Back to Journals » Cancer Management and Research » Volume 11
Dihydrodiosgenin inhibits endothelial cell-derived factor VIII and platelet-mediated hepatocellular carcinoma metastasis
Authors Zhuang M, Xin G, Wei Z, Li S, Xing Z, Ji C, Du J, Niu H, Huang W
Received 19 January 2019
Accepted for publication 20 April 2019
Published 31 May 2019 Volume 2019:11 Pages 4871—4882
DOI https://doi.org/10.2147/CMAR.S202225
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Xueqiong Zhu
Manjiao Zhuang,1 Guang Xin,1 Zeliang Wei,1 Shiyi Li,1 Zhihua Xing,1 Chengjie Ji,1,2 Junrong Du,1 Hai Niu,1,3 Wen Huang1
1Laboratory of Ethnopharmacology, West China School of Pharmacy, West China Hospital, Sichuan University, Chengdu, Sichuan, People’s Republic of China; 2Department of Laboratory Medicine, Sichuan Academy of Medical Sciences & Sichuan Provincial People’s Hospital, Chengdu, Sichuan, People’s Republic of China; 3College of Mathematics, Sichuan University, Chengdu, Sichuan, People’s Republic of China
Background: Our previous studies have demonstrated that diosgenin and diosgenin derivatives exhibit excellent antithrombotic activity via regulating platelet function and coagulation factor level. Platelets and blood coagulation system are highly associated with tumor hematogenous metastasis. Therefore, the purpose of this study was to evaluate whether dihydrodiosgenin (dydio) mediated-platelet inhibition or coagulation factor level modulation is involved in hepatocellular carcinoma cell (HCC) metastasis.
Methods: Cell viability was examined by MTT and colony formation assays. Platelet aggregation text and morphology were used to assess dydio’s role on tumor cell-induced platelet activation (TCIPA). Scratch assay, adhesion assay and Western blot were used to evaluate dydio’s role on platelet-mediated metastasis. Western blot and fluorescence detection were performed to clarify dydio’s role on endothelial cell (EC) function. The mice lung metastasis model was constructed to investigated dydio’s function on coagulation factor and platelet-mediated metastasis.
Results: This study found that pretreatment with dydio caused a significant inhibition of TCIPA. Platelets exposed to dydio significantly inhibited their adhesion to tumor cells, meanwhile, releasates of platelets that pretreated with dydio led to diminished cancer cell proliferation and migration along with the increase of epithelial markers E-cadherin and loss of mesenchymal phenotype. Additionally, ECs pretreated with dydio suppressed factor VIII (FVIII) level which in turn restrained the activation of platelets and the adhesion of cancer cells or platelets to ECs. Interestingly, our study demonstrated that FVIII could promote HCC proliferation. In vivo study revealed that mice intragastrical (i.g.) administration with dydio significantly inhibited the lung metastasis of hepal-6 cells which is highly correlated with the altered platelet function and coagulation level.
Conclusion: Taken together, these results demonstrated that dydio altered platelet function and coagulation FVIII level, resulting in decreased metastatic potential of HCC. Thus, our study reveals that dydio exerts novel mechanisms of antitumor action beside its direct antitumor activity.
Keywords: dihydrodiosgenin, hepatocellular carcinoma, platelet, endothelial cell, tumor microenvironment, FVIII
Introduction
Hepatocellular carcinoma (HCC) is the fifth most common cancer and the third most frequent cause of cancer death worldwide, with its global incidence appears to be increasing sharply.1 Venous thromboembolism is frequently found in patients with cancer that contributes to patients’ morbidity.2,3 Approximately 50–80% of hepatocellular carcinoma patients are accompanied by portal or hepatic vein invasion and tumor thrombus is recognized as a poor prognosis in hepatocellular carcinoma patients.4–7 Despite advances in therapeutic strategies of hepatocellular carcinoma, once tumor advanced along with tumor thrombus, it may result in death.8 Therefore, it is critical to develop novel therapeutic strategies on hepatocellular carcinoma and related thrombosis treatment and explore its mechanism.
Aberrant platelet activation or aggregation frequently occurs in the vasculature of cancer patients, especially in patients with metastatic tumors.9 Extensive evidence indicated that tumor cells could activate platelets to form tumor thrombus—a process identified as tumor cell-induced platelet activation (TCIPA).10,11 Platelets within tumor microenvironment can regulate cancer cell survival as well as hematogenous metastasis.12 They surround tumor cells and assist tumor metastasis by protecting circulating tumor cells from sheer forces or elimination by the immune system, facilitating their adhesion to vascular endothelial cells (ECs).13 It is worth noting that platelets might also contribute to the development of HCC.14–16 Lisman and Luyendyk demonstrated that platelets played an important role in HCC growth and metastasis.17 Meanwhile, platelet lysate has been found to stimulate proliferation of four different HCCs.18 A recent report from Zhang et al showed that elevated platelet activation and circulating ADP levels inhibited HCC differentiation and promoted tumor progression via platelet-tumor cell binding,19 supporting a tight connection between platelet and HCC. Therefore, platelet is a promising therapeutic target for HCC treatment.
ECs exhibit essential role in the control of vascular function, especially in blood coagulation function. It secretes a variety of molecules involved in regulating platelet function and blood coagulation which is highly associated with thrombosis.20,21 It is notable to mention that ECs are the major source of factor VIII (FVIII).22 While high FVIII has been observed in patients with malignancy.23,24 Therefore, improve coagulation abnormalities through targeting ECs may be a potential strategy in the treatment of cancer. Additionally, the ECs that line blood vessels are the first cells in contact with tumors, thus possessing important role in suppressing metastasis of tumors.25 The study by Chouaib et al showed that ECs are the interface between circulating blood cells, tumor cells and the extracellular matrix, thereby playing a central role in controlling tumor cell behavior and metastasis formation.26 Through paracrine signals or direct contact, ECs regulate various aspects of cancer cell function, including proliferation as well as metastasis.27,28 Therefore, platelets and ECs within the tumor microenvironment are recognized as the main targets for the treatment of tumor hematogenous metastasis as well as tumor thrombus. It is critical to develop a novel therapeutic strategy targeting platelets or ECs on HCC treatment.
Diosgenin is a phytosteroid sapogenin, extracted from Dioscorea zingiberensis.29 It displays broad pharmacological activities such immunity-regulating, antitumor, anti-inflammation as well as antithrombotic.30–32 Extensive studies have well demonstrated that diosgenin exerts direct antitumor action.33 Additionally, various reports have demonstrated that diosgenin exhibited a specific activity to induce apoptosis on HCC. For example, Li et al demonstrated that diosgenin induced G2/M cell cycle arrest and apoptosis in human HCC.34 Kim et al showed that diosgenin induced HepG2 cells apoptosis via ROS generation and mitochondrial dysfunction.35 Our previous studies demonstrated that diosgenin and relevant derivatives exerted the antithrombosis activity on arterial and venous thrombosis by inhibiting platelet aggregation and improving the anticoagulation function as characterized by a significant decrease of coagulation FVIII level.36–38
Dihydrodiosgenin (dydio), a parent aglycone of diosgenyl saponin, was synthesized by opening spiroacetal ring of diosgenin (Figure 1).39 In this study, we aim to investigate the pharmacological effects of dydio on coagulation and platelet-mediated metastasis. Our study demonstrated that dydio constrained TCIPA and substantially dampened platelet adhesion to tumor cell as well as prometastatic effects of platelets. Meanwhile, dydio treatment decreased endothelial-derived FVIII level which probably resulted in an inhibition of platelet activation and abrogation of tumor cell and ECs interaction. Taken together, our study demonstrates that dydio exerts novel mechanisms of antitumor action.
 | Figure 1 Structure of dydio. Abbreviation: dydio, dihydrodiosgenin. |
Materials and methods
Materials
DMEM, FBS, penicillin and streptomycin were purchased from GIBCO (Carlsbad, CA, USA). Cell cycle detection kit, 3,3′-dioctadecyloxacarbocyanine perchlorate (DiO), 3,3′-dioctadecyloxacarbocyanine perchlorate (Dil), BCA protein assay kit and rapamycin were purchased from Beyotime Institute of Biotechnology (Shanghai, China). FVIII was purchased from Prospec (St Louis, MO, USA). Unless indicated otherwise, the other reagents were purchased from Sigma (St Louis, MO, USA).
Cell culture
Murine hepatocellular carcinoma cell line Hepal-6, human hepatocellular carcinoma cell line HepG2 and human umbilical vein cell (HUVEC) were received from Regenerative Medicine Research Center, West China Hospital, and were approved for use in experimental research by the Ethics Committee of West China Center of Medical Sciences, Sichuan University. All cell lines were maintained in DMEMsupplemented with 10% FBS, penicillin (100 U/mL) and streptomycin (0.1 mg/mL). Cells were sustained at 37°C in a humidified atmosphere with 5% CO2.
Cell viability assay
The HepG2 and Hepal-6 cells were cultured in 96-well plates at a density of 5×103 per well. Once the confluence reached to 80%, cells were incubated with different concentrations of dydio (0, 12.5, 25, 50, 100 and 200 μmol/L) or releasates of platelets pretreated with dydio or DMSO for 24 hours. Then cell viability was measured by MTT assay (Sigma) according to the manufacturer’s protocol.
Colony formation
Colony formation assay was performed as previously described.40 HepG2 or Hepal-6 were seeded into 6-well plates at 1,000 cells/mL and allowed to attach for 24 hours. After that cells were treated with 35 μmol/L dydio. Cells were incubated with fresh medium once every 3 days for additional 12 days and stained with 0.5% crystal violet after 4% paraformaldehyde fixing. Then, the morphology of cells was observed by a microscope (uX71; Olympus Corp., Tokyo, Japan) and the survival fraction of the clones was used to evaluate the effects of different treatments.
Preparation of platelets
Mouse whole blood was collected as described previously,41 mce were anesthetized with intraperitoneal pentobarbital sodium (40 mg/kg), and whole blood was collected by inferior vena and anticoagulated with 3.8% trisodium citrate (9:1, v/v). Platelet-rich plasma (PRP) was prepared by centrifugation at 100 g for 10 minutes. Platelet-poor plasma (PPP) was obtained by centrifugation at 160 g for 10 min. Platelets was obtained by centrifugation at 1,000 g for 5 min from PRP. Final samples were resuspended in modified Tyrode buffer. Platelet purity was confirmed with CD41a expression by flow cytometric measurement. Platelets were washed with Tyrode’s buffer for future experiment.
Platelets aggregation assay
The platelet pre-incubated with 35 mmol/L dydio for 3 hours at 37°C, aggregation assay was performed at 37 °C by an LBY-NJ4 aggregometer (Pulisheng, Beijing, China). Aggregation was initiated by exposure to 1x105/mL Hepal-6 cells as previous study.42 Aggregation was recorded for 5 minutes, and data were expressed as the change in light transmission.
For in vivo platelets aggregation assay, whole blood was collected into tubes with citrate anticoagulant from anesthetized experiment mice in lung metastasis model. The platelet aggregation assay was carried out at 37°C by a LBY-NJ4 aggregometer (Pulisheng, Beijing, China). Aggregation was initiated by the addition of ADP solution (final concentration, 20 μm).
Platelet morphology
For microscopic observation, platelets were fixed with 1% paraformaldehyde for 1 hour and loaded on a coverslip chamber. Platelets morphology was observed using an inverted microscope (uX71; Olympus Corp., Tokyo, Japan).
Scratch assay
The Hepal-6 cells were seeded in six-well plates and incubated for 12 hours in starvation medium. The cellular monolayer was wounded with a sterile 1,000 μL-pipette tip and washed with starvation medium to remove detached cells from the plates. The cells were incubated with platelets releasates and photographed by using a phase-contrast microscope. Quantification of relative “scratch” was carried out by using the Image J software (NIH, MD, USA).
Adhesion assay
The adhesion assay was performed as previous study.10 Hepal-6 cells were incubated in 6-well plates until the cellular confluence reached 90%. Platelets pretreated with dydio or not were co-incubated with the Hepal-6 culture plate at 37°C for 4 hours. The nonadherent platelets were washed and the adhesion status was detected under a fluorescence microscope.
In some condition, HUVECs were plated in 6-well plates until the cellular confluence reached 70% and then incubated with 35 μmol/L dydio for 24 hours. Thereafter, dil-labeled platelets and dio-labeled Hepal-6 or HepG2 cells were co-incubated with the HUVEC culture plate at 37°C for 4 hours in the absence or presence of 2 U/mL FVIII (platelets: HUVECs 1,000: 1) under static condition. The nonadherent platelets and tumor cells were washed by PBS. The adhesion status was detected under a fluorescence microscope.
Western blot
Western blot was performed as previously described.43 Antibody E-cadherin and FVIII were purchased from Santa Cruz Biotechnology Inc. (Dallas, TX, USA); Quantifications of relative protein expressions were carried out by using the Image Lab 3.0 software (Bio-Rad Laboratories Inc., Hercules, CA, USA).
Bleeding time
The bleeding time was measured as previously described.44 Briefly, the mouse tail vein was severed 2 mm from its tip, and the tail was immersed in normal saline (37°C) immediately. Bleeding time from the incision to the cessation was recorded. A bleeding time of 900 seconds was used as the cutoff time for the purpose of statistical analysis.
Anticoagulation activity assay
FVIII activity was detected as previous study.45 Onestage clotting assay was used for measuring coagulation FVIII activities. Mouse plasma was ten times diluted in human factor deficient plasma, and the activities of FVIII was detected by a SYSMEX CA-7000 automatic Coagulometer (Sysmex, Kobe, Japan).
Experimental tumor metastasis model
Male C57BL/6J mice (4 weeks old, 18–22 g) were purchased from Chengdu Dossy experimental animal Co. Ltd. (Chengdu, China). After 1 week of acclimatization, mice were injected intravenously via the tail vein with 1×106 Hepal-6 cells to construct experimental lung metastasis. The mice were randomly divided into two groups (N=6 per group), model group, dydio group. Sixmale C57Bl/6J mice without Hepal-6 cells injection were considered as vehicle control. Dydio group were treated with 100 mg/kg/d dydio by oral administration, model or vehicle control group were administrated with same volume of saline. All animals were sacrificed on the 21st day, followed by a comprehensive visual examination of all organs. Lungs were collected and fixed in 4% paraformaldehyde and embedded and sectioned at 4 μm for H&E. Ethical approvals for all experiments have been obtained from the Ethics Committee of West China Center of Medical Sciences, Sichuan University (Approval 2018121A), and were performed in accordance with ARRIVE guidelines.46
Statistical analysis
Data are presented as means ±SD. The statistical analysis of results was performed by Student’s paired t-tests and ANOVA. A value of P<0.05 was considered statistically significant.
Results
Dydio treatment inhibits the cell viability of HCC and TCIPA
The effect of dydio on the cell viability of HCCs was determined by using MTT (3-(4,5-dimethyl-2-thia-zolyl)-2, 5-diphenyl 2H-tetrazolium bromide) assay. Dydio inhibited both HepG2 and Hepal-6 cells viability in a concentration dependent manner, with IC50 values of 35.83 μm and 31.26 μm respectively (Figure 2A). Results of colony formation assay revealed that cells treated with 35 μm/L dydio displayed much smaller and fewer colonies (Figure 2B). According to the IC50 value and more than 50% of viable cell number after dydio treatment, 35 μm was employed as the optimal concentration for treatment in the following experiments.
 | Figure 2 Effect of dydio on HCC viability and TCIPA. (A) Effect of increasing concentration of dydio on HepG2 or Hepal-6 cell viability. (B) 35 μmol/L dydio on HepG2 or Hepal-6 cell colony formation. (C) Effect of dydio on platelet aggregation induced by Hepal-6 tumor cells. (D) Effect of dydio on morphology change of platelet induced by Hepal-6 tumor cells (magnification ×100).*p<0.05, **p<0.01 vs 0 μmol/L or TC group.Abbreviations: Ctr, control; dydio, dihydrodiosgenin; TC, tumor cells; HCC, hepatocellular carcinoma; TCIPA, tumor cell-induced platelet activation. |
To investigate inhibitory potential of dydio in cancer metastasis, we next studied dydio on TCIPA. When platelets were exposed to Hepal-6 cells, platelet aggregation was significantly inhibited by additional dydio (Figure 2C), demonstrating that dydio may exert potential antiplatelet activity. Pseudopodium was an essential structure during the platelet adhesion to tumor cells.47 As depicted in Figure 2D, tumor cells stimulated platelets to flatten, protrude pseudopodium and cluster, and the morphological transformation was associated with platelet activation. By comparison, dydio significantly inhibited filopodia extension of adherent platelets, confirming that the platelets inhibited activity of dydio.
Dydio suppresses HCC viability and migration via inhibiting platelet activation
Platelet-cancer cell interactions play a key role in successful haematogenous metastasis. Distant metastases mainly occur via the blood circulation, and platelets were suggested to be the first blood cells to interact with tumor cells.48 Therefore, we investigated the effect of dydio on platelet adhesion to the tumor cells. The results demonstrated that dydio pretreatment significantly decreased platelet adhesion to Hepal-6 cells (Figure 3A).
 | Figure 3 Dydio treatment inhibits platelet-mediated tumor cells migration. (A) Representative epifluorescence microcopy images of dil-labeled platelets (red) adhering to tumor cells (magnification ×100). (B) Platelets were pretreated with 0 or 35 μmol/L dydio, washed, and activated with Hepal-6 tumor cells to generate releasates. (C) Cell viability was assessed after 24 hours of exposure to platelet releasates. (D) Tumor cell migration was assessed after 24 hours of exposure to platelet releasates (magnification ×100). (E) E-cadherin expression was detected after 24 hours of exposure to platelet releasates. (F) Phase-contrast micrographs of Hepal-6 cell after 24 hours of exposure to platelet releasates (magnification ×200).**p<0.01 vs Plt releasates group. Abbreviations: Ctr, control; dydio, dihydrodiosgenin; TC, tumor cells; Plt, platelets. |
To establish the role of dydio on platelet-mediated Hepal-6 cell proliferation or migration, platelets from mice were pretreated with dydio or PBS, washed, stimulated with Hepal-6 tumor cells, and then platelet releasates were collected and incubated with Hepal-6 cells (Figure 3B, schematic). Treatment with platelets releasates resulted in a significant increase in both Hepal-6 cells viability and migration compared with that in the control group. While releasates of platelet that pretreated with dydio exhibited less influence on Hepall-6 cells viability and migration (Figure 3C and D). Platelets in the blood are capable of initiating the induction of epithelial-mesenchymal transition (EMT) in cancer cells through releasing a variety of growth factors, thus promoting the metastasis of tumor cells.49,50 To test whether dydio was involved in platelet induced-EMT, the expression of E-cadherin was detected. As depicted in Figure 3E, tumor cell-activated platelet releasates significantly decreased E-cadherin expression in Hepal-6 cells in comparison with controls (Figure 3E), whereas releasates of platelet pretreated with dydio did not affect E-cadherin expression. Moreover, upon exposure to platelets releasates, Hepal-6 cells adopted a mesenchymal phenotype with disappearance of defined cell–cell contacts, elongated morphology and growing individually, often crossing over each other, while Hepal-6 cells did not acquire a mesenchymal morphology when exposure to dydio-treated platelets releasates (Figure 3F).
Collectively, these data demonstrate that dydio treatment inhibits tumor cells migration via altering platelet function.
Dydio impairs platelet, tumor cell and ECs interaction via suppressing EC-derived FVIII level
Our previous studies demonstrated that diosgenin derivatives could significantly reduce FVIII level in animal model.36,37 FVIII is mainly produced in ECs, therefore we investigated FVIII expression during dydio treatment in HUVEC. Figure 4A showed that dydio decreased the expression of FVIII which in consistence with our previous studies. Meanwhile, dydio has also been shown to suppress secretion of FVIII which resulted in a lower FVIII level in the HUVEC culture medium (Figure 4B).
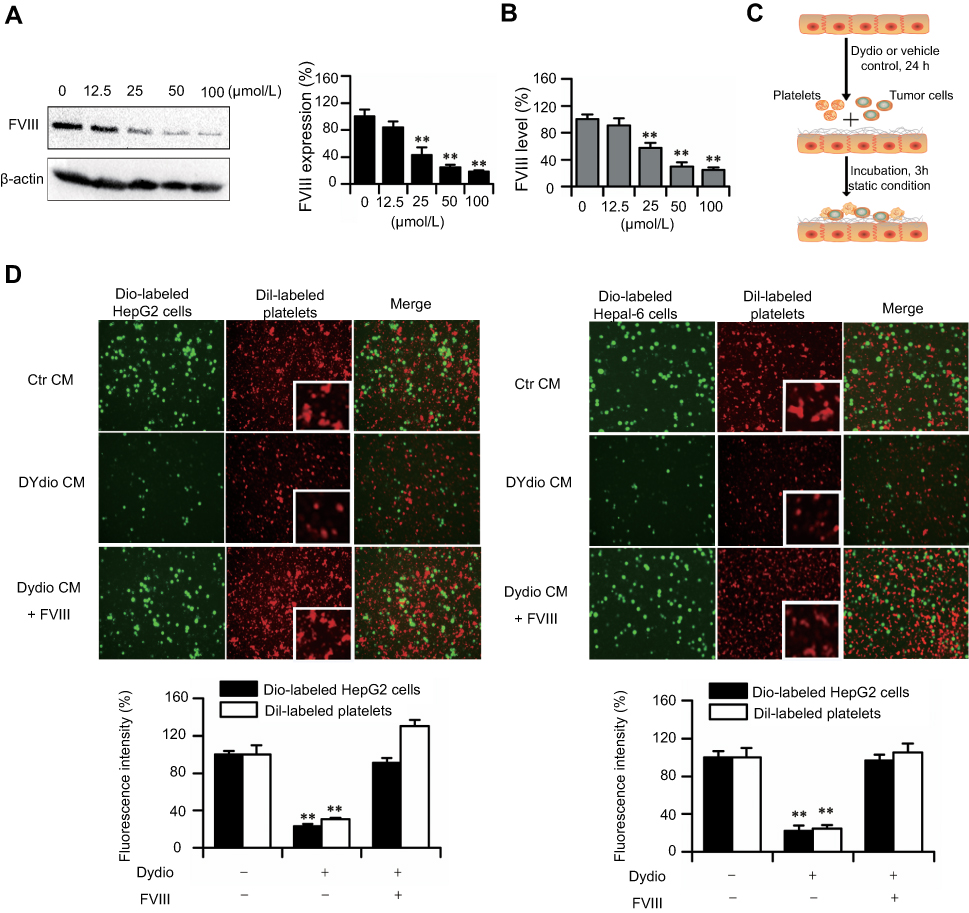 | Figure 4 Dydio treatment abrogates platelet, tumor cell and ECs interaction via decreasing EC-derived FVIII release. (A) Dydio decreases the expression of FVIII in HUVEC. (B) Dydio reduce the FVIII secretion. (C) Schematic of dydio treatment on ECs. ECs were pretreated with 35 μmol/L dydio or vehicle control, washed, and then tumor cells or platelets were added and incubated for 3 hours in the static condition, washed to observe cells adhesion. (D) Representative epifluorescence microcopy images of dio–labeled HCC (green; left, HepG2; right, Hepal-6) and dil-labeled platelets (red) adhering to HUVEC (magnification ×100). **p<0.01 vs 0 μmol/L or Ctr CM.Abbreviations: Ctr, control; dydio, dihydrodiosgenin; TC, tumor cells; Plt, platelets; CM, culture media; HUVEC, human umbilical vein cell; ECs, endothelial cells; FVIII, factor VIII. |
Kellie et al demonstrated that elevated FVIII level is capable of accelerating platelet activation.51 To determine whether dydio affect platelet-tumor-EC interaction via modulating endothelial-derived FVIII secretion, dil-labeled platelets or dio-labeled tumor cells were allowed to adhere to ECs under static conditions (Figure 4C). As depicted in Figure 4D, treatment HUVEC with dydio resulted in a significant reduction in washed mice platelets or HepG2/Hepal-6 cells recruitment to ECs and inhibited morphological activation of platelets (see larger image) as compared with controls. It can also note from the fluorescence images that interaction of cancer cells and platelets were significantly decreased after treating with dydio. While additional FVIII restored dydio-inhibited morphological activation of platelets (see larger image) and the interaction between tumor cells, platelets and ECs.
Therefore, it can be concluded that dydio abrogates platelet activation as well as the adhesion of cancer cells and platelets to ECs via decreasing FVIII secretion.
FVIII promotes HCC proliferation
Liver diseases including liver metastasis of carcinoma are associated with markedly elevated plasma FVIII level.52 Liver sinusoidal endothelial cells are one of the main sources of FVIII. Therefore, we speculated dydio might regulate HCC progression via modulating FVIII level (Figure 5A). To elevated FVIII on HCC progression, MTT assay was carried out. FVIII accelerated the proliferation of HepG2 and Hepal-6 cells in a concentration dependent manner (Figure 5B and C). Flow cytometer was conducted to observe the effect of FVIII on cell cycle distribution. FVIII could promote the proliferation of Hepal-6 cell which reflected by influencing the distribution of cell cycle (Figure 5D). The G1 phase was significantly decreased from 54.82% to 48.14%, while the number of cells in the S phase gradually increased from 27.52% to 34.49%.
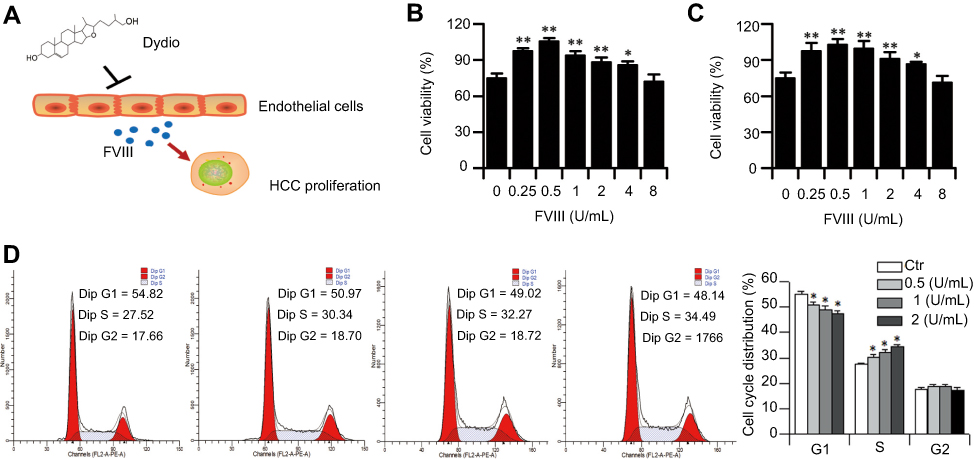 | Figure 5 FVIII promotes HCC proliferation. (A) Schematic overview of the role of FVIII in dydio-mediated tumor progression. (B and C) Cell viability of HepG2 or Hepal-6 was detected after 24-hour treatment with increasing concentrations of FVIII (0, 0.25, 0.5, 1, 2, 4 and 8 U/mL). (D) Number of DNA cell cycle rate in Hepal-6 cells were analyzed by using flow cytometry.*p < 0.05, **p < 0.01 vs 0 μmol/L or Ctr group.Abbreviations: Ctr, control; HCC, hepatocellular carcinoma; dydio, dihydrodiosgenin; FVIII, factor VIII; Dip G1, diploid first gap; Dip S, diploid synthesis; Dip G2, diploid second gap. |
Dydio inhibits hepal-6 lung metastasis highly correlated with the altered platelet function and coagulation level
To determine whether dydio-inhibited platelets activation is involved in tumor metastasis and related thrombosis in vivo, a well-characterized Hepal-6 hepatocellular carcinoma cell model of tumor metastasis was employed. Histologic analyses of the lung tissue revealed that the number of spontaneous lung metastases was reduced in mice treated with dydio when compared with model group (Figure 6A). Furthermore, the weights of freshly harvested lungs from mice treated with dydio were lower than those harvested from model mice (Figure 6B). The increased lung weight in the control mice is probably due to the metastatic burden of the tumors. In addition to less spontaneous lung metastases, mice treated with dydio also have significantly fewer and smaller thrombi in their lungs and pulmonary vasculature than those found in the model mice. (Figure 6C and D).
 | Figure 6 Dydio inhibits hepal-6 tumor cells lung metastasis highly correlated with the altered platelet function and coagulation level. (A) Representative photographs obtained from lungs of Hepal-6-injected mice. (B) The weight of lungs in each group. (C) H&E staining for lung tissues demonstrated tumor-induced thrombi formation within lung tissue. The stars indicate the tumor-induced thrombi. (D) Quantification of thrombi formation within lung tissue. (E) Platelets aggregation induced by ADP in vivo. (F) Platelets count in each group. (G) Blood clotting time in each group. (H) FVIII activities in each group.*p<0.05, **p<0.01 vs Model group.Abbreviations: Ctr, control; dydio, dihydrodiosgenin; FVIII, factor VIII. |
To clarify the role of dydio on thrombosis, in vivo platelet aggregation assay was carried out. As shown in Figure 6E, platelet from tumor-bearing mice treated with dydio exhibited a lower platelet aggregation as compared with model group. While there was no significant change in platelet counts among different groups (Figure 6F). Bleeding time is a classic index of a study of antithrombotic effect in vivo. It may be noted that the tumor-bearing mice treated with dydio exert shorter blood clotting time than the model group (Figure 6G). Moreover, our data showed that tumor-bearing mice displayed a higher FVIII level when compared with the control group. While dydio showed noticeable effects in reducing FVIIII level (Figure 6H). Taken together, these data suggested dydio exhibits ideal anticancer activity which is highly correlated with the modulation of platelet function and coagulation level.
Discussion
Interactions of tumor cells with its microenvironment are sufficient to prime tumor cells for initial metastasis. Platelets and ECs within tumor microenvironment are characterized as vital therapeutic targets for the treatment of hematogenous metastasis as well as tumor thrombus. Our study demonstrated that dydio inhibited HCC metastasis via inhibiting platelet activation as well as decreasing ECs-derived FVIII. Overall, our study reveals that dydio exhibits novel mechanisms of antitumor activity.
Diosgenin, a steroidal saponin presents in a variety of plants, was found to affect various phases of tumorigenesis including proliferation inhibition, apoptosis induction, migration and angiogenesis suppression.31,33,53 The mechanism of diosgenin and its derivatives involving in tumor metastasis has been well elucidated. The direct antitumor activities of diosgenin are possibly involved in PI3K/Akt, MAPKs and NF-κB signaling pathways.33 Meanwhile, various studies have demonstrated that diosgenin could induce HCC cell apoptosis.34,35 Our results are in accordance with the previous studies, confirming that dydio significantly inhibits HCC viability with its IC50 around 35 μmol/L.
Beside the direct anticarcinogenic action, our previous studies have demonstrated that diosgenin and its derivatives exert antithrombosis activity on the thrombosis model via improving the anticoagulation function as well as inhibiting platelet aggregation.36–38,45 It is widely accepted that the formation of tumor thrombus involving platelets is one of the most essential processes in tumor hematogenous metastasis. This physical interaction might allow platelets to shield cancerous cells from high shear forces or immune response, thus facilitating metastasis. Recently, a growing number of studies have focused on antiplatelet agent as cancer therapeutics. For example, extensive literatures have demonstrated that aspirin-mediated inactivation of platelets could suppress tumor metastasis.54 Our data demonstrated that dydio inhibited TCIPA as well as morphological activation of platelets which were characterized by less pseudopodium. Consistently, platelets treated with dydio displayed a weaker adhesion capacity to Hepal-6 tumor cells.
Moreover, dydio was demonstrated to alter the releasates of platelets turnover to inhibit tumor progression. TCIPA triggers secretion of pro-angiogenic and protumorigenic factors from platelets which display essential role in tumor progression.55,56 For example, platelet-derived nucleotides promote tumor-cell transendothelial migration and metastasis via P2Y2 receptor.57 Our study showed that releasates from platelets treated with dydio significantly suppressed Hepal-6 cell viability and migration. The EMT is a crucial cellular program during tumor migration, invasion and metastasis, during which tumor cells lose epithelial markers and gain mesenchymal traits.58 It is notable to mention that platelet secretion is able to induce EMT, thus resulting in an enhanced tumor metastasis.50 Our results demonstrated dydio could inhibit platelet-induced HCC EMT which were characterized by increased E-cadherin expression and loss of mesenchymal phenotype. These data strongly suggest that dydio may suppress HCC by limiting the proliferative and prometastatic effects of platelets releasates. However, to clarify the role of dydio in HCC progression, future studies are needed to examine altered factors within platelet releasates pretreated by dydio.
In addition to inhibiting platelet activation, dydio inhibited the release of FVIII from ECs which in turn restrained the activation of platelets and the adhesion of cancer cells or platelets to ECs. Beside the unique function in hemostasis, ECs that line blood vessels are the first cells in contact with tumor cells and platelets, thus normally acting as a physiological barrier in suppressing cancer cell dissemination to distant sites.27 Subtle changes in ECs can be easily transmitted to the tumors with profound effects on cancer fate. Meanwhile, EC is the main source of an essential blood-clotting protein FVIII.59 While FVIII is capable of accelerating platelet activation.51 Our study demonstrated that dydio inhibited the release of FVIII from ECs which in turn restrained platelet activation and the interaction between tumor cells, platelets and ECs. These altered interactions lead to disruption of the physical shield around tumor cells which further destroys tumor cell adhesion to the endothelium.
In 1865, Armand Trousseau first described the relationship between activation of the blood coagulation system and carcinogenesis. Thereafter many studies, both human and animal, have suggested a link between cancer development and blood coagulation disorders.60 Jae et al havedemonstrated that coagulation facilitates tumor cell spreading in the pulmonary vasculature during early metastatic colony formation.61 Liver metastasis of carcinoma are highly closely related to elevated plasma FVIII level.52 Interestingly, our study demonstrated that FVIII could significantly induced Hepal-6 proliferation via affecting cell cycle distribution. Thus, FVIII may represent a new molecular target or assistant index for diagnosis of HCC therapy. Taken together, it can be concluded that dydio may control tumor metastasis via improving coagulation abnormalities. However, an in-depth study is needed to investigate FVIII role of HCC progression.
Thromboembolism disease is a frequent complication of malignant tumor, which is closely associated with the progression, therapy and prognosis of cancer. Thus, it is urgent to develop novel therapeutic strategies targeting both tumor and tumor microenvironment. Traditional tumor chemical therapy is to kill and wound cancer cells directly with numerous side effects. Compared with them, our study demonstrated that dydio not only possesses ideal antitumor activity, but also exerts excellent activity in improving tumor thrombus through targeting tumor microenvironment.
Conclusion
Taken together, dydio displayed antimetastasis of HCC both in vivo and in vitro via modulating platelet function and FVIIII level (Figure 7). Thus, in addition to direct action toward tumor cells, our study demonstrated that dydio exerts novel mechanisms of antitumor action. Moreover, our study supports the idea of utilizing targeted platelet therapies to inhibit platelet-mediated malignancy.
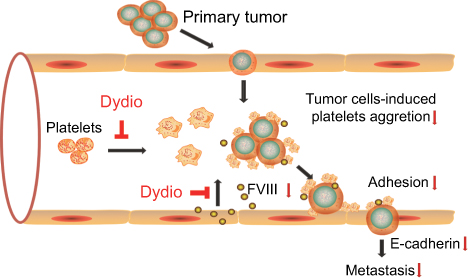 | Figure 7 Schematic overview of the role of dydio in tumor metastasis. Abbreviations: dydio, dihydrodiosgenin; FVIII, factor VIII. |
Acknowledgments
This work was financed by grant-in-aid for scientific research from the National Natural Science Foundation of China (Grant No. 81673710), the National Science Foundation for Young Scientists of China (Grant No. 81803866), the project funded by China Postdoctoral Science Foundation (Grant No. 2018M640932) and the Applied Basic Research Programs of Department of Science and Technology of Sichuan Province (Grant No. 19YYJC2473).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Chiba T, Iwama A, Yokosuka O. Cancer stem cells in hepatocellular carcinoma: therapeutic implications based on stem cell biology. Hepatol Res. 2016;46(1):50–57. doi:10.1111/hepr.12548
2. Khorana AA, Francis CW, Culakova E, et al. Thromboembolism is a leading cause of death in cancer patients receiving outpatient chemotherapy. J Thromb Haemost. 2010;5(3):632–634. doi:10.1111/j.1538-7836.2007.02374.x
3. Sørensen HT, Mellemkjaer L, Olsen JH, Baron JA. Prognosis of cancers associated with venous thromboembolism. N Engl J Med. 2000;343(25):1846–1850. doi:10.1056/NEJM200012213432504
4. Minagawa M, Makuuchi M, Takayama T, Ohtomo K. Selection criteria for hepatectomy in patients with hepatocellular carcinoma and portal vein tumor thrombus. Ann Surg. 2001;233(3):379–384.
5. Chen XP, Qiu FZ, Wu ZD, et al. Effects of location and extension of portal vein tumor thrombus on long-term outcomes of surgical treatment for hepatocellular carcinoma. Ann Surg Oncol. 2006;13(7):940–946. doi:10.1245/ASO.2006.08.007
6. Llovet JM, Bustamante J, Castells C, et al. Natural history of untreated nonsurgical hepatocellular carcinoma: rationale for the design and evaluation of therapeutic trials. Hepatology. 2010;29(1):62–67. doi:10.1002/hep.510290145
7. Lv WF, Liu KC, Lu D, et al. Transarterial chemoembolization for hepatocellular carcinoma combined with portal vein tumor thrombosis. Cancer Manag Res. 2018;10:4719–4726. doi:10.2147/CMAR.S166527
8. Vishwanath IP, Zakiah SS, Qi CP, et al. Development and novel therapeutics in hepatocellular carcinoma: a review. Therap Clin Risk Manag. 2016;12:445–455.
9. Mezouar S, Frère C, Darbousset R, et al. Role of platelets in cancer and cancer-associated thrombosis: experimental and clinical evidences. Thromb Res. 2016;139:65–76. doi:10.1016/j.thromres.2016.01.006
10. Zhang W, Dang SY, Hong T, et al. A humanized single-chain antibody against beta 3 integrin inhibits pulmonary metastasis by preferentially fragmenting activated platelets in the tumor microenvironment. Blood. 2012;120(14):2889–2898. doi:10.1182/blood-2012-04-425207
11. Zhao L, Thorsheim CL, Suzuki A, et al. Phosphatidylinositol transfer protein-α in platelets is inconsequential for thrombosis yet is utilized for tumor metastasis. Nat Commun. 17;8(1):1216. doi:10.1038/s41467-017-01181-4
12. Yan MJ, Jurasz P. The role of platelets in the tumor microenvironment: from solid tumors to leukemia. BBA-Mol Cell Res. 2016;1863(3):392–400.
13. Gay LJ, Feldinghabermann B. Contribution of platelets to tumour metastasis. Nat Rev Cancer. 2011;11(2):123–134. doi:10.1038/nrc3004
14. Franco AT, Corken A, Ware J. Platelets at the interface of thrombosis, inflammation, and cancer. Blood. 2015;126(5):582–588. doi:10.1182/blood-2014-08-531582
15. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19(11):1423–1437. doi:10.1038/nm.3394
16. Menter DG, Tucker SC, Kopetz S, Sood AK, Crissman JD, Honn KV. Platelets and cancer: a casual or causal relationship: revisited. Cancer Metastasis Rev. 2014;33(1):231–269. doi:10.1007/s10555-014-9498-0
17. Lisman T, Luyendyk JP. Platelets as modulators of liver diseases. Semin Thromb Hemost. 2018;44(2):114–125. doi:10.1055/s-0037-1604091
18. Carr BI, Cavallini A, D‘Alessandro R. Platelet extracts induce growth, migration and invasion in human hepatocellular carcinoma in vitro. BMC Cancer. 2014;14:43.
19. Zhang R, Guo H, Xu J, et al. Activated platelets inhibit hepatocellular carcinoma cell differentiation and promote tumor progression via platelet-tumor cell binding. Oncotarget. 2016;7(37):60609–60622. doi:10.18632/oncotarget.11300
20. Gong N, Chatterjee S. Platelet endothelial cell adhesion molecule in cell signaling and thrombosis. Mol Cell Biochem. 2003;253(1–2):151–158.
21. Jacquemin M, Neyrinck A, Hermanns MI, et al. FVIII production by human lung microvascular endothelial cells. Blood. 2006;108(2):515–17. doi:10.1182/blood-2005-11-4571
22. Van den Biggelaar M, Bouwens EAM, Kootstra NA, et al. Storage and regulated secretion of factor VIII in blood outgrowth endothelial cells. Haematologica. 2009;94(5):670–678. doi:10.3324/haematol.13427
23. Yigit E, Gönüllü G, Yücel İ, et al. Relation between hemostatic parameters and prognostic/predictive factors in breast cancer. Eur J Intern Med. 2008;19(8):602–607. doi:10.1016/j.ejim.2007.06.036
24. Auwerda JJA, Pieter S, De Maat MPM, et al. Prothrombotic coagulation abnormalities in patients with newly diagnosed multiple myeloma. Haematologica. 2007;92(2):279–280.
25. Van Beijnum JR, Rousch M, Castermans K, van der Linden E, Griffioen AW. Isolation of endothelial cells from fresh tissues. Nat Protoc. 2008;3(6):1085–1091.
26. Chouaib S, Kieda C, Benlalam H, Noman MZ, Mami-Chouaib F, Rüegg C. Endothelial cells as key determinants of the tumor microenvironment: interaction with tumor cells, extracellular matrix and immune killer cells. Crit Rev Immunol. 2010;30(6):529–545.
27. Franses JW, Baker AB, Chitalia VC, Edelman ER. Stromal endothelial cells directly influence cancer progression. Sci Transl Med. 2011;3(66):66ra5. doi:10.1126/scitranslmed.3001542
28. Zervantonakis IK, Hughesalford SK, Charest JL, Condeelis JS, Gertler FB, Kamm RD. Three-dimensional microfluidic model for tumor cell intravasation and endothelial barrier function. Proc Natl Acad Sci USA. 2012;109(34):13515–13520. doi:10.1073/pnas.1210182109
29. Jung DH, Park HJ, Byun HE, et al. Diosgenin inhibits macrophage-derived inflammatory mediators through downregulation of CK2, JNK, NF-κB and AP-1 activation. Int Immunopharmacol. 2010;10(9):1047–1054. doi:10.1016/j.intimp.2010.06.004
30. He Z, Chen H, Li G, et al. Diosgenin inhibits the migration of human breast cancer MDA-MB-231 cells by suppressing Vav2 activity. Phytomedicine. 2014;21(6):871–876. doi:10.1016/j.phymed.2014.02.002
31. Yang HP, Yue L, Jiang WW, Liu Q, Kou J-P, Yu B-Y. Diosgenin inhibits tumor necrosis factor-induced tissue factor activity and expression in THP-1 cells via down-regulation of the NF-κB, Akt, and MAPK signaling pathways. Chin J Nat Med. 2013;11(6):608–6015. doi:10.1016/S1875-5364(13)60070-9
32. Ma HD, Deng YR, Tian Z, Lian Z-X. Traditional Chinese medicine and immune regulation. Clin Rev Allergy Immunol. 2013;44(3):229–241. doi:10.1007/s12016-012-8332-0
33. Chen Y, Tang YM, Yu SL, et al. Advances in the pharmacological activities and mechanisms of diosgenin. Chin J Nat Med. 2015;13(8):578–587. doi:10.1016/S1875-5364(15)30053-4
34. Li YJ, Wang XR, Chen S, et al. Diosgenin induces G2/M cell cycle arrest and apoptosis in human hepatocellular carcinoma cells. Oncol Rep. 2015;33(2):693–698. doi:10.3892/or.2014.3629
35. Kim DS, Jeon BK, Lee YE, et al. Diosgenin induces apoptosis in HepG2 cells through generation of reactive oxygen species and mitochondrial pathway. Evid-Based Compl Alt. 2012;2012:981675.
36. Zheng H, Wei Z, Xin G, et al. Preventive effect of a novel diosgenin derivative on arterial and venous thrombosis in vivo. Bioorg Med Chem Lett. 2016;26(14):3364–3369. doi:10.1016/j.bmcl.2016.05.032
37. Wei Z, Xin G, Wang H, et al. The diosgenin prodrug nanoparticles with pH-responsive as a drug delivery system uniquely prevents thrombosis without increased bleeding risk. Nanomedicine. 2018;14(3):637–684.
38. Gong G, Qin Y, Huang W. Anti-thrombosis effect of diosgenin extract from Dioscorea zingiberensis C.H. Wright in vitro and in vivo. Phytomedicine. 2011;18(6):458–463. doi:10.1016/j.phymed.2010.08.015
39. Fueyo MCD, Dansey MV, Paolo LS, Pecci A, Veleiro AS, Burton G. C(16)-C(22) oxygen-bridged analogues of ce DAF-12 and LXR ligands. Steroids. 2016;112:109–114. doi:10.1016/j.steroids.2016.05.009
40. Kim HS, Kim MJ, Kim EJ, Yang Y, Lee M-S, Lim J-S. Berberine-induced AMPK activation inhibits the metastatic potential of melanoma cells via reduction of ERK activity and COX-2 protein expression. Biochem Pharmacol. 2012;83(3):385–394. doi:10.1016/j.bcp.2011.11.008
41. Kamiyama M, Shirai T, Tamura S, et al. ASK1 facilitates tumor metastasis through phosphorylation of an ADP receptor P2Y12 in platelets. Cell Death Differ. 17;24(12):2066–2076. doi:10.1038/cdd.2017.114
42. Johnson KE, Forward JA, Tippy MD, et al. Tamoxifen directly inhibits platelet angiogenic potential and platelet-mediated metastasis. Arterioscler Thromb Vasc Biol. 17;37(4):664–674. doi:10.1161/ATVBAHA.116.308791
43. Ma L, Yao X, Wei Z, et al. Deoxyarbutin displays antitumour activity against melanoma in vitro and in vivo through a p38-mediated mitochondria associated apoptotic pathway. Sci Rep. 17;7:1.
44. Broze GJ, Yin ZF, Lasky N. A tail vein bleeding time model and delayed bleeding in hemophiliac mice. Thromb Haemost. 2001;86(04):747–748.
45. Zhang R, Huang B, Du D, et al. Anti-thrombosis effect of diosgenyl saponins in vitro and in vivo. Steroids. 2013;78(11):1064–1070. doi:10.1016/j.steroids.2013.07.003
46. Kilkenny C, Browne WJ, Cuthill IC, et al. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 2014;4(1):e1000412.
47. Ward Y, Lake R, Faraji F, et al. Platelets promote metastasis via binding tumor CD97 leading to bidirectional signaling that coordinates transendothelial migration. Cell Rep. 2018;23(3):808–822.
48. Labelle M, Hynes RO. The initial hours of metastasis: the importance of cooperative host-tumor cell interactions during hematogenous dissemination. Cancer Discov. 2012;2(12):1091–1099. doi:10.1158/2159-8290.CD-12-0329
49. Möhle R, Green D, Moore MA, Nachman RL, Rafii S. Constitutive production and thrombin-induced release of vascular endothelial growth factor by human megakaryocytes and platelets. Proc Natl Acad Sci USA. 1997;94(2):663–668.
50. Myriam L, Shahinoor B, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell. 2011;20(5):576–590. doi:10.1016/j.ccr.2011.09.009
51. Machlus KR, Feng-Chang L, Wolberg AS. Procoagulant activity induced by vascular injury determines contribution of elevated factor VIII to thrombosis and thrombus stability in mice. Blood. 2011;118(14):3960–3968. doi:10.1182/blood-2011-06-362814
52. Hollestelle MJ, Geertzen HGM, Straatsburg IH, van Gulik TM, van Mourik JA. Factor VIII expression in liver disease. Thromb Haemost. 2004;91(2):267–275. doi:10.1160/TH03-05-0310
53. Chen PS, Shis YW, Huang HC, Cheng H-W, Means RE. Diosgenin, a steroidal saponin, inhibits migration and invasion of human prostate cancer PC-3 cells by reducing matrix metalloproteinases expression. PLoS One. 2011;6(5):e20164. doi:10.1371/journal.pone.0020164
54. Goel A, Chang DK, Ricciardiello L, Gasche C, Boland CR. A novel mechanism for aspirin-mediated growth inhibition of human colon cancer cells. Clin Cancer Res. 2003;9(1):383–390.
55. Italiano JE, Richardson JL, Sunita PH, et al. Angiogenesis is regulated by a novel mechanism: pro- and antiangiogenic proteins are organized into separate platelet alpha granules and differentially released. Blood. 2008;111(3):1227–1233. doi:10.1182/blood-2007-09-113837
56. Haemmerle M, Stone RL, Menter DG, Afshar-Kharghan V, Sood AK. The platelet lifeline to cancer: challenges and opportunities. Cancer Cell. 2018;33(6):965–983. doi:10.1016/j.ccell.2018.03.002
57. Schumacher D, Strilic B, Sivaraj KK, Wettschureck N, Offermanns S. Platelet-derived nucleotides promote tumor-cell transendothelial migration and metastasis via P2Y2 receptor. Cancer Cell. 2013;24(1):130–137. doi:10.1016/j.ccr.2013.05.008
58. Creighton CJ, Gibbons DL, Kurie JM. The role of epithelial- mesenchymal transition programming in invasion and metastasis: a clinical perspective. Cancer Manag Res. 2013;5:187–195. doi:10.2147/CMAR.S35171
59. Storb R, Marchioro TL, Graham TC, Willemin M, Hougie C, Thomas ED. Canine hemophilia and hemopoietic grafting. Blood. 1972;40(2):234–238.
60. Lima LG, Monteiro RQ. Activation of blood coagulation in cancer: implications for tumour progression. Biosci Rep. 2013;33(5):e00064. doi:10.1042/BSR20130057
61. Im JH, Fu WH, Bhatia SK, et al. Coagulation facilitates tumor cell spreading in the pulmonary vasculature during early metastatic colony formation. Cancer Res. 2004;64(23):8613–8619. doi:10.1158/0008-5472.CAN-04-2078
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.