Back to Archived Journals » Hypoxia » Volume 2
Direct phosphorylation events involved in HIF-α regulation: the role of GSK-3β
Authors Mennerich D, Dimova E ![]() , Kietzmann T
, Kietzmann T
Received 15 January 2014
Accepted for publication 24 February 2014
Published 30 April 2014 Volume 2014:2 Pages 35—45
DOI https://doi.org/10.2147/HP.S60703
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Daniela Mennerich,* Elitsa Y Dimova,* Thomas Kietzmann
Faculty of Biochemistry and Molecular Medicine and Biocenter Oulu, University of Oulu, Oulu, Finland
*These authors contributed equally to this work
Abstract: Hypoxia-inducible factors (HIFs), consisting of α- and β-subunits, are critical regulators of the transcriptional response to hypoxia under both physiological and pathological conditions. To a large extent, the protein stability and the recruitment of coactivators to the C-terminal transactivation domain of the HIF α-subunits determine overall HIF activity. The regulation of HIF α-subunit protein stability and coactivator recruitment is mainly achieved by oxygen-dependent posttranslational hydroxylation of conserved proline and asparagine residues, respectively. Under hypoxia, the hydroxylation events are inhibited and HIF α-subunits stabilize, translocate to the nucleus, dimerize with the β-subunits, and trigger a transcriptional response. However, under normal oxygen conditions, HIF α-subunits can be activated by various growth and coagulation factors, hormones, cytokines, or stress factors implicating the involvement of different kinase pathways in their regulation, thereby making HIF-α-regulating kinases attractive therapeutic targets. From the kinases known to regulate HIF α-subunits, only a few phosphorylate HIF-α directly. Here, we review the direct phosphorylation of HIF-α with an emphasis on the role of glycogen synthase kinase-3β and the consequences for HIF-1α function.
Keywords: HIF-1, phosphorylation, GSK-3β, kinase, hypoxia, ubiquitinylation, tumor suppressor
Introduction
Aerobic life is dependent on an adequate supply of oxygen. The ability of mammals to respond to an inadequate O2 supply, commonly termed hypoxia, is crucial for their survival. Although a proper response to changed O2 tensions triggers adaptation, a number of pathological conditions or failures in the O2 response are associated with various diseases such as anemia, myocardial infarction, thrombosis, atherosclerosis, or cancer.
When exposed to hypoxia or even anoxic conditions, mammalian organisms initiate a variety of responses in different organs, aiming to increase the delivery of oxygen to the tissues. In addition to the switch from an aerobic to an anaerobic metabolism and the suppression of energy-using reactions, the carotid body chemoreceptor cells stimulate the brain stem center controlling the respiratory and cardiovascular systems to enhance ventilation, heart rate, and blood pressure (reviewed by Prabhakar1). In addition, neuroepithelial cells in the lung contribute to adjusting pulmonary perfusion and gas exchange. Moreover, organs and cells switch their gene expression profile: the kidneys produce erythropoietin, which increases red blood cell production in the bone marrow, and vascular cells produce vascular endothelial growth factor to promote angiogenesis and flow of enhanced blood volume (reviewed by Semenza2). In addition to the expression of erythropoietin and vascular endothelial growth factor, the expression of more than 500 genes, products of which are involved in glycolysis, angiogenesis, erythropoiesis, cell death, and differentiation, is also changed on exposure to hypoxia (reviewed by Wenger and Stiehl3 and Semenza4).
In mammals, the hypoxia-dependent changes on the level of gene expression are mainly mediated by the α-subunits of hypoxia-inducible transcription factors (HIFs). HIF α-subunits are tightly regulated, and posttranslational hydroxylations in response to hypoxia appear to be of major importance. In addition to hypoxia, HIF α-subunits were also found to respond to various growth and coagulation factors, hormones, cytokines, or stress factors already under normoxia. These signals are often mediated by different protein kinases. Indeed, different kinases, among them glycogen synthase kinase 3β (GSK-3β), have been identified to directly phosphorylate HIF-α proteins. This review discusses the regulation of HIF-α by GSK-3β and compares it with hydroxylase-dependent HIF-α protein regulation.
HIFs: basic aspects
In their active form, HIFs are heterodimeric transcription factors consisting of an α- and β-subunit. The HIF β-subunit represents the stable nuclear subunit primarily represented by the ubiquitously found ARNT (arylhydrocarbon receptor-nuclear translocator) protein; however, ARNT2 or ARTN3, although to a lesser extent, also appear to be able to take part in the formation of HIF dimers (reviewed by Semenza5). In contrast, the α-subunits represent the O2-sensitive dimerization partner. So far, three α-subunit proteins, HIF-1α, HIF-2α (also known as EPAS,6 HLF,7 HRF,8 or MOP29), and HIF-3α have been identified. Together, the different HIF α- and β-subunits may give rise to the formation of several combinations of HIF dimers.5,10 HIF-1α and HIF-2α are the best-studied HIF-α isoforms. Although they share structural and functional similarities, it appears that differences in the cell-type expression pattern, the target genes, the embryonic deletion phenotypes, and the effects on tumorigenesis exist between HIF-1α and HIF-2α.11–14 The function of HIF-3α, from which several splice variants exist in humans,15,16 is largely unknown, although some human HIF-3α variants and a mouse splice variant termed inhibitory PAS protein (IPAS) appear to act as negative regulators of the hypoxic response.16–19
Similar to the ARNT proteins, the HIF α-proteins belong to the basic helix-loop-helix PAS (Per-ARNT-Sim) protein family. In particular, HIF-1α and HIF-2α show the highest degree of sequence identity in the basic helix-loop-helix (85%), PAS-A (68%), and PAS-B (73%) domains. Both also contain two nuclear localization sequences responsible for translocation to the nucleus under hypoxia; they are localized in the N terminus (amino acids 17–33 in HIF-1α and amino acids 1–50 in HIF-2α) and in the C terminus (amino acids 718–721 in HIF-1α and amino acids 689–870 in HIF-2α).20,21 With the exception of HIF-3α, which does not contain a C-terminal transactivation domain (C-TAD),22,23 HIF α-subunits also contain N- and C-terminal transcriptional activation domains (N-TAD and C-TAD). A unique oxygen-dependent degradation domain (ODDD, amino acids 401–603 in HIF-1α and amino acids 517–682 in HIF-2α) overlaps N-TAD. The residues between N-TAD and C-TAD represent an inhibitory domain (amino acids 604–785 in HIF-1α and amino acids 683–825 in HIF-2α).24,25
Oxygen-dependent regulation of HIF α-subunits: role of hydroxylation
HIF α-subunit activation under hypoxia is mainly the result of an increased protein stability and coactivator recruitment, although transcriptional and translational mechanisms also were shown to be involved in HIF α-subunit activation.22,26–30 As a result, HIF-α proteins accumulate, translocate to the nucleus, and dimerize with HIF-β to form a functional transcription factor.31 Thus, in the presence of oxygen (ie, normoxia), HIF-α proteins become degraded. This is primarily achieved by oxygen-dependent hydroxylations at the ODDD.32 Under normoxia, prolyl hydroxylase domain proteins (PHDs),33,34 in particular PHD2, hydroxylate two crucial residues in the ODDD of HIF α-subunits (P402 and P564 in HIF-1α and P405 and P531 in HIF-2α).25,32,35 Prolyl hydroxylation is required for binding the von Hippel-Lindau protein (VHL),36,37 which represents the substrate recognition subunit of an E3 ubiquitin–protein ligase consisting of elongin C, elongin B, RING box 1, cullin 2, and an E2 ubiquitin-conjugating enzyme (Figure 1). The prolyl hydroxylation and ubiquitination can be further promoted by the binding of PHD2 to OS938 and that of HIF-1α, VHL, and elongin C to SSAT2, respectively.39,40 In addition to prolyl hydroxylation, a conserved asparagine residue (N803 in HIF-1α and N852 in HIF-2α) in the C-TAD is hydroxylated by the factor-inhibiting HIF in an oxygen-dependent manner. This hydroxylation prevents interaction with the coactivator proteins CBP/p300.41–44 Thus, the major posttranslational modification appears to be the oxygen-dependent hydroxylation.36,37
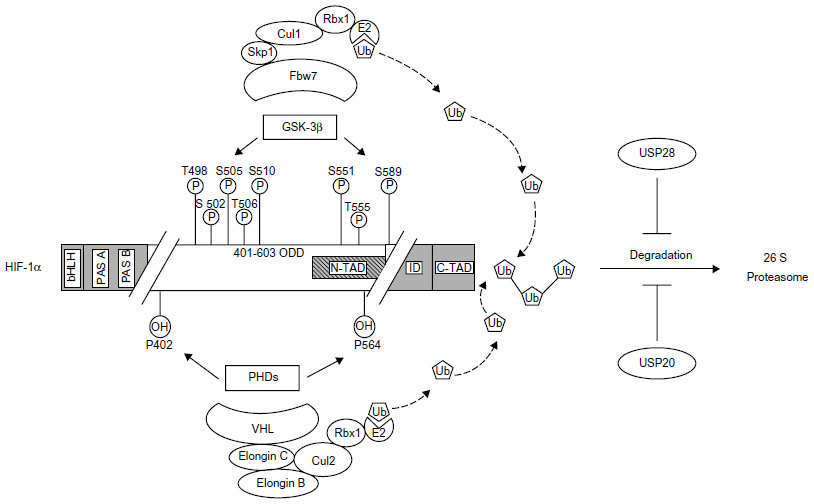 | Figure 1 Phosphorylation- and hydroxylation-mediated proteasomal degradation of hypoxia-inducible factor 1α (HIF-1α). |
Regulation of HIF α-subunits by phosphorylation
In addition to hydroxylation, HIF-α transcriptional activity and protein stability appear also to be dynamically regulated by other posttranslational modifications such as acetylation, S-nitrosylation, SUMOylation, and phosphorylation (for review, see Dimova and Kietzmann45). Phosphorylation appears to be of special importance under normoxic conditions, mediating the response of HIF-α to various growth and coagulation factors, hormones, cytokines, or stress factors (reviewed by Dimova et al46) under normoxia. Indeed, a panel of protein kinases is reported to be involved in HIF-1α phosphorylation, either directly (Table 1) or indirectly.47–52 Although the individual action of certain kinases on HIF-1α regulation was mainly studied in vitro (Table 1), the in vivo mechanisms are likely much more complex. At least the extent to which the kinases can be involved in HIF-α phosphorylation may vary according to the signal, cell type, or tissue. Given the different developmental and/or differentiation status of a cell or tissue, the expression of various growth factors, their receptors, and respective signaling components and the composition of the extracellular surroundings differ. Thus, it seems not to be surprising that phosphorylation of HIF-α by different kinases or after modulation of signaling pathways may be a highly cell type-specific event. Although direct proof is currently lacking, it is plausible that the phosphorylation pattern of HIF-α in a certain cell may be explained by different layers of regulations that affect kinases depending on the cellular context.
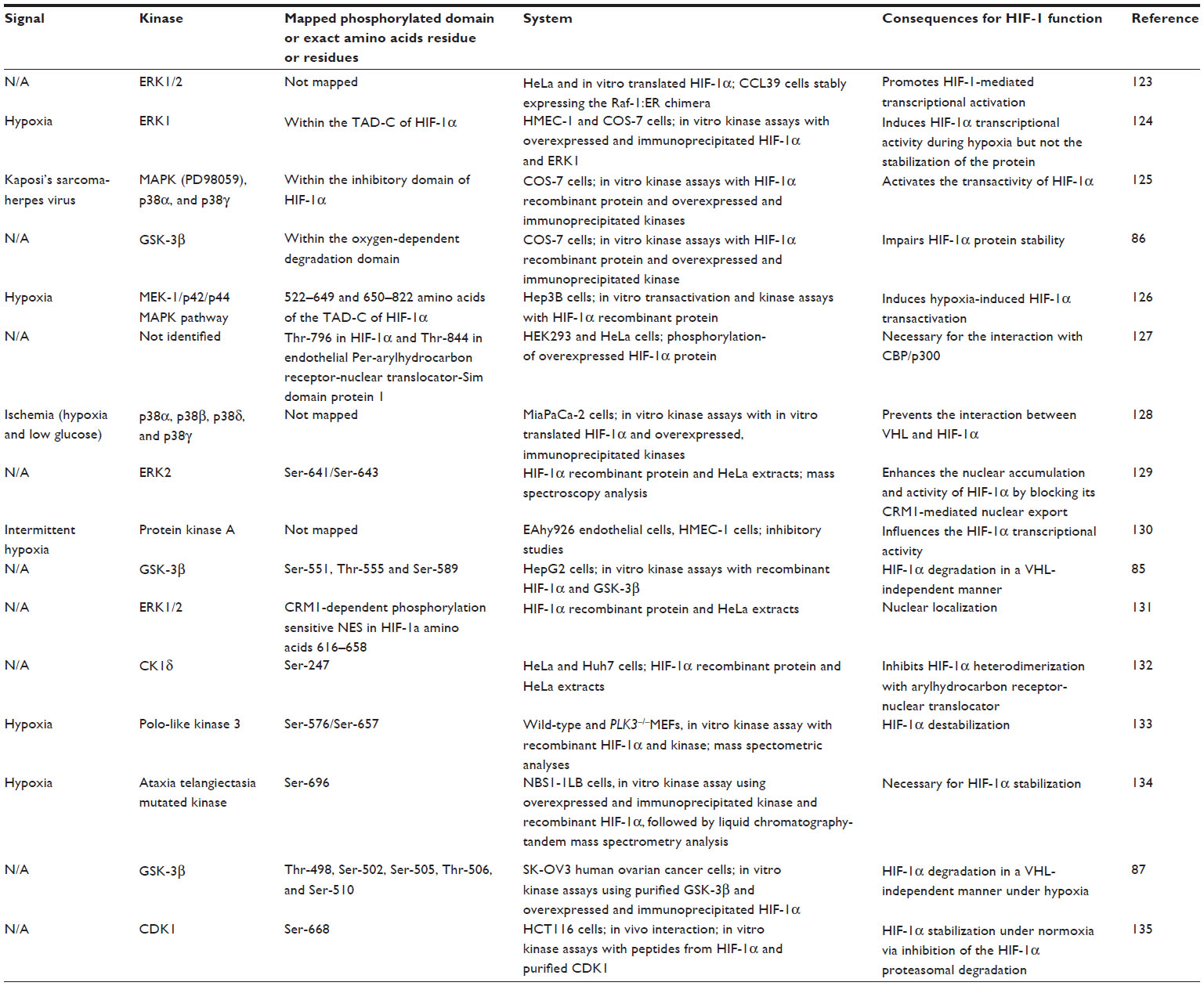 | Table 1 HIF-α as a direct phosphorylation target |
In addition to being activated by a variety of extracellular signals, the PI3K/Akt cascade appeared also to be regulated by hypoxia, thus integrating hypoxia signaling with extracellular signals affecting multiple cellular processes such as apoptosis, metabolism, cell proliferation, and cell growth (for review, see Braccini et al53). The PI3K/Akt pathway is considered to control HIF-1α within the cell via regulation of HIF-1α protein synthesis and stability. However, it appeared that HIF-α proteins are not directly phosphorylated by PKB/Akt but, rather, by a PKB/Akt target. The PKB/Akt targets HDM2,54,55 mammalian target of rapamycin (mTOR),56 and GSK-357 were shown to contribute to changes in HIF-α protein levels; however, only GSK-3 was shown to do this directly, ie, by phosphorylating HIF-α proteins.
GSK-3 and HIF-α regulation
GSK-3 is a serine/threonine kinase that was first identified as a negative regulator of glycogen synthesis; inhibition is achieved through phosphorylation of glycogen synthase.58,59 Since its initial discovery, GSK-3 has been found to be involved in numerous signaling pathways initiated by diverse stimuli and to contribute to the regulation of cell proliferation, stem cell renewal, apoptosis, and development, which are processes often associated with hypoxia. Because of these multiple involvements, dysregulation of GSK-3 has been implicated in the pathogenesis of human diseases, including type 2 diabetes, bipolar disorders, inflammation, Alzheimer’s disease, and cancer (reviewed by Frame and Cohen,60 Grimes and Jope,61 and Woodgett62). Two isoforms, GSK-3α (51 kDa) and GSK-3β (47 kDa), have been identified in mammals. Despite their homology in the catalytic domain (98%), they significantly differ in their N- and C-terminal parts63,64 and do not have entirely overlapping roles in metabolism (reviewed in Force and Woodgett65). Moreover, GSK-3β (GSK-3β−/−) homozygous knockout mice showed an embryonic lethal phenotype around day 16 because of hepatic apoptosis or a cardiac pattern defect,66,67 whereas homozygous GSK-3α (GSK-3α−/−) knockout mice are viable and fertile.68,69 GSK-3 is a target of PKB/Akt, which can phosphorylate both GSK-3 isoforms (serine 21 of GSK-3α and serine 9 of GSK-3β), leading to an inhibition of GSK-3 activity.70 Interestingly, these serine residues can also be phosphorylated by other kinases such as ERK1/2,71 p70 ribosomal S6 kinase 1,72 cAMP (cyclic adenosine monophosphate)-dependent protein kinase A (PKA),73 and protein kinase C (PKC).74 In contrast, an autophosphorylation event leading to phosphorylation of tyrosine 279 in GSK-3α and of tyrosine 216 in GSK-3β increases GSK-3 activity.75,76 Neurons seem to possess a spliced GSK-3β variant called GSK-3β2 that contains a 13 amino acid residue insert within the kinase domain, leading to reduced kinase activity.77,78
Although GSK-3 is mostly known in the insulin field as a regulator of glycogen synthesis, it has been shown that early hypoxia enhanced PI3K/Akt activity and increased HIF-1α protein levels.57 Similarly, hypoxia was capable of inhibiting GSK-3β by phosphorylation in different cell types, such as PC-12 (rat pheochromocytoma cell line) cells,79 HT1080 (human fibrosarcoma cell line) cells,80 and HepG2 (human liver hepatocellular cell line) cells,57 as well as in vivo.81 Although this effect was not observed in other cell types, including some breast cancer cell lines,82 PC-3 prostate cancer cells,83 and 3T3 cells,84 it was considered to have a cell type-specific component. However, the findings that GSK-3 inhibition57 and small interfering RNA-mediated depletion induced HIF-1α, whereas GSK-3β overexpression reduced HIF-1α protein levels,85 suggested that HIF-1α is a direct target of GSK-3β.
Indeed, the ODDD86 and three sites, S-551, T-555, and S-589, located within the ODDD overlapping the N-TAD of HIF-1α were found to be directly phosphorylated by GSK-3β.85 Another study reported five sites, T-498, S-502, S-505, T-506, and S-510, within the N-TAD of HIF-1α as GSK-3β phosphorylation sites.87 The disparity of the different phosphorylation sites is difficult to explain, but the different oxygen concentrations (8% O2 compared with 2% O2) used in these studies may contribute to the differences. It is possible that different oxygen levels may induce variable signaling pathways that have unequal effects on HIF-1α and its ability to act as a substrate for GSK-3β. Another possibility could be the different cell types (HepG2 compared with SK-OV-3 [human ovarian cancer cells]) that were used in the studies. Despite the differences in the phosphorylation sites, both studies demonstrated that the regulation of HIF-1α by GSK-3β is independent of O2, hydroxylation, and recruitment of the VHL-containing E3 ubiquitin ligase. Experiments with VHL-deficient cells showed that GSK-3β-dependent HIF-1α degradation occurred independent of VHL, indicating that the phosphorylation of HIF-1α by GSK-3β target HIF-1α for proteasomal degradation in an oxygen-independent manner.85 This suggested involvement and recruitment of another so-far-unknown E3 ubiquitin ligase to GSK-3β-phosphorylated HIF-1α.
Indeed, two groups demonstrated that the F-box and WD protein Fbw7 (also known as hCdc4 in yeast, hSel10 in Caenorhabditis elegans, or Ago in Drosophila) acted as the substrate-recognition component of a multisubunit E3 ubiquitin ligase, which was crucial for the proteasomal degradation of GSK-3β phosphorylated HIF-1α.85,87 In this E3 ligase, Fbw7 interacts with SKP1 (S-phase kinase-associated protein 1), CUL1 (cullin 1), and RBX1, forming the so-called SCF complex. Similar to VHL, Fbw7 is considered to serve as a tumor suppressor, and three Fbw7 isoforms (Fbw7α, Fbw7β, and Fbw7γ) are known to be produced by alternative splicing. They are found in the nucleoplasm, cytoplasm, and nucleolus, respectively.88 In addition to HIF-1α, Fbw7 was shown to be involved in the degradation of various oncogenic proteins, including cyclin E,89 c-Myc,90,91 c-Jun,92,93 and Notch.94
Several studies have shown that loss of the fbw7 gene is associated with malignant transformation, especially in ovarian cells and T cells,95 in breast cancer cells,96 and later also in human colorectal cancers,97 which leads then to chromosomal instability and some types of malignancy. Furthermore, investigation of more than 1,500 human tumors revealed that approximately 6% of those tumors showed mutations in the Fbw7 coding region. Specifically, cholangiocarcinomas (35%), T-cell acute lymphocytic leukemia (31%), and endometrial (9%), colon (9%), and stomach (6%) cancer98 had the highest mutation rates. Strikingly, nearly half (43%) of these were missense mutations that resulted in amino acid substitutions within the WD40 domain (Arg465 and Arg479), which are shared by all three Fbw7 isoforms, suggesting that all Fbw isoforms might collectively contribute to the tumor-suppressor function.98
With respect to HIF-1α, all three Fbw7 isoforms were able to induce HIF-1α degradation, and the loss of the Fbw7 WD domain abolishes GSK-3-initiated degradation, leading to higher HIF-1α levels, which has been found to be associated with several tumors.99–101 The finding that HIF α subunits can be targeted for degradation by two different E3 substrate recognition proteins indicates that the system is highly dynamic.
Important in this context is that ubiquitinylation of proteins is a reversible posttranslational modification. The removal of ubiquitin is mediated by a family of deubiquitylating enzymes. The human genome encodes nearly 100 deubiquitylating enzymes that are predicted to be active and that oppose the function of around 600 E3 ligases.102,103 Similar to E3s, deubiquitylating enzymes have a central role in cell cycle regulation and DNA damage response and, depending on the context, can act either as a tumor promoter or suppressor (see references in Love et al104). With respect to VHL, two different deubiqiutinating enzymes, VDU1 (USP33) and VDU2 (USP20), were suggested to oppose the VHL-E3 ubiquitin ligase.105,106 Later, it was shown that VDU2 but not VDU1 can interact with HIF-1α.107 Experiments with cycloheximide and hypoxia showed that the half-life of HIF-1α was significantly increased upon overexpression of VDU2, whereas a catalytic inactive VDU2 C154A mutant had no effect. In addition, it was shown that only VDU2, not VDU1, deubiquitinated HIF-1α, resulting in the stabilization of HIF-1α protein107 (Figure 1). Experiments with GSK-3β- and Fbw7-deficient cells revealed that GSK-3β- and Fbw7-dependent HIF-1α degradation can be antagonized by ubiquitin-specific protease 28 (Figure 1).99 These findings suggest that the GSK-3β-dependent degradation of HIF-1α is not limited by the presence of oxygen and is therefore independent of VHL. Together, these results demonstrate that HIF-1α protein stability is regulated in a dynamic manner involving different ubiquitin ligases and deubiquitinases. As such, the hydroxylation- and VHL-dependent ubiquitination and degradation of HIF-1α under normoxia is opposed by the deubiquitinase VDU2. In contrast, the oxygen-independent but phosphorylation-dependent ubiquitination of HIF-1α is counteracted by ubiquitin-specific protease 28-mediated deubiquitination. The latter process allows the integration of the HIF system into the cellular response to various physiologic and pathophysiologic signals independent of the oxygen tension.
Interconnection among the GSK-3, hypoxia/HIF-α, and Wnt/β-catenin pathways
The finding that GSK-3β is involved in the degradation of HIF-1α indicated similarities with the destruction of β-catenin in the canonical Wnt signaling pathway. In this pathway, GSK-3β and β-catenin are part of a “destructive complex” in which binding of GSK-3β and β-catenin promotes phosphorylation of β-catenin by GSK-3β, which requires priming phosphorylation by casein kinase 1, α-isoform. The phosphorylated β-catenin is recognized by the F-box/WD protein β-TrCP and subsequently ubiquitylated and targeted for proteasomal degradation (for review, see Cohen and Frame108 and Metcalfe and Bienz109). When this phosphorylation event is blocked, β-catenin accumulates and binds to the T-cell-specific transcription factor/lymphoid enhancer-binding factor 1 family of transcriptional activators to activate numerous target genes (reviewed by Reya and Clevers110) contributing to embryonic development and adult tissue homeostasis (reviewed by Clevers111). Similarly, GSK-3β-mediated phosphorylation of HIF-1α recruits Fbw7, and thus targets HIF-1α for ubiquitylation and proteasomal degradation.99 Those very similar scenarios imply interference or interconnection of both the Wnt/β-catenin and hypoxia/HIF-1 signaling on the level of GSK-3. Actually, crosstalk between the hypoxia and/or HIF-1α and Wnt/β-catenin pathway was reported and appears to be quite complex because of controversial and likely cell/tissue/differentiation-stage specific data.112–117 Indeed, it was reported that hypoxia and/or HIF-1α can inhibit Wnt/β-catenin signaling. Several mechanisms, such as binding of HIF-1α to hARD1 (human arrest-defective-1 protein) with subsequent interference with acetylation of β-catenin,112 blocking processing and secretion of Wnt proteins,113 down-regulating β-catenin via p53-dependent activation of Siah-1 (seven in absentia homolog 1),118 or direct interaction between HIF-1α and β-catenin119,120 were proposed to contribute to these effects.
In contrast, hypoxia was also shown to activate Wnt/β-catenin signaling in undifferentiated cells and in vivo.115,117 In hypoxic embryonic stem cells, this occurred via HIF-1α-mediated expression of lymphoid enhancer-binding factor 1 and T-cell-specific transcription factor, followed subsequently by increased interaction of β-catenin with lymphoid enhancer-binding factor 1/T-cell-specific transcription factor, and thus activating Wnt/β-catenin targets.115 In addition, hypoxia was able to activate β-catenin via GSK-3β inactivation116,121 in different human cell lines such as HT-29 (human colorectal adenocarcinoma cell line) and HepG2; this activation contributed to an endothelial mesenchymal transition program, leading to significantly increased invasiveness,121 and in renal tubular cells, this process impaired wound healing.116 Together, the reported findings indicate that complex interconnections between hypoxia and/or the HIF-1α and Wnt/β-catenin pathway exist and that cell-, tissue-, and differentiation-specific aspects contribute to their functional consequences.
Conclusion
Hypoxia and HIFs play important roles in many critical aspects of physiological and pathological processes. Most solid cancers contain hypoxic areas, and clinical data demonstrate that overexpression of HIF-1α is associated with an increased risk for patient mortality. In line, downregulation of HIFs interferes with tumor growth, vascularization, invasion, and metastasis, as well as radiation and chemotherapy. Activation of multiple oncogenic pathways including growth factor signaling coupled with enhanced kinase signaling is a common event in tumors, thus making it likely that kinases are involved in the modulation of HIF-α function. Because regulation of HIF-α protein stability is critical for its activation, identification of kinases contributing to HIF-α stability may provide a link explaining normoxic HIF-α stabilization by extracellular stimuli. In light of this, dysregulation of GSK-3β is thought to underlie the pathogenesis of various diseases that are also associated with hypoxia and changed HIF-α levels, such as type 2 diabetes mellitus, Alzheimer’s disease, mood disorders, cardiovascular diseases, and cancer.122 Thus, given that GSK-3 upstream regulation leads to inhibition of GSK-3 and HIF-1α accumulation, this raises the question whether it is an option to target GSK-3 in those diseases and disregard the adverse effects.
Although research from the last decade has demonstrated that a number of kinase pathways contribute to HIF-1α regulation, data for HIF-2α or HIF-3α are limited. Taking into consideration the overlapping, but different, roles of the HIF-α proteins, more knowledge about the phosphorylation-dependent regulation of HIF-2α and HIF-3α is necessary to better understand both already-observed general and different effects.
Acknowledgments
We apologize to all researchers who excellently contributed to the field and whose work was not cited because of space limitations. This work was supported by grants from the Biocenter Oulu, Finnish Academy of Science, and the Sigrid Juselius Foundation.
Disclosure
The authors have no conflicts of interest in this work.
References
Prabhakar NR. Sensing hypoxia: physiology, genetics and epigenetics. J Physiol. 2013 1;591(Pt 9):2245–2257. | |
Semenza GL. Involvement of oxygen-sensing pathways in physiologic and pathologic erythropoiesis. Blood. 2009;114(10):2015–2019. | |
Wenger RH, Stiehl DP, Camenisch G. Integration of oxygen signaling at the consensus HRE. Sci STKE. 2005;2005(306):re12. | |
Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148(3):399–408. | |
Semenza GL. Life with oxygen. Science. 2007;318(5847):62–64. | |
Tian H, McKnight SL, Russell DW. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997;11(1):72–82. | |
Ema M, Taya S, Yokotani N, Sogawa K, Matsuda Y, Fujii-Kuriyama Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1alpha regulates the VEGF expression and is potentially involved in lung and vascular development. Proc Natl Acad Sci U S A. 1997;94(9):4273–4278. | |
Flamme I, Fröhlich T, von Reutern M, Kappel A, Damert A, Risau W. HRF, a putative basic helix-loop-helix-PAS-domain transcription factor is closely related to hypoxia-inducible factor-1 alpha and developmentally expressed in blood vessels. Mech Dev. 1997;63(1):51–60. | |
Hogenesch JB, Chan WK, Jackiw VH, et al. Characterization of a subset of the basic-helix-loop-helix-PAS superfamily that interacts with components of the dioxin signaling pathway. J Biol Chem. 1997;272(13):8581–8593. | |
Wenger RH Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression. FASEB J. 2002;16(10):1151–1162. | |
Scortegagna M, Ding K, Oktay Y, et al. Multiple organ pathology, metabolic abnormalities and impaired homeostasis of reactive oxygen species in Epas1−/− mice. Nat Genet. 2003;35(4):331–340. | |
Scortegagna M, Morris MA, Oktay Y, Bennett M, Garcia JA. The HIF family member EPAS1/HIF-2alpha is required for normal hematopoiesis in mice. Blood. 2003;102(5):1634–1640. | |
Hu CJ, Wang LY, Chodosh LA, Keith B, Simon MC. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol Cell Biol. 2003;23(24):9361–9374. | |
Sowter HM, Raval RR, Moore JW, Ratcliffe PJ, Harris AL. Predominant role of hypoxia-inducible transcription factor (Hif)-1alpha versus Hif-2alpha in regulation of the transcriptional response to hypoxia. Cancer Res. 2003;63(19):6130–6134. | |
Pasanen A, Heikkilä M, Rautavuoma K, Hirsilä M, Kivirikko KI, Myllyharju J. Hypoxia-inducible factor (HIF)-3alpha is subject to extensive alternative splicing in human tissues and cancer cells and is regulated by HIF-1 but not HIF-2. Int J Biochem Cell Biol. 2010;42(7):1189–200. | |
Tanaka T, Wiesener M, Bernhardt W, Eckardt KU, Warnecke C. The human HIF (hypoxia-inducible factor)-3alpha gene is a HIF-1 target gene and may modulate hypoxic gene induction. Biochem J. 2009;424(1):143–151. | |
Makino Y, Cao R, Svensson K, et al. Inhibitory PAS domain protein is a negative regulator of hypoxia-inducible gene expression. Nature. 2001;414(6863):550–554. | |
Augstein A, Poitz DM, Braun-Dullaeus RC, Strasser RH, Schmeisser A. Cell-specific and hypoxia-dependent regulation of human HIF-3α: inhibition of the expression of HIF target genes in vascular cells. Cell Mol Life Sci. 2011;68(15):2627–2642. | |
Heikkilä M, Pasanen A, Kivirikko KI, Myllyharju J. Roles of the human hypoxia-inducible factor (HIF)-3α variants in the hypoxia response. Cell Mol Life Sci. 2011;68(23):3885–3901. | |
Kallio PJ, Okamoto K, O’Brien S, et al. Signal transduction in hypoxic cells: inducible nuclear translocation and recruitment of the CBP/p300 coactivator by the hypoxia-inducible factor-1alpha. EMBO J. 1998;17(22):6573–6586. | |
Luo JC, Shibuya M. A variant of nuclear localization signal of bipartite-type is required for the nuclear translocation of hypoxia inducible factors (1alpha, 2alpha and 3alpha). Oncogene. 2001;20(12):1435–1444. | |
Kietzmann T, Cornesse Y, Brechtel K, Modaressi S, Jungermann K. Perivenous expression of the mRNA of the three hypoxia-inducible factor alpha-subunits, HIF1alpha, HIF2alpha and HIF3alpha, in rat liver. Biochem J. 2001;354(Pt 3):531–537. | |
Hara S, Hamada J, Kobayashi C, Kondo Y, Imura N. Expression and characterization of hypoxia-inducible factor (HIF)-3alpha in human kidney: suppression of HIF-mediated gene expression by HIF-3alpha. Biochem Biophys Res Commun. 2001;287(4):808–813. | |
Jiang BH, Zheng JZ, Leung SW, Roe R, Semenza GL. Transactivation and inhibitory domains of hypoxia-inducible factor 1alpha. Modulation of transcriptional activity by oxygen tension. J Biol Chem. 1997; 272(31):19253–19260. | |
Fedele AO, Whitelaw ML, Peet DJ. Regulation of gene expression by the hypoxia-inducible factors. Mol Interv. 2002;2(4):229–243. | |
Wang GL, Jiang BH, Semenza GL. Effect of altered redox states on expression and DNA-binding activity of hypoxia-inducible factor 1. Biochem Biophys Res Commun. 1995;212(2):550–556. | |
Görlach A, Camenisch G, Kvietikova I, Vogt L, Wenger RH, Gassmann M. Efficient translation of mouse hypoxia-inducible factor-1alpha under normoxic and hypoxic conditions. Biochim Biophys Acta. 2000;1493(1–2):125–134. | |
Gross J, Rheinländer C, Fuchs J, et al. Expression of hypoxia-inducible factor-1 in the cochlea of newborn rats. Hear Res. 2003;183(1–2):73–83. | |
Heidbreder M, Fröhlich F, Jöhren O, Dendorfer A, Qadri F, Dominiak P. Hypoxia rapidly activates HIF-3alpha mRNA expression. FASEB J. 2003;17(11):1541–1543. | |
Pascual O, Denavit-Saubié M, Dumas S, et al. Selective cardiorespiratory and catecholaminergic areas express the hypoxia-inducible factor-1alpha (HIF-1alpha) under in vivo hypoxia in rat brainstem. Eur J Neurosci. 2001;14(12):1981–1991. | |
Kaelin WG Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30(4):393–402. | |
Huang LE, Gu J, Schau M, Bunn HF. Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci U S A. 1998;95(14):7987–7992. | |
Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294(5545):1337–1340. | |
Epstein AC, Gleadle JM, McNeill LA, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107(1):43–54. | |
O’Rourke JF, Tian YM, Ratcliffe PJ, Pugh CW. Oxygen-regulated and transactivating domains in endothelial PAS protein 1: comparison with hypoxia-inducible factor-1alpha. J Biol Chem. 1999;274(4):2060–2071. | |
Ivan M, Kondo K, Yang H, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292(5516):464–468. | |
Jaakkola P, Mole DR, Tian YM, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292(5516):468–472. | |
Baek JH, Mahon PC, Oh J, et al. OS-9 interacts with hypoxia-inducible factor 1alpha and prolyl hydroxylases to promote oxygen-dependent degradation of HIF-1alpha. Mol Cell. 2005;17(4):503–512. | |
Baek JH, Liu YV, McDonald KR, et al. Spermidine/spermine-N1-acetyltransferase 2 is an essential component of the ubiquitin ligase complex that regulates hypoxia-inducible factor 1alpha. J Biol Chem. 2007;282(32):23572–23580. | |
Baek JH, Liu YV, McDonald KR, Wesley JB, Zhang H, Semenza GL. Spermidine/spermine N(1)-acetyltransferase-1 binds to hypoxia-inducible factor-1alpha (HIF-1alpha) and RACK1 and promotes ubiquitination and degradation of HIF-1alpha. J Biol Chem. 2007;282(46):33358–33366. | |
Ema M, Hirota K, Mimura J, et al. Molecular mechanisms of transcription activation by HLF and HIF1alpha in response to hypoxia: their stabilization and redox signal-induced interaction with CBP/p300. EMBO J. 1999;18(7):1905–1914. | |
Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002;16(12):1466–1471. | |
Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science. 2002;295(5556):858–861. | |
Mahon PC, Hirota K, Semenza GL. FIH-1: a novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001;15(20):2675–2686. | |
Dimova EY, Kietzmann T. Hypoxia-inducible factors: post-translational crosstalk of signaling pathways. Methods Mol Biol. 2010;647:215–236. | |
Dimova EY, Michiels C, Kietzmann T. Kinases as upstream regulators of the HIF system: their emerging potential as anti-cancer drug targets. Curr Pharm Des. 2009;15(33):3867–3877. | |
Stiehl DP, Jelkmann W, Wenger RH, Hellwig-Bürgel T. Normoxic induction of the hypoxia-inducible factor 1alpha by insulin and interleukin-1beta involves the phosphatidylinositol 3-kinase pathway. FEBS Lett. 2002;512(1–3):157–162. | |
Sang N, Stiehl DP, Bohensky J, Leshchinsky I, Srinivas V, Caro J. MAPK signaling up-regulates the activity of hypoxia-inducible factors by its effects on p300. J Biol Chem. 2003;278(16):14013–14019. | |
Comerford KM, Cummins EP, Taylor CT. c-Jun NH2-terminal kinase activation contributes to hypoxia-inducible factor 1alpha-dependent P-glycoprotein expression in hypoxia. Cancer Res. 2004;64(24):9057–9061. | |
Datta K, Li J, Bhattacharya R, Gasparian L, Wang E, Mukhopadhyay D. Protein kinase C zeta transactivates hypoxia-inducible factor alpha by promoting its association with p300 in renal cancer. Cancer Res. 2004;64(2):456–462. | |
Mottet D, Ruys SP, Demazy C, Raes M, Michiels C. Role for casein kinase 2 in the regulation of HIF-1 activity. Int J Cancer. 2005;117(5):764–774. | |
Sakamoto T, Weng JS, Hara T, et al. Hypoxia-inducible factor 1 regulation through cross talk between mTOR and MT1-MMP. Mol Cell Biol. 2014;34(1):30–42. | |
Braccini L, Ciraolo E, Martini M, et al. PI3K keeps the balance between metabolism and cancer. Adv Biol Regul. 2012;52(3):389–405. | |
Bárdos JI, Chau NM, Ashcroft M. Growth factor-mediated induction of HDM2 positively regulates hypoxia-inducible factor 1alpha expression. Mol Cell Biol. 2004;24(7):2905–2914. | |
Skinner HD, Zheng JZ, Fang J, Agani F, Jiang BH. Vascular endothelial growth factor transcriptional activation is mediated by hypoxia-inducible factor 1alpha, HDM2, and p70S6K1 in response to phosphatidylinositol 3-kinase/AKT signaling. J Biol Chem. 2004;279(44):45643–45651. | |
Treins C, Giorgetti-Peraldi S, Murdaca J, Semenza GL, Van Obberghen E. Insulin stimulates hypoxia-inducible factor 1 through a phosphatidylinositol 3-kinase/target of rapamycin-dependent signaling pathway. J Biol Chem. 2002;277(31):27975–27981. | |
Mottet D, Dumont V, Deccache Y, et al. Regulation of hypoxia-inducible factor-1alpha protein level during hypoxic conditions by the phosphatidylinositol 3-kinase/Akt/glycogen synthase kinase 3beta pathway in HepG2 cells. J Biol Chem. 2003;278(33):31277–31285. | |
Embi N, Rylatt DB, Cohen P. Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur J Biochem. 1980;107(2):519–527. | |
Woodgett JR, Cohen P. Multisite phosphorylation of glycogen synthase. Molecular basis for the substrate specificity of glycogen synthase kinase-3 and casein kinase-II (glycogen synthase kinase-5). Biochim Biophys Acta. 1984;788(3):339–347. | |
Frame S, Cohen P. GSK-3 takes centre stage more than 20 years after its discovery. Biochem J. 2001;359(Pt 1):1–16. | |
Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol. 2001;65(4):391–426. | |
Woodgett JR. Judging a protein by more than its name: GSK-3. Sci STKE. 2001;2001(100):re12. | |
Woodgett JR. Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 1990;9(8):2431–2438. | |
Woodgett JR. cDNA cloning and properties of glycogen synthase kinase-3. Methods Enzymol. 199200:564–577. | |
Force T, Woodgett JR. Unique and overlapping functions of GSK-3 isoforms in cell differentiation and proliferation and cardiovascular development. J Biol Chem. 2009;284(15):9643–9647. | |
Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature. 2000;406(6791):86–90. | |
Kerkela R, Kockeritz L, Macaulay K, et al. Deletion of GSK-3beta in mice leads to hypertrophic cardiomyopathy secondary to cardiomyoblast hyperproliferation. J Clin Invest. 2008;118(11):3609–3618. | |
MacAulay K, Doble BW, Patel S, et al. Glycogen synthase kinase 3alpha-specific regulation of murine hepatic glycogen metabolism. Cell Metab. 2007;6(4):329–337. | |
Kaidanovich-Beilin O, Woodgett JR. GSK-3: Functional Insights from Cell Biology and Animal Models. Front Mol Neurosci. 2011;4:40. | |
Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378(6559):785–789. | |
Brady MJ, Bourbonais FJ, Saltiel AR. The activation of glycogen synthase by insulin switches from kinase inhibition to phosphatase activation during adipogenesis in 3T3-L1 cells. J Biol Chem. 1998;273(23):14063–14066. | |
Armstrong JL, Bonavaud SM, Toole BJ, Yeaman SJ. Regulation of glycogen synthesis by amino acids in cultured human muscle cells. J Biol Chem. 2001;276(2):952–956. | |
Li M, Wang X, Meintzer MK, Laessig T, Birnbaum MJ, Heidenreich KA. Cyclic AMP promotes neuronal survival by phosphorylation of glycogen synthase kinase 3beta. Mol Cell Biol. 2000;20(24):9356–9363. | |
Ballou LM, Tian PY, Lin HY, Jiang YP, Lin RZ. Dual regulation of glycogen synthase kinase-3beta by the alpha1A-adrenergic receptor. J Biol Chem. 2001;276(44):40910–40916. | |
Wang QM, Fiol CJ, DePaoli-Roach AA, Roach PJ. Glycogen synthase kinase-3 beta is a dual specificity kinase differentially regulated by tyrosine and serine/threonine phosphorylation. J Biol Chem. 1994;269(20):14566–14574. | |
Cole AR, Knebel A, Morrice NA, et al. GSK-3 phosphorylation of the Alzheimer epitope within collapsin response mediator proteins regulates axon elongation in primary neurons. J Biol Chem. 2004;279(48):50176–50180. | |
Mukai F, Ishiguro K, Sano Y, Fujita SC. Alternative splicing isoform of tau protein kinase I/glycogen synthase kinase 3beta. J Neurochem. 2002;81(5):1073–1083. | |
Saeki K, Machida M, Kinoshita Y, Takasawa R, Tanuma S. Glycogen synthase kinase-3β2 has lower phosphorylation activity to tau than glycogen synthase kinase-3β1. Biol Pharm Bull. 2011;34(1):146–149. | |
Beitner-Johnson D, Rust RT, Hsieh TC, Millhorn DE. Hypoxia activates Akt and induces phosphorylation of GSK-3 in PC12 cells. Cell Signal. 2001;13(1):23–27. | |
Chen EY, Mazure NM, Cooper JA, Giaccia AJ. Hypoxia activates a platelet-derived growth factor receptor/phosphatidylinositol 3-kinase/Akt pathway that results in glycogen synthase kinase-3 inactivation. Cancer Res. 2001;61(6):2429–2433. | |
Roh MS, Eom TY, Zmijewska AA, De Sarno P, Roth KA, Jope RS. Hypoxia activates glycogen synthase kinase-3 in mouse brain in vivo: protection by mood stabilizers and imipramine. Biol Psychiatry. 2005;57(3):278–286. | |
Blancher C, Moore JW, Robertson N, Harris AL. Effects of ras and von Hippel-Lindau (VHL) gene mutations on hypoxia-inducible factor (HIF)-1alpha, HIF-2alpha, and vascular endothelial growth factor expression and their regulation by the phosphatidylinositol 3′-kinase/Akt signaling pathway. Cancer Res. 2001;61(19):7349–7355. | |
Zhong H, Chiles K, Feldser D, et al. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res. 2000;60(6):1541–1545. | |
Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol Cell Biol. 2001;21(12):3995–4004. | |
Flügel D, Görlach A, Michiels C, Kietzmann T. Glycogen synthase kinase 3 phosphorylates hypoxia-inducible factor 1alpha and mediates its destabilization in a VHL-independent manner. Mol Cell Biol. 2007;27(9):3253–3265. | |
Sodhi A, Montaner S, Miyazaki H, Gutkind JS. MAPK and Akt act cooperatively but independently on hypoxia inducible factor-1alpha in rasV12 upregulation of VEGF. Biochem Biophys Res Commun. 2001;287(1):292–300. | |
Cassavaugh JM, Hale SA, Wellman TL, Howe AK, Wong C, Lounsbury KM. Negative regulation of HIF-1α by an FBW7-mediated degradation pathway during hypoxia. J Cell Biochem. 2011;112(12):3882–3890. | |
Welcker M, Clurman BE. Fbw7/hCDC4 dimerization regulates its substrate interactions. Cell Div. 2007;2:7. | |
Koepp DM, Schaefer LK, Ye X, et al. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science. 2001;294(5540):173–177. | |
Welcker M, Orian A, Jin J, et al. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc Natl Acad Sci U S A. 2004;101(24):9085–9090. | |
Yada M, Hatakeyama S, Kamura T, et al. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J. 2004;23(10):2116–2125. | |
Nateri AS, Riera-Sans L, Da Costa C, Behrens A. The ubiquitin ligase SCFFbw7 antagonizes apoptotic JNK signaling. Science. 2004;303(5662):1374–1378. | |
Wei W, Jin J, Schlisio S, Harper JW, Kaelin WG Jr. The v-Jun point mutation allows c-Jun to escape GSK-3-dependent recognition and destruction by the Fbw7 ubiquitin ligase. Cancer Cell. 2005;8(1):25–33. | |
Tetzlaff MT, Yu W, Li M, et al. Defective cardiovascular development and elevated cyclin E and Notch proteins in mice lacking the Fbw7 F-box protein. Proc Natl Acad Sci U S A. 2004;101(10):3338–3345. | |
Moberg KH, Bell DW, Wahrer DC, Haber DA, Hariharan IK. Archipelago regulates Cyclin E levels in Drosophila and is mutated in human cancer cell lines. Nature. 2001;413(6853):311–316. | |
Strohmaier H, Spruck CH, Kaiser P, Won KA, Sangfelt O, Reed SI. Human F-box protein hCdc4 targets cyclin E for proteolysis and is mutated in a breast cancer cell line. Nature. 2001;413(6853):316–322. | |
Rajagopalan H, Jallepalli PV, Rago C, et al. Inactivation of hCDC4 can cause chromosomal instability. Nature. 2004;428(6978):77–81. | |
Akhoondi S, Sun D, von der Lehr N, et al. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res. 2007;67(19):9006–9012. | |
Flügel D, Görlach A, Kietzmann T. GSK-3β regulates cell growth, migration, and angiogenesis via Fbw7 and USP28-dependent degradation of HIF-1α. Blood. 2012;119(5):1292–1301. | |
Hirota K, Semenza GL. Regulation of hypoxia-inducible factor 1 by prolyl and asparaginyl hydroxylases. Biochem Biophys Res Commun. 2005;338(1):610–616. | |
Brahimi-Horn MC, Pouysségur J. Oxygen, a source of life and stress. FEBS Lett. 2007;581(19):3582–3591. | |
Scheel H, Hofmann K. Prediction of a common structural scaffold for proteasome lid, COP9-signalosome and eIF3 complexes. BMC Bioinformatics. 2005;6:71. | |
Nijman SM, Luna-Vargas MP, Velds A, et al. A genomic and functional inventory of deubiquitinating enzymes. Cell. 2005;123(5):773–786. | |
Love KR, Catic A, Schlieker C, Ploegh HL. Mechanisms, biology and inhibitors of deubiquitinating enzymes. Nat Chem Biol. 2007;3(11):697–705. | |
Li Z, Na X, Wang D, Schoen SR, Messing EM, Wu G. Ubiquitination of a novel deubiquitinating enzyme requires direct binding to von Hippel-Lindau tumor suppressor protein. J Biol Chem. 2002;277(7):4656–4662. | |
Li Z, Wang D, Na X, Schoen SR, Messing EM, Wu G. Identification of a deubiquitinating enzyme subfamily as substrates of the von Hippel-Lindau tumor suppressor. Biochem Biophys Res Commun. 2002;294(3):700–709. | |
Li Z, Wang D, Messing EM, Wu G. VHL protein-interacting deubiquitinating enzyme 2 deubiquitinates and stabilizes HIF-1alpha. EMBO Rep. 2005;6(4):373–378. | |
Cohen P, Frame S. The renaissance of GSK-3. Nat Rev Mol Cell Biol. 2001;2(10):769–776. | |
Metcalfe C, Bienz M. Inhibition of GSK-3 by Wnt signalling – two contrasting models. J Cell Sci. 2011;124(Pt 21):3537–3544. | |
Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434(7035):843–850. | |
Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127(3):469–480. | |
Lim JH, Chun YS, Park JW. Hypoxia-inducible factor-1alpha obstructs a Wnt signaling pathway by inhibiting the hARD1-mediated activation of beta-catenin. Cancer Res. 2008;68(13):5177–5184. | |
Verras M, Papandreou I, Lim AL, Denko NC. Tumor hypoxia blocks Wnt processing and secretion through the induction of endoplasmic reticulum stress. Mol Cell Biol. 2008;28(23):7212–7224. | |
Liu L, Zhu XD, Wang WQ, et al. Activation of beta-catenin by hypoxia in hepatocellular carcinoma contributes to enhanced metastatic potential and poor prognosis. Clin Cancer Res. 2010;16(10):2740–2750. | |
Mazumdar J, O’Brien WT, Johnson RS, et al. O2 regulates stem cells through Wnt/β-catenin signalling. Nat Cell Biol. 2010;12(10):1007–1013. | |
Peng J, Ramesh G, Sun L, Dong Z. Impaired wound healing in hypoxic renal tubular cells: roles of hypoxia-inducible factor-1 and glycogen synthase kinase 3β/β-catenin signaling. J Pharmacol Exp Ther. 2012;340(1):176–184. | |
Medley TL, Furtado M, Lam NT, et al. Effect of oxygen on cardiac differentiation in mouse iPS cells: role of hypoxia inducible factor-1 and Wnt/beta-catenin signaling. PLoS One. 2013;8(11):e80280. | |
Wang D, Wang Y, Kong T, Fan F, Jiang Y. Hypoxia-induced β-catenin downregulation involves p53-dependent activation of Siah-1. Cancer Sci. 2011;102(7):1322–1328. | |
Kaidi A, Williams AC, Paraskeva C. Interaction between beta-catenin and HIF-1 promotes cellular adaptation to hypoxia. Nat Cell Biol. 2007;9(2):210–217. | |
Zhang Q, Bai X, Chen W, et al. Wnt/β-catenin signaling enhances hypoxia-induced epithelial-mesenchymal transition in hepatocellular carcinoma via crosstalk with hif-1α signaling. Carcinogenesis. 2013;34(5):962–973. | |
Cannito S, Novo E, Compagnone A, et al. Redox mechanisms switch on hypoxia-dependent epithelial-mesenchymal transition in cancer cells. Carcinogenesis. 2008;29(12):2267–2278. | |
Gao C, Liu Y, Li L, Hölscher C. New animal models of Alzheimer’s disease that display insulin desensitization in the brain. Rev Neurosci. 2013;24(6):607–615. | |
Richard DE, Berra E, Gothié E, Roux D, Pouysségur J. p42/p44 mitogen-activated protein kinases phosphorylate hypoxia-inducible factor 1alpha (HIF-1alpha) and enhance the transcriptional activity of HIF-1. J Biol Chem. 1999;274(46):32631–32637. | |
Minet E, Arnould T, Michel G, et al. ERK activation upon hypoxia: involvement in HIF-1 activation. FEBS Lett. 2000;468(1):53–58. | |
Sodhi A, Montaner S, Patel V, et al. The Kaposi’s sarcoma-associated herpes virus G protein-coupled receptor up-regulates vascular endothelial growth factor expression and secretion through mitogen-activated protein kinase and p38 pathways acting on hypoxia-inducible factor 1alpha. Cancer Res. 2000;60(17):4873–4880. | |
Lee E, Yim S, Lee SK, Park H. Two transactivation domains of hypoxia-inducible factor-1alpha regulated by the MEK-1/p42/p44 MAPK pathway. Mol Cells. 2002;14(1):9–15. | |
Gradin K, Takasaki C, Fujii-Kuriyama Y, Sogawa K. The transcriptional activation function of the HIF-like factor requires phosphorylation at a conserved threonine. J Biol Chem. 2002;277(26):23508–23514. | |
Kwon SJ, Song JJ, Lee YJ. Signal pathway of hypoxia-inducible factor-1alpha phosphorylation and its interaction with von Hippel-Lindau tumor suppressor protein during ischemia in MiaPaCa-2 pancreatic cancer cells. Clin Cancer Res. 2005;11(21):7607–7613. | |
Mylonis I, Chachami G, Samiotaki M, et al. Identification of MAPK phosphorylation sites and their role in the localization and activity of hypoxia-inducible factor-1alpha. J Biol Chem. 2006;281(44):33095–33106. | |
Toffoli S, Feron O, Raes M, Michiels C. Intermittent hypoxia changes HIF-1alpha phosphorylation pattern in endothelial cells: unravelling of a new PKA-dependent regulation of HIF-1alpha. Biochim Biophys Acta. 2007;1773(10):1558–1571. | |
Mylonis I, Chachami G, Paraskeva E, Simos G. Atypical CRM1-dependent nuclear export signal mediates regulation of hypoxia-inducible factor-1alpha by MAPK. J Biol Chem. 2008;283(41):27620–27627. | |
Kalousi A, Mylonis I, Politou AS, Chachami G, Paraskeva E, Simos G. Casein kinase 1 regulates human hypoxia-inducible factor HIF-1. J Cell Sci. 2010;123(Pt 17):2976–2986. | |
Xu D, Yao Y, Lu L, Costa M, Dai W. Plk3 functions as an essential component of the hypoxia regulatory pathway by direct phosphorylation of HIF-1alpha. J Biol Chem. 2010;285(50):38944–38950. | |
Cam H, Easton JB, High A, Houghton PJ. mTORC1 signaling under hypoxic conditions is controlled by ATM-dependent phosphorylation of HIF-1α. Mol Cell. 2010;40(4):509–520. | |
Warfel NA, Dolloff NG, Dicker DT, Malysz J, El-Deiry WS. CDK1 stabilizes HIF-1α via direct phosphorylation of Ser668 to promote tumor growth. Cell Cycle. 2013;12(23):3689–3701. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.