Back to Journals » Therapeutics and Clinical Risk Management » Volume 12
Dimethyl fumarate in the management of multiple sclerosis: appropriate patient selection and special considerations
Authors Prosperini L ![]() , Pontecorvo S
, Pontecorvo S
Received 13 November 2015
Accepted for publication 20 January 2016
Published 2 March 2016 Volume 2016:12 Pages 339—350
DOI https://doi.org/10.2147/TCRM.S85099
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Luca Prosperini, Simona Pontecorvo
Department of Neurology and Psychiatry, Sapienza University, Rome, Italy
Abstract: Delayed-release dimethyl fumarate (DMF), also known as gastroresistant DMF, is the most recently approved oral disease-modifying treatment (DMT) for relapsing multiple sclerosis. Two randomized clinical trials (Determination of the Efficacy and Safety of Oral Fumarate in Relapsing–Remitting MS [DEFINE] and Comparator and an Oral Fumarate in Relapsing-Remitting Multiple Sclerosis [CONFIRM]) demonstrated significant efficacy in reducing relapse rate and radiological signs of disease activity, as seen on magnetic resonance imaging. The DEFINE study also indicated a significant effect of DMF on disability worsening, while the low incidence of confirmed disability worsening in the CONFIRM trial rendered an insignificant reduction among the DMF-treated groups when compared to placebo. DMF also demonstrated a good safety profile and acceptable tolerability, since the most common side effects (gastrointestinal events and flushing reactions) are usually transient and mild to moderate in severity. Here, we discuss the place in therapy of DMF for individuals with relapsing multiple sclerosis, providing a tentative therapeutic algorithm to manage newly diagnosed patients and those who do not adequately respond to self-injectable DMTs. Literature data supporting the potential role of DMF as a first-line therapy are presented. The possibility of using DMF as switching treatment or even as an add-on strategy in patients with breakthrough disease despite self-injectable DMTs will also be discussed. Lastly, we argue about the role of DMF as an exit strategy from natalizumab-treated patients who are considered at risk for developing multifocal progressive leukoencephalopathy.
Keywords: multiple sclerosis, dimethyl fumarate, oral drugs, therapeutic algorithm
Introduction
Multiple sclerosis (MS) is a chronic immunomediated inflammatory disease of the central nervous system (CNS) that typically affects young adults.1 The risk of MS is greater in women than in men; female:male ratios are between 1.5:1 and 3.5:1 in most populations, with a trend toward greater values in the most recent studies.2
The etiology of MS is still not completely known, but presumably involves interaction among genetic, environmental, and other factors triggering an aberrant autoimmune attack, resulting in damage to myelin and axons.3 The pathogenesis of MS involves immune attack against CNS antigens mediated through activated CD4+ myelin-reactive T cells, with contribution by B cells.4 Therefore, MS is believed to be an autoimmune disorder, but the antigen specificity of the immune response is still unknown.4 Pathologically, MS is characterized by perivascular infiltrates of mononuclear cells, demyelination, remyelination, oligodendrocyte depletion, astrocytosis, axonal loss, and neuronal degeneration that result in the formation of MS plaques in the brain and spinal cord.5
The clinical course of MS may be considered the expression of two clinical phenomena: relapses of acute neurological symptoms, which end with a partial or complete remission, and progression, which refers to the steady and irreversible worsening of symptoms.6 This analysis brings into the equation the interplay between two biological activities: inflammation and degeneration.7
Historically, four clinical MS phenotypes were defined: relapsing remitting, secondary progressive, primary progressive, and progressive relapsing.8 However, more recently there was a reexamination of MS disease phenotypes that included a consideration of inflammatory activity (based on the clinical relapse rate and imaging findings), disability worsening (as a consequence of relapses), and disease progression.9 Accordingly, “progressive relapsing” was merged with the primary-progressive phenotype.9
Moreover, the therapy landscape in MS over the last few years has dramatically broadened, thus increasing the complexity of treatment decisions.10 Therefore, the new classification has provided a framework not only for communication, prognostication, and clinical trial design but also to guide treatment decision-making in daily clinical setting.9
Currently available disease-modifying treatments for relapsing–remitting multiple sclerosis
At the time of writing, 12 disease-modifying treatments (DMTs) are available for the treatment of relapsing MS (R-MS; Table 1): self-injectable drugs, including four IFNβ formulations and two glatiramer acetate (GA) formulations; an immunosuppressive agent (mitoxantrone); two humanized monoclonal antibodies (natalizumab and alemtuzumab); and three oral compounds (fingolimod, teriflunomide, and dimethyl fumarate [DMF]).11–29
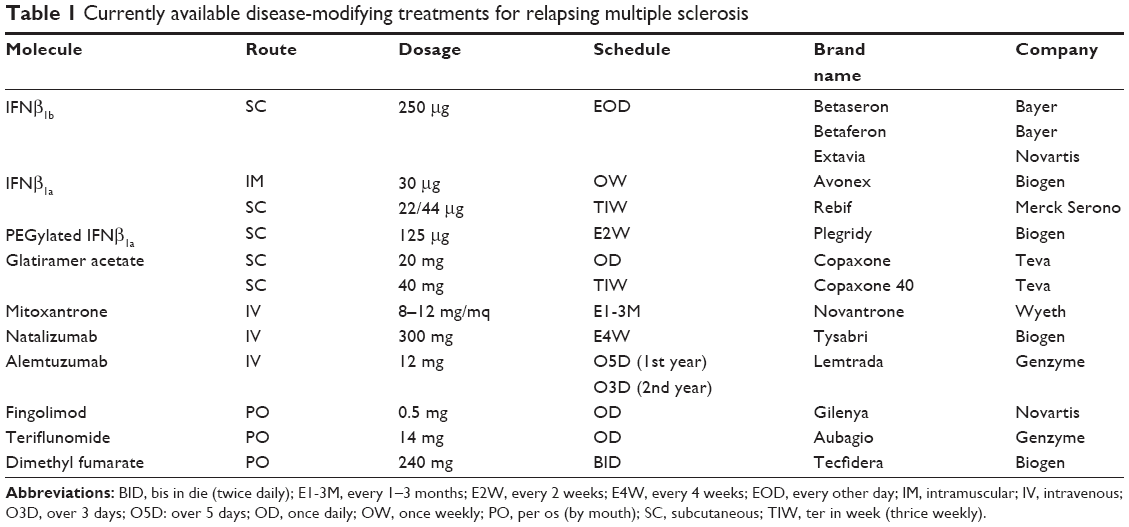 | Table 1 Currently available disease-modifying treatments for relapsing multiple sclerosis |
Historically, self-injectable DMTs have been the mainstay of MS therapy, and are still widely used in patients with R-MS. Based on data from the head-to-head randomized controlled trials (RCTs) INCOMIN and EVIDENCE, high-dose, high-frequency IFNβ formulations are considered more efficacious than low-dose subcutaneous (SC) IFNβ1a, especially in the short term.30,31 However, a Danish RCT and some postmarketing studies did not fully support this observation, providing conflicting results.32–37 According to findings from three head-to-head RCTs (BECOME, REGARD, and BEYOND), the efficacy of GA is comparable to that of IFNβ1a and IFNβ1b,38–40 even though IFNβ appeared better than GA in controlling radiological disease activity, as seen on brain magnetic resonance imaging (MRI).41,42 By contrast, the CombiRx study found GA significantly better than IFNβ in reducing the risk of relapse; however, this RCT was not specifically designed to compare IFNβ and GA.43 More recently, a large multicenter real-world study showed slightly lower relapse incidence among patients treated with GA or SC IFNβ1a relative to intramuscular IFNβ1a and SC IFNβ1b, without any difference in terms of disability worsening.44
Despite their excellent safety profile, IFNβ and GA may be poorly tolerated, due to systemic or injection-related adverse events, leading to discontinuation or lack of adherence in a relevant number of patients.45,46 Moreover, self-injectable DMTs are only partially effective in preventing clinical and MRI activity.47 Mitoxantrone was approved for rapidly worsening R-MS or secondary-progressive MS, and has proven its efficacy in several trials;18,19,48 however, the increased incidence of cardiotoxicity and treatment-related leukemia has discouraged its more widespread use.49,50 According to European Medicines Agency criteria, monoclonal antibodies, such as natalizumab and alemtuzumab, are currently used in adult patients with R-MS who do not adequately respond to a self-injectable DMT or in those presenting rapidly evolving disease activity, defined by clinical or imaging features.51,52
There is evidence that alemtuzumab is superior to SC IFNβ1a in reducing relapse rate, disability worsening, and MRI activity.21–23 Moreover, nonrandomized postmarketing observational studies support the use of natalizumab rather than self-injectable DMTs or fingolimod in nonresponders to a first-line treatment course.53–55 Despite the two drugs’ impressive efficacy in controlling disease activity, and even in reducing disability (in some cases), there are several safety concerns, due to the increased risk of opportunistic infections and autoimmunomediated conditions in patients treated with natalizumab and alemtuzumab.56,57
Orally administered DMTs (fingolimod, teriflunomide, and DMF) represent a significant therapeutic advance, since the oral administration route represents an attractive opportunity for patients and may potentially promote enhanced therapeutic adherence.58 Moreover, data from RCTs has shown that these oral drugs are at least equivalent or even superior to self-injectable DMTs in terms of efficacy.25,29,59 Fingolimod demonstrated higher efficacy than intramuscular IFNβ1a on both clinical and MRI end points; however, there were no between-group differences in terms of disability outcomes.25 Although CONFIRM was not designed as a head-to-head RCT, its post-hoc analysis showed a significantly greater treatment effect of DMF versus GA in reducing relapse rate and MRI activity, but not disability worsening.29 A similar risk of treatment failure (defined as first occurrence of confirmed relapse or permanent treatment discontinuation for any cause) was reported between teriflunomide 14 mg and SC IFNβ1a 44 μg in the TENERE study.59 However, despite these encouraging data, even oral DMTs may have safety and tolerability issues, especially when compared with the good and well-known long-term safety profile of self-injectable platform therapies.60 Table 2 shows the main findings in terms of efficacy, safety, and tolerability regarding oral DMTs currently used in R-MS.
 | Table 2 Main features of the currently approved oral disease-modifying treatments for relapsing multiple sclerosis (only data relative to currently approved dose presented) |
Dimethyl fumarate for relapsing–remitting multiple sclerosis: mechanisms of action
Fumaric acid (FA) and its derivatives have been used for decades as topical and oral long-term treatments for psoriasis, a long-lasting inflammatory skin disease. In 1994, Germany approved an oral compound of FA esters (FAEs) containing DMF and ethyl hydrogen fumarate as mixed salts for psoriasis treatment, but it is also used off-license in many other countries.61 Evidence shows that FAEs provided long-term safety and effectiveness in patients with psoriasis.62,63 Based on prolonged experience, and combined with the positive results obtained in the animal model of MS,64 the prospect of utilizing DMF for autoimmune conditions other than psoriasis was envisioned.65
In 2003, Biogen purchased licensing rights to develop a second-generation DMF for treating psoriasis and MS,66 two conditions sharing an autoimmune T-cell-mediated pathogenetic pathway.67 DMF is a lipophilic ester of FA (a key intermediate in the citric acid cycle used by cells to produce energy) that after oral assumption is rapidly metabolized into its active metabolite monomethyl fumarate (MMF) by intestinal esterases.68,69 The currently used oral delayed-release DMF formulation allows the compound to bypass the stomach and be released at intestinal level. After administration, the blood concentration of MMF peaks at approximately 2–2.5 hours (Tmax), but it is delayed with coingestion of a fat-rich meal. This latter phenomenon underpins the observed lower incidence of metabolism-related side effects of DMF when the drug is taken with high-calorie food.70,71
Preclinical studies have demonstrated that DMF has immunomodulating and antioxidant properties, and similarly to other DMTs, such as IFNβ, GA, and fingolimod, DMF exerts a pleiotropic effect.72 The immunomodulating properties of DMF mainly encompass the induction of a shift from a proinflammatory Th1- to an anti-inflammatory Th2-cytokine pattern.73 Due to its lipophilic nature, the DMF derivative MMF is able to translocate into blood cells, where it interacts with glutathione and promotes glutathione depletion.74,75 The rapid decrease of intracellular glutathione leads to a rebound increase in concentration that in turn suppresses the translocation of nuclear NFκB into the nucleus, thereby preventing the transcription of inflammatory cytokines and adhesion molecules.75
The antioxidant potential of DMF appears to be mediated by two different molecular pathways involving the nuclear factor Nrf276 and the high-affinity receptor for nicotinic acid/niacin and β-hydroxybutyrate HCAR2.77,78 Under normal conditions, Nrf2 is sequestered in the cytosol via Keap1; DMF/MMF irreversibly bind to Keap1, leading to translocation of Nrf2 into the nucleus, thus imitating the physiological oxidative stress-response pathway that activates cell-defense mechanisms.76,79
HCAR2 is a G-protein-coupled membrane receptor; its activation results in reduced cAMP levels, which in turn leads to neutrophil apoptosis. After crossing the blood–brain barrier (BBB), MMF binds to HCAR2 and switches microglia and astrocytes to an anti-inflammatory phenotype, thereby indirectly affecting neuronal survival and function, including restoration of synaptic alterations occurring in experimental MS models.77,78 The potential for a direct effect on CNS-resident cells is supported by the evidence that upon oral administration, DMF is rapidly metabolized to MMF, which crosses the BBB and achieves detectable levels in the CNS.79
This latter pathway is also implicated in the flushing reaction, a well-recognized side effect of DMF due to increased prostaglandin levels; indeed, by binding HCAR2, MMF induces prostanoid-forming enzymes in different epidermal cell types.80 Although the proposed mechanism of action does not include effects on lymphocytes, DMF shifts the immunophenotypes of circulating T cells, leading to a reduction of memory cells and a relative expansion of naïve cells, regardless of the absolute lymphocyte count (ALC).81 In addition, ALC reduction of about a third has been observed in RCTs and in a recent post-marketing experience,28,29 with the most relevant reduction for CD8 and CD19 cells (40% and 48%, respectively).82,83
Dimethyl fumarate for relapsing multiple sclerosis: efficacy data
In 2006, an 18-week open-label prospective study in ten patients with R-MS demonstrated that 720 mg FAEs (240 mg thrice weekly) induced significant reduction in number and volume of contrast-enhancing lesions (CELs) compared with baseline (P<0.05); this effect was sustained over a further 48-week treatment phase with 360 mg FAEs (120 mg thrice weekly) and even after a 4-week washout period.84
A 48-week, Phase IIB, randomized, placebo-controlled, double-blind study of DMF was subsequently conducted on 257 patients with R-MS.85 When compared with placebo, patients treated with DMF 240 mg TID (ter in die [thrice daily]) had a 69% reduction in the mean cumulative number of CELs (P=0.002), 48% reduction in the mean number of new or newly enlarging T2-hyperintense lesions (P<0.001), and 53% reduction in the mean number of new T1-hypointense lesions (P=0.01). The annualized relapse rate (ARR) also decreased by 32% in the active group versus placebo (P<0.05). Subsequent data analysis also showed that the percentage of CELs evolving to T1-hypointense lesions (“black holes”) was 34% lower with DMF treatment versus placebo.86
Following these seminal studies, two pivotal Phase III randomized double-blind clinical trials were conducted in patients with R-MS and at least one relapse in the previous year or at least one CEL within 6 weeks prior to randomization: DEFINE and CONFIRM.28,29
In the 2-year DEFINE study, the ARR decreased by 53% and 48% in the DMF 240 mg BID (bis in die [twice daily]) and 240 mg TID groups, respectively, when compared with placebo (P<0.001). The risk of confirmed disability worsening was also reduced by 38% and 34% in the DMF 240 mg BID and 240 mg TID groups, respectively, when compared with placebo (P≤0.01). Moreover, both DMF doses significantly reduced the numbers of CELs and new or enlarging T2-hyperintense lesions (P<0.001).28 A potential cytoprotective and remyelinating effect of DMF was also suggested by a subset analysis of the DEFINE study in patients who underwent unconventional MRI investigation based on magnetization-transfer ratio.87
The 2-year CONFIRM trial, which included an active-comparator group of patients treated with GA, demonstrated ARR reductions of 44%, 51%, and 29% in the DMF 240 mg BID, DMF 240 mg TID, and GA groups, respectively, compared to the placebo group (P≤0.01).29 Reductions in disability worsening versus placebo were not significant either in the DMF groups and in the GA group, probably due to the low proportion of patients with disability worsening in the placebo group. Compared with placebo, DMF 240 mg BID, DMF 240 mg TID, and GA significantly reduced the numbers of CELs, new or enlarging T2-hyperintense lesions, and new T1-weighted hypointense lesions (P<0.001). Although CONFIRM was not specifically designed as a head-to-head study, a post hoc direct evaluation showed a stronger effect of both DMF 240 mg BID and DMF 240 mg TID in reducing the number of new or enlarging T2-hyperintense lesions when compared to GA (P<0.01). Moreover, DMF 240 mg TID but not DMF 240 mg BID was superior to GA in reducing ARR and new T1-hypointense lesions (P<0.05). No significant differences were found in proportion of relapsing patients with, or time to, disability worsening.29 A more recent post hoc analysis showed that DMF significantly reduced the risk of inflammatory disease activity (defined as such an event as relapse, CELs, or new/enlarging T2-hyperintense lesions) over 2 years compared with GA; the differential treatment effect was seen by 24 weeks.88
A subsequent MRI analysis of CONFIRM also showed that DMF BID and TID produced significant and consistent reductions versus placebo of lesion volumes, but reductions in brain atrophy and magnetization-transfer ratio changes did not reach statistical significance.89
An integrated analysis of the two pivotal Phase III trials showed that at 2 years, DMF 240 mg BID and TID significantly reduced the ARR and the risks of relapses, with 12-week and 24-week confirmed disability worsening in respect to placebo.90 Although the efficacy of DMF is generally consistent across patient subgroups, the most relevant effect of DMF is observed in younger treatment-naïve individuals with low Expanded Disability Status Scale (EDSS) score.90 Moreover, patients treated with both DMF doses improved in some patient-reported outcomes referring to quality of life with respect to placebo.91
ENDORSE (NCT00835770) is an ongoing 8-year extension phase including patients enrolled in both pivotal trials to evaluate the long-term efficacy and safety of DMF.92 An interim analysis of this extension study supports the use of DMF as a long-term treatment option for patients with R-MS, since early treatment with DMF was associated with lower relapse rates and reduced risk of disability worsening over 5 years when compared with patient groups originally randomized to GA or placebo.93
According to a recent Cochrane review, there is moderate-quality evidence to support the use of DMF for reducing both the proportion of relapsing patients and the ARR, while there is no evidence to support the benefit on disability outcome, and the quality of available data is too low to evaluate the benefit on MRI outcomes.94
Dimethyl fumarate for relapsing multiple sclerosis: safety and tolerability data
Overall, the short-term safety profile for DMF in patients with R-MS is highly favorable,28,29 and long-term safety analysis from ENDORSE study sustains a good benefit:risk ratio for this oral compound.95
The commonest adverse events observed in patients receiving DMF include flushing, gastrointestinal (GI) events (eg, diarrhea, nausea, abdominal pain, and vomiting), proteinuria, and pruritus.28,29 Flushing and GI events are usually highest in the first few weeks of treatment and decrease thereafter.71 Flushing is a prostaglandin-mediated phenomenon that may be mitigated by pretreatment with aspirin,96 but the appropriate dose and schedule of administration of aspirin is not yet established for this purpose. While the exact mechanisms leading to GI events are not well known, the observation of transient eosinophilia in the gastric mucosa of patients treated with DMF suggested that the leukotriene-receptor antagonist montelukast may help to control some GI symptoms.97 Moreover, it has been observed that consuming a high-fat and -protein meal just before DMF administration may reduce GI and flushing side effects by effectively delaying intestinal absorption.98 In the daily clinical setting, dose titration with DMF 120 mg BID for 7 days, and then increased to 240 mg BID, is recommended to reduce the incidence of flushing and GI events. In some patients, a slower increase may be tried to enhance tolerability, based on weekly increases by 120 mg, as follows: first week, 120 mg OD; second week, 120 mg BID; third week, 120 mg and 240 mg daily; fourth week, 240 mg BID.99
According to Phase III trials, the incidences of flushing and GI events range from 28% to 35% and 10% to 17%, respectively, and are of mild or moderate severity.28,29 Although the overall incidence of adverse events leading to treatment discontinuation was similar across the study groups (DEFINE, 13% in the placebo group and 16% in each of the DMF groups; CONFIRM, 10% in the placebo group, 12% in each of the DMF groups, and 10% in the GA group), discontinuations due to flushing and GI events occurred more frequently in patients who received DMF than in those randomized to placebo. Discontinuations due to flushing and GI events were 2%–4% and 2%–6%, respectively, in patients treated with DMF versus ≤1% in placebo group.28,29
According to Phase III RCTs, at 1 year, white-cell and lymphocyte counts decrease by approximately 10%–12% and 28%–32%, respectively, in respect to baseline values. The incidences of grade 2 leukopenia (<3×109/L) and grade 3 lymphopenia (<0.5×109/L) were 4%–10% and 4%–7% in the DMF groups, respectively, versus ≤1% in the placebo groups.28,29 A 35% reduction in ALC has also been reported after 3–6 months of DMF treatment in a real-world retrospective study. In this latter, grade 3 (ALC <0.8×109) and grade 2 lymphopenia occurred in 24% and 5% of treated patients, respectively.83
A higher risk of lymphopenia has been noted in adults older than 55 years, in those with lower baseline lymphocyte counts, and in those switching from natalizumab.100 Although incidences of infections and serious infections in RCTs are similar for patients on DMF and placebo, a theoretical increased risk for developing opportunistic John Cunningham virus (JCV)-mediated progressive multifocal leukoencephalopathy (PML) has been postulated in DMF-treated patients with MS.82
To the best of our knowledge, six cases of PML have been reported so far in patients receiving FAEs for psoriasis.101–107 Notably, each of these patients had well-known risk factors for PML, including lymphocytopenia, sarcoidosis, cancer history, and prior efalizumab use.
One recent article assumes that there is no increased incidence of PML in DMF-treated patients with MS, and provides a mathematical model to estimate the risk of PML under immunotherapy assuming a constant incidence rate of three cases per million person-years.108 However, at the time of that publication, only one case of PML had been reported. To date, four cases of PML (one fatal) have occurred in DMF-treated patients with MS who had not been treated before with other DMTs known to be associated with an increased PML risk. All cases occurred in subjects older than 50 years. Three of four patients had prolonged grade 3 lymphopenia prior to PML development. The remaining case had prolonged ALC 0.6×109 (corresponding to a grade 2 lymphopenia) for more than 6 months before PML diagnosis.109
According to the European Medicines Agency, a baseline brain MRI should be available as a reference, and a complete blood count is recommended prior to initiation of DMF and should be repeated every 3 months during treatment for safety monitoring. Grade 3 lymphopenia should be monitored at even shorter intervals. Many clinicians advocate dosage reduction with grade 3 lymphopenia. DMF should be discontinued in cases of persistent grade 3 lymphopenia for 6 or more months, and ALC monitoring should be provided until lymphopenia resolution. When PML is suspected, treatment should be withdrawn immediately and appropriate diagnostic workup performed.109
Increased liver enzymes were transiently observed in the first few months of treatment, and in all cases were not accompanied by concurrent increase of bilirubin levels.28,29 DMF is not recommended for use in pregnant women (pregnancy category C).110 However, gestational exposure to DMF seems to be not associated with increased risk of fetal abnormalities or adverse pregnancy outcomes, as indicated by both preclinical data from animal reproductive toxicology studies and the outcomes of pregnancies occurring during the clinical development program.111
Selection of appropriate drug for individual patients
Before starting any DMT, neurologists must consider the patient’s views, requirements, and potential adherence difficulties. Treatment history (if applicable), psychological aspects potentially affecting treatment adherence, pregnancy desire, and coping strategies (especially in the newly diagnosed) should be carefully evaluated. The oral administration route of DMF is an attractive opportunity for patients, and may potentially provide an enhanced therapeutic adherence compared with self-injectable DMTs.58
Despite the efficacy of DMF 240 mg BID and TID being broadly comparable, DMF is authorized at 240 mg BID as first-line therapy for patients with an established diagnosis of R-MS. This “place in therapy” should allow neurologists to prescribe early over the course of the disease, before irreparable damage has occurred and when the chance to obtain a good therapeutic response is highest. This latter notion is supported by post hoc analyses combining data from DEFINE and CONFIRM showing that early treatment with DMF (defined as within 1 year from MS diagnosis or EDSS score ≤2) resulted in a stronger effect when compared to the whole intention-to-treat combined cohort.112,113 Consistently, subgroup analyses of the two pivotal RCTs showed that the treatment effect of DMF seems to be particularly great in patients who were treatment-naïve and in those with a lower baseline EDSS.114,115 However, DMF may be still effective in patients with suboptimal response to prior treatment with IFNβ, as suggested by another integrated analysis of DEFINE and CONFIRM.90
The possibility of using DMF as an add-on treatment in patients who do not adequately respond to IFNβ or GA was investigated in the Phase II open-label EXPLORE study.116 In this study, 108 patients with R-MS who experienced disease activity (defined as either one or more relapses within 12 months or one or more CELs on brain MRI within 6 weeks prior to study enrollment) while under established treatment with IFNβ or GA for at least 1 year were submitted to receive a 6-month course of DMF 240 mg TID in addition to their prescribed self-injectable DMT. Although the study was designed to test the safety and tolerability of add-on therapy, MRI activity was also investigated. Though DMF was used at a higher dosage (720 mg daily), the safety profile of its combination with IFNβ or GA was consistent with the known safety profile of DMF monotherapy. The exploratory MRI analysis showed a decrease in mean number of CELs and new T2-hyperintense lesions. Despite these promising results on the control of MRI activity, the EXPLORE study suffers from several weaknesses, including a very short follow-up and lack of randomization and a control group.116 Therefore, future efforts should investigate the efficacy and longer-term safety profile of combining DMF with injectable DMTs in cases of suboptimal response to GA or IFNβ.
Use in JCV-positive patients
The possibility of using DMF as an effective exit strategy from natalizumab is under investigation.117 Other than natalizumab exposure, previous immunosuppression and to a greater extent seropositivity to JCV are the main risk factors for developing PML.118 As a result, the majority of patients at high risk of PML are advised to stop treatment.119 Natalizumab discontinuation may induce the clinical and radiological disease reactivation that in some cases may lead to the accumulation of fixed disability.120 Currently, there are no evidence-based data or established guidelines driving specific treatment switching to control following natalizumab disease reactivation.
A recently published case report suggests that DMF may not be able to halt clinical and radiological post-natalizumab disease reactivation in patients with highly active MS.121 However, preliminary results on 530 patients suggested that initiation of DMF within 90 days of washout after natalizumab withdrawal may represent a valid treatment option with only minor safety concerns.117 In fact, the increased risk for PML in natalizumab is explained by the specific mode of action of this drug, which decreases immunosurveillance of the brain through its effect on the adhesion molecule located at the BBB level. By contrast, the mechanism of action of DMF does not seem to explain an increased risk for PML, unless severe lymphocyte depletion occurs.100 Another proposed mechanism by which DMF may potentially induce PML encompasses its capacity to reduce binding of peripheral blood mononuclear cells to vascular cell-adhesion molecules.122 Potentially, this may not only preclude effective neuroimmunosurveillance for JCV infected cells but also may induce the release of premature B cells from marrow stores, which has been suggested to be implicated in viral expression and transformation.123
However, screening for anti-JCV antibody status is not recommended for DMF treatment. A possible diagnosis of preclinical PML should be ruled out in patients who discontinue natalizumab before starting DMF. In light of the increased risk of DMF-induced lymphopenia in patients previously treated with natalizumab,100 stringent monitoring of differential blood count and prompt DMF discontinuation in the event of a 6-month sustained grade 3 lymphopenia are recommended to reduce the risk of PML.109 Special attention should also be paid to older patients changing treatment from natalizumab to DMF.100
What if treatment fails?
As required for all other first-line DMTs, once the therapy is started, the response to DMF should be carefully assessed to early identify those patients who experience breakthrough disease.47 If relapses and MRI activity occur in the first few months from DMF beginning, the possibility of a delayed treatment effect should be considered and therapy with DMF continued. This suggestion comes from findings from a Phase IIB RCT showing that the effect of DMF on the control of BBB breakdown was delayed up to 8–12 weeks from treatment start (when compared to placebo).85 In cases of disease activity 6–12 months after treatment start, a prompt switch to an alternative and more effective drug should be considered to avoid further relapses and the accumulation of disability over time. In this scenario, we recommend not trying all available first-line DMTs, but an escalation to second-line DMTs, such as natalizumab and alemtuzumab, is warranted.53,55
Based on a recent systematic review with mixed-treatment comparisons, DMF seems to have no significant difference in terms of efficacy (ARR) when compared to fingolimod, and even to be superior to teriflunomide.124 However, several methodological concerns have been raised for carrying out indirect comparisons of treatment arms across RCTs as done by Hutchinson et al,124 and thus further studies based on more advanced statistical approaches (eg, network meta-analysis) are warranted to better compare the efficacy and safety of different DMTs.125 In the absence of head-to-head comparison RCTs, the best way to indirectly assess the benefit of DMTs across different independent studies is absolute risk reduction, which is very similar for DMF, fingolimod, and teriflunomide, ranging from 17% to 22%.
Other aspects should also be taken into consideration. Despite its supposed inferiority in reducing the ARR, teriflunomide is the only oral agent that met the disability-worsening end point in two independent trials.26,27 In addition, fingolimod is the only oral agent that demonstrated a significant reduction in brain-volume loss compared to placebo,24 while DMF and teriflunomide did not meet the brain-atrophy end point.26,89
With regard to tolerability, discontinuation due to side effects has been reported to be higher with DMF than fingolimod.126 Therefore, we might suggest switching from DMF to fingolimod (or teriflunomide) predominantly in cases of intolerable GI events or other side effects. However, teriflunomide may be associated with teratogenicity, thus contraindicating its use in potentially childbearing women.110
Conclusion
A treatment algorithm based on the efficacy and safety profile of DMF is proposed in Figure 1. For newly-diagnosed patients with R-MS, we suggest considering first-line treatment with DMF over self-injectable therapies, due to its demonstrated efficacy, simplicity of treatment initiation, and administration route. Phase III RCTs have demonstrated its success as an efficacious oral DMT with a favorable tolerability and safety profile.28,29 The early side effects of DMF, such as GI and flushing reactions, tend to decrease with time on therapy and are generally self-limited.71
 | Figure 1 Proposed treatment algorithm for treating R-MS with DMF. |
Patients experiencing breakthrough disease with self-injectable DMTs may be favorably switched to DMF,124 but in cases of relapse with residual disability might be recommended to escalate to high-efficacy monoclonal antibodies (natalizumab, alemtuzumab).21–23,53–55 This latter suggestion also comes from subgroup analysis of RCTs showing that the treatment effect of oral drugs seems to be particularly great in patients with a lower EDSS.114,115 An escalation strategy, rather than a switch to other oral drugs (fingolimod or teriflunomide), is even suggested in patients who have failed DMF treatment,124 while there are still no recommendations or suggestions to manage patients with subclinical disease activity while on DMF.
Based on its putative antioxidative cytoprotective effect, DMF may also be considered as a therapeutic option for progressive forms of MS.127 However, a Phase III RCT aimed at investigating whether DMF (compared with placebo) slows disability progression not related to relapses in secondary progressive MS has recently been terminated by sponsor decision.128
In conclusion, DMF is a promising candidate for newly diagnosed treatment-naïve patients with R-MS, and a possible switching option in some selected cases (especially in cases of poor tolerability or safety concerns with other DMTs).
Disclosure
LP and SP have received consulting and/or lecture fees and travel grants from Biogen, Genzyme, Novartis, and Teva. The authors report no other conflicts of interest in this work.
References
Compston A, Coles A. Multiple sclerosis. Lancet. 2002;359(9313):1221–1231. | ||
Trojano M, Lucchese G, Graziano G, et al. Geographical variations in sex ratio trends over time in multiple sclerosis. PLoS One. 2012;7(10):e48078. | ||
Brück W, Stadelmann C. The spectrum of multiple sclerosis: new lessons from pathology. Curr Opin Neurol. 2005;18(3):221–224. | ||
Garg N, Smith TW. An update on immunopathogenesis, diagnosis, and treatment of multiple sclerosis. Brain Behav. 2015;5(9):e00362. | ||
Kutzelnigg A, Lassmann H. Pathology of multiple sclerosis and related inflammatory demyelinating diseases. Handb Clin Neurol. 2014;122: 15–58. | ||
Tremlett H, Zhao Y, Rieckmann P, Hutchinson M. New perspectives in the natural history of multiple sclerosis. Neurology. 2010;74(24):2004–2015. | ||
Confavreux C, Vukusic S. Natural history of multiple sclerosis: a unifying concept. Brain. 2006;129(Pt 3):606–616. | ||
Lublin FD, Reingold SC. Defining the clinical course of multiple sclerosis: results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology. 1996;46(4):907–911. | ||
Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83(3):278–286. | ||
Ransohoff RM, Hafler DA, Lucchinetti CF. Multiple sclerosis – a quiet revolution. Nat Rev Neurol. 2015;11(3):134–142. | ||
[No authors listed]. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. I. Clinical results of a multicenter, randomized, double-blind, placebo-controlled trial. The IFNB Multiple Sclerosis Study Group. Neurology. 1993;43(4):655–661. | ||
Paty DW, Li DK. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. II. MRI analysis results of a multicenter, randomized, double-blind, placebo-controlled trial. UBC MS/MRI Study Group and the IFNB Multiple Sclerosis Study Group. Neurology. 1993;43(4):662–667. | ||
Jacobs LD, Cookfair DL, Rudick RA, et al. Intramuscular interferon beta-1a for disease progression in relapsing multiple sclerosis. The Multiple Sclerosis Collaborative Research Group (MSCRG). Ann Neurol. 1996;39(3):285–294. | ||
Rudick RA, Goodkin DE, Jacobs LD, et al. Impact of interferon beta-1a on neurologic disability in relapsing multiple sclerosis. The Multiple Sclerosis Collaborative Research Group (MSCRG). Neurology. 1997;49(2):358–363. | ||
[No authors listed]. Randomised double-blind placebo-controlled study of interferon beta-1a in relapsing/remitting multiple sclerosis. PRISMS (Prevention of Relapses and Disability by Interferon beta-1a Subcutaneously in Multiple Sclerosis) Study Group. Lancet. 1998;352(9139):1498–1504. | ||
Li DK, Paty DW. Magnetic resonance imaging results of the PRISMS trial: a randomized, double-blind, placebo-controlled study of interferon-beta1a in relapsing-remitting multiple sclerosis. Prevention of relapses and disability by interferon-beta1a subcutaneously in multiple sclerosis. Ann Neurol. 1999;46(2):197–206. | ||
Johnson KP, Brooks BR, Cohen JA, et al. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind placebo-controlled trial. The Copolymer 1 Multiple Sclerosis Study Group. Neurology. 1995;45(7):1268–1276. | ||
Millefiorini E, Gasperini C, Pozzilli C, et al. Randomized placebo-controlled trial of mitoxantrone in relapsing-remitting multiple sclerosis: 24-month clinical and MRI outcome. J Neurol. 1997;244(3): 153–159. | ||
Edan G, Miller D, Clanet M, et al. Therapeutic effect of mitoxantrone combined with methylprednisolone in multiple sclerosis: a randomised multicentre study of active disease using MRI and clinical criteria. J Neurol Neurosurg Psychiatry. 1997;62(2):112–118. | ||
Polman CH, O’Connor PW, Havrdova E, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;354(9):899–910. | ||
CAMMS223 Trial Investigators, Coles AJ, Compston DA, et al. Alemtuzumab vs. interferon beta-1a in early multiple sclerosis. N Engl J Med. 2008;359(17):1786–1801. | ||
Cohen JA, Coles AJ, Arnold DL, et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet. 2012;380(9856):1819–1828. | ||
Coles AJ, Twyman CL, Arnold DL, et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. Lancet. 2012;380(9856): 1829–1839. | ||
Kappos L, Radue EW, O’Connor P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362(5):387–401. | ||
Cohen JA, Barkhof F, Comi G, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362(5):402–415. | ||
O’Connor P, Wolinsky JS, Confavreux C, et al. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med. 2011;365(14):1293–1303. | ||
Confavreux C, O’Connor P, Comi G, et al. Oral teriflunomide for patients with relapsing multiple sclerosis (TOWER): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 2014;13(3):247–256. | ||
Gold R, Kappos L, Arnold DL, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. 2012;367(12):1098–1107. | ||
Fox RJ, Miller DH, Phillips JT, et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med. 2012;367(12):1087–1097. | ||
Durelli L, Verdun E, Barbero P, et al. Every-other-day interferon beta-1b versus once-weekly interferon beta-1a for multiple sclerosis: results of a 2-year prospective randomised multicentre study (INCOMIN). Lancet. 2002;359(9316):1453–1460. | ||
Panitch H, Goodin DS, Francis G, et al. Randomized, comparative study of interferon beta-1a treatment regimens in MS: the EVIDENCE trial. Neurology. 2002;59(10):1496–1506. | ||
Koch-Henriksen N, Sørensen PS, Christensen T, et al. A randomized study of two interferon-beta treatments in relapsing-remitting multiple sclerosis. Neurology. 2006;66(7):1056–1060. | ||
Milanese C, La Mantia L, Palumbo R, et al. A post-marketing study on interferon beta 1b and 1a treatment in relapsing-remitting multiple sclerosis: different response in drop-outs and treated patients. J Neurol Neurosurg Psychiatry. 2003;74(12):1689–1692. | ||
Trojano M, Liguori M, Paolicelli D, et al. Interferon beta in relapsing-remitting multiple sclerosis: an independent postmarketing study in southern Italy. Mult Scler. 2003;9(5):451–457. | ||
Río J, Tintoré M, Nos C, Téllez N, Galán I, Montalban X. Interferon beta in relapsing-remitting multiple sclerosis: an eight years experience in a specialist multiple sclerosis centre. J Neurol. 2005;252(7):795–800. | ||
Patti F, Pappalardo A, Florio C, et al. Effects of interferon beta-1a and -1b over time:6-year results of an observational head-to-head study. Acta Neurol Scand. 2006;113(4):241–247. | ||
Limmroth V, Malessa R, Zettl UK, et al. Quality Assessment in Multiple Sclerosis Therapy (QUASIMS): a comparison of interferon beta therapies for relapsing-remitting multiple sclerosis. J Neurol. 2007;254(1):67–77. | ||
Cadavid D, Wolansky LJ, Skurnick J, et al. Efficacy of treatment of MS with IFNβ-1b or glatiramer acetate by monthly brain MRI in the BECOME study. Neurology. 2009;72(23):1976–1983. | ||
Mikol DD, Barkhof F, Chang P, et al. Comparison of subcutaneous interferon beta-1a with glatiramer acetate in patients with relapsing multiple sclerosis (the REbif vs Glatiramer Acetate in Relapsing MS Disease [REGARD] study): a multicentre, randomised, parallel, open-label trial. Lancet Neurol. 2008;7(10):903–914. | ||
O’Connor P, Filippi M, Arnason B, et al. 250 μg or 500 μg Interferon beta-1b versus 20 mg glatiramer acetate in relapsing-remitting multiple sclerosis: a prospective, randomised, multicentre study. Lancet Neurol. 2009;8(10):889–897. | ||
Cadavid D, Cheriyan J, Skurnick J, et al. New acute and chronic black holes in patients with multiple sclerosis randomised to interferon beta-1b or glatiramer acetate. J Neurol Neurosurg Psychiatry. 2009;80(12):1337–1343. | ||
Filippi M, Rocca MA, Camesasca F, et al. Interferon β-1b and glatiramer acetate effects on permanent black hole evolution. Neurology. 2011;76(14):1222–1228. | ||
Lublin FD, Cofield SS, Cutter GR, et al. Randomized study combining interferon and glatiramer acetate in multiple sclerosis. Ann Neurol. 2013;73(3):327–340. | ||
Kalincik T, Jokubaitis V, Izquierdo G, et al. Comparative effectiveness of glatiramer acetate and interferon beta formulations in relapsing-remitting multiple sclerosis. Mult Scler. 2015;21(9):1159–1171. | ||
Clerico M, Barbero P, Contessa G, Ferrero C, Durelli L. Adherence to interferon-beta treatment and results of therapy switching. J Neurol Sci. 2007;259(1–2):104–108. | ||
Balak DM, Hengstman GJ, Çakmak A, Thio HB. Cutaneous adverse events associated with disease-modifying treatment in multiple sclerosis: a systematic review. Mult Scler. 2012;18(12):1705–1717. | ||
Rudick RA, Polman CH. Current approaches to the identification and management of breakthrough disease in patients with multiple sclerosis. Lancet Neurol. 2009;8(6):545–559. | ||
Hartung HP, Gonsette R, König N, et al. Mitoxantrone in progressive multiple sclerosis: a placebo-controlled, double-blind, randomised, multicentre trial. Lancet. 2002;360(9350):2018–2025. | ||
Martinelli V, Cocco E, Capra R, et al. Acute myeloid leukemia in Italian patients with multiple sclerosis treated with mitoxantrone. Neurology. 2011;77(21):1887–1895. | ||
Mulroy E, Joyce E, Scott J, et al. Long-term risk of leukaemia or cardiomyopathy after mitoxantrone therapy for multiple sclerosis. Eur Neurol. 2012;67(1):45–47. | ||
European Medicines Agency. Tysabri 300 mg [summary of product characteristics]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000603/WC500044686.pdf. Accessed January 26, 2016. | ||
European Medicines Agency. Lemtrada 12 mg concentrate for solution for infusion [summary of product characteristics]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/003718/WC500150521.pdf. Accessed January 26, 2016. | ||
Prosperini L, Giannì C, Leonardi L, et al. Escalation to natalizumab or switching among immunomodulators in relapsing multiple sclerosis. Mult Scler. 2012;18(1):64–71. | ||
Kalincik T, Horakova D, Spelman T, et al. Switch to natalizumab versus fingolimod in active relapsing-remitting multiple sclerosis. Ann Neurol. 2015;77(3):425–435. | ||
Spelman T, Kalincik T, Zhang A, et al. Comparative efficacy of switching to natalizumab in active multiple sclerosis. Ann Clin Transl Neurol. 2015;2(4):373–387. | ||
Berger JR, Houff S. Opportunistic infections and other risks with newer multiple sclerosis therapies. Ann Neurol. 2009;65(4):367–377. | ||
Williamson EM, Berger JR. Infection risk in patients on multiple sclerosis therapeutics. CNS Drugs. 2015;29(3):229–244. | ||
Utz KS, Hoog J, Wentrup A, et al. Patient preferences for disease-modifying drugs in multiple sclerosis therapy: a choice-based conjoint analysis. Ther Adv Neurol Disord. 2014;7(6):263–275. | ||
Vermersch P, Czlonkowska A, Grimaldi LM, et al. Teriflunomide versus subcutaneous interferon beta-1a in patients with relapsing multiple sclerosis: a randomised, controlled phase 3 trial. Mult Scler. 2014;20(6):705–716. | ||
Rommer PS, Zettl UK, Kieseier B, et al. Requirement for safety monitoring for approved multiple sclerosis therapies: an overview. Clin Exp Immunol. 2014;175(3):397–407. | ||
Mrowietz U, Christophers E, Altmeyer P. Treatment of severe psoriasis with fumaric acid esters: scientific background and guidelines for therapeutic use. The German Fumaric Acid Ester Consensus Conference. Br J Dermatol. 1999;141(3):424–429. | ||
Hoefnagel JJ, Thio HB, Willemze R, Bouwes Bavinck JN. Long-term safety aspects of systemic therapy with fumaric acid esters in severe psoriasis. Br J Dermatol. 2003;149(2):363–369. | ||
Reich K, Thaci D, Mrowietz U, Kamps A, Neureither M, Luger T. Efficacy and safety of fumaric acid esters in the long-term treatment of psoriasis – a retrospective study (FUTURE). J Dtsch Dermatol Ges. 2009;7(7):603–611. | ||
Schilling S, Goelz S, Linker R, Luehder F, Gold R. Fumaric acid esters are effective in chronic experimental autoimmune encephalomyelitis and suppress macrophage infiltration. Clin Exp Immunol. 2006;145(1):101–107. | ||
Moharregh-Khiabani D, Linker RA, Gold R, Stangel M. Fumaric acid and its esters: an emerging treatment for multiple sclerosis. Curr Neuropharmacol. 2009;7(1):60–64. | ||
Wakkee M, Thio HB. Drug evaluation: BG-12, an immunomodulatory dimethylfumarate. Curr Opin Investig Drugs. 2007;8(11):955–962. | ||
Kwok T, Loo WJ, Guenther L. Psoriasis and multiple sclerosis: is there a link? J Cutan Med Surg. 2010;14(4):151–155. | ||
Litjens NH, Burggraaf J, van Strijen E, et al. Pharmacokinetics of oral fumarates in healthy subjects. Br J Clin Pharmacol. 2004;58(4):429–432. | ||
Litjens NH, van Strijen E, van Gulpen C, et al. In vitro pharmacokinetics of anti-psoriatic fumaric acid esters. BMC Pharmacol. 2004;4:22. | ||
Dubey D, Kieseier BC, Hartung HP, et al. Dimethyl fumarate in relapsing-remitting multiple sclerosis: rationale, mechanisms of action, pharmacokinetics, efficacy and safety. Expert Rev Neurother. 2015;15(4):339–346. | ||
Phillips JT, Selmaj K, Gold R, et al. Clinical significance of gastrointestinal and flushing events in patients with multiple sclerosis treated with delayed-release dimethyl fumarate. Int J MS Care. 2015;17(5):236–243. | ||
Albrecht P, Bouchachia I, Goebels N, et al. Effects of dimethyl fumarate on neuroprotection and immunomodulation. J Neuroinflammation. 2012;9:163. | ||
Zoghi S, Amirghofran Z, Nikseresht A, et al. Cytokine secretion pattern in treatment of lymphocytes of multiple sclerosis patients with fumaric acid esters. Immunol Invest. 2011;40(6):581–596. | ||
Werdenberg D, Joshi R, Wolffram S, Merkle HP, Langguth P. Presystemic metabolism and intestinal absorption of antipsoriatic fumaric acid esters. Biopharm Drug Dispos. 2003;24(6):259–273. | ||
Lin SX, Lisi L, Dello Russo C, et al. The anti-inflammatory effects of dimethyl fumarate in astrocytes involve glutathione and haem oxygenase-1. ASN Neuro. 2011;3(2):e00055. | ||
Scannevin RH, Chollate S, Jung M, et al. Fumarates promote cytoprotection of central nervous system cells against oxidative stress via the nuclear factor (erythroid-derived 2)-like 2 pathway. J Pharmacol Exp Ther. 2012;341(1):274–284. | ||
Chen H, Assmann JC, Krenz A, et al. Hydroxycarboxylic acid receptor 2 mediates dimethyl fumarate’s protective effect in EAE. J Clin Invest. 2014;124(5):2188–2192. | ||
Parodi B, Rossi S, Morando S, et al. Fumarates modulate microglia activation through a novel HCAR2 signaling pathway and rescue synaptic dysregulation in inflamed CNS. Acta Neuropathol. 2015;130(2):279–295. | ||
Linker RA, Lee DH, Ryan S, et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain. 2011;134(Pt 3):678–692. | ||
Hanson J, Gille A, Zwykiel S, et al. Nicotinic acid- and monomethyl fumarate-induced flushing involves GPR109A expressed by keratinocytes and COX-2-dependent prostanoid formation in mice. J Clin Invest. 2010;120(8):2910–2919. | ||
Longbrake EE, Ramsbottom MJ, Cantoni C, Ghezzi L, Cross AH, Piccio L. Dimethyl fumarate selectively reduces memory T cells in multiple sclerosis patients. Mult Scler. Epub 2015 Oct 12. | ||
Spencer CM, Crabtree-Hartman EC, Lehmann-Horn K, Cree BA, Zamvil SS. Reduction of CD8(+) T lymphocytes in multiple sclerosis patients treated with dimethyl fumarate. Neurol Neuroimmunol Neuroinflamm. 2015;2(3):e76. | ||
Berkovich R, Weiner LP. Effects of dimethyl fumarate on lymphocyte subsets. Mult Scler Relat Disord. 2015;4(4):339–341. | ||
Schimrigk S, Brune N, Hellwig K, et al. Oral fumaric acid esters for the treatment of active multiple sclerosis: an open-label, baseline-controlled pilot study. Eur J Neurol. 2006;13(6):604–610. | ||
Kappos L, Gold R, Miller DH, et al. Efficacy and safety of oral fumarate in patients with relapsing-remitting multiple sclerosis: a multicentre, randomised, double-blind, placebo-controlled phase IIB study. Lancet. 2008;372(9648):1463–1472. | ||
MacManus DG, Miller DH, Kappos L, et al. BG-12 reduces evolution of new enhancing lesions to T1-hypointense lesions in patients with multiple sclerosis. J Neurol. 2011;258(3):449–456. | ||
Arnold DL, Gold R, Kappos L, et al. Magnetization transfer ratio in the delayed-release dimethyl fumarate DEFINE study. J Neurol. 2014;261(12):2429–2437. | ||
Kremenchutzky M, Fox RJ, Phillips JT, et al. Efficacy of delayed-release dimethyl fumarate vs glatiramer acetate on a novel composite outcome measure of inflammatory disease activity: post-hoc analysis of the CONFIRM study. 2015. Available from: http://onlinelibrary.ectrims-congress.eu/ectrims/2015/31st/116024/undefined. Accessed January 26, 2016. | ||
Miller DH, Fox RJ, Phillips JT, et al. Effects of delayed-release dimethyl fumarate on MRI measures in the phase 3 CONFIRM study. Neurology. 2015;84(11):1145–1152. | ||
Viglietta V, Miller D, Bar-Or A, et al. Efficacy of delayed-release dimethyl fumarate in relapsing-remitting multiple sclerosis: integrated analysis of the phase 3 trials. Ann Clin Transl Neurol. 2015;2(2):103–118. | ||
Kita M, Fox RJ, Phillips JT, et al. Effects of BG-12 (dimethyl fumarate) on health-related quality of life in patients with relapsing-remitting multiple sclerosis: findings from the CONFIRM study. Mult Scler. 2014;20(2):253–257. | ||
Biogen. Monotherapy safety and efficacy extension study in multiple sclerosis (MS) (ENDORSE). Available from: https://clinicaltrials.gov/ct2/show/NCT00835770?term=bg-12&rank=6. NLM identifier: NCT00835770. Accessed January 26, 2016. | ||
Hutchinson M, Gold R, Fox RJ, et al. Six-year follow-up of delayed-release dimethyl fumarate in RRMS: integrated clinical efficacy data from the DEFINE, CONFIRM, and ENDORSE studies. 2015. Available from: http://onlinelibrary.ectrims-congress.eu/ectrims/2015/31st/115447/michael.hutchinson.six-year.follow-up.of.delayed-release.dimethyl.fumarate.in.html. Accessed January 26, 2016. | ||
Xu Z, Zhang F, Sun F, Gu K, Dong S, He D. Dimethyl fumarate for multiple sclerosis. Cochrane Database Syst Rev. 2015;4:CD011076. | ||
Pozzilli C. Long-term follow-up of the safety of delayed-release dimethyl fumarate in RRMS: interim results from the ENDORSE extension study. 2014. Available from: http://onlinelibrary.ectrims-congress.eu/ectrims/2014/ACTRIMS-ECTRIMS2014/63773/carlo.pozzilli.long-term.follow-up.of.the.safety.of.delayed-release.dimethyl.html. Accessed January 26, 2016. | ||
O’Gorman J, Russell HK, Li J, Phillips G, Kurukulasuriya NC, Viglietta V. Effect of aspirin pretreatment or slow dose titration on flushing and gastrointestinal events in healthy volunteers receiving delayed-release dimethyl fumarate. Clin Ther. 2015;37(7):1402–1419.e5. | ||
Tornatore C, Amjad F. Attenuation of dimethyl fumarate-related gastrointestinal symptoms with montelukast. In:(P7.251). Neurology. 2014;82(10 Suppl):P7.251. | ||
Phillips JT, Hutchinson M, Fox R, Gold R, Havrdova E. Managing flushing and gastrointestinal events associated with delayed-release dimethyl fumarate: experiences of an international panel. Mult Scler Relat Disord. 2014;3(4):513–519. | ||
Phillips TJ, Erwin AA, Agrella S, et al. Consensus management of gastrointestinal events associated with delayed-release dimethyl fumarate: a Delphi study. Neurol Ther. 2015;4(2):137–146. | ||
Longbrake EE, Naismith RT, Parks BJ, Wu GF, Cross AH. Dimethyl fumarate-associated lymphopenia: risk factors and clinical significance. Mult Scler J Exp Transl Clin. 2015;1: doi:10.1177/2055 217315596994. | ||
Buttmann M, Stoll G. Case reports of PML in patients treated for psoriasis. N Engl J Med. 2013;369(11):1081. | ||
Ermis U, Weis J, Schulz JB. PML in a patient treated with fumaric acid. N Engl J Med. 2013;368(17):1657–1658. | ||
Van Oosten BW, Killestein J, Barkhof F, Polman CH, Wattjes MP. PML in a patient treated with dimethyl fumarate from a compounding pharmacy. N Engl J Med. 2013;368(17):1658–1659. | ||
Stoppe M, Thomä E, Liebert UG, et al. Cerebellar manifestation of PML under fumarate and after efalizumab treatment of psoriasis. J Neurol. 2014;261(5):1021–1024. | ||
Dammeier N, Schubert V, Hauser TK, Bornemann A, Bischof F. Case report of a patient with progressive multifocal leukoencephalopathy under treatment with dimethyl fumarate. BMC Neurol. 2015;15:108. | ||
Nieuwkamp DJ, Murk JL, van Oosten BW, et al. PML in a patient without severe lymphocytopenia receiving dimethyl fumarate. N Engl J Med. 2015;372(15):1474–1476. | ||
Rosenkranz T, Novas M, Terborg C. PML in a patient with lymphocytopenia treated with dimethyl fumarate. N Engl J Med. 2015;372(15):1476–1478. | ||
Van Schependom J, Gielen J, Laton J, Nagels G. Assessing PML risk under immunotherapy: if all you have is a hammer, everything looks like a nail. Mult Scler. Epub 2015 Jul 21. | ||
European Medicines Agency. Updated recommendations to minimise the risk of the rare brain infection PML with Tecfidera [press release]. 2015. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/news/2015/10/news_detail_002423.jsp&mid=WC0b01ac058004d5c1. Accessed January 26, 2016. | ||
Coyle PK. Multiple sclerosis in pregnancy. Contin Minneap Minn. 2014;20(1):42–59. | ||
Li J, Rox RJ, Phillips JT, et al. Delayed-release dimethyl fumarate and pregnancy: preclinical studies and pregnancy outcomes from clinical trials and postmarketing experience. 2015. Available from: http://onlinelibrary.ectrims-congress.eu/ectrims/2015/31st/115441/jie.li.delayed-release.dimethyl.fumarate.and.pregnancy.preclinical.studies.and.html. Accessed February 10, 2016. | ||
Gold R, Giovannoni G, Phillips JT, et al. Efficacy of delayed-release dimethyl fumarate in early multiple sclerosis: post-hoc analysis of the phase 3 DEFINE and CONFIRM studies according to baseline disability. 2015. Available from: http://onlinelibrary.ectrims-congress.eu/ectrims/2015/31st/115562/ralf.gold.efficacy.of.delayed-release.dimethyl.fumarate.in.early.multiple.html. Accessed January 26, 2016. | ||
Marantz J, Gold R, Giovannoni G, Phillips JT, Fox RJ, Zhang A. Longer-term follow-up of the efficacy of delayed-release dimethyl fumarate in newly diagnosed patients with RRMS: an integrated analysis of DEFINE, CONFIRM, and ENDORSE. 2015. Available from: http://onlinelibrary.ectrims-congress.eu/ectrims/2015/31st/115560/undefined. Accessed January 26, 2016. | ||
Bar-Or A, Gold R, Kappos L, et al. Clinical efficacy of BG-12 (dimethyl fumarate) in patients with relapsing-remitting multiple sclerosis: subgroup analyses of the DEFINE study. J Neurol. 2013;260(9):2297–2305. | ||
Hutchinson M, Fox RJ, Miller DH, et al. Clinical efficacy of BG-12 (dimethyl fumarate) in patients with relapsing-remitting multiple sclerosis: subgroup analyses of the CONFIRM study. J Neurol. 2013;260(9):2286–2296. | ||
Calkwood J, Vollmer T, Fox RJ, Zhang R. Safety and tolerability of delayed-release dimethyl fumarate (DMF) when administered in combination with interferon beta or glatiramer acetate in relapsing-remitting MS. Int J MS Care. Epub 2015 Oct 30. | ||
Cohan S, Moses H, Calkwood J, et al. Real-world clinical outcomes in relapsing-remitting multiple sclerosis patients who switch from natalizumab to delayed-release dimethyl fumarate: a multicenter, retrospective, observational study (STRATEGY). 2015. Available from: http://onlinelibrary.ectrims-congress.eu/ectrims/2015/31st/115559/stanley.cohan.real-world.clinical.outcomes.in.relapsing-remitting.multiple.html?f=m3. Accessed January 26, 2016. | ||
Plavina T, Subramanyam M, Bloomgren G, et al. Anti-JC virus antibody levels in serum or plasma further define risk of natalizumab-associated progressive multifocal leukoencephalopathy. Ann Neurol. 2014;76(6):802–812. | ||
Havla J, Kleiter I, Kümpfel T. Bridging, switching or drug holidays – how to treat a patient who stops natalizumab? Ther Clin Risk Manag. 2013;9:361–369. | ||
Prosperini L, Annovazzi P, Capobianco M, et al. Natalizumab discontinuation in patients with multiple sclerosis: profiling risk and benefits at therapeutic crossroads. Mult Scler. 2015;21(13):1713–1722. | ||
Patti F, Leone C, Zappia M. Clinical and radiologic rebound after discontinuation of natalizumab therapy in a highly active multiple sclerosis patient was not halted by dimethyl-fumarate: a case report. BMC Neurol. 2015;15:252. | ||
Wallbrecht K, Drick N, Hund AC, Schön MP. Downregulation of endothelial adhesion molecules by dimethylfumarate, but not monomethylfumarate, and impairment of dynamic lymphocyte-endothelial cell interactions. Exp Dermatol. 2011;20(12):980–985. | ||
Rubant SA, Ludwig RJ, Diehl S, et al. Dimethylfumarate reduces leukocyte rolling in vivo through modulation of adhesion molecule expression. J Invest Dermatol. 2008;128(2):326–331. | ||
Hutchinson M, Fox RJ, Havrdova E, et al. Efficacy and safety of BG-12 (dimethyl fumarate) and other disease-modifying therapies for the treatment of relapsing-remitting multiple sclerosis: a systematic review and mixed treatment comparison. Curr Med Res Opin. 2014;30(4):613–627. | ||
Nixon R, Capkun-Niggli G, Bergvall N, Dias S. Re: Hutchinson M, Fox RJ, Havrdova E, et al. Efficacy and safety of BG-12 (dimethyl fumarate) and other disease modifying therapies for the treatment of relapsing-remitting multiple sclerosis: a systematic review and mixed treatment comparison. Curr Med Res Opin. 2015;31(1):51–52. | ||
Vollmer BL, Nair K, Sillau SH, et al. Comparison of fingolimod and dimethyl fumarate in the treatment of multiple sclerosis: one year experience. 2015. Available from: http://onlinelibrary.ectrims-congress.eu/ectrims/2015/31st/115882/undefined. Accessed January 26, 2016. | ||
Strassburger-Krogias K, Ellrichmann G, Krogias C, Altmeyer P, Chan A, Gold R. Fumarate treatment in progressive forms of multiple sclerosis: first results of a single-center observational study. Ther Adv Neurol Disord. 2014;7(5):232–238. | ||
Biogen. BG00012 and Disability Progression in Secondary Progressive Multiple Sclerosis (SPMS) (INSPIRE). Available from: https://clinicaltrials.gov/ct2/show/NCT02430532. NLM identifier: NCT02430532. Accessed February 8, 2016. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.