissues. Various selection and testing schemes that have been applied to representatives of four RNA virus families—Reoviridae, Paramyxoviridae, Picornaviridae, and Rhabdoviridae are presented in this review.
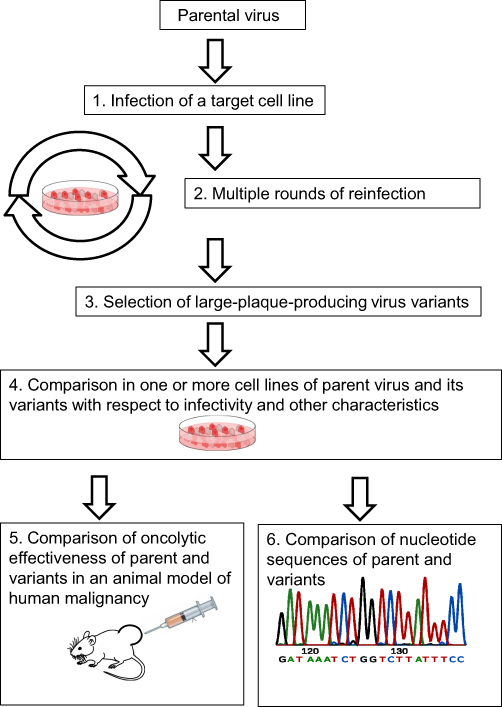 |
Figure 1 Selection process for broadening virus infectivity. Steps 5 or/and 6 might be omitted in some experiments. |
Reoviruses
Reoviridae is a family of non-enveloped double-stranded RNA viruses with a very wide host range that includes animals, plants, and fungi.8 The genome length of reoviruses ranges from 18.2 to 30.5 kb.8,9 The name “reovirus” stands for Respiratory Enteric Orphan virus.10 The abbreviation resulted from the observation that the virus usually could be isolated from the mammalian respiratory and enteric tracts, but it causes very few, if any clinical symptoms (hence “orphan”). Antibodies to reoviruses have been observed in almost all human subjects.8,11 The family includes Orthoreovirus among its genera. Orthoreovirus infection is common in humans, but in most cases, it causes very mild symptoms. For simplicity, Orthoreovirus representatives are referred to as reoviruses in the text that follows.
Prompted by observations that reoviruses have oncolytic potential, viral modifications that may improve reovirus oncolytic potency and/or specificity were studied and described.12,13 Four mammalian orthoreovirus serotypes, T1, T2, T3, and T4, are known. For each serotype, representative strains have been isolated: for type 1, Lang (T1L); for type 2, Jones (T2J); for type 3, Abney (T3A) and Dearing (T3D)9 and for type 4, Ndelle (T4N). Among reovirus strains, T3D has been the most studied. One of its derivative strains was given the commercial name Pelareorep (REOLYSIN®) by Oncolytics Biotech, the company that is developing anticancer therapeutic agents using this reovirus.10,14
Reoviruses can enter cells using Junctional Adhesion Molecule-A (JAM-A) as the cell’s entry receptor.9,15 JAM-A protein in humans is encoded by the F11R gene. Reoviruses are capable of using glycans as co-receptors; for example, some serotype T3 viruses use sialic acid as a co-receptor.15
Extensive preclinical cell-based and animal studies showed that T3D reovirus and related strains are capable, as many other OVs, of killing a broad range of cancer cells both directly and also indirectly through the activation of the immune system.16,17 Subsequent clinical trials have shown promise for REOLYSIN efficacy. Intra-lesion REOLYSIN injections generated one complete response, two partial responses and four stable diseases among 19 patients with variable solid tumors. Remaining patients from 19 were non-responders.18 In clinical trials, multiple intravenous REOLYSIN injections were combined with simultaneous administration of chemotherapeutic agents. Such treatment schemes failed to show any efficacy for recurrent ovarian, tubal, peritoneal19 or pancreatic20 adenocarcinomas. However, overall survival time of patients with metastatic breast cancer who were treated with a combination of REOLYSIN and chemotherapy was seven months longer than that of patients treated by chemotherapy alone.21 Side effects of the combined REOLYSIN-chemotherapy treatment were comparatively mild (grade 3 or less) and included pyrexia, chills, myalgia, pain, fatigue, and nausea.21 Rare severe adverse events (grade 4) were also registered. They included neutropenia and severe respiratory problems.19
Adaptation of reoviruses
Adaptation studies have been directed toward enhancing and broadening reovirus infectivity as well as increasing its safety (Table 1). Theoretically, these two goals could be mutually exclusive because increased infectivity could compromise safety. Therefore, virus strains that have been adapted for higher infectivity should receive extra scrutiny for safety.
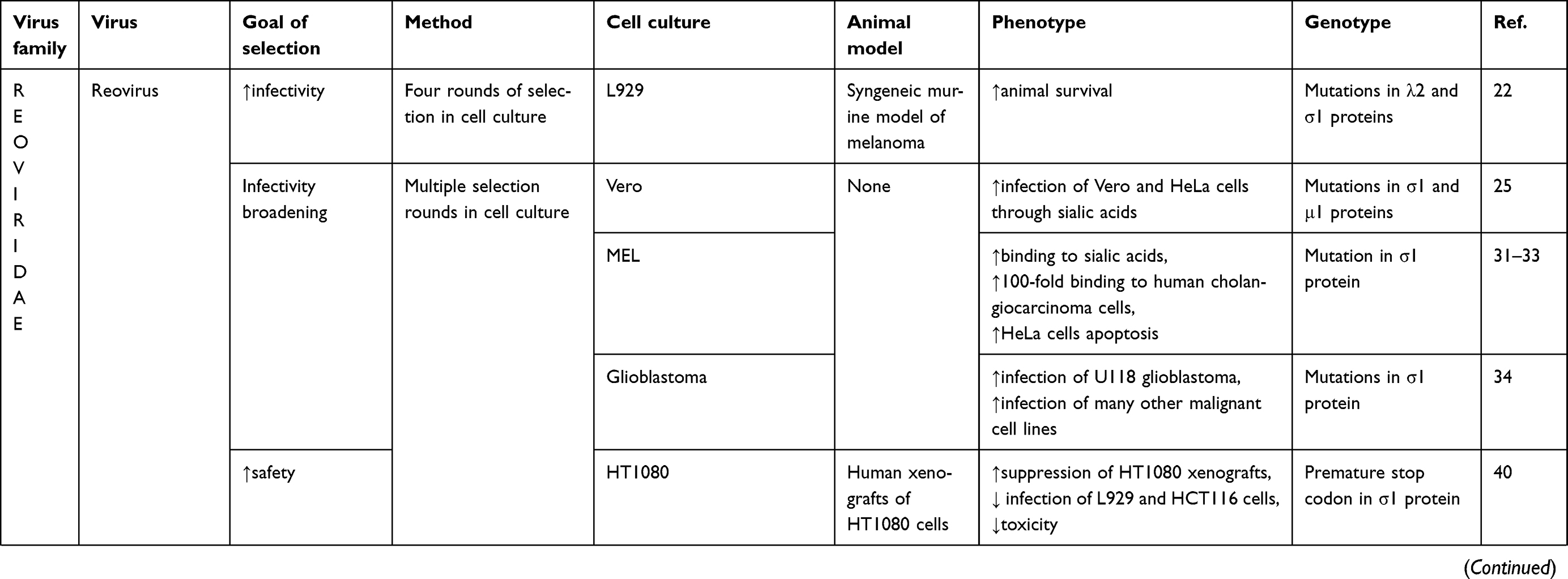 |
Table 1 Adaptation studies of oncolytic viruses |
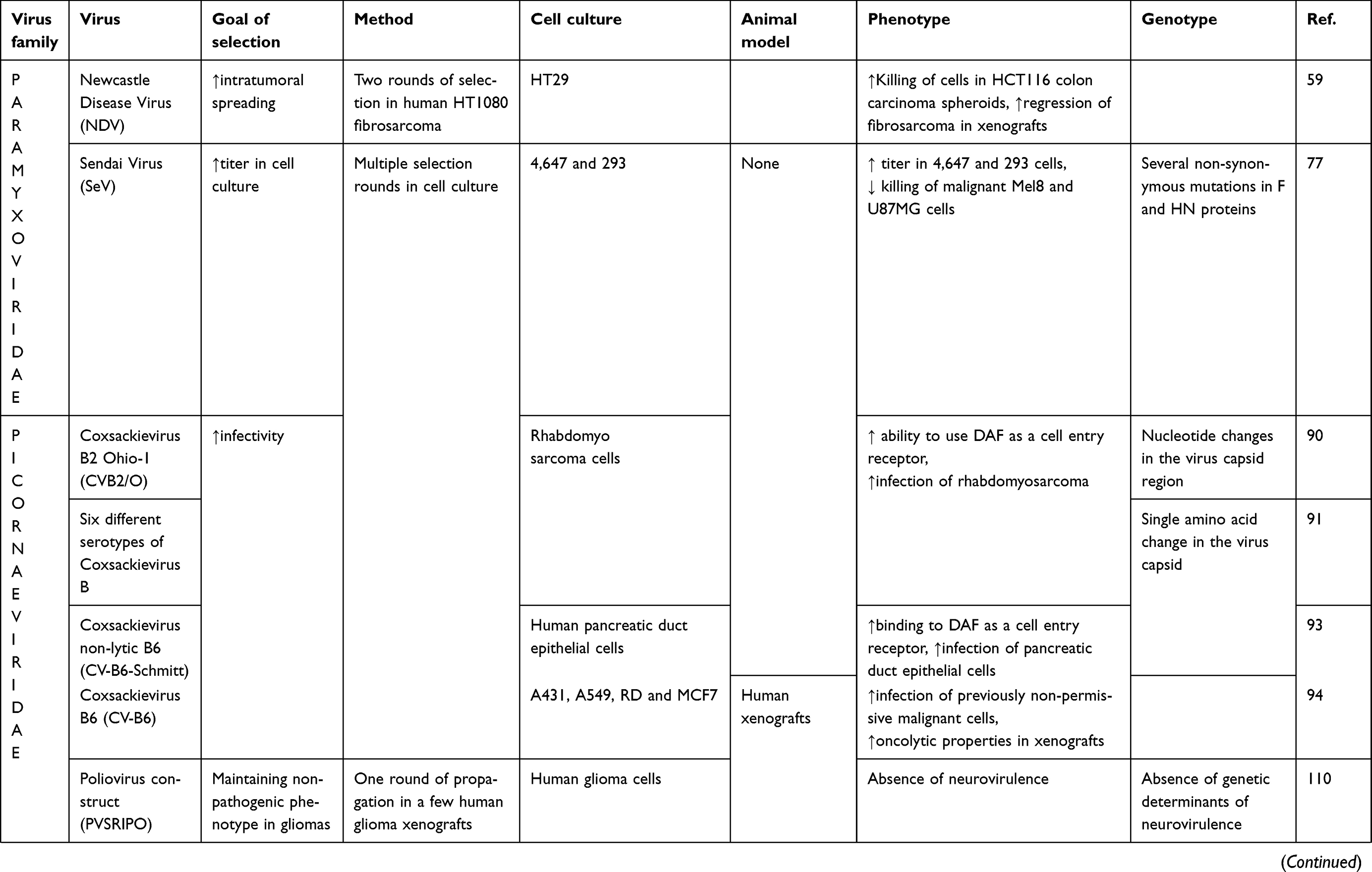 |
Table 1 (Continued). |
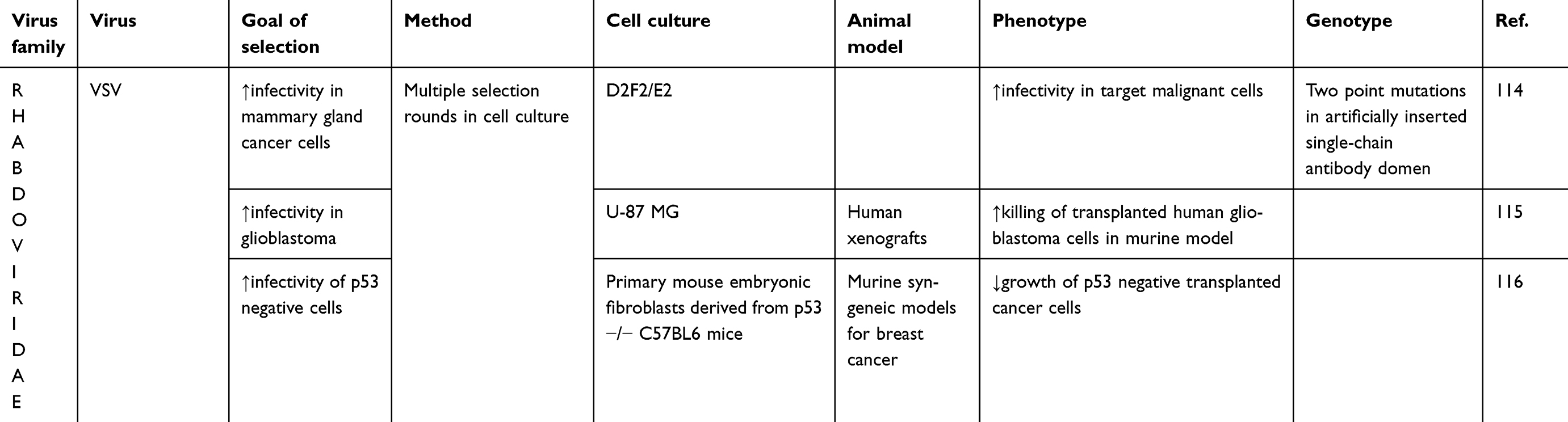 |
Table 1 (Continued). |
Infectivity enhancement
Shmulevitz’s team managed to enhance reovirus infectivity in a few steps.22 First, three strains of reovirus (T1L, T2J, and T3D) were used in combination to infect murine L929 cells. (L929 cells are highly permissive to reovirus infection and are used for its propagation.) Second, four rounds of virus selection in L929 cells were performed; the largest plaques were selected in each round. Third, after all the selection rounds, two variants were isolated that consistently formed plaques larger than those formed by all parental virus strains. These reovirus variants produced larger plaques on several human and mouse cell lines with moderate susceptibility to reovirus, including human colorectal carcinoma cells, pancreatic ductal carcinoma cells, and murine ovarian cancer cells. Mutations in λ2 vertex and σ1 cell attachment proteins were identified in these strains. Authors of the study concluded that these mutations were responsible for an increase in the proportion of infectious progeny virus particles produced by the selected strains. Later they showed that reduction of the reovirus virion-associated σ1 protein results in earlier depletion of σ1 during viral uncoating and promotes the establishment of productive virus infection in malignant cells.23
Finally, the oncolytic efficacy of these two reovirus variants was tested in a syngeneic murine model of melanoma. Animals treated with both selected viral variants survived significantly longer than animals treated with parental virus strains.22 Thus, the study demonstrated that it is possible to select reovirus variants with better oncolytic infectivity and therapeutic potential without compromising safety. It would be interesting to learn if these preclinical observations can be translated into beneficial effects in clinical studies.
Infectivity broadening
In the Vero cell line, infectivity of wild-type T3D reovirus is not as good as in the L929 cell line. However, through infection persistence establishment, a virus variant was selected that was capable of infecting Vero cells efficiently.24 This variant had amino acid substitutions in two reovirus proteins, σ1 and μ1.24,25 The first protein, σ1, participates in virus attachment to the cell surface; the other protein, μ1, participates in virus disassembly (uncoating) from its outer capsid. As a result of the amino acid changes in the σ1 and μ1 proteins, three phenotypic differences between the selected reovirus variant and the parental strain were observed.24,25 First, the virus binding to the Vero cell surface became five times stronger. Moreover, binding became sensitive to neuraminidase treatment, indicating the involvement of sialic acid residues attached to the cell outer membrane in the binding process. Second, the rate of disassembly of the virions inside infected cells increased. Third, the virus became very sensitive to interferon (IFN).26 Later this sensitivity was attributed to a unique mutation in the S1 gene, overlapping the σ1 and σ1 s reading frames.26 The adapted virus’s oncolytic efficiency and its safety profile remain to be tested in animal models.
Wild-type T3 reovirus (along with T1 reovirus) uses JAM-A as a receptor.15,27 Human gliomas, melanomas, ovarian, prostate, and some other cancers express little or no JAM-A.28–30 Low or no JAM-A expression makes many cancer cells resistant to reovirus T3. Virus variants that enter cancer cells in a JAM-A-independent manner could overcome this resistance. An adaptation study was performed in pursuit of this goal.31,32 It included establishment of persistent reovirus infection of a murine erythroleukemia (MEL) cell line which does not express JAM-A. Analysis of reassorted viruses created from various combinations of parental and MEL adapted strains demonstrated that the σ1 protein is responsible for the phenotypic change. Viral variants with a point mutation in the σ1 protein have an increased ability to bind sialic acids located on the cell surface. Their binding to human cholangiocarcinoma cells was improved 100-fold31–33 compared with the T3 parent. In addition, selected viral variants were capable of causing much higher levels of apoptosis of HeLa cells than the parental virus strain.31,32 However, the safety profile of the parental virus was better than that of the sialic acid binding mutants.33
A similar goal of adapting reovirus to infect a broader range of cancer cells was pursued by another research team.34 This team performed selection rounds of T3D virus variants in glioblastoma cells, which do not express JAM-A. Three viral variants were isolated that acquired the ability to infect the previously reovirus resistant U118 glioblastoma cell line.34 It was demonstrated that all three selected virus variants rely on sialic acids for cell entry. Not surprisingly, this viral phenotypic change was associated with mutations located close to the sialic acid binding motif of σ1 cell attachment protein. One of the virus mutants gained the ability to infect a wide range of new cell lines that were not permissive to T3D reovirus infection. These cell lines included chicken hepatocellular carcinoma LMH, murine endothelioma, human bone osteosarcoma U2OS, and Ewing sarcoma STA-ET2.34 As such, the goal of broadening the range of cancer cells that could be infected by reovirus was achieved. However, virus dissemination specificity and safety profiles were not reported.34 The possibility that broadening the reovirus infectivity compromised its safety, by analogy with the observations reported earlier,33 could not be ruled out.
Safety improvement
IFN sensitivity increase
IFN pathways are frequently defective in malignant cells, making them more sensitive than normal cells to OVs.35 However, viruses, in general, can inhibit some components of the IFN defensive system of their host cells, whether malignant or not. Consequently, the virus ability to inhibit IFN defenses in normal cells could promote infection of these cells and decrease OV safety.
So, for the best discrimination between malignant and normal cells, OVs should be sensitive to IFN. Such reovirus variants were obtained by chemical mutagenesis and tested in normal parental and Ras-transformed mouse NIH-3T3 cells. As expected, a correlation was found between increased virus IFN sensitivity and decreased virus ability to infect normal cells. An IFN-hypersensitive reovirus variant was highly dependent on Ras activation, and it discriminated between normal and transformed cells much better than the parental strain.36 A single amino acid substitution in one region of λ2 methyltransferase was found to be the major determinant of IFN sensitivity in this variant.37 The results of the study suggest that optimized reoviruses with improved safety could be selected by modification of virus IFN sensitivity.
Attenuation of JAM-A receptor binding
In a separate study, Kim and coauthors created attenuated versions of reovirus by establishing persistent infection of wild-type T3D variant in human HT1080 fibrosarcoma cells.38–40 Attenuated reovirus kept its oncolytic efficiency but demonstrated reduced pathogenicity in the animal model.40 It was able to infect three tested lymphoma cell lines, HBL-2, Granta, and Z138C, and was able to suppress the growth of HCT116 colon carcinoma cells. This reovirus variant maintained its ability to kill parental HT1080 cells in vitro but developed reduced ability to infect L929 and HCT116 cells. In murine xenografts, attenuated reovirus suppressed HT1080 cell growth as efficiently as its parental T3D strain. However, in contrast to the parental strain, mice injected with the mutant virus showed no visible toxicity after 1 month post-treatment and only began to develop black tail syndrome after 3–7 months. It was shown that the attenuated virus has a premature stop codon in the σ1 cell surface-attachment protein, resulting in a truncated translational product. The stop codon prevents translation of the σ1 head, thereby preventing binding to JAM-A.40
Genomic changes that affect infectivity and safety
Almost all studies cited above demonstrated that critical genotype changes were responsible for increase or broadening of reovirus infectivity. The changes typically occurred in viral gene for cell attachment protein σ1. Moreover, these studies also indicated that genotype changes, which were responsible for the improvement of the safety profile of reoviruses, could also be mapped to the same protein. Collectively, these data suggest that small changes in reovirus genes cause gain or loss of σ1 ability to bind sialic acids or JAM-A located on the host cell surface. Gaining the ability to bind new cell entry receptors increases and broadens reovirus infectivity, while losing it most likely increases virus safety. Achievement of the optimal interplay between this gain or loss as well as the creation of relevant optimal reovirus variants is challenging and critical tasks for future research.
Paramyxoviruses
Newcastle disease virus (NDV)
NDV is a negative-sense, single-stranded RNA virus that belongs to the genus Orthoavulavirus in the family Paramyxoviridae.41 Its genome size is approximately 15 kb. NDV causes highly contagious and serious disease in birds42 but only mild conjunctivitis in humans.43 The virus’ oncolytic properties are well studied.44–49 It infects a broad spectrum of cancer cell lines, including fibrosarcoma, osteosarcoma, cervical and bladder carcinomas, neuroblastoma, and Wilm’s tumor.50 Among all viral proteins, one (F protein) is of special interest for approaches that aim to improve viral oncolysis. This glycoprotein is responsible for viral fusion with the cell membrane and viral spread from cell to cell through syncytia formation. F protein is synthesized as an inactive precursor (F0) and is activated through proteolytic cleavage by cellular proteases. The amino acid sequence of the cleavage site determines NDV virulence. F0 proteins of low pathogenicity viruses have monobasic cleavage sites that are susceptible to trypsin-like proteases only, resulting in restricted infection. In contrast, F0 proteins of the most pathogenic virus strains have polybasic cleavage sites that are susceptible to a larger spectrum of proteases, resulting in systemic infection. Genetic modification of the F0 cleavage site could increase virus fusogenic potential and improve its spread among malignant cells, thereby increasing its oncolytic potential.45
In animal models, NDV promotes both direct and immuno-modulated cancer cell death.44,46,48 When injected intratumorally or peritumorally in syngeneic animals with transplantable colon carcinomas, the virus caused significant slowing of tumor growth, which prolonged animal survival. Intravenous NDV injection in the same animal model did not retard the growth of the tumors but did promote significant animal weight loss.51 Systemic virus application was also shown to be much less effective than local in treatment of murine metastatic melanoma as well as in treatment of colon and renal carcinomas.52
NDV-based oncolytic therapy has been reported to be beneficial in several clinical trials that included treatment of melanomas, glioblastomas, head, and neck squamous cell carcinomas and other malignant diseases. Descriptions of these trials are compiled in summary tables in recently published reviews.46,49 Two larger trials deserve special mentioning. One was a Phase III trial that involved approximately 50 colorectal cancer patients.53 The trial tested efficacy of injections of patients’ with their autologous tumor cells infected with NDV, following resection of patients’ liver metastases. In the subgroup of colon cancer patients, statistically significant improvement in 10-year overall survival was observed.53 Another trial was an NDV dose escalation study performed with approximately 100 patients with different advanced malignancies. In this trial variable dosages of NDV were injected intravenously. A few responses to the therapy were observed at the highest dose levels.54–57
Because NDV can cause epidemic disease in poultry, its further development as oncolytic therapy must address concerns about potential environmental contamination, which could cause viral outbreaks in domestic birds. Hopefully, genetic modifications such as those introduced into an NDV strain by Cheng and coauthors58 will address this issue by attenuating the virus sufficiently to make it nonpathogenic to birds.
Improving intratumoral spreading ability
Inside tumors, viral infection spread could be limited due to tissue barriers that do not exist in a cell culture. For example, the human fibrosarcoma HT1080 cell line is highly sensitive to NDV infection, but NDV spread within the tumor formed by HT1080 cells is restricted.59 Because of this limitation, elimination of HT1080 tumors by virotherapy is usually incomplete.59 Beier and coauthors59 hypothesized that virus selection could improve NDV intratumoral spreading ability. To confirm their hypothesis, the authors performed two rounds of virus selection in tumor xenografts of HT1080 fibrosarcoma (Figure 2). In the first round, they injected NDV into a number of HT1080 fibrosarcoma xenograft carrying animals, and identified an animal with a tumor, which was most affected by the virus treatment. They excised this tumor from the euthanized animal, dissected it into fragments and incubated them on top of HCT116 carcinoma cells’ monolayer. NDV variants were plaque purified from this monolayer and further amplified in the allantoic fluid of embryonated chicken eggs. In the second round of selection, the virus harvested from the eggs was used for reinfection of the tumors of a new set of animals with HT1080 fibrosarcoma xenografts. The virus variants from the animal with the smallest tumor were again plaque purified in a HCT116 colon carcinoma cell culture and further characterized.
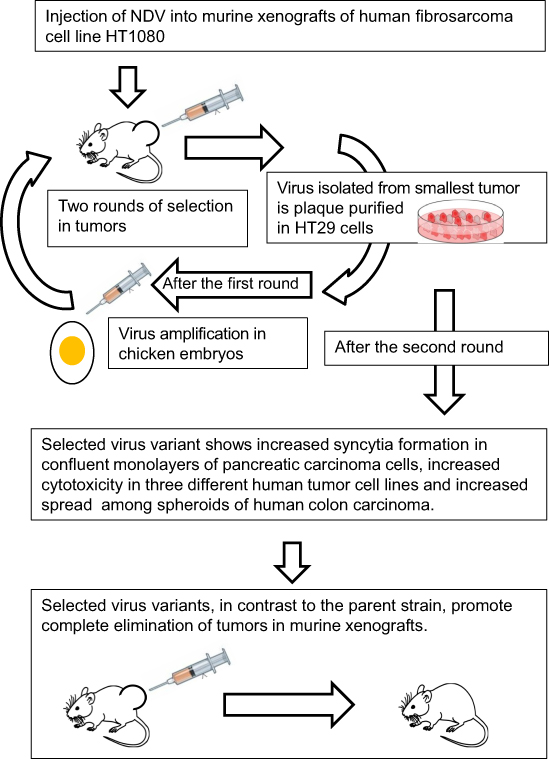 |
Figure 2 Selection process for increasing Newcastle Disease Virus (NDV) intratumoral spreading ability. |
The characterization included viral infection of various malignant cell types’ monolayers and of colon carcinoma’s spheroids. The latter represents a more clinically relevant three-dimensional cancer model than monolayers of malignant cells. The authors59 concluded that through the selection steps phenotypically distinct virus clones were produced. Their fusogenicity, viral spread, and growth rate distinguished them from the parental strain. Some of the selected virus variants also exhibited stronger cytotoxicity in a variety of cancer cell monolayers. Besides, a few of the selected clones were able to kill spheroids from HCT116 carcinoma cells completely, while their parental strain was capable of only partial killing.59
However, the most clinically relevant test was done by comparing the virus variants’ antitumor effects in HT1080 fibrosarcoma murine-human xenografts. Tumor growth inhibition was stronger in animals that were injected with the virus variants than in animals that were injected with a parental strain. Moreover, the selected NDV variants not only slowed tumors growth but also led to their complete elimination in a few animals, an effect which had not been observed with the parental virus strain.59 The animals that were treated with the selected virus variants did not lose weight or demonstrate any other signs of viral toxicity. The authors concluded that NDV intratumoral spreading ability could be improved by rounds of viral adaptation without compromising viral safety.59
The study described above shows that multiple viral progenies of the same viral strain are diverse with respect to their spreading capability, and rapid selection of clones with increased spreading capability is possible. However, this study did not connect viral phenotypic changes with any genomic changes. It left unanswered the question if the selected virus strains had changes in the gene region that corresponds to the proteolytic cleavage site of the F0. Therefore, the mechanism of improved intra-tumor spreading remains a mystery. The study also raises many other questions. How stable is the acquired phenotype? How universal is the spreading ability of selected clones for different tumor types? If the clones were selected using xenografts, tumors of which were formed by one type of malignant cells, could the selected viruses equally efficiently spread in tumors formed by other types of malignant cells? Hopefully, future research will provide answers to these pressing questions.
Sendai virus (SeV)
SeV is a negative-sense single-stranded RNA virus with a genome of 15.3 kb. It belongs to the genus Respirovirus, family Paramyxoviridae.8 SeV causes respiratory infections in mice, hamsters, guinea pigs, rats, and other rodents.60 It spreads through aerosols or direct animal contact. The virus is a very common murine pathogen; it can be isolated from mice colonies worldwide60 including the US.61 SeV and human parainfluenza virus type 1 (HPIV-1), which causes human disease, induce production of cross-reactive antibodies. Thus, in the US, SeV has been used in clinical trials of immunization of both adults and children against HPIV-1. SeV administration was well tolerated and it triggered the production of neutralizing antibodies towards HPIV-1.62,63 These studies represent essential proof of SeV safety for humans.
SeV causes severe murine disease, so a genetically modified strain was created to be nonpathogenic for experimental mice to study this virus’ oncolytic properties.64–67 This recombinant strain suppressed or eradicated tumor growth in a variety of human xenograft tumors, including sarcoma, melanoma, neuroblastoma, pancreas, colon, hepatocellular, and prostate carcinomas.64–67 Complete eradication of established brain tumors was also observed in a few animals as a result of treatment with another recombinant variant of SeV.68
Wild-type SeV is infectious and is immuno-suppressive for rodents. However, UV-inactivated SeV virions have immune-stimulating properties: they are capable of promoting immuno-modulated tumor regression of colon,69,70 bladder,71 and kidney72 cancers in syngeneic mice. UV-inactivated SeV virions also promote human prostate cancer eradication in murine xenografts.73
In the 1990s, the Moscow strain of SeV was tested as an anticancer agent in a few dozen patients affected by various malignancies with metastatic growth.74 In the majority of patients, SeV was ineffective or caused transient improvement. However, a few patients achieved long-term remission even when the virus was used as a monotherapy. In these cases, complete disappearance of metastatic disease was observed without signs of its recurrence for 5–10 years or more after viral treatment. Short descriptions of these cases are presented in the patent.74
Adaptation of Sendai virus to grow in cell culture
High titers of SeV can be produced in the allantoic fluid of embryonated chicken eggs.75,76 However, such virus growth depends on a supply of specific pathogen-free eggs, which are expensive and not readily available in all countries. SeV propagation in cell culture might be a cheaper alternative to virus growth in eggs. In a study by Zainutdinov and colleagues77 two cell lines were chosen for culture of SeV variants, 4,647 (African green monkey kidney cells) and HEK293 (human embryonic kidney cells). Both cell lines are certified for viral vaccine production in Russia. Figure 3 shows the scheme of adaptation experiment that included multiple (20–25 times) passaging of SeV in both cell cultures. The passaging caused numerous non-synonymous nucleotide substitutions.77 Most of these substitutions were in the F and HN genes, which encode surface proteins of the SeV virion.77 Mutation accumulation was associated with a significant decrease of SeV oncolytic activity toward melanoma (Mel8) and glioma (U87MG) cells.77 Reverse passaging in embryonated chicken eggs of SeV, which had been adapted to grow in the 4,647-cell line, partially restored its oncolytic activity.77 The reverse changes in the virus phenotype corresponded to reverse changes in its genotype. Some of the acquired non-synonymous SeV nucleotide substitutions were changed back to those in the parental genotype. Similar reverse passaging of SeV, which was adapted to grow in the HEK293-cell line, did not lead to recovery of its oncolytic parental phenotype or genotype.77 The results of this study highlight the challenging problem that an adaptation towards one desirable virus characteristic might be accompanied by loss of another.
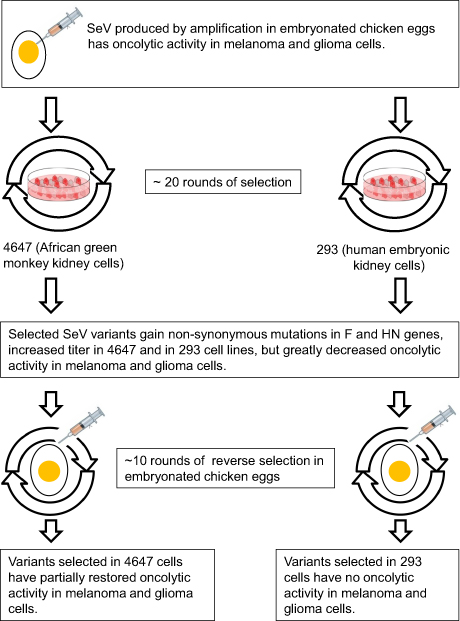 |
Figure 3 Changes in Sendai virus phenotype and genotype during adaptation to cell culture. SeV strains adapted to grow in 4,647 or HEK293-cell cultures lost their oncolytic properties. They were subjected to reverse passaging in embryonated chicken eggs for evaluation of their ability to restore their oncolytic potentials. |
Picornaviruses
Coxsackieviruses
Coxsackieviruses belong to the genus Enterovirus in the family Picornaviridae.8 This family consists of non-enveloped, linear, positive-sense single-stranded RNA viruses. Their genomes are approximately 7 kb long. Coxsackieviruses are widely distributed in humans. They are most frequently transmitted by the fecal-oral route and less frequently by respiratory aerosols. The genus Enterovirus also includes poliovirus and echovirus.8 Coxsackieviruses were discovered in the late 1940s. Subclinical infection of children with these viruses seemed to interfere with poliovirus infection.78 Coxsackieviruses are divided into group A and group B based on their pathogenicity in newborn mice. At least 23 serotypes of group A and six serotypes of group B are recognized.79 Coxsackievirus A13, A15, A18, A21 (CVA21),80–85 and Coxsackievirus B3 (CVB3)86,87 are being studied as oncolytic agents.
Human melanoma cells implanted as xenografts into immuno-deficient mice demonstrated high susceptibility to viral oncolysis by CVA21.80 Tumor burden in these animals was rapidly reduced after a single viral administration.80 Coxsackieviruses A13, A15, and A18 demonstrated similar anti-melanoma efficacy.83 It is interesting that intratumoral, intraperitoneal, or intravenous administrations of CVA21 were equally effective in reducing the tumor volume of melanoma xenografts implanted into immuno-deficient mice.81
CVB3 demonstrated oncolytic activity against nine human non-small cell lung cancer cell lines.86 This activity positively correlated with the expression of viral receptors such as coxsackievirus and adenovirus receptor (CAR) as well as decay-accelerating factor (DAF).86 Moreover, CVB3 injection into one implanted tumor resulted in its durable regression along with the regression of another identical uninjected tumor in the same animal. Furthermore, viral ability to spread from tumor to tumor in an animal was demonstrated by detection of replication-competent CVB3 in the uninjected tumor.86 This preclinical research drives interest in Coxsackievirus adaptation studies.
Adaptation of Coxsackieviruses through infectivity broadening
Adaptation studies of Coxsackieviruses, which are summarized in Table 1, have been directed toward broadening the range of different malignancies that could be infected. This process frequently involves a change in receptor usage. Thus, for productive infection, CVA21 needs simultaneously two cell entry receptors: DAF and intercellular cell adhesion molecule 1 (ICAM-1).88 Some rhabdomyosarcomas, express DAF but not ICAM-1.89 Consequently, for targeting such tumors, it would be beneficial to have a CVA21 variant that could use DAF alone without ICAM-1. Such a virus variant was created by rounds of selection in a DAF-expressing, ICAM-1-non-expressing rhabdomyosarcoma cell line. The selected virus variant retained its binding ability to ICAM-1 but acquired the ability to infect an ICAM-1-negative rhabdomyosarcoma and one additional ICAM-1-negative cancer cell line.89 It was shown that the selected CVA21 variant binds DAF with higher affinity than its parental strain. Virus genome sequencing revealed two amino acid substitutions in viral capsid protein VP3, which are believed to be responsible for the acquired virus phenotype.89
Coxsackievirus B2 strain Ohio-1 (CVB2/O) uses CAR as a cell entry receptor and is infectious for HeLa cells that express this receptor. However, CVB2/O is unable to infect rhabdomyosarcoma cell lines, even though some other coxsackievirus B strains can do so using DAF as a cell entry receptor.90 Multiple rounds of selection in a rhabdomyosarcoma cell line promoted CVB2/O ability to infect this cell line using DAF.90 It is interesting that the selected virus variant retained the CAR binding ability that characterized its parental strain. Nucleotide sequencing revealed a few mutations in viral capsid regions, which most likely were responsible for the acquired virus phenotype.90
In another study six different immunotypes of group B Coxsackievirus were passaged using the same rhabdomyosarcoma cell line. After multiple blind rounds of selection, followed by plaque purification, a single virus variant (CVB3) was isolated. This virus variant gained the ability to use DAF as a cell entry receptor. Sequence analysis revealed that a single amino acid change in the virus capsid was responsible for this acquired phenotype.91,92
A similar result was achieved with Coxsackievirus non-lytic B6 strain (CV-B6-Schmitt). The relevant study included multiple blind passages in human pancreatic duct epithelial cells. The selected viral strain gained lytic capability toward these cells and acquired the ability to use DAF as a cell entry receptor. The substitution of a single amino acid in the virus capsid protein VP1 was responsible for the new phenotype.93
Another strain of Coxsackievirus B6 (CV-B6) demonstrated a high ability to infect one set of malignant human cell lines (C33A, DU145, AsPC-1, SK-Mel28) and a low or no ability to infect others (A431, A549, RD, and MCF7). To broaden CV-B6 infection ability, the parental virus strain was subjected to up to 15 rounds of selection in cell lines, which were not permissive for viral infection. After such passaging, the new selected virus variants acquired the ability to infect the previously non-permissive cells in which they were passaged, without losing their high replication ability in the original cancer cell lines.94 Several experiments with murine xenografts demonstrated that the selected viral variants showed significantly improved oncolytic properties in comparison with the parental strain.94
Changes in capsid proteins affect infectivity of Coxsackieviruses
Almost all the studies cited above demonstrate that the main changes responsible for the virus ability to bind the DAF receptor are in genomic regions that code for viral capsid proteins. Thus, these changes seem to be responsible for broadening Coxsackievirus infectivity.
Poliovirus
Polioviruses, like Coxsackieviruses, belong to the Picornaviridae family, and thus they are also represented by non-enveloped, linear, positive-sense single-stranded RNA viruses. The poliovirus genome is about 7.5 kb long.95 While wild-type poliovirus virus variants cause serious human disease, their derivative attenuated vaccine strains usually do not cause disease in individuals with healthy immune systems. The oral polio vaccine strains (Sabin 1, Sabin 2, and Sabin 3) were selected as spontaneous mutants of wild-type isolates following repeated passages in simian kidney cells.96,97 The procedure introduced mutations in the viral internal ribosome entry site (IRES) and attenuated the ability of the virus to infect human nervous tissue.96,97
PVSRIPO
PVSRIPO is a recombinant strain of poliovirus Sabin type 1, in which the IRES part of the genome is replaced with the IRES from human rhinovirus type 2. It was shown that this construct has oncolytic properties.98 Poliovirus and its derivative construct PVSRIPO both enter a host cell using poliovirus receptor (PVR), also called Nectin-like molecule 5 (Necl5), or CD155 cell receptor. The expression of this molecule correlates with virus infection ability in glioblastoma multiforme (GBM) cells.98 Thus, GBM expressing PVR is a target for PVSRIPO treatment. GBM is a rare but very aggressive brain malignancy. Treatment options are very limited, while patients have a median survival time of only 15 months.99 However, patients’ survival rate measured two and three years after PVSRIPO intratumoral injections was higher than that of the historical controls.100 Other brain tumors also express CD155 at high levels and this expression in cell cultures correlated with PVSRIPO oncolytic efficiency.101 The PVSRIPO construct demonstrated oncolytic efficacy in human breast and prostate cancer xenograft models.102 Like many other OVs, PVSRIPO kills cancer cells both directly (through infection or apoptosis) and indirectly (through broad activation of the immune system).103–106
Testing genotype stability of recombinant poliovirus (PVSRIPO)
Experimental evolution can help in assessing the risk of a virus reversion to its virulent state during cancer treatment.107 Reversion of vaccine attenuated poliovirus to the parental wild-type neurovirulent phenotype does occur but it is extremely rare.108,109 Is it possible that PVSRIPO virus could gain neurovirulence during glioblastoma treatment due to its amplification in brain tissue? Dobrikova and coauthors addressed this question by examining the genetic stability of PVSRIPO during its propagation in human glioma xenografts (Figure 4).110 In the relevant study intratumoral virus inoculations caused tumor regression in all animals with implanted gliomas. The virus isolated from the tumors of sacrificed animals and further amplified in human glioma cell culture retained the attenuated neurovirulence phenotype. Sequence analyses confirmed that the genetic determinants for this attenuated phenotype were not changed upon in vivo passaging in gliomas.110 This study demonstrated that PVSRIPO did not gain neurovirulence during its propagation in glioma tissue.
 |
Figure 4 Testing phenotype and genotype stabilities of attenuated poliovirus recombinant. |
Rhabdoviruses
Improved infectivity and selectivity of Vesicular Stomatitis Virus (VSV)
VSV belongs to the family Rhabdoviridae. It is a negative-sense, single-stranded RNA virus with a genome of approximately 11 kb. VSV infects some mammals including domestic animals; it usually causes a non-lethal illness, accompanied by fever and ulceration of the epithelium of oral and nasal cavities, feet, and teats.111 Insects are vectors for VSV, so infection incidence depends on insect activity. VSV infection is usually asymptomatic in humans.111
VSV and VSV-based constructs demonstrated oncolytic properties in multiple cell lines and animal models.112,113 However, in animal models, VSV can be neurotoxic.112 Numerous attenuated virus constructs were created to avoid this problem and work on further improvement of these constructs is ongoing.112,113 Higher oncoselectivity and oncoinfectivity are needed.
An interesting adaptation study was performed with one VSV recombinant construct in pursuit of these goals. The construct had one substituted gene, which expressed a chimeric Sindbis virus glycoprotein and a single-chain antibody directed toward the human Her2/neu receptor. Such substitution allows for very specific targeting of human breast cancer cells expressing ErbB-2 (Her2). However, the recombinant virus demonstrated poor growth characteristics in its target cell line. The problem was easily overcome by using the directed evolution approach. Fifteen passages in the targeted breast cancer cell line generated an adapted virus with significantly improved growth characteristics. Sequencing of the genome of this adapted virus revealed only two mutations, both of which occurred in the gene region that encoded the single-chain antibody. Surprisingly, an additional N-glycosylation site was created by one of the mutations. Along with improved infectivity, the adapted virus showed a higher density of glycoprotein on the viral envelope.114 Thus, two adaptive mutations made the VSV construct more infections toward one ERB-2 positive breast cancer cell line. How helpful are these mutations for infecting other ERB-2 positive cell lines? How will these mutations affect virus safety? These questions await further study.
A similar study was done with VSV and glioblastoma cells. As a result of multiple passaging of wild-type VSV, a selected variant, VSV-rp30, was isolated with much higher selectivity and lytic capability toward glioblastoma cells than the wild-type virus. The variant VSV-rp30 was very effective at replicating, spreading within and selectively killing transplanted human glioblastoma cells in a murine model.115
Because p53 deficiency frequently characterizes malignant cells, directed VSV evolution was also used to obtain a virus variant with increased selectivity toward p53-deficient cells. After approximately 40 passages, the variant virus became much more infective and cytotoxic against target cells in comparison with the parental virus. Moreover, the virus infectivity increase was specific even for p53-deficient cells that were not used for virus selection. Syngeneic animal models demonstrated that the selected VSV variant significantly delayed the growth of p53-deficient mammary gland tumors compared with its parental strain.116 This study suggests that gene-specific virus adaptation is possible.
Conclusion remarks
The directed evolution/adaptation studies described in this review are summarized in Table 1. They demonstrate that oncolytic RNA viruses can acquire desirable characteristics through rounds of selection. The viruses can be selected to have higher infectivity toward a particular type of tumor cell or to infect a broader spectrum of malignant cells. They can be also selected for better intratumoral spreading ability or a better safety profile. Mapping of genotypic changes responsible for the selection acquired phenotype provides valuable insights into the molecular mechanisms that are responsible for viral infection of cancer cells. However, this type of research has problems and limitations.
An adaptation toward one desirable virus characteristic might be accompanied by a loss of another one. It is not always straightforward to map changes of viral phenotype to genotype. Culture conditions for maintaining acquired desirable traits of selected virus variants are not always easy to define. The stability of the viral phenotype acquired through directed evolution is questionable. Theoretically, without selection pressure any new viral trait might be lost through reverse evolution.
The directed evolution studies described in this review involved RNA viruses, whose replication is accompanied by high mutation rates, and which demonstrate major genetic variability. Virus populations are usually not made of a single variant with a defined nucleic acid sequence. Rather, they represented by a broad spectrum of non-identical but related mutants. These mutants called quasispecies and they collectively contribute to the characteristics of the population.117,118 High mutation rates of RNA viruses and rapid increase in the diversity of quasispecies can both be explained by the low replication fidelity of the RNA-dependent RNA polymerase (RdRp) that replicates RNA virus genomes.119 The low replication fidelity of RdRp causes its high error rate. On the one hand, the high error rate of RdRp is beneficial for directed evolution because it allows performing a comparatively low number of selection cycles for rapid achievement of the bioselection goal. On the other hand, the low fidelity of RdRp could promote a high rate of reverse evolution which can compromise the ultimate bioselection goal.
However, a high mutation rate and corresponding rapid genomic change could represent an advantage for cancer patient treatment. Perhaps oncolytic RNA virus adaptation could occur naturally, within heterogeneous tumors and metastases inside a patient’s body. May be such “personalized” targeted evolution could produce multiple optimized virus variants that would be capable of causing complete tumor clearance despite antiviral immunity.
Both directed evolution and genetic engineering are approaches that could generate viral variants optimized for oncolytic applications. Despite the drawbacks of directed evolution, the approach has certain advantages over genetic engineering because it allows achieving a desirable virus phenotype without knowing in advance the genetic determinants, which are responsible for the phenotype. These two approaches complement each other and their combination will pave the way toward more safe and effective OV treatment.
Acknowledgments
The study was funded by the Russian Foundation for Basic Research, research project N 18-34-00286 mol_a and by the grant from the Center of Strategic Planning of The Ministry of Healthcare of the Russian Federation, project code 1.1096. The authors are grateful to Dr. Anton Komar and to Dr. Judith Miller for their constructive comments and corrections that significantly improved the manuscript.
Disclosure
Mr. Sergei S Zainutdinov reports grants from the Russian Foundation for Basic Research, during the conduct of the study. The authors report no other conflicts of interest in this work.
References
1. Donnelly O, Harrington K, Melcher A, Pandha H. Live viruses to treat cancer. J R Soc Med. 2013;106(8):310–314. doi:10.1177/0141076813494196
2. Altinoz MA, Guloksuz S, Elmaci I. Rabies virus vaccine as an immune adjuvant against cancers and glioblastoma: new studies may resurrect a neglected potential. Clin Transl Oncol. 2017;19(7):785–792. doi:10.1007/s12094-017-1613-6
3. Kelly E, Russell SJ. History of oncolytic viruses: genesis to genetic engineering. Mol Ther. 2007;15(4):651–659. doi:10.1038/sj.mt.6300108
4. Lichty BD, Breitbach CJ, Stojdl DF, Bell JC. Going viral with cancer immunotherapy. Nat Rev Cancer. 2014;14(8):559–567. doi:10.1038/nrc3770
5. Liu TC, Kirn D. Systemic efficacy with oncolytic virus therapeutics: clinical proof-of-concept and future directions. Cancer Res. 2007;67(2):429–432. doi:10.1158/0008-5472.CAN-06-2871
6. Dolgin E. Oncolytic viruses get a boost with first FDA-approval recommendation. Nat Rev Drug Discov. 2015;14(6):369–371. doi:10.1038/nrd4643
7. Conry RM, Westbrook B, McKee S, Norwood TG. Talimogene laherparepvec: first in class oncolytic virotherapy. Hum Vaccin Immunother. 2018;14(4):839–846. doi:10.1080/21645515.2017.1412896
8. International Committee on Taxonomy of Viruses. Virus taxonomy: 2018 release. Available from: https://talk.ictvonline.org/taxonomy/.
9. Coombs KM. Reovirus Structure and Morphogenesis. In: Polly Roy, ed. Reoviruses: Entry, Assembly and Morphogenesis. Berlin-Heidelberg, Germany: Springer-Verlag; 2006: 117–167. doi:10.1007/3-540-30773-7
10. Stevenson A, Chandranesan J. Reoviruses. In: Wallace MR, eds. Drugs and Diseases, Infectious Diseases. Medscape eMedicine; 2017. Available from: https://emedicine.medscape.com/article/227348-overview. Accessed February 1, 2019.
11. Nibert LM., Structure of Mammalian Reovirus particles. In: Kenneth KL, Oldstone MBA, eds. Reoviruses I, Structure, Proteins, and Geneticsc (Current Topics in Microbiology and Immunology). Vol. 1. Berlin-Heidelberg, Germany: Springer; 1998:1–30. doi:10.1007/978-3-642-72092-5
12. Mohamed A, Johnston RN, Shmulevitz M. Potential for improving potency and specificity of reovirus oncolysis with next-generation reovirus variants. Viruses. 2015;7(12):6251–6278. doi:10.3390/v7122936
13. Kemp V, Hoeben RC, van den Wollenberg DJ. Exploring reovirus plasticity for improving its use as oncolytic virus. Viruses. 2015;8(1). doi:10.3390/v8010004
14. Comins C, Simpson GR, Relph K, Harrington KJ, Melcher A, Pandha H. Reoviral Therapy for Cancer: Strategies for Improving Antitumor Efficacy Using Radio- and Chemotherapy. In: Lattime E, Gerson S,eds.Gene Therapy of Cancer.
15. Forrest JC, Dermody TS. Reovirus receptors and pathogenesis. J Virol. 2003;77(17):9109–9115. doi:10.1128/jvi.77.17.9109-9115.2003
16. Gong J, Sachdev E, Mita AC, Mita MM. Clinical development of reovirus for cancer therapy: an oncolytic virus with immune-mediated antitumor activity. World J Methodol. 2016;6(1):25–42. doi:10.5662/wjm.v6.i1.25
17. Bourhill T, Mori Y, Rancourt DE, Shmulevitz M, Johnston RN. Going (Reo)Viral: factors promoting successful reoviral oncolytic infection. Viruses. 2018;10(8):421. doi:10.3390/v10080421
18. Morris DG, Feng X, DiFrancesco LM, et al. REO-001: A phase I trial of percutaneous intralesional administration of reovirus type 3 dearing (Reolysin(R)) in patients with advanced solid tumors. Invest New Drugs. 2013;31(3):696–706. doi:10.1007/s10637-012-9865-z
19. Cohn DE, Sill MW, Walker JL, et al. Randomized phase IIB evaluation of weekly paclitaxel versus weekly paclitaxel with oncolytic reovirus (Reolysin(R)) in recurrent ovarian, tubal, or peritoneal cancer: an NRG Oncology/Gynecologic Oncology Group study. Gynecol Oncol. 2017;146(3):477–483. doi:10.1016/j.ygyno.2017.07.135
20. Noonan AM, Farren MR, Geyer SM, et al. Randomized phase 2 trial of the oncolytic virus pelareorep (Reolysin) in upfront treatment of metastatic pancreatic adenocarcinoma. Mol Ther. 2016;24(6):1150–1158. doi:10.1038/mt.2016.66
21. Bernstein V, Ellard SL, Dent SF, et al. A randomized phase II study of weekly paclitaxel with or without pelareorep in patients with metastatic breast cancer: final analysis of Canadian Cancer Trials Group IND.213. Breast Cancer Res Treat. 2018;167(2):485–493. doi:10.1007/s10549-017-4538-4
22. Shmulevitz M, Gujar SA, Ahn DG, Mohamed A, Lee PW. Reovirus variants with mutations in genome segments S1 and L2 exhibit enhanced virion infectivity and superior oncolysis. J Virol. 2012;86(13):7403–7413. doi:10.1128/JVI.00304-12
23. Mohamed A, Teicher C, Haefliger S, Shmulevitz M. Reduction of virion-associated sigma1 fibers on oncolytic reovirus variants promotes adaptation toward tumorigenic cells. J Virol. 2015;89(8):4319–4334. doi:10.1128/JVI.03651-14
24. Jabre R, Sandekian V, Lemay G. Amino acid substitutions in sigma1 and mu1 outer capsid proteins are selected during mammalian reovirus adaptation to Vero cells. Virus Res. 2013;176(1–2):188–198. doi:10.1016/j.virusres.2013.06.007
25. Sandekian V, Lemay G. Amino acids substitutions in sigma1 and mu1 outer capsid proteins of a Vero cell-adapted mammalian orthoreovirus are required for optimal virus binding and disassembly. Virus Res. 2015;196:20–29. doi:10.1016/j.virusres.2014.11.002
26. Lanoie D, Cote S, Degeorges E, Lemay G. A single mutation in the mammalian orthoreovirus S1 gene is responsible for increased interferon sensitivity in a virus mutant selected in Vero cells. Virology. 2018;528:73–79. doi:10.1016/j.virol.2018.12.010
27. Barton ES, Forrest JC, Connolly JL, et al. Junction adhesion molecule is a receptor for reovirus. Cell. 2001;104(3):441–451.
28. Human Protein Atlas. Available from: https://www.proteinatlas.org/.
29. Uhlen M, Bjorling E, Agaton C, et al. A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol Cell Proteomics. 2005;4(12):1920–1932. doi:10.1074/mcp.M500279-MCP200
30. Uhlen M, Oksvold P, Fagerberg L, et al. Towards a knowledge-based Human Protein Atlas. Nat Biotechnol. 2010;28(12):1248–1250. doi:10.1038/nbt1210-1248
31. Chappell JD, Gunn VL, Wetzel JD, Baer GS, Dermody TS. Mutations in type 3 reovirus that determine binding to sialic acid are contained in the fibrous tail domain of viral attachment protein sigma1. J Virol. 1997;71(3):1834–1841.
32. Barton ES, Connolly JL, Forrest JC, Chappell JD, Dermody TS. Utilization of sialic acid as a coreceptor enhances reovirus attachment by multistep adhesion strengthening. J Biol Chem. 2001;276(3):2200–2211. doi:10.1074/jbc.M004680200
33. Barton ES, Youree BE, Ebert DH, et al. Utilization of sialic acid as a coreceptor is required for reovirus-induced biliary disease. J Clin Invest. 2003;111(12):1823–1833. doi:10.1172/JCI16303
34. van den Wollenberg DJ, Dautzenberg IJ, van den Hengel SK, Cramer SJ, de Groot RJ, Hoeben RC. Isolation of reovirus T3D mutants capable of infecting human tumor cells independent of junction adhesion molecule-A. PLoS One. 2012;7(10):e48064. doi:10.1371/journal.pone.0048064
35. Matveeva OV, Chumakov PM. Defects in interferon pathways as potential biomarkers of sensitivity to oncolytic viruses. Rev Med Virol. 2018;28(6):e2008. doi:10.1002/rmv.v28.6
36. Rudd P, Lemay G. Correlation between interferon sensitivity of reovirus isolates and ability to discriminate between normal and Ras-transformed cells. J Gen Virol. 2005;86(Pt 5):1489–1497. doi:10.1099/vir.0.80628-0
37. Sandekian V, Lemay G. A single amino acid substitution in the mRNA capping enzyme lambda2 of a mammalian orthoreovirus mutant increases interferon sensitivity. Virology. 2015;483:229–235. doi:10.1016/j.virol.2015.04.020
38. Kim M, Egan C, Alain T, et al. Acquired resistance to reoviral oncolysis in Ras-transformed fibrosarcoma cells. Oncogene. 2007;26(28):4124–4134. doi:10.1038/sj.onc.1210189
39. Kim M, Williamson CT, Prudhomme J, et al. The viral tropism of two distinct oncolytic viruses, reovirus and myxoma virus, is modulated by cellular tumor suppressor gene status. Oncogene. 2010;29(27):3990–3996. doi:10.1038/onc.2010.137
40. Kim M, Garant KA, zur Nieden NI, et al. Attenuated reovirus displays oncolysis with reduced host toxicity. Br J Cancer. 2011;104(2):290–299. doi:10.1038/sj.bjc.6606053
41. de Leeuw O, Peeters B. Complete nucleotide sequence of Newcastle disease virus: evidence for the existence of a new genus within the subfamily Paramyxovirinae. J Gen Virol. 1999;80(Pt 1):131–136. doi:10.1099/0022-1317-80-1-131
42. Dimitrov KM, Ramey AM, Qiu X, Bahl J, Afonso CL. Temporal, geographic, and host distribution of avian paramyxovirus 1 (Newcastle disease virus). Infect Genet Evol. 2016;39:22–34. doi:10.1016/j.meegid.2016.01.008
43. Nelson CB, Pomeroy BS, Schrall K, Park WE, Lindeman RJ. An outbreak of conjunctivitis due to Newcastle disease virus (NDV) occurring in poultry workers. Am J Public Health Nations Health. 1952;42(6):672–678. doi:10.2105/ajph.42.6.672
44. Fournier P, Bian H, Szeberenyi J, Schirrmacher V. Analysis of three properties of Newcastle disease virus for fighting cancer: tumor-selective replication, antitumor cytotoxicity, and immunostimulation. Methods Mol Biol. 2012;797:177–204. doi:10.1007/978-1-61779-340-0_13
45. Zamarin D, Palese P. Oncolytic Newcastle disease virus for cancer therapy: old challenges and new directions. Future Microbiol. 2012;7(3):347–367. doi:10.2217/fmb.12.4
46. Matveeva OV, Guo ZS, Shabalina SA, Chumakov PM. Oncolysis by paramyxoviruses: multiple mechanisms contribute to therapeutic efficiency. Mol Ther Oncolytics. 2015;2:15011. doi:10.1038/mto.2015.11
47. Matveeva OV, Guo ZS, Senin VM, Senina AV, Shabalina SA, Chumakov PM. Oncolysis by paramyxoviruses: preclinical and clinical studies. Mol Ther Oncolytics. 2015;2:15017. doi:10.1038/mto.2015.17
48. Schirrmacher V. Oncolytic Newcastle disease virus as a prospective anti-cancer therapy. A biologic agent with potential to break therapy resistance. Expert Opin Biol Ther. 2015;15(12):1757–1771. doi:10.1517/14712598.2015.1088000
49. Matveeva OV, Kochneva GV, Zainutdinov SS, Ilyinskaya GV, Chumakov PM. [Oncolytic Paramyxoviruses: mechanism of Action, Preclinical and Clinical Studies]. Mol Biol (Mosk). 2018;52(3):360–379. doi:10.7868/S0026898418030023
50. Reichard KW, Lorence RM, Cascino CJ, et al. Newcastle disease virus selectively kills human tumor cells. J Surg Res. 1992;52(5):448–453.
51. Apostolidis L, Schirrmacher V, Fournier P. Host mediated anti-tumor effect of oncolytic Newcastle disease virus after locoregional application. Int J Oncol. 2007;31(5):1009–1019.
52. Schirrmacher V, Griesbach A, Ahlert T. Antitumor effects of Newcastle Disease Virus in vivo: local versus systemic effects. Int J Oncol. 2001;18(5):945–952. doi:10.3892/ijo.18.5.945
53. Schulze T, Kemmner W, Weitz J, Wernecke KD, Schirrmacher V, Schlag PM. Efficiency of adjuvant active specific immunization with Newcastle disease virus modified tumor cells in colorectal cancer patients following resection of liver metastases: results of a prospective randomized trial. Cancer Immunol Immunother. 2009;58(1):61–69. doi:10.1007/s00262-008-0526-1
54. Lorence RM, Pecora AL, Major PP, et al. Overview of phase I studies of intravenous administration of PV701, an oncolytic virus. Curr Opin Mol Ther. 2003;5(6):618–624.
55. Laurie SA, Bell JC, Atkins HL, et al. A phase 1 clinical study of intravenous administration of PV701, an oncolytic virus, using two-step desensitization. Clin Cancer Res. 2006;12(8):2555–2562. doi:10.1158/1078-0432.CCR-05-2038
56. Lorence RM, Roberts MS, O’Neil JD, et al. Phase 1 clinical experience using intravenous administration of PV701, an oncolytic Newcastle disease virus. Curr Cancer Drug Targets. 2007;7(2):157–167.
57. Hotte SJ, Lorence RM, Hirte HW, et al. An optimized clinical regimen for the oncolytic virus PV701. Clin Cancer Res. 2007;13(3):977–985. doi:10.1158/1078-0432.CCR-06-1817
58. Cheng X, Wang W, Xu Q, et al. Genetic modification of oncolytic newcastle disease virus for cancer therapy. J Virol. 2016;90(11):5343–5352. doi:10.1128/JVI.00136-16
59. Beier R, Hermiston T, Mumberg D. Isolation of more potent oncolytic paramyxovirus by bioselection. Gene Ther. 2012;23(10):13.
60. Institute of Laboratory Animal Resources (U.S.). Committee on Infectious Diseases of Mice and Rats. Infectious Diseases of Mice and Rats. Washington (DC): National Academy Press; 1991.
61. Parker JC, Whiteman MD, Richter CB. Susceptibility of inbred and outbred mouse strains to Sendai virus and prevalence of infection in laboratory rodents. Infect Immun. 1978;19(1):123–130.
62. Slobod KS, Shenep JL, Lujan-Zilbermann J, et al. Safety and immunogenicity of intranasal murine parainfluenza virus type 1 (Sendai virus) in healthy human adults. Vaccine. 2004;22(23–24):3182–3186. doi:10.1016/j.vaccine.2004.01.053
63. Adderson E, Branum K, Sealy RE, et al. Safety and immunogenicity of an intranasal Sendai virus-based human parainfluenza virus type 1 vaccine in 3- to 6-year-old children. Clin Vaccine Immunol. 2015;22(3):298–303. doi:10.1128/CVI.00618-14
64. Kinoh H, Inoue M, Washizawa K, et al. Generation of a recombinant Sendai virus that is selectively activated and lyses human tumor cells expressing matrix metalloproteinases. Gene Ther. 2004;11(14):1137–1145. doi:10.1038/sj.gt.3302272
65. Yonemitsu Y, Ueda Y, Kinoh H, Hasegawa M. Immunostimulatory virotherapy using recombinant Sendai virus as a new cancer therapeutic regimen. Front Biosci. 2008;13:1892–1898.
66. Kinoh H, Inoue M. New cancer therapy using genetically-engineered oncolytic Sendai virus vector. Front Biosci. 2008;13:2327–2334.
67. Tatsuta K, Tanaka S, Tajiri T, et al. Complete elimination of established neuroblastoma by synergistic action of gamma-irradiation and DCs treated with rSeV expressing interferon-beta gene. Gene Ther. 2009;16(2):240–251. doi:10.1038/gt.2008.161
68. Iwadate Y, Inoue M, Saegusa T, et al. Recombinant Sendai virus vector induces complete remission of established brain tumors through efficient interleukin-2 gene transfer in vaccinated rats. Clin Cancer Res. 2005;11(10):3821–3827. doi:10.1158/1078-0432.CCR-04-1485
69. Kurooka M, Kaneda Y. Inactivated Sendai virus particles eradicate tumors by inducing immune responses through blocking regulatory T cells. Cancer Res. 2007;67(1):227–236. doi:10.1158/0008-5472.CAN-06-1615
70. Kawano H, Komaba S, Kanamori T, Kaneda Y. A new therapy for highly effective tumor eradication using HVJ-E combined with chemotherapy. BMC Med. 2007;5:28. doi:10.1186/1741-7015-5-28
71. Kawano H, Komaba S, Yamasaki T, et al. New potential therapy for orthotopic bladder carcinoma by combining HVJ envelope with doxorubicin. Cancer Chemother Pharmacol. 2008;61(6):973–978. doi:10.1007/s00280-007-0553-1
72. Fujihara A, Kurooka M, Miki T, Kaneda Y. Intratumoral injection of inactivated Sendai virus particles elicits strong antitumor activity by enhancing local CXCL10 expression and systemic NK cell activation. Cancer Immunol Immunother. 2008;57(1):73–84. doi:10.1007/s00262-007-0351-y
73. Kawaguchi Y, Miyamoto Y, Inoue T, Kaneda Y. Efficient eradication of hormone-resistant human prostate cancers by inactivated Sendai virus particle. Int J Cancer. 2009;124(10):2478–2487. doi:10.1002/ijc.24234
74. Senina A, Matveeva O, Senin V, Inventors. Method for cancer immunotherapy and pharmaceutical compositions based on oncolytic non-pathogenic Sendai virus.
75. Tatsumoto N, Arditi M, Yamashita M. Sendai virus propagation using chicken eggs. Bio Protoc. 2018;8(18). doi:10.21769/BioProtoc.3009
76. Giles RE, Ruddle FH. Production of Sendai virus for cell fusion. In Vitro. 1973;9(2):103–107.
77. Zainutdinov SS, Tikunov AY, Matveeva OV, Netesov SV, Kochneva GV. Complete genome sequence of the oncolytic sendai virus strain Moscow. Genome Announc. 2016;4(4). doi:10.1128/genomeA.00933-16
78. Dalldorf G, Gifford R. Clinical and epidemiologic observations of Coxsackie-virus infection. N Engl J Med. 1951;244(23):868–873. doi:10.1056/NEJM195106072442302
79. Mahy BWJ. Classification and general propeties. In: Bendinelli M, Friedman H, eds. Coxsackieviruses. A General Update. New York City, USA: Springer US; 1988: 1–18.doi:10.1007/978-1-4757-0247-7
80. Shafren DR, Au GG, Nguyen T, et al. Systemic therapy of malignant human melanoma tumors by a common cold-producing enterovirus, coxsackievirus a21. Clin Cancer Res. 2004;10(1 Pt 1):53–60.
81. Au GG, Lindberg AM, Barry RD, Shafren DR. Oncolysis of vascular malignant human melanoma tumors by Coxsackievirus A21. Int J Oncol. 2005;26(6):1471–1476. doi:10.3892/ijo.26.6.1471
82. Skelding KA, Barry RD, Shafren DR. Enhanced oncolysis mediated by Coxsackievirus A21 in combination with doxorubicin hydrochloride. Invest New Drugs. 2012;30(2):568–581. doi:10.1007/s10637-010-9614-0
83. Au GG, Beagley LG, Haley ES, Barry RD, Shafren DR. Oncolysis of malignant human melanoma tumors by Coxsackieviruses A13, A15 and A18. Virol J. 2011;8:22. doi:10.1186/1743-422X-8-22
84. Bradley S, Jakes AD, Harrington K, Pandha H, Melcher A, Errington-Mais F. Applications of coxsackievirus A21 in oncology. Oncolytic Virother. 2014;3:47–55. doi:10.2147/OV.S56322
85. Agarwala SS. The role of intralesional therapies in Melanoma. Oncology (Williston Park). 2016;30(5):436–441.
86. Miyamoto S, Inoue H, Nakamura T, et al. Coxsackievirus B3 is an oncolytic virus with immunostimulatory properties that is active against lung adenocarcinoma. Cancer Res. 2012;72(10):2609–2621. doi:10.1158/0008-5472.CAN-11-3185
87. Miyamoto S, Sagara M, Kohara H, Tani K. Oncolytic coxsackievirus therapy as an immunostimulator. Rinsho Ketsueki. 2017;58(8):977–982. doi:10.11406/rinketsu.58.977
88. Shafren DR, Dorahy DJ, Ingham RA, Burns GF, Barry RD. Coxsackievirus A21 binds to decay-accelerating factor but requires intercellular adhesion molecule 1 for cell entry. J Virol. 1997;71(6):4736–4743.
89. Johansson ES, Xing L, Cheng RH, Shafren DR. Enhanced cellular receptor usage by a bioselected variant of coxsackievirus a21. J Virol. 2004;78(22):12603–12612. doi:10.1128/JVI.78.22.12603-12612.2004
90. Polacek C, Ekstrom JO, Lundgren A, Lindberg AM. Cytolytic replication of coxsackievirus B2 in CAR-deficient rhabdomyosarcoma cells. Virus Res. 2005;113(2):107–115. doi:10.1016/j.virusres.2005.04.021
91. Pan J, Narayanan B, Shah S, et al. Single amino acid changes in the virus capsid permit coxsackievirus B3 to bind decay-accelerating factor. J Virol. 2011;85(14):7436–7443. doi:10.1128/JVI.00503-11
92. Reagan KJ, Goldberg B, Crowell RL. Altered receptor specificity of coxsackievirus B3 after growth in rhabdomyosarcoma cells. J Virol. 1984;49(3):635–640.
93. Smura T, Natri O, Ylipaasto P, et al. Enterovirus strain and type-specific differences in growth kinetics and virus-induced cell destruction in human pancreatic duct epithelial HPDE cells. Virus Res. 2015;210:188–197. doi:10.1016/j.virusres.2015.08.003
94. Svyatchenko VA, Ternovoy VA, Kiselev NN, et al. Bioselection of coxsackievirus B6 strain variants with altered tropism to human cancer cell lines. Arch Virol. 2017;162(11):3355–3362. doi:10.1007/s00705-017-3492-0
95. Montmayeur AM, Ng TF, Schmidt A, et al. High-throughput next-generation sequencing of polioviruses. J Clin Microbiol. 2017;55(2):606–615. doi:10.1128/JCM.02121-16
96. Sabine AB, Boulger LR. History of Sabin attenuated poliovirus oral live vaccine strains. J Biol Stand. 1973;1(2):115–118. doi:10.1016/0092-1157(73)90048-6
97. Bandyopadhyay AS, Garon J, Seib K, Orenstein WA. Polio vaccination: past, present and future. Future Microbiol. 2015;10(5):791–808. doi:10.2217/fmb.15.19
98. Brown MC, Dobrikova EY, Dobrikov MI, et al. Oncolytic polio virotherapy of cancer. Cancer. 2014;120(21):3277–3286. doi:10.1002/cncr.28862
99. Bleeker FE, Molenaar RJ, Leenstra S. Recent advances in the molecular understanding of glioblastoma. J Neurooncol. 2012;108(1):11–27. doi:10.1007/s11060-011-0793-0
100. Desjardins A, Gromeier M, Herndon JE
101. Thompson EM, Brown M, Dobrikova E, et al. Poliovirus receptor (CD155) expression in pediatric brain tumors mediates oncolysis of medulloblastoma and pleomorphic xanthoastrocytoma. J Neuropathol Exp Neurol. 2018;77(8):696–702. doi:10.1093/jnen/nly045
102. Holl EK, Brown MC, Boczkowski D, et al. Recombinant oncolytic poliovirus, PVSRIPO, has potent cytotoxic and innate inflammatory effects, mediating therapy in human breast and prostate cancer xenograft models. Oncotarget. 2016;7(48):79828–79841. doi:10.18632/oncotarget.12975
103. Brown MC, Gromeier M. Cytotoxic and immunogenic mechanisms of recombinant oncolytic poliovirus. Curr Opin Virol. 2015;13:81–85. doi:10.1016/j.coviro.2015.05.007
104. Brown MC, Holl EK, Boczkowski D, et al. Cancer immunotherapy with recombinant poliovirus induces IFN-dominant activation of dendritic cells and tumor antigen-specific CTLs. Sci Transl Med. 2017;9:408. doi:10.1126/scitranslmed.aan4220
105. Walton RW, Brown MC, Sacco MT, Gromeier M. Engineered oncolytic poliovirus PVSRIPO subverts MDA5-dependent innate immune responses in cancer cells. J Virol. 2018;92(19). doi:10.1128/JVI.02230-17
106. Gromeier M, Nair SK. Recombinant poliovirus for cancer immunotherapy. Annu Rev Med. 2018;69:289–299. doi:10.1146/annurev-med-050715-104655
107. Sanjuan R, Grdzelishvili VZ. Evolution of oncolytic viruses. Curr Opin Virol. 2015;13:1–5. doi:10.1016/j.coviro.2015.01.014
108. Kew OM, Nottay BK, Hatch MH, Nakano JH, Obijeski JF. Multiple genetic changes can occur in the oral poliovaccines upon replication in humans. J Gen Virol. 1981;56(Pt 2):337–347. doi:10.1099/0022-1317-56-2-337
109. Famulare M, Chang S, Iber J, et al. Sabin vaccine reversion in the field: a comprehensive analysis of sabin-like poliovirus isolates in Nigeria. J Virol. 2016;90(1):317–331. doi:10.1128/JVI.01532-15
110. Dobrikova EY, Broadt T, Poiley-Nelson J, et al. Recombinant oncolytic poliovirus eliminates glioma in vivo without genetic adaptation to a pathogenic phenotype. Mol Ther. 2008;16(11):1865–1872. doi:10.1038/mt.2008.184
111. Lyles DS, Kusmin IV, Rupprecht CE. Rhabdoviridae. In: Fields BN, Knipe DM, Howley PM, eds. Fields Virology, Vol. 1, Chapter 31,
112. Felt SA, Grdzelishvili VZ. Recent advances in vesicular stomatitis virus-based oncolytic virotherapy: a 5-year update. J Gen Virol. 2017. doi:10.1099/jgv.0.000980
113. Melzer MK, Lopez-Martinez A, Altomonte J. Oncolytic vesicular stomatitis virus as a viro-immunotherapy: defeating cancer with a “Hammer” and “Anvil”. Biomedicines. 2017;5(1). doi:10.3390/biomedicines5010008
114. Gao Y, Whitaker-Dowling P, Watkins SC, Griffin JA, Bergman I. Rapid adaptation of a recombinant vesicular stomatitis virus to a targeted cell line. J Virol. 2006;80(17):8603–8612. doi:10.1128/JVI.00142-06
115. Wollmann G, Tattersall P, van den Pol AN. Targeting human glioblastoma cells: comparison of nine viruses with oncolytic potential. J Virol. 2005;79(10):6005–6022. doi:10.1128/JVI.79.10.6005-6022.2005
116. Garijo R, Hernandez-Alonso P, Rivas C, Diallo JS, Sanjuan R. Experimental evolution of an oncolytic vesicular stomatitis virus with increased selectivity for p53-deficient cells. PLoS One. 2014;9(7):e102365. doi:10.1371/journal.pone.0102365
117. Lauring AS, Andino R. Quasispecies theory and the behavior of RNA viruses. PLoS Pathog. 2010;6(7):e1001005. doi:10.1371/journal.ppat.1000975
118. Borderia AV, Rozen-Gagnon K, Vignuzzi M. Fidelity variants and RNA quasispecies. Curr Top Microbiol Immunol. 2016;392:303–322. doi:10.1007/82_2015_483
119. Duffy S. Why are RNA virus mutation rates so damn high? PLoS Biol. 2018;16(8):e3000003. doi:10.1371/journal.pbio.3000003
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.