Back to Journals » International Journal of Nanomedicine » Volume 14
Dimeric c(RGD) peptide conjugated nanostructured lipid carriers for efficient delivery of Gambogic acid to breast cancer
Authors Kebebe D ![]() , Wu Y, Zhang B, Yang J, Liu Y
, Wu Y, Zhang B, Yang J, Liu Y ![]() , Li X, Ma Z, Lu P, Liu Z, Li J
, Li X, Ma Z, Lu P, Liu Z, Li J
Received 21 January 2019
Accepted for publication 6 July 2019
Published 2 August 2019 Volume 2019:14 Pages 6179—6195
DOI https://doi.org/10.2147/IJN.S202424
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Dereje Kebebe,1–3,* Yumei Wu,1,2,* Bing Zhang,1,2 Jian Yang,1,2 Yuanyuan Liu,1,2 Xinyue Li,1,2 Zhe Ma,1,2 Peng Lu,1,2 Zhidong Liu,1,2 Jiawei Li1,2
1Tianjin State Key Laboratory of Modern Chinese Medicine, Institute of Traditional Chinese Medicine, Tianjin University of Traditional Chinese Medicine, Tianjin 301617, People’s Republic of China; 2Engineering Research Center of Modern Chinese Medicine Discovery and Preparation Technique, Ministry of Education, Tianjin University of Traditional Chinese Medicine, Tianjin 301617, People’s Republic of China; 3Department of Pharmaceutics, School of Pharmacy, Institute of Health Sciences, Jimma University, Jimma, Ethiopia
*These authors contributed equally to this work
Background and purpose: Gambogic acid (GA) is a natural compound that exhibited a promising multi-target antitumor activity against several types of cancer. However, the clinical application of this drug is limited due to its poor solubility and low tumor cell-specific delivery. In this study, the monomeric and dimeric Cyclo (Arg-Gly-Asp) c(RGD) tumor targeting peptides (c(RGDfK) and E-[c(RGDfK)2]) were used to modify GA loaded nanostructured lipid carriers (NLC) to reduce the limitations associated with GA and improve its antitumor activity.
Methods: GA-NLC was prepared by emulsification and solvent evaporation methods and the surface of the NLC was conjugated with the c(RGD) peptides via an amide bond. The formulations were characterized for particle size, morphology and zeta potential, encapsulation efficiency and drug loading. The in-vitro cytotoxicity and cell uptake studies were conducted using 4T1 cell. Furthermore, the in-vivo antitumor activity and bio-distribution study were performed on female BALB/c nude mice.
Results: The c(RGD) peptides modified GA-NLC was successfully prepared with the particles size about 20 nm. The HPLC analysis, FT-IR and 1H-NMR spectra confirmed the successful conjugation of the peptides with the NLC. The in-vitro cytotoxicity study on 4T1 cells revealed that c(RGD) peptides modified GA-NLCs showed significantly higher cytotoxicity at 0.25 and 0.5 μg/mL as compared to unmodified GA-NLC. Furthermore, the cell uptake study demonstrated that better accumulation of E-[c(RGDfK)2] peptides modified NLC in 4T1 cell after 12 h incubation. Moreover, the in-vivo study showed that c(RGD)s functionalized GA-NLC exhibited better accumulation in tumor tissue and tumor growth inhibition. In contrast to the monomeric c(RGD) peptide, the dimeric c(RGD) peptide (E-[c(RGDfK)2]) conjugated GA-NLC showed the improved antitumor activity and tumor targeting ability of GA-NLC.
Conclusion: These data provide further support for the potential clinical applications of E-[c(RGDfK)2]-GA-NLC in breast cancer therapy.
Keywords: c(RGD) peptides, nanostructured lipid carriers, Gambogic acid, breast cancer, cell uptake, antitumor
Introduction
Natural plant ingredients have always been major sources of chemotherapeutic agents, and they are considered to have less toxic effects compared to the currently used chemotherapy.1 Gambogic acid (GA) is a natural anticancer compound obtained from resin gamboges, a traditional Chinese medicinal herb Garcinia hanburyi Hook. F.2 GA has been proven to exhibit anti-proliferative effect, induce apoptosis, reverse multidrug resistance and possess anti-angiogenic properties on certain tumors, such as breast, prostate, lung and pancreatic cancer cells.3–8 GA has a unique caged xanthone structure, and has multiple targets, including the Bcl-2 family proteins, redox regulatory proteins, NF-ᴋB, and integrin pathways.9 Interestingly, selective induction of apoptosis by GA in cancer cells compared with normal cells, suggests that GA might be an effective anticancer drug candidate with low toxicity to normal cells.6,10 Currently, GA is in the pipeline of clinical trials in China.11 Although GA has a promising anti-tumor activity, the clinical application of this agent has been restricted due to its low solubility, short half-life and insufficient selective accumulation in a cancer cell.12,13
So far, some GA loaded nano-carriers have been developed to improve the solubility and delivery of GA to cancer cells. For instance, albumin-based NPs,14 TiO2 Nanofibers,15 folate decorated arginine based poly(ester-urea-urethane) NPs,16 magnetic Fe3O4 NPs,17 PEG-PCL NPs,18 PEGylated liposome19 and Titania-coated gold nanorods.20 However, various drawbacks are noticed with these drug delivery systems such as poor degradation and accumulation in a normal issue,21 long-term stability problem and rapid elimination by the mononuclear phagocyte system22 and intrinsic cytotoxicity and low encapsulation efficiency.15 Therefore, it is essential to develop other alternative strategies for efficient delivery of GA into a cancer cell.
Reports revealed that NLC display advantages over other colloidal carriers such as liposomes, nanoemulsions, and polymeric NPs due to an entire set of unique benefits such as enhanced drug loading capacity, prevention of drug expulsion, greater flexibility in modulating drug release which in turn make NLCs a very versatile drug delivery system via different routes of administration.23 Recently, surface modification of NPs with the specific tumor-homing ligands are known to significantly raise retention and accumulation of NPs in the tumor vasculature, and improve the selective and efficient internalization by target tumor cells.24 To date, several kinds of targeting moieties have been invented to modify the surface of NPs, among these, tumor targeting and cell penetrating peptides have attained much attention due to their several unique advantages. Currently, studies demonstrated that tumor homing peptides decorated nan-carriers could enhance the tumor-specific targeting and increase tumor cell penetration of cancer therapeutic agents like antibodies, thereby improve the anticancer efficacy of the chemotherapeutic agents.25,26 RGD is an excellent tumor-targeting ligand and RGD-conjugated nano-carriers could target tumors efficiently.27 Surface modification of NLC with integrin targeting ligand (RGD peptides) is an attractive approach due to the fact that the integrin receptors are known to be overexpressed in most type of cancer cells.28
Taking these facts into consideration, in our previous study,29 we demonstrated the potentiality of some tumor penetrating peptide (CRGDRGPDC and RGERPPR) in improving cell accumulation and tumor growth inhibition of GA. To further investigate the potentiality of this strategy, in present work we explored the monomeric and dimeric c(RGD) peptides namely c(RGDfK), and E-[c(RGDfK)2] that are known to have high selectivity to integrin and stability as a model targeting ligands to modify the surface of GA-NLC. The c(RGD) peptides are selected as active targeting ligands owing to its high selectivity to tumor cells, lower immunogenicity, high stability, easy manufacturability in comparison to antibodies and high specificity to the breast cancer as compared to folate receptor.30
To the best our knowledge, no research reports on using of these peptides for the surface decoration of GA-NLC. More importantly, though E-[c(RGDfK)2] is well known as the tumor targeting peptides and has been used extensively for targeting of imaging agents, its potential of targeting chemotherapeutic agent loaded nanoparticles has not been fully exploited.
Materials and methods
Materials
Gambogic acid (GA) (HPLC >98%) was purchased from Weikeqi Bio-Tech Co. Ltd, China; Compritol 888 ATO (Glycerol behenate) and Myrj 52 (Polyethylene Glycol (40) Monostearate) were obtained from Saint-Priest Cedex, France; MCT 812 (Miglyol® 812) was purchased from Beijing Feng Li Jing Qi Trading Co., Ltd. China; LIPOID S-100 (Soybean Lecithin) was obtained from Shanghai Dong Shang Industrial Co., Ltd, China; cRGDfK and E-[c(RGDfK)2] were synthesized by ChinaPeptide Biotech Co. Ltd Shanghai, China; DSPE-PEG2000-COOH was obtained from Nansoft Biotechnology LLC NC, USA; EDC (1-ethyl-3-(3-dimethylaminopropyl) carbodiimide) was achieved from CIVI CHEM, China; N-hydroxysuccimide (NHS) was obtained from Acros Organics, USA; Methanol (HPLC grade), chloroform and ethanol (Analytical grade) were procured from Thermo Fisher Scientific (China) Co., Ltd.; 0.22 μm syringe microporous membrane was achieved from Millex-GP, Millipore USA. PBS, RMPI-60 medium and 96-well white basal cell culture plate were obtained from Corning, NY; CCK-8 kit was obtained from Dojindo Lab, Japan; and DMSO, Coumarin-6, cisplatin, and L-arginine were purchased from Sigma-Aldrich, China.
1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide (DiR) was purchased from AAT Bioquest, Inc. USA; and Fetal Bovine Serum FBS, 0.25% trypsin +0.02% EDTA and double antibiotics (10000U Penicillin streptomycin) were purchased from Gibco, USA; and paraformaldehyde (4%) was obtained from Solarbio Science and Technology Co Ltd (Beijing, China). Xylene and hydrogen peroxide were achieved from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Primary antibodies (anti-Ki67 and anti-CD31) were purchased from Abcam (Cambridge, UK). Hematoxylin was acquired from Wuhan Biotechnology Co., Ltd (Wuhan, China).
Cell culture
Mouse breast cancer 4T1 cells were obtained from Shanghai Baili Biotechnology Co., Ltd. The cells were maintained in complete medium consisted of 90% basal medium (RPMI 1640), 10% fetal bovine serum, streptomycin (100 units/mL) and penicillin (100 units/mL) in a humidified incubator at 37 °C under 5% CO2 atmosphere.
Animals
Healthy female BALB/c nude mice, 6–8 weeks, weight 20±2 g, were purchased from the Institute of Radiation Medicine, Chinese Academy of Medical Sciences (Tianjin, China) and housed in the animal compartment with temperature: 25±2 °C, a relative humidity of 50±2%. After a week acclimation period, the animals were used for the experiments. All experimental procedure and animals care were performed in accordance with the protocol of animal experiments handling issued by the Animal Research Center of Tianjin University of Traditional Chinese Medicine. The experimental procedure was approved by the Animal Ethics Committee of Tianjin University of Traditional Chinese Medicine (Document number: TCM-LAE2018021).
Preparation GA-NLC
GA loaded NLC (GA-NLC) was prepared by the emulsification and solvent evaporation method. Briefly, GA and lecithin were dissolved in ethanol; Compritol 888 ATO and MCT 812 were dissolved in chloroform, the two organic solutions were mixed together to form the oil phase. An aqueous phase was prepared by dissolving Myrj 52 in deionized water. The oil phase was quickly added to the aqueous phase under magnetic stirring at 75 °C. The formed NLC was kept under stirring to evaporate the organic solvent and placed in the refrigerator at 4 °C followed by characterization for size, zeta potential, and encapsulation efficiency.
Conjugation of GA-NLC with c(RGDfK) and E-[c(RGDfK)2] peptides
GA-NLC containing DSPE–PEG2000–COOH was prepared by the same method mentioned in the above section, except that DSPE–PEG2000–COOH was dissolved in the chloroform. Conjugation of the peptides to NLC was achieved through an amide bond between a carboxylic group of DSPE–PEG2000–COOH on the surface of NLC and amine group of the peptides using EDC/NHS activation chemistry as per previously reported by our group with minor modification.29 The molar ratio of DSPE–PEG2000–COOH:peptide:EDC:NHS was 1:1:2:2. Briefly, EDC/NHS was dissolved in PBS (pH 7.4), and added into GA-NLC, stirred for 0.5 hr. Then, a solution of the peptides (c(RGDfK) and E-[c(RGDfK)2]) in PBS (pH 7.4) was added to the NLC in a drop-wise manner with gentle stirring, and the resulting mixture was kept under gentle stirring in the dark for 24 h at room temperature. The conjugated GA-NLC was purified from unconjugated and by-products by membrane dialysis bag (MW cut off 2000 Da, Shanghai Yuanye Bio-Technology Co., Ltd) against distilled water for 36 h. The c(RGD)-GA-NLC were kept at 4 °C until use.
Characterization of GA-NLC, c(RGDfK)-GA-NLC and E-[c(RGDfK)2])-GA-NLC
Determination of conjugation efficiency
The extent of coupling of c(RGD) peptides with the DSPE-PEG2000 on the surface of NLC was evaluated indirectly by the HPLC (Agilent 1260) determination of the unconjugated free peptide fraction after ultrafiltration of the sample (Amicon ultra, MWCO 3 kDa; Millipore Company). The HPLC system: a C18 column (Kromasil 100-5-C-18, 250 mm x 4. 6 mm i.d, 5 µm) was used with a mobile phase of 0.1% trifluoroacetic acid in water (eluent A) and 0.1% trifluoroacetic acid in acetonitrile (eluent B) (Supplementary material).
FT-IR and 1H-NMR spectra
The conjugation of c(RGD) to NLC was achieved through the amide bond between the carboxylic group of DSPE-PEG2000-COOH present on the surface of NLC and –NH2 group of c(RGD) by using EDC/NHS. The peptide conjugation was confirmed by FT-IR analysis and 1H-NMR. The FT-IR spectra of the c(RGD) peptides, DPSE-PEG2000-COOH, and freeze-dried DPSE-PEG-c(RGD) modified GA-NLC were recorded using Fourier-transform infrared spectrometer (Nicolet 6700 FTIR, Madison, USA) in KBr disks in the wave number range of 4000 to 400 cm−1.
1H NMR spectra were also performed to further confirm the conjugation between c(RGD) peptides and the NLC through DSPE-PEG2000-COOH. The appropriate amount of DSPE-PEG2000-COOH, c(RGD) peptides, and the freeze-dried DSPE-PEG-c(RGD) modified GA-NLC was weighed and dissolved in DMSO-d6 for H-NMR analysis (Varian Inova 500 MHz, California USA).
Determination of particle size and zeta potential
Particle size, polydispersity index (PDI), and zeta potential (ZP) of the GA-NLC and c(RGD)-GA-NLC were determined by a Zeta-sizer (Nano-ZS; Malvern Instruments Ltd., UK). The samples were diluted with ultra-purified water (1:5) to a weak opalescence before the measurement. Analyses were performed at 25±0.2 °C. All determinations were performed in triplicate.
Differential scanning calorimetry (DSC)
DSC of the samples was performed using a thermal analyzer (Jade DSC, PerkinElmer Inc. USA) in order to characterize the physical state of GA in NLC. DSC was conducted for the samples: GA, Compritol 888 ATO, the physical mixture of GA and Compritol 888 ATO, GA-NLC, blank NLC, c(RGDfK)-GA-NLC and E-[c(RGDfK)2]-GA-NLC. The samples were heated from ambient temperature to 350 °C at a constant heating rate of 10 °C/min under a nitrogen atmosphere.
Transmission electron microscopy (TEM)
The morphology of GA-NLC and c(RGD) coupled NLC were studied by TEM (JEOL, Japan). Briefly, the samples were diluted with deionized water, and dropped on a copper grid as a film followed by negative staining with 2% phosphotungstic acid. Then, the samples were dried and observed under TEM.
Determination of encapsulation efficiency
GA-NLC formulations were transferred to the upper chamber of a centrifuge tube fitted with an ultrafilter (Amicon ultra, MWCO 10 kDa; Millipore Company) and centrifuged at 4000 rpm for 20 min. The ultrafiltrate containing unencapsulated GA was determined by HPLC method. To determine the total content of the drug, GA formulations was dissolved in methanol and sonicated for 10 mins, and diluted appropriately by methanol. The obtained dispersion was filtered through a Millipore filter (MillexHV; Millipore Company) and analyzed with the validated HPLC method. Encapsulation Efficiency (EE%) was calculated by the following formula:
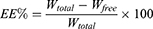
Where, Wtotal, Wfree and are the total weight of GA in the NLC, and the weight of free GA, respectively.
The concentration of GA in NLC formulations was determined by Agilent HPLC system with variable wavelength ultraviolet absorbance detector (VWD) and a reverse phase C18 column (250 mm × 4. 6 mm i.d, 5 µm) operating at 30 °C. The mobile phase composed of methanol and 0.1% phosphoric acid (95:5 (v/v)) was allowed to flow at 1 mL/min. The detection wavelength was set at 360 nm and the injected volume of the sample was 20 µL. The method was linear over the concentration range of 1–200 μg/mL. The linear regression equation was Y=25.341X-12.569 with a correlation coefficient (R2) of 1. The % RSDs for intraday and inter-day precision and accuracy study of the method were below 2%.
In vitro anticancer activity study
Cytotoxicity studies
The cytotoxicity of GA solution, blank NLC, GA-NLC, c(RGDfK)-GA-NLC, and E-[c(RGDfK)2]-GA-NLC against 4T1 cells was evaluated using CCK-8 assay. The cells were seeded into a 96-well plate at a density of 5×104 cells per well and incubated for 24 h. Then, the medium containing the formulations with different concentrations of GA (0.1, 0.25, 0.5, 1, 2.5 µg/mL) was added to each well and incubated for a further 24 h. After the incubation for 24 h, and the medium containing the formulations was aspirated and washed with 100 μL PBS. Then, 100 μL 10% CCK-8 was added to each well and incubated for a further 1 h. Finally, the plates were subjected to microplate reading (Multiskan MK3; Thermo Scientific, Atlanta, GA) for cell viability assays at a wavelength of 450 nm. Cell viability was calculated as:

Where, As, Ac and Ab refer to the absorbance of the experimental wells, the control wells, and the blank wells, respectively.
In vitro cellular uptake study
Uptake of the formulations by 4T1 cells was evaluated by High Content Cell Imaging Analysis System (GE InCell Analyzer 2000, Fairfield, CT) using Coumarin-6 (C-6) as a fluorescent probe. The C-6 labeled formulations (C-6-NLC, c(RGDfK)-C-6-NLC and E-[c(RGDfK)2]-C-6-NLC) were prepared by the same method mentioned above for GA-NLC except that GA was replaced by C-6. The concentration of C-6 in each formulation was determined by using HPLC. In addition, a free C-6 solution with a similar concentration of C-6-NLCs was prepared in DMSO and diluted in the similar manner of C-6-NLC, and quantitative cellular uptake properties of the preparations were studied at non-toxic concentration. In order to predict a non-toxic concentration, cytotoxicity test was conducted for all C-6 formulations in different concentrations.
To evaluate the cellular uptake, 4T1 cell were seeded at a density of 4×105 cells/well in 96-well culture plates. After 24 h of incubation, the cells were treated with free C-6, C-6-NLC, c(RGDfK)-C-6-NLC and E-[c(RGDfK)2]-C-6-NLC in their non-toxic concentration and further incubated for 1, 2, 4 and 12 hrs. Next, 100 μL of cold PBS was used to stop the cellular uptake and washed three times with PBS. The cells were fixed with 4% paraformaldehyde for 20 min, and then washed once every 5 mins with PBS, and 20 μL of 0.20 μg/mL DAPI per well was added to stain the nuclei, then washed 3 times with PBS for 10 min, and 150 μL of PBS was added to each well. The whole process was conducted in a dark room. Quantitative and qualitative analysis of cell uptake was performed using a high-content imaging system (GE InCell Analyzer 2000, Fairfield, CT).
In-vivo antitumor effects
Establishing the breast cancer model
The mouse mammary carcinoma 4T1-Luc cells (4T1-Luc, luciferase) were taken from the exponential growth phase and resuspended in RPMI 1640, centrifuged and washed 3 times to remove the serum. The cell concentration was adjusted to 2×107 cells/mL with RPMI 1640. The right armpit of female BALB/c nude mice was wiped with alcohol-cotton, and the cell suspension (0.1 mL/mouse) was inoculated using a 1 mL syringe. When the tumor volume grew up to 100 mm3 (tumor volume V = ab2/2; a: tumor length, b: tumor width,), the mice were randomly divided into six group (n=6 per group): saline group, positive drug group, free GA sol, GA-NLC, c(RGDfK)-GA-NLC, and E-[c(RGDfK)2]-NLC. For the saline group, 100 μL of normal saline was injected. The cisplatin (1 mg/kg) was given for the positive drug group, and the free GA sol, GA-NLC, c(RGDfK)-GA-NLC, and E-[c(RGDfK)2]-GA-NLC (GA: 2 mg/kg) were injected into a tail vein. 100 μL/mice, once every 3 days, 5 times.
In-vivo antitumor activity study
The tumor volume was determined every three days using a Vernier caliper and their weights were measured using an electronic scale. Every day, their diet, activity, mental state, death, and other general conditions were observed. Finally, 24 h after the last dose of the formulations, the tumor tissue, and the major organs were collected and weighed for the determination of the rate of tumor inhibition and viscera indices, and histological examination and immunohistochemistry.
The rate of tumor inhibition (ROI) and tumor growth inhibition (TGI) were calculated as a measure for in-vivo antitumor efficacy. In addition, the change in body weight as a function time of the treatment was calculated as the inhibition rate of body weight (IRBW). Each parameter was calculated by the following formula:

Where MC and MT are the mean tumor weight of the model control group and the mean tumor weight of the treatment group, respectively.
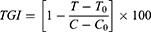
Where T and T0 are the mean tumor volumes of the treated group on the final day of the treatment and before the treatment, respectively. C and Co are mean tumor volume of the control group after and before the treatment, respectively.
Histological examination
In order to better observe the microscopic changes of tumor tissues and organs, the tumor tissue and the major organs were collected at the end of the in-vivo antitumor activity study (mentioned in the above section). The tissues were soaked in 10% formalin, and subjected to hematoxylin and eosin (HE) staining. Specifically, the tumor tissue and major organs were isolated, added into 10% formalin for fixation, and embedding was done in paraffin. Then, tissue sections of 4 μm thickness were prepared. The sections were placed on glass slides, deparaffined and dehydrated in xylene and graded alcohols, respectively. Then, the slides were stained with hematoxylin and eosin. Later, all the slides were observed under a microscope (Microscope: NIKON Eclipse Ci, Imaging System: NIKON digital sight DS-FI2, JAPAN, Multiplier: 100×, 200×).
Immunohistochemical analysis
Immunohistochemical staining was performed to detect the expressions of Ki67 and CD31. The tumor sections were incubated with anti-Ki67 and anti-CD31 (1:100) at 4 °C overnight. Then, the sections were stained with a streptavidin-peroxidase system, the signal was visualized using diaminobenzidine substrate, and counterstaining was done with hematoxylin. For quantitative data, the microvessel counts (MVCs) was used to express CD31, and integrated optical density (IOD) of Ki67 was measured using Image-Pro Plus 6.0 (Media Cybernetics, Inc., Rockville, MD, USA).
In vivo biosafety evaluation of blank NLC and its c(RGD) peptides conjugates
In order to evaluate the possible toxicity arise from the materials used to formulate NLC and conjugated peptides in-vivo, the repeated dose toxicity study of blank NLC (B-NLC) and c(RGD)-B-NLC was conducted on normal BALB/c nude mice at the equivalent dose that had been used for the treatment using histological examination.
In-vivo bio-distribution study
In-vivo bio-distribution and tumor targeting efficiency of the c(RGD)s modified NLC formulations were evaluated with an In-Vivo Imaging System (IVIS) in BALB/c nude mice with 4T1-Luc using DiR as a fluorescence probe. The breast cancer model was established by the similar methods mentioned above and IVIS was done when the tumor volume grew up to 300 mm3.
DiR-labeled NLC and c(RGD)s modified NLC were prepared according to the method of preparation mentioned for c(RGD) modified GA-NLC, except, the GA was replaced with DiR to obtain DiR-labeled NLC and c(RGD)-modified NLC for the study of tissue distribution. The free DiR was prepared by dissolving the DiR into ethanol to form 2.5 mg/mL, and diluted with normal saline to 100 μg/mL.
BALB/c nude mice were randomly divided into four groups (n=6) comprising DiR-Sol, DiR-NLC, c(RGDfK)-DiR-NLC, and E-[c(RGDfK)2]-DiR-NLC group. The formulations were given at the dose of 0.5 mg/kg.
At the time of 2 h, 3 h, 6 h, 18, 21 and 24 h after the administration of DiR labeled NLC formulations, each mouse was placed in a living body imaging system to observe the fluorescence intensity of each organ at an excitation wavelength of 710 and emission wavelength of 790 nm. During the imaging acquiring process, the animals were anesthetized by 4% chloral hydrate. After 24 h of imaging, the animals were sacrificed, and the main organs and tumor were excised. The fluorescence intensity of each organ and tumor were measured.
Statistical analysis
All numerical parameters were expressed as means ± standard deviation. The Student’s t-test and IC50 determination were performed by Origin 2018 and GraphPad Prism 8.0., respectively. The p-value <0.05 was taken as statistically significant.
Results
Preparation and characterization of c(RGD)s modified GA-NLC
Conjugation of c(RGD)s with GA-NLC
The c(RGD)s functionalized GA-NLC was successfully developed using DSPE-PEG2000-COOH as a linker between the peptides and NLC by carbodiimide reaction. The conjugation of c(RGD) to DSPE-PEG2000-COOH on the surface of NLC was confirmed by HPLC, FTIR and 1H-NMR analysis.
The HPLC method was utilized for estimating the amount of free c(RGD) left in the formulation after the coupling reaction. The HPLC analysis of standard free c(RGDfK) and E-[c(RGDfK)2] showed that the peaks were observed at a retention time of 9.0 and 7.2 mins, respectively. As shown in Figure S1, there was no significant peak for the free c(RGD) around their respective retention time in ultra-filtrate samples, indicating that there was no considerable amount of free c(RGD) left unreacted in the formulation. The appearance of the peak of free c(RGD) peptides at their respective retention time in sample mixed with free c(RGD) peptides clearly indicates that the HPLC method is sensitive to detect free c(RGD) in the presence of the test samples. Therefore, almost all of the c(RGD) peptides added to the formulations had been conjugated on the surface of the NLC, suggesting nearly 100% coupling efficiency.
The FT-IR spectra of DSPE-PEG2000-COOH exhibited the absorption of C=O stretching of the carboxylic acid group at 1736 cm−1. This carbonyl band disappeared in DSPE-PEG conjugated with c(RGD) peptides, and the new peak appeared at 1651 cm−1, corresponding to C=O stretching of the amide bond formed between the –COOH group of DSPE-PEG2000-COOH and the –NH2 of c(RGD). In addition, the peak at 1560 cm−1, which attribute to the N-H bending vibration of the peptide (Figure S2).
The 1H-NMR spectra were obtained for DSPE-PEG2000-COOH, both c(RGD) peptides and DSPE-PEG-cRGD in deuterated DMSO. As shown in Figure S3A–D, and S3E, the proton peaks at 1.23 and 3.52 are corresponding to the DSPE and methylene protons of PEG unit of DSPE-PEG-COOH, respectively. The peak at 2.5 ppm was attributed to the solvent (DMSO-d6). Chemical shift as a benzene ring and secondary amide (C-NH) of the peptides are observed between 7.1 and 8.6 ppm, indicating the successful conjugation of the c(RGD) peptide to DSPE-PEG2000-COOH on the surface of GA-NLC.
Particle size, zeta potential, and morphology
As shown in Table 1 and Figure 1A and C, the mean particle size of the NLC was about 19 to 26 nm and with zeta potential ranges −3 to −7.6 mV (Figure 1B and D), and the PDI was between 0.2 and 0.4, suggesting an acceptable particle size and distribution of nanoparticles for cancer targeting application. The morphological characteristics of the NLC were evaluated by TEM. Morphologically the NPs were spherical (Figure 1E).
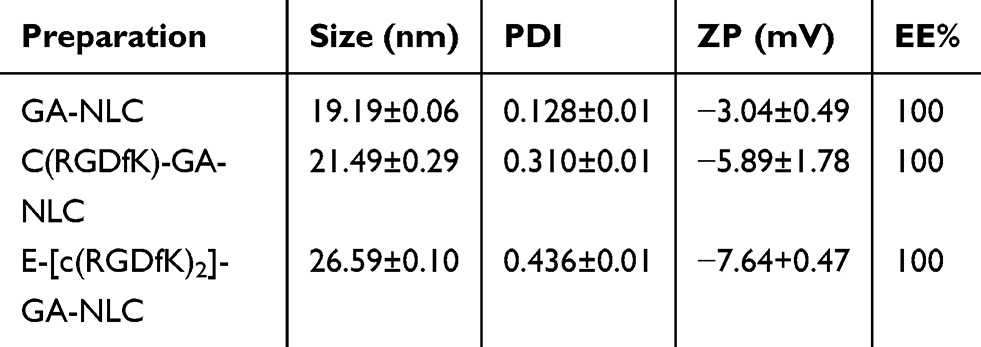 |
Table 1 Mean particle size, PDI, ZP, and EE% of GA-NLC, cRGDfK-GA-NLCand E-[c(RGDfK)2]-GA-NLC (Mean ± SD, n=3) |
 |
Figure 1 The size and morphology of the formulations: (A and B) are particle size distribution and zeta potential of GA-NLC, (C and D) are particle size distribution and zeta potential of c(RGD) peptides modified GA-NLC as determined by dynamic light scattering. E represents the TEM images of c(RGD) peptide modified GA-NLC (scale bar=200 nm). |
The drug encapsulation efficiency
The GA encapsulation efficiency (EE %) and drug loading capacity (DL %) were evaluated by HPLC method. The result showed that GA was encapsulated in the NLC with EE% of 100%, this could be due to the high lipid solubility of GA. No significant difference in the EE% was noticed between c(RGD) peptide modified GA-NLC and unmodified GA-NLC, which indicate no effect of the c(RGD) surface modification on EE% of GA-NLC.
Differential scanning calorimetry (DSC)
The DSC thermograms of GA, a physical mixture of the ingredients, blank NLC, GA-NLC, and c(RGD) modified GA-NLCs are shown in Figure S4. The thermogram showed that the melting peak of GA was 73.4 °C and the melting peak was also detected in the physical mixture of Compritol 888 ATO and GA. However, no melting peak of GA was observed in NLC. This may suggest that the GA was successfully encapsulated in its amorphous form, and no leakage of GA from the NLC system.
Cytotoxicity study
The in-vitro antitumor activity of GA-NLC and c(RGD) peptides modified GA-NLC were evaluated on 4T1 cell after 24 h incubation. The cell viability was measured using the CCK-8 assay. As can be seen from Figure 2, the cytotoxicity of the formulation was concentration-dependent. The IC50 values of free GA, GA-NLC, c(RGDfK)-GA-NLC, and E-[c(RGDfK)2]-GA-NLC were 2.22, 0.26, 0.17, and 0.12 µg/mL, respectively. The IC50 values of all NLC formulations were about 10-fold less than that of free GA, suggesting the cytotoxicity is highly influenced by the nano-carriers. No considerable cytotoxicity was observed at the lowest concentration (0.1 µg/mL) of all formulations. However, at 0.25, 0.5. 1 and 2.5 µg/mL, all NLC group exhibited a significantly higher (p<0.001) cytotoxicity than that of free GA (Figure 2), which primarily attributed to the enhanced cellular uptake of nanoparticles through endocytosis pathways, and the improved stability of GA in the NLC, which could protect the drug from epimerization and hydrolysis, thereby enhancing the therapeutic activity. More importantly, c(RGD) decorated GA-NLC particularly, c(RGDfK)-GA-NLC and E-[c(RGDfK)2]-GA-NLC demonstrated a higher cytotoxicity at lower concentration of GA (0.25 and 0.5 µg/mL) as compared to undecorated GA-NLC, indicating the conjugating peptides enhanced the cell uptake by 4T1 cell which may further improve the cytotoxicity due to the potential to increase the intracellular concentration of GA. The free GA could exhibit cytotoxicity only at the highest concentration (2.5 µg/mL). This could be attributed to the MDR effect, the free drug out-fluxing through p-glycoprotein which leads to the reduced the intracellular concentration of the drug. Moreover, the blank NLC showed no considerable cytotoxicity, suggesting the safety of the excipient used to formulate the NLC and could be useful as a drug carrier. Generally, based on IC50 and % cell viability, it can be suggested that E-[c(RGDfK)2] modified GA-NLC exhibited superior in-vitro anticancer activity against 4T1 cell.
 |
Figure 2 4T1 cell viability after 24 h incubation with free GA (solution), blank NLC, GA-NLC, c(RGDfK)-GA-NLC, and E-[c(RGDfK)2]-GA-NLC to 4T1 cells. At lower concentration of GA, c(RGD) peptides modified GA-NLC showed significantly lower cell viability (%). Free GA was able to inhibit the growth of 4T1 cell only at higher concentration (*p-value <0.05 represent vs GA-NLC). |
Cell uptake study
In order to select the appropriate concentration of C-6 for cell uptake study, the cytotoxicity of different concentration of C-6 labeled NLC was determined. Accordingly, as it can be shown in Figure S5, C-6 at the concentration of 0.5 μg/mL C-6 found to exhibit cytotoxicity to some extent. However, at 0.05 μg/mL and lower concentration, no considerable cytotoxicity was observed. Therefore, C-6 at the concentration of 0.05 μg/mL was used in cell uptake study.
Fluorescence intensity (FI) of C-6 was used to evaluate the uptake efficiencies. As shown in Figure 3A and B, the FI increase with the incubation time for all formulations under investigation. At all-time points, all C-6-NLC formulations exhibited greater FI than that of C-6-Sol, suggesting drug accumulate better in nanocarrier drug delivery system than in free drug form, which could be due to the fact that nanoparticulate systems are capable of entering live cells, often through several endocytic pathways.31 After 12 h of incubation, the E-[c(RGDfK)2] peptide modified C-6-NLC demonstrated significantly higher FI as compared to the c(RGDfK) and unmodified C-6-NLC. This indicates that the E-[c(RGDfK)2] enhanced the cell internalization of the loaded drug considerably. Therefore, dimeric c(RGD) (E-[c(RGDfK)2]) could exhibit better accumulation of C-6 inside the cell in comparison to monomeric c(RGD) (c(RGDfK)).
 |
Figure 3 Cell uptake of C-6 labeled formulations by 4T1 cells: (A) the image of cell uptake of C-6 labeled formulations by 4T1 cell after 12 h incubation. Green color belongs to C-6 and blue is DAPI. (B) the FI of C-6 labeled NLC and c(RGD)s modified C-6 labeled NLC at different incubation time interval (1 h, 4 h, and 12 h); (***p-value <0.001 indicate in comparison to C-6-NLC). |
In-vivo antitumor activity study
The in-vivo antitumor activity study was evaluated in BALB/c nude mice inoculated with 4T1-Luc cells. After eight days of the tumor cell inoculation, the mice developed a tumor volume in the range of 28.1–269 mm3. The mice with too small or large tumor volume had been excluded from the experiment. The tumor growth inhibition potential was assessed by measuring the changes in tumor volume after the administration of the formulations. As shown in Figure 4A, fast tumor growth was observed in control group treated with NS, while tumor volume growth in GA-NLC and peptides modified GA-NLC groups were significantly inhibited as compared to the GA-sol group (p<0.05). Particularly, the mice treated with E-[c(RGDfK)2]-GA-NLC and Cisplatin displayed the strongest anti-tumor effect as compared to c(RGDfK) modified GA-NLC, and unconjugated GA-NLC. The tumor growth inhibition (TGI) rate of the mice treated by the peptides modified GA-NLC and positive control drug (Cisplatin) on the last day of the treatment were >50%. As shown in Table S1, the E-[c(RGDfK)2]-GA-NLC demonstrated higher TGI as compared to that of other formulations. According to the weight of tumor mass excised from the xenograft mice after the last day of treatments (Figure 4B), relatively lower weight of the tumor was obtained from the mice treated with c(RGD) peptides (c(RGDfK) and E-[c(RGDfK)2]) functionalized GA-NLC and Cisplatin as compared to GA-NLC and GA-sol group. Based on the weight of a tumor, the ROI was calculated (Table S1) and slightly higher ROI (>50%) was exhibited by c(RGD) peptides (c(RGDfK) and E-[c(RGDfK)2]) decorated GA-NLC and Cisplatin in comparison with GA-NLC and GA sol.
 |
Figure 4 In-vivo antitumor effect of the formulations. (A) Changes in tumor volume after cell inoculation (day 0–20) and over the course of treatment (day 10–20). At the last day of the treatment the mice treated with [c(RGDfK)2]-GA-NLC and Cisplatin showed considerably lower tumor volume in comparison to free GA, GA-NLC and control group. (B) Weight of tumor at the end of treatment excised from mice treated with different formulations of GA. (***p-value <0.001 and *p-value <0.05 in relation to GA-NLC). |
The change in body weight of tumor-bearing mice as a function of time is commonly used as one of the safety markers with inhibition rate of body weight (IRBW %) considered as an index.32 There was no significant change in body weight in all treatment group over the course of the treatment, suggesting that both the peptide modified and unmodified GA-NLC were well tolerated. However, the body weight of the mice treated with NS showed slightly increased with IRBW of −6.64%. On other hands, a slightly decreased body weight was observed in E-[c(RGDfK)2]-GA-NLC group with IRBW of 7.17% (Table S1 and Figure S6). Furthermore, the visceral indexes (liver, spleen, lung, and kidney) were calculated and compared with the control group, no significant difference was observed (Table S2). However, large spleen index was observed in all group of mice. This could be associated with splenomegaly induced following inoculation of 4T1 cell into the mammary fat pads of female BALB⁄c mice.33
Histological examination
The result of HE showed that no significantly serious histological change was observed in main organs, however, a large number of neutrophils were seen in spleen tissue in both treatment and control groups. The HE of tumor from the normal saline group showed that no tumor cell necrosis was observed in the tissue, but, extensively necrotic, and the necrotic cells was seen in tumor tissues of all treatment group (Figure 5).
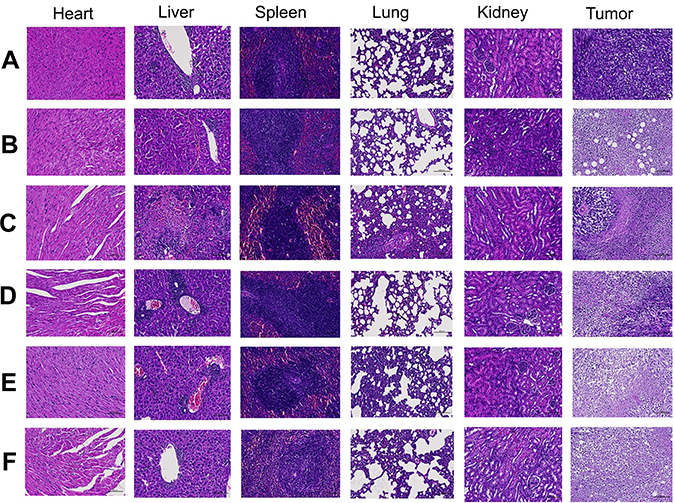 |
Figure 5 Histological examination of major organ and tumor tissue after treatment with (A) Normal Saline, (B) Cisplatin, (C) GA Sol, (D) GA-NLC, (E) c(RGDfK)-GA-NLC, (F) E-[c(RGDfK)2]-GA-NLC (Magnification 200×). No significantly serious histological change was observed in main organs, however extensively necrotic, and the necrotic cells in tumor tissues of all treatment group. |
Immunohistochemistry analysis
In order to further investigate the antitumor efficacy of c(RGD)-modified GA NLC, IHC staining was performed to detect the expressions of Ki67and CD31 on tumor tissues after the treatment. The expression level of Ki67 and CD31 were evaluated quantitatively using the Image Pro Plus 6.0 analysis system (Media Cybernetics, Silver Spring, MD). The IHC staining of Ki-67 was used to evaluate the proliferation of tumor cells in vivo. As it can be seen from the Figure 6A and C), both c(RGD) peptides conjugated GA NLC demonstrated a significantly lower (p<0.001) level of Ki67 expression than unmodified GA-NLC, and free GA, indicating the excellent antiproliferative activity of c(RGD) functionalized GA-NLC. Furthermore, CD31 is the most specific and sensitive endothelial marker. To confirm the antiangiogenesis, microvessel counts (MVCs) was conducted according to Weidner.34 The results showed a significantly decreased MVCs in E-[c(RGDfK)2]-GA-NLC as compared to remaining treatment group which suggest E-[c(RGDfK)2] improved significantly antiangiogenic effect GA-NLC (Figure 6B and C).
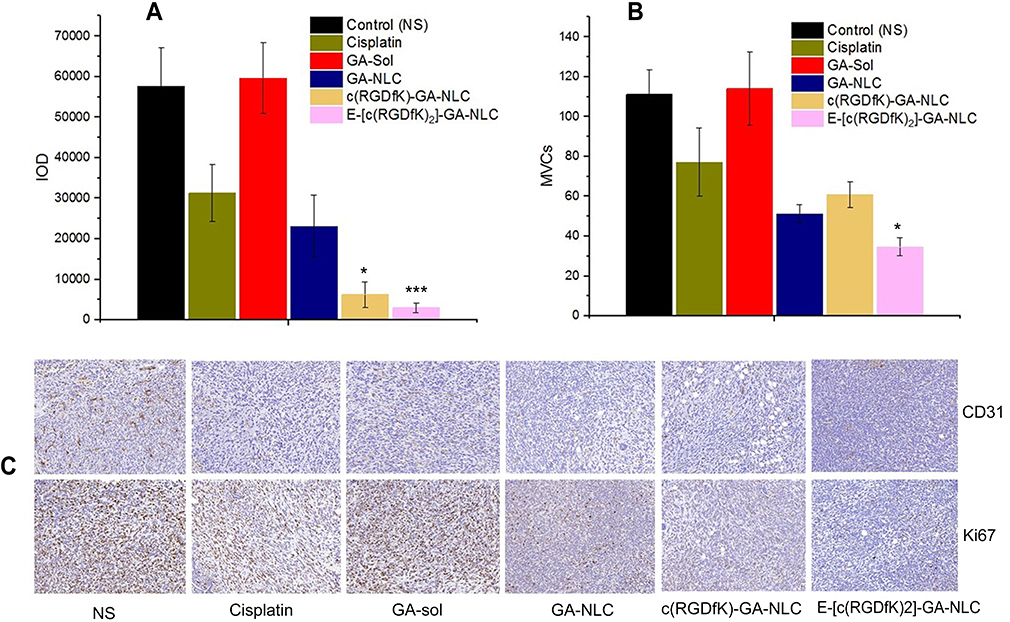 |
Figure 6 Immunohistochemical staining results of tumor tissue after treatment with different formulations of GA. (A) Integrated optical density (IOD) of Ki67 expression. (B) Microvessel counts (MVCs) of CD31. (C) IHC staining images of both Ki67 and CD31. (*p<0.05 vs GA-NLC, **p<0.01 vs GA-NLC). |
In vivo biosafety evaluation of blank NLC and its c(RGD) peptides conjugates
The possible toxicity due to the NLC itself was assessed by treating the mice with blank NLC and c(RGD) peptides decorated blank NLC. Accordingly, the mean weights of the control and all treatment groups remained similar over the treatment period without significantly increased or decreased (Figure 7A). In addition, no mortality was observed during these studies. Additionally, no abnormal clinical signs or behaviors were observed in all groups of mice. Figure 7B showed the liver, heart, lung, spleen and kidney indices of mice treated with normal saline, blank NLC, and c(RGDfK)-NLC, and E-[c(RGDfK)2]-NLC. As it can be seen, no significant differences (p>0.05) in these visceral index among all groups.
 |
Figure 7 (A) Change in body weight of BALB/c nude during the treatment period with blank NLC and its c(RGD) peptides conjugate. (B) Organ indices calculated from the major organs removed after 24 h of the last day treatment. |
The possible toxicity of the B-NLC and c(RGD)-B-NLC to organs including heart, liver, spleen, lung and kidney were investigated using histopathological examination. The result of HE showed that no noticeable histopathological changes or lesions were detected in main organs of all group (Figure 8), suggesting that the bare NLC and c(RGD) modified NLC are well tolerable and has excellent biocompatibility at the dose used for the treatment.
 |
Figure 8 HE staining of major organ after treatment with (A) Normal Saline, (B) Blank-NLC (B-NLC), (C) c(RGDfK)-B-NLC, (D) E-[c(RGDfK)2]-GA-NLC (magnification 100×). |
In-vivo bio-distribution
In this study, the IVIS was used to evaluate the in-vivo bio-distribution and targeting efficiency of peptides modified NLC on BALB/c nude mice bearing 4T1 tumors. DiR was used as a fluorescent probe encapsulated in the NLC. In contrast, free DiR sol exhibited no DiR accumulation at the tumor site over a period of imaging due to rapid clearance and nonspecific distribution after intravenous administration (Figure 9A). The fluorescence in the free DiR group mainly concentrated on the liver tissues throughout the imaging periods, and gradually decreased after 6 h. In general, within the first 3 h, no distribution of DiR in tumor area for NLC groups, which attribute to the longer circulation time due to the presence of PEG on the surface of NLC. The strong FI was observed in tumor sites for all NLC group after 6 h, which is attributed to the accumulation of NLC in tumor tissue via enhanced permeability and retention (EPR) effect. Furthermore, fluorescent signals in tumor sites were highest at 24 h and the fluorescence degree of the c(RGD) peptides modified groups (c(RGDfK)-GA-NLC and E-[c(RGDfK)2]-GA-NLC) was slightly stronger than that of NLC group (GA-NLC). Accordingly, it can be suggested that c(RGD) peptide modified NLC could facilitate the delivery of drugs into the tumor with long-term retention, which would enhance the antitumor effect, this could be due to the high affinity of c(RGD) peptides to αvβ3 receptor that is overexpressed on the surface of most of the cancer cells and mediates the internalization of the nano-carrier into the cell.
 |
Figure 9 Bio-distribution study of c(RGD) modified DiR-NLC in tumor bearing mice. A: Free DiR (A), DiR-NLC (B), c(RGDfK)-DiR-NLC (C), and E-[c(RGDfK)2]-DiR-NLC (D) fluorescence images of mice treated with different DiR formulations in-vivo at different time points (2, 3, 6, 18, 21, and 24 h); B: Ex-vivo fluorescence images of mice organs and tumor tissues excised from mice treated with different DiR formulations; and C: fluorescence signal of liver and tumor obtained from mice treated with different DiR formulations (*p<0.05 vs DiR-NLC). |
To clearly observe the fluorescence signals, the tumors and major organs were excised and collected at 24 h after injection. The Ex-Vivo imaging results of excised organs and tumor are shown in Figure 9B and C. As can be seen in the figure, no fluorescence signals were observed in the tumor for DiR-Sol group. However, the strong signals in the tumor were observed in all NLC groups. The result also showed that higher accumulation of DiR in the tumor of the group treated with E-[c(RGDfK)2]-GA-NLC than c(RGDfK)-GA-NLC and GA-NLC. No obvious fluorescence uptake was observed in the hearts, lungs, spleens, and kidneys. However, the DiR was accumulated considerably in the liver in all groups.
Discussion
In this study, GA loaded NLC was developed in order to circumvent the poor solubility of GA, and to passively target it into the tumor tissue via EPR effect. However, the passive targeting of NPs is not able to accumulate the drug into cancer cells in sufficient concentration. Therefore, to enhance the targeting ability and cell accumulation of GA-NLC, we functionalized the surface of the NLC with dimeric and monomeric c(RGD) peptides. These c(RGD) peptides are known to be an efficient tumor targeting ligand via the integrin receptor because integrin αvβ3 is overexpressed not only on tumoral endothelium but also on cancer cells in a number of cancer cell lines. Therefore, nano-carriers decorated with the c(RGD) peptide could be considered as a dual targeting system.35 The c(RGD) conjugation to the surface of GA-NLC was performed via a carbodiimide reaction using DSPE-PEG2000-COOH as a linker. The DSPE-PEG2000-COOH offers an excellent anchor for conjugation of ligands. Reports showed that linear chains of PEG, attached to the surface of NP are able to create a steric hindrance, leading to a remarkable inhibition of protein adsorption and less recognition by macrophages. However, extreme PEGylation can result in a strong inhibition of cellular uptake and low capability of binding with protein targets, reducing the potential of the delivery system. Consequently, the nano-carrier with intermediate PEGylation (PEG2000) is known to display a better cellular uptake in cancer cells as compared with that of long-chain PEGylation (PEG5000).36–38 Accordingly, we selected DSPE-PEG2000 among the DSPE-PEG with different molecular weight.
The size of NLC is an important parameter in the targeting of a drug by NPs. NPs with <100 nm are known to accumulate in tumor tissue by ERP effect. On other hands, larger NPs (>100 nm) are usually cleared from the circulation by phagocytosis. Similarly, very small NPs (<10 nm) have a high rate of clearance.39 The particle size of GA-NLC and both c(RGD) functionalized GA-NLC were found within the acceptable range, thus facilitating potentially passive targeting. In addition, surface modification of GA-NLC did not affect the size of the particles significantly.
The cell viability study of the present work suggests the c(RGD) decorated GA-NLC demonstrated a higher anti-proliferative effect in comparison to unmodified GA-NLC at a lower concentration of GA. Furthermore, as biocompatibility is a great concern for the drug delivery, the cytotoxicity of the blank NLC was found non-toxic to the cell, indicating the excellent biocompatibility and could be used for cancer drug delivery system efficiently.
Cellular uptake study is an essential aspect in the evaluation of a potential drug delivery system. Such study would shed light on the effectiveness of the system into in-vivo condition. In this work, we used C-6 as a fluorescent probe to investigate the qualitative and quantitative cellular uptake characteristic of NLCs. The nuclei of the cells with blue fluorescence (DAPI) was used to determine the position of C-6 labeled NLC (green fluorescence) indicating the accumulation site of nanoparticulate systems within the cell. According to the result of this study, the dimeric c(RGD) (E-[c(RGDfK)2]) modified C-6-NLC showed higher FI at 12 h of incubation, which could be attributed to the fact that the higher affinity of dimeric c(RGD) for the αvβ3 integrin receptors, which are the most promising target for cancer therapy because they are upregulated on the actively proliferating endothelium of tumor tissues and are responsible for angiogenesis, metastatic activity, and cell migration and invasion.40 Studies showed that dimer c(RGD) peptide E-[c(RGDfK)2] has more affinity to αvβ3 integrins than monomeric c(RGD) (c(RGDfK).41 Thus, E-[c(RGDfK)2] modification on the surface of NLC could assist the formulation uptake by 4T1 cells via integrin-mediated endocytosis.
The in vivo antitumor study of this work revealed that all GA-NLC formulation had better tumor growth inhibition in contrast to GA-solution. This could be due to the fact that the appropriate particle size of the NLC permeates for a higher accumulation of drug-loaded carriers in tumor tissue by the EPR effect. As expected, E-[c(RGDfK)2GA-NLC exhibited a superior tumor growth inhibition rate among all formulation. This could be attributed to the high specific binding affinity of E-[c(RGDfK)2] to integrin αvβ3 receptor that is known to be overexpressed in cancer cells, which could mediate E-[c(RGDfK)2]-GA-NLC efficiently home to the tumors, facilitate their intracellular uptake through integrin avβ3 meditated endocytosis and enhance the suppression effect of GA on 4T1 cell growth and proliferation.
Evaluation of the histopathological changes resulted in preclinical animals by novel drugs represents the basis of their safety evaluation before they can be translated into clinical studies. Studies demonstrated GA is widely distributed in all of the main tissues; heart, liver, spleen, lung, and kidney tissue.42 Therefore, morphological alteration induced by the administered the formulations should be assessed. Accordingly, there were no obvious lesions in the heart and kidney tissues of all formulations. However, a large number of neutrophils were seen in spleen tissue in both treatment and control groups. This could be associated with splenomegaly induced following inoculation of 4T1 cell into the mammary fat pads of female BALB⁄c mice.33
Immunohistochemical (IHC) staining is currently considered as the gold standard for the quantitative determination of protein biomarkers, particularly when biomarker numbers are increased in proliferative tumor tissues.43 In the present study, in order to further investigate the antitumor effect of the c(RGD)-GA-NLC, IHC staining for the expression of Ki67 and CD31 were conducted. Immune staining with Ki67, is a reliable means of estimating the growth fraction of neoplastic cell populations quickly. Similarly, CD31 is the important markers of angiogenesis, and used to determine the anti-angiogenesis effect of therapeutic agents. The result of IHC staining of our study provides another evidence for the antitumor effect improving the ability of E-[c(RGDfK)2] decorated GA-NLC.
Safety and biocompatibility are the primary goal of research in nanoparticles drug delivery system.44 In order to exploit the potential of nanotechnology in nanomedicine, the issue of safety and toxicity should be given great attention. Most of the time, a nanoparticle that exhibited both biocompatibility and capability to deliver drugs to a tissue of interest is regarded as optimal without considering its potential harmful effect.45 In general, most of the study report the toxicity of the whole formulation and the toxic effect due to the nanoparticles itself are not mentioned. Consequently, discrimination between toxicity due to drug or nanoparticle has not been made. Therefore, the specific emphasis should be given to the toxicity of the blank nanoparticles (non-drug loaded particles).44 Generally, lipid nanoparticles are considered biocompatible,46 and our in-vitro cytotoxicity study of blank NLC demonstrated that no significant toxicity. However, since there are a variety of cells affected by nanoparticle exposure in the different route of administration, cytotoxicity assays using single-cell studies is not sufficient.47 As a result, we aimed to evaluate the possible toxicity of the materials used to formulate NLC in-vivo. Among many possible methods to study toxicity, we performed repeated dose toxicity study at the therapeutic dose using histological examination, organ indices and body weight variation. Accordingly, during the entire experimental period, no mortality, abnormal clinical sign or behavior were observed in both control and treatment groups. In addition, the blank NLC as well as c(RGD) conjugated NLC did not show any significant changes in body weight over the course of therapy. Organ indices could be used as an indicator in toxicity study.48 As can be seen from Figure 7, no remarkable changes in weight of organ among the groups. More importantly, no abnormal histological changes were observed in major organs collected from all groups of the mice, which suggest the safety of the NLC.
Tumor-specific targeting the chemotherapeutic agents is a key approach to accumulate the drug into the tumor cell, thereby enhancing efficacy and reducing toxicity to the normal cell. To evaluate the targeting ability of the c(RGD)-GA-NLC, we performed the real-time in vivo imaging technique using DiR as the fluorescent probe owing to its NIR excitation and emission wavelength, which could efficiently minimize the interference of animal auto-fluorescence.49 The result of the imaging study also suggests that the dimeric c(RGD) peptide modified DiR-NLC accumulated in the tumor tissue remarkably in comparison to other groups after 24 h of injection.
Conclusion
In the present study, we successfully developed monomeric and dimeric c(RGD) peptides modified GA-NLC to achieve improved drug accumulation, targeting, and antitumor efficiency of GA in the breast cancer cell. The in-vitro cytotoxicity study revealed that c(RGD) peptides decorated GA-NLCs showed significantly higher anticancer activity as compared to unmodified GA-NLC in the lower concentration of GA. Furthermore, the cell uptake study confirmed the better accumulation of c(RGD) peptides conjugated NLC in the 4T1 cell. The in-vivo antitumor studies suggest that c(RGD)-conjugated NLC exhibited enhanced tumor growth inhibition. More importantly, E-[c(RGDfK)2]-GA-NLC showed superior in vivo tumor growth inhibition in contrast to c(RGDfK) modified NLC. The in-vivo bio-distribution study demonstrated that all NLC formulation showed a targeted ability in the tumor site as compared to DiR sol. Furthermore, the ex-vivo imaging of excised organs suggests higher FI was observed in E-[c(RGDfK)2 modified NLC in comparison to unmodified NLC. Finally, considering all findings altogether, it can be concluded that the c(RGD) particularly, E-[c(RGDfK)2]-modified GA-NLC could be a promising approach for targeted therapy of breast cancer.
Acknowledgments
This study was financially supported by the Tianjin Natural Science Foundation (16JCYBJC28200). We would like to acknowledge Mr Joel Wake for his help in improving the language of the manuscript.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. AlQathama A, Prieto J. Natural products with therapeutic potential in melanoma metastasis. Nat Prod Rep. 2015;32:1170–1182. doi:10.1039/c4np00130c
2. Liang L, Zhang Z. Gambogic acid inhibits malignant melanoma cell proliferation through mitochondrial p66shc/ROS-p53/bax-mediated apoptosis. Cell Physiol Biochem. 2016;38(4):1618–1630. doi:10.1159/000443102
3. Wang S, Wang L, Chen M, Wang Y. Gambogic acid sensitizes resistant breast cancer cells to doxorubicin through inhibiting P-glycoprotein and suppressing survivin expression. Chem Biol Interact. 2015;235:76–84. doi:10.1016/j.cbi.2015.03.017
4. Lü L, Tang D, Wang L, et al. Gambogic acid inhibits TNF-α-induced invasion of human prostate cancer PC3 cells in vitro through PI3K/Akt and NF-κB signaling pathways. Acta Pharmacol Sin. 2012;33(4):531–541. doi:10.1038/aps.2011.180
5. Qi Q, Lu N, Li C, et al. Involvement of RECK in gambogic acid induced anti-invasive effect in A549 human lung carcinoma cells. Mol Carcinog. 2015;54(Suppl 1):E13–E25. doi:10.1002/mc.22138
6. Yang Y, Yang L, You Q, et al. Differential apoptotic induction of gambogic acid, a novel anticancer natural product, on hepatoma cells and normal hepatocytes. Cancer Lett. 2007;256(2):259–266. doi:10.1016/j.canlet.2007.06.014
7. Youns M, ElKhoely A, Kamel R. The growth inhibitory effect of gambogic acid on pancreatic cancer cells. Naunyn Schmiedebergs Arch Pharmacol. 2018;391(5):551–560. doi:10.1007/s00210-018-1485-5
8. Li D, Yang H, Li R, et al. Antitumor activity of gambogic acid on NCI-H1993 xenografts via MET signaling pathway downregulation. Oncol Lett. 2015;10(5):2802–2806. doi:10.3892/ol.2015.3719
9. Wang X, Chen W. Gambogic acid is a novel anti-cancer agent that inhibits cell proliferation, angiogenesis and metastasis. Anticancer Agents Med Chem. 2012;12(8):994–1000.
10. Qi Q, You Q, Gu H, et al. Studies on the toxicity of gambogic acid in rats. J Ethnopharmacol. 2008;117(3):433–438. doi:10.1016/j.jep.2008.02.027
11. Chi Y, Zhan XK, Yu H, et al. An open-labeled, randomized, multicenter phase IIa study of gambogic acid injection for advanced malignant tumors. Chin Med J (Engl). 2013;126(9):1642–1646.
12. Lyu L, Huang LQ, Huang T, Xiang W, Yuan JD, Zhang CH. Cell-penetrating peptide conjugates of gambogic acid enhance the antitumor effect on human bladder cancer EJ cells through ROS-mediated apoptosis. Drug Des Devel Ther. 2018;12:743–756. doi:10.2147/DDDT.S161821
13. Liu L, Qi X, Zhong Z, Zhang E. Nanomedicine-based combination of gambogic acid and retinoic acid chlorochalcone for enhanced anticancer efficacy in osteosarcoma. Biomed Pharmacother. 2016;83:79–84. doi:10.1016/j.biopha.2016.06.001
14. Zhang Y, Yang Z, Tan X, Tang X, Yang Z. Development of a more efficient albumin-based delivery system for gambogic acid with low toxicity for lung cancer therapy. AAPS PharmSciTech. 2016;18(6):1987–1997. doi:10.1208/s12249-016-0670-4
15. Li J, Wang X, Shao Y, Lu X, Chen B. A novel exploration of a combination of gambogic acid with TiO2 nanofibers: the photodynamic effect for HepG2 cell proliferation. Materials. 2014;7(9):6865–6878. doi:10.3390/ma7096865
16. He M, Ro L, Liu J, Chu CC. Folate-decorated arginine-based poly(ester urea urethane) nanoparticles as carriers for gambogic acid and effect on cancer cells. J Biomed Mater Res A. 2017;105(2):475–490. doi:10.1002/jbm.a.35924
17. Chen B, Cailian W, Zhang H. Gambogic acid-loaded magnetic Fe3O4 nanoparticles inhibit Panc-1 pancreatic cancer cell proliferation and migration by inactivating transcription factor ETS1. Int J Nanomedicine. 2012;781. doi:10.2147/IJN
18. Zhang D, Zou Z, Ren W, et al. Gambogic acid-loaded PEG-PCL nanoparticles act as an effective antitumor agent against gastric cancer. Pharm Dev Technol. 2018;23(1):33–40. doi:10.1080/10837450.2017.1295068
19. Doddapaneni R, Patel K, Owaid I, Singh M. Tumor neovasculature-targeted cationic PEGylated liposomes of gambogic acid for the treatment of triple-negative breast cancer. Drug Deliv. 2016;23:1232–1241. doi:10.3109/10717544.2015.1124472
20. Wan H, Chen J, Yu X, Zhu X. Titania-coated gold nanorods as an effective carrier for gambogic acid. RSC Adv. 2017;7(78):49518–49525. doi:10.1039/C7RA08560E
21. Mandal A, Bisht R, Pal D, et al. Diagnosis and Drug Delivery to the Brain: Novel Strategies. In: Mitra A, Cholkar K, editors.Emerging nanotechnologies for diagnostics, drug delivery and medical devices. Amesterdam: Elsevier; 2017;355–374.
22. Yokoyama M. Polymeric micelles as drug carriers: their lights and shadows. J Drug Target. 2014;22(7):576–583. doi:10.3109/1061186X.2014.934688
23. Pasquarelli N, Porazik C, Bayer H, et al. Contrasting effects of selective MAGL and FAAH inhibition on dopamine depletion and GDNF expression in a chronic MPTP mouse model of Parkinson’s disease. Neurochem Int. 2017;110:14–24. doi:10.1016/j.neuint.2017.08.003
24. Petros RA, DeSimone JM. Strategies in the design of nanoparticles for therapeutic applications. Nat Rev Drug Discov. 2010;9:615–627. doi:10.1038/nrd2591
25. Lin C, Zhang X, Chen H, et al. Dual-ligand modified liposomes provide effective local targeted delivery of lung-cancer drug by antibody and tumor lineage-homing cell-penetrating peptide. Drug Deliv. 2018;25(1):256–266. doi:10.1080/10717544.2018.1425777
26. Majumder P, Bhunia S, Chaudhuri A. A lipid-based cell penetrating nano-assembly for RNAi-mediated anti-angiogenic cancer therapy. Chem Commun (Camb). 2018;54(12):1489–1492. doi:10.1039/c7cc08517f
27. Zhang Q, Lu L, Zhang L, et al. Dual-functionalized liposomal delivery system for solid tumors based on RGD and a pH-responsive antimicrobial peptide. Sci Rep. 2016;6:19800. doi:10.1038/srep19800
28. White DE, Muller WJ. Multifaceted roles of integrins in breast cancer metastasis. J Mammary Gland Biol Neoplasia. 2007;12(2–3):135–142. doi:10.1007/s10911-007-9045-5
29. Huang R, Li J, Kebebe D, Wu Y, Zhang B, Liu Z. Cell penetrating peptides functionalized gambogic acid-nanostructured lipid carrier for cancer treatment. Drug Deliv. 2018;25:757–765. doi:10.1080/10717544.2018.1446474
30. Rashidi LH, Homayoni H, Zou X, Liu L, Chen W. Investigation of the strategies for targeting of the afterglow nanoparticles to tumor cells. Photodiagnosis Photodyn Ther. 2016;13:244–254. doi:10.1016/j.pdpdt.2015.08.001
31. Salatin S, Yari Khosroushahi A. Overviews on the cellular uptake mechanism of polysaccharide colloidal nanoparticles. J Cell Mol Med. 2017;21(9):1668–1686. doi:10.1111/jcmm.13110
32. Zhang ZH, Wang XP, Ayman WY, Munyendo WL, Lv HX, JP Z. Studies on lactoferrin nanoparticles of gambogic acid for oral delivery. Drug Deliv. 2013;20:86–93. doi:10.3109/10717544.2013.766781
33. DuPre SA, Redelman D, Hunter KW
34. Weidner N. Current pathologic methods for measuring intratumoral microvessel density within breast carcinoma and other solid tumors. Breast Cancer Res Treat. 1995;36:169–180.
35. Danhier F, Le Breton A, Préat V. RGD-based strategies to target alpha(v) beta(3) integrin in cancer therapy and diagnosis. Mol Pharm. 2012;9:2961–2973. doi:10.1021/mp3002733
36. Pozzi D, Colapicchioni V, Caracciolo G, et al. Effect of polyethyleneglycol (PEG) chain length on the bio–nano-interactions between PEGylated lipid nanoparticles and biological fluids: from nanostructure to uptake in cancer cells. Nanoscale. 2014;6:2782. doi:10.1039/c3nr05559k
37. Hada T, Sakurai Y, Harashima H. Optimization of a siRNA carrier modified with a pH-sensitive cationic lipid and a Cyclic RGD peptide for efficiently targeting tumor endothelial cells. Pharmaceutics. 2015;7:320–333. doi:10.3390/pharmaceutics7030320
38. Estelrich J, Busquets M, Morán M. Effect of PEGylation on ligand-targeted magnetoliposomes: a missed goal. ACS Omega. 2017;2:6544−6555. doi:10.1021/acsomega.7b01318
39. Bahrami B, Hojjat-Farsangi M, Mohammadi H, et al. Nanoparticles and targeted drug delivery in cancer therapy. Immunol Lett. 2017;190. doi:10.1016/j.imlet.2017.06.004
40. Hynes R. A reevaluation of integrins as regulators of angiogenesis. Nat Med. 2002;8:918–921. doi:10.1038/nm0902-918
41. Wester HJ, Kessler H. Molecular targeting with peptides or peptide- polymer conjugates: just a question of size? J Nucl Med. 2005;46:1552–1560.
42. Yin D, Yang Y, Cai H, Wang F, Peng D, He L. Gambogic acid-loaded electrosprayed particles for site-specific treatment of hepatocellular carcinoma. Mol Pharm. 2014;11(11):4107–4117. doi:10.1021/mp500214a
43. Dunstan R, Wharton KJ, Quigley C, et al. The use of immunohistochemistry for biomarker assessment - can it compete with other technologies? Toxicol Patho. 2011;139(6):988–1002. doi:10.1177/0192623311419163
44. De Jong W, Borm P. Drug delivery and nanoparticles: applications and hazards. Int J Nanomedicine. 2008;3:133–149. doi:10.2147/IJN.S596
45. Wolfram J, Zhu M, Yang Y. Safety of nanoparticles in medicine. Curr Drug Targets. 2015;16:1671–1681. doi:10.2174/1389450115666140804124808
46. Kim M, Kwon S-H, Choi J, Lee A. A promising biocompatible platform: lipid-based and bio-inspired smart drug delivery systems for cancer therapy. Int J Mol Sci. 2018;19(12):3859. doi:10.3390/ijms19123859
47. Kong B, Seog JH, Graham LM, Lee SB. Experimental considerations on the cytotoxicity of nanoparticles. Nanomedicine. 2011;6(5):929–941. doi:10.2217/nnm.11.77
48. Hong T-K, Tripathy N, Son H-J, et al. A comprehensive in vitro and in vivo study of ZnO nanoparticles toxicit. J Mater Chem B. 2013;1:2985. doi:10.1039/c3tb20251h
49. Sharma P, Brown S, Walter G, et al. Nanoparticles for bioimaging. Adv Colloid Interface Sci. 2006;123:471–485. doi:10.1016/j.cis.2006.05.026
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.