Back to Journals » Journal of Inflammation Research » Volume 7
Differential NF-κB and MAPK activation underlies fluoride- and TPA-mediated CXCL8 (IL-8) induction in lung epithelial cells
Authors Refsnes M, Skuland T, Låg M, Schwarze P, Øvrevik J
Received 18 June 2014
Accepted for publication 30 August 2014
Published 12 December 2014 Volume 2014:7 Pages 169—185
DOI https://doi.org/10.2147/JIR.S69646
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Ning Quan
Magne Refsnes, Tonje Skuland, Marit Låg, Per E Schwarze, Johan Øvrevik
Department of Air Pollution and Noise, Division of Environmental Medicine, Norwegian Institute of Public Health, Oslo, Norway
Abstract: Different toxic agents have a varying potential to induce the production of the proinflammatory chemokine, CXCL8 (interleukin [IL]-8), in lung cells. A critical question is which mechanisms determine the magnitude and persistence of the CXCL8 responses to different stimuli. To approach this, we compared the potential of the phorbol ester, 12-O-tetradecanoylphorbol-13-acetate (TPA), and sodium fluoride (NaF) to induce CXCL8 responses in A549 cells, with emphasis on the importance of nuclear factor kappa B (NF-κB)- and mitogen-activated protein kinase (MAPK) signaling. Notably, TPA induced a greater release of CXCL8 than did NaF. Furthermore, TPA induced a strong, rapid, but transient upregulation of CXCL8 messenger (m)RNA, whereas NaF induced a weaker, more delayed, but persistent upregulation. With respect to signaling, TPA led to an early, strong, and relatively transient extracellular signal-regulated kinase (ERK)1/2 phosphorylation, and a less marked and even more transient phosphorylation of c-jun-N-terminal kinases (JNK1/2) and p38. In contrast, NaF elicited a lower, but relatively sustained increase in phosphorylation of ERK1/2, and a marked phosphorylation of p38 and JNK1/2, with the JNK1/2 response as most transient. Only ERK1/2 inhibition affected the TPA response, whereas inhibition of all the three MAPK cascades reduced NaF-induced CXCL8 release. TPA also induced an early, marked phosphorylation/translocation of p65 (NF-κB), whereas NaF induced slower, less pronounced effects on p65. The CXCL8 responses by TPA and NaF were reduced by p65-siRNA. In conclusion, all MAPK cascades were involved in NaF-induced CXCL8 release, whereas only ERK1/2 activation was involved in response to TPA. Furthermore, NF-κB activation appeared to be indispensable for CXCL8 induction. The early response, magnitude, and persistency of MAPK and NF-κB signaling seemed to be critical determinants for the potential to induce CXCL8. These findings underscore that a strong, rapid, and relatively transient activation of ERK1/2 in combination with NF-kB may be sufficient for a strong induction of CXCL8, which may exceed the effects of a more moderate ERK1/2 activation in combination with activation of p38, JNK1/2, and NF-κB.
Keywords: TPA, sodium fluoride, CXCL8, MAPK, NF-κB, A549 cells
Introduction
The chemokine CXCL8 (interleukin [IL]-8) plays a crucial role in lung inflammation by recruiting neutrophils to the sites of injury or infection.1 Elevated CXCL8 levels are typical in airway diseases such as chronic obstructive pulmonary disease, cystic fibrosis, and severe asthma, and are believed to be an important factor in the pathogenesis of such disorders.2–4 In addition, CXCL8 also has mitogenic, motogenic, and angiogenic properties and may contribute to cancer development.5 Thus, elucidating the mechanisms of CXCL8 regulation is of considerable therapeutic interest.
Activation of CXCL8 production is known to be mediated via several signal pathways, including different mitogen-activated protein kinases (MAPKs) and the nuclear factor kappa B (NF-κB) transcription factor. The MAPKs are members of a family of serine-threonine kinases, to which many externally activated signaling pathways converge.6 Both extracellular signal-regulated kinase-1 and -2 (ERK1/2), the c-jun-N-terminal kinases (JNK1/2), and the p38 MAPKs seem to influence CXCL8 synthesis, the contribution of each depending on the cell type and stimulant.7–13 The central element of the classical NF-κB pathway is a homo- or heterodimer, most typically consisting of the p65/p50 dimer, which within resting cells is inactivated in the cytosol due to binding to IκB. Upon stimulation, IκB is phosphorylated, ubiquitinated, and degraded, thus allowing p65/p50 to translocate to the nucleus and bind to κB-sites in promoters of target genes.14 NF-κB signaling is regarded as indispensable for inducible CXCL8 regulation. The effect of NF-κB activation alone on the CXCL8 response is, however, normally marginal. Therefore, a combined activation of NF-κB and at least one of the main MAPK cascades are required to stimulate any considerable increase in CXCL8 production. Maximal CXCL8 responses are believed to require a combined activation of NF-κB along with all the three main MAPK cascades.15 In particular, post-transcriptional stabilization of CXCL8 messenger (m)RNA by p38 has been suggested to be essential to achieve high levels of CXCL8 release, as CXCL8 mRNA otherwise is highly unstable and degrades rapidly in the absence of p38 activity.15 The MAPK and NF-κB cascades are usually regarded as separate pathways. However, it has also been reported that MAPK activation may be localized upstream to the activation of NF-κB, possibly depending on both the stimulator and cell type.12,16–19
A large range of toxic agents are known to interact with the lung epithelial cell layer, triggering innate immune responses and the release of proinflammatory cytokines, including CXCL8.20 However, the ability to stimulate CXCL8 production varies considerably. Proinflammatory cytokines, such as IL-1 and tumor necrosis factor (TNF)-α, may upregulate CXCL8 release by 100-fold, while other agents cause more moderate 5–10-fold increases.15 An important question is which mechanisms determine the magnitude and persistence of the observed CXCL8 responses. We have previously reported varying potentials to induce CXCL8 responses in lung cells by different toxic agents.21–26 Of note, the phorbol ester, 12-O-tetradecanoylphorbol-13-acetate (TPA), induced a strong CXCL8 response in A549 cells that markedly exceeded the effect of sodium fluoride (NaF),9 and with a different time course of response. TPA, a classical promoting agent in studies of cancer development, is a rapid and strong activator of protein kinase C (PKC), and also a potent MAPK activator.27 TPA usually seems to exert its cellular effects via ERK1/2 activation,18,19 although it has also been reported that TPA may activate p38.28 Fluoride exposure in the form of hydrogen fluoride is involved in airway disease, as reflected by an association with occupational asthma in epidemiologic studies,29 and with respiratory inflammation in experimental human clinical studies.30,31 To investigate how these effects might be elicited, mechanistic studies of fluoride responses are of essence. In addition to our previous in vitro studies on fluoride exposure in lung epithelial cells,8,9,26,32–34 several mechanistic studies on fluoride exposure have emerged the recent years, including studies on the role of MAPK in COX-2 upregulation in lung epithelial cells,35 on oxidative stress and lipoxygenase activity in monocytes/macrophages,36,37 and on MAPK and oxidative stress in microglia cells.38 NaF has been reported to mediate its effect via a persistent G-protein activation and/or phosphatase inhibition.39,40 In A549 cells, NaF induced a weak, delayed, but persistent PKC activation, in contrast to TPA that elicited a strong, rapid, but transient effect. Upon PKC inhibition, the stimulatory effect of TPA on CXCL8 release was nearly abolished, whereas the response of NaF on CXCL8 release was only partially reduced.9 Furthermore, NaF elicited a persistent activation/phosphorylation of MAPKs, involving both EGF receptor and Src-dependent processes in A549 cells. The NaF effect was partially reduced by the inhibition of the MAPKs ERK1/2, p38, and JNK1/2, respectively, whereas the combined inhibition of different MAPKs (p38/JNK, p38/ERK1/2, and ERK1/2/JNK1/2) completely abolished the response.8,9,33 In the present study, we have compared the CXCL8 responses induced by TPA and NaF with respect to the involvement of MAPK and NF-κB pathways, with emphasis on the magnitude and duration of the response.
Materials and methods
Reagents
Nutrition mixture F12 HAM Kaighn’s modification culture medium was obtained from Sigma-Aldrich Co. (St Louis, MO, USA). Fetal bovine serum (FBS) was from EuroClone SpA (Milano, Italy). Ampicillin and fungizone were purchased from Bristol-Myers Squibb (New York, NY, USA) and penicillin/streptomycin from BioWhittaker® (VWR International, Radnor, PA, USA). NaF was obtained from Honeywell International, Inc. (Morristown, NJ, USA). TPA, SB202190, PD98059, SP600125, curcumin, MG132, and BAY11-7082 were purchased from Calbiochem-Novabiochem Corporation (San Diego, CA, USA); phenylmethylsulfonyl fluoride and pyrrolidine dithiocarbamate (PDTC) were from Sigma-Aldrich Co. JNK2 SMARTpool small interfering (si)RNA reagent against JNK2 and nonspecific control pool (negative control) were from Upstate Biotechnology, Inc. (Lake Placid, NY, USA). HiPerFect transfection reagent was from Qiagen NV (Venlo, the Netherlands). SiRNA against p65 and nonsense siRNA were from Cell Signaling Technology, Inc. (Danvers, MA, USA). Cytokine ELISA assay for human CXCL8 (IL-8) (Cyto-Set® CHC1304) was supplied by Thermo Fisher Scientific (Waltham, MA, USA). Acrylamide/Bis-acrylamide mix (30%), TEMED, and Bio-Rad DC™ Protein Assay were from Bio-Rad Laboratories Inc. (Hercules, CA, USA). The Compartment Protein Extraction Kit was from BioChain Institute, Inc. (Newark, CA, USA). The Absolutely RNA™ reverse transcription (RT) polymerase chain reaction (PCR) Miniprep kit was from Stratagene California (La Jolla, CA, USA). The predesigned TaqMan® Gene Expression Assays, TaqMan Universal PCR Master Mix, and the High-Capacity cDNA Archive Kit were purchased from Thermo Fisher Scientific. SuperSignal® West Dura chemoluminiscence system was obtained from Thermo Fisher Scientific, and the stripping solution (Mild Antibody Stripping Solution®) was obtained from Chemicon International, Inc. (Billerica, MA, USA). All other chemicals were purchased from commercial sources at the highest purity available.
Antibodies
Specific antibodies against phospho-p38 (Thr180/Tyr182), p38, phospho-JNK1/2 (Thr183/Tyr185), JNK 1/2, IκBα, phospho-IκBα (Ser32), p65, phopho-p65 (Ser536), glyceraldehyde 3-phosphate dehydrogenase (GAPDH), and histon H1 were obtained from Cell Signaling Technology, Inc. Antibodies against phospho-ERK1/2 (Tyr-204) were obtained from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). The antibody against JNK2 was from Upstate Biotechnology, Inc., and the antibody against β-actin was obtained from Sigma-Aldrich Co.
Cell cultures and exposure
A549 cells, a human epithelial lung cell line, from the American Type Culture Collection (Manassas, VA, USA) were cultured in F12 HAM Kaighn’s modification medium, supplemented with ampicillin (0.1 mg/mL), penicillin (0.1 mg/mL), streptomycin (0.1 mg/mL), fungizone (0.25 μg/mL), and 10% heat-inactivated FBS. The cells (passage number 79–100) were plated in 35 mm six-well culture dishes (2×104 cells/well) and grown to near confluency at 37°C in a humidified atmosphere of 5% CO2 in air. The cells were exposed to varying concentrations of NaF (1.0–3.75 mM) and TPA (1–100 nM) for the indicated periods of time. When used, pharmacological inhibitors were added 1 hour prior to NaF or TPA exposure and kept in the culture medium during the whole incubation period. The concentrations of MAPK inhibitors PD98059 (25 μM), SB202190 (10 μM), and SP600125 (20 μM) were based on previous studies of concentration–response relationships for the respective inhibitors.9 For the NF-κB inhibitors, different concentrations were examined: curcumin (0–100 μM); MG132 (0–10 μM); BAY11-7082 (0–10 μM); or PDTC (0–75 μM).
Separation of nuclei and cytosol
For the extraction of nuclei and cytosol, we used a Compartment Protein Extraction Kit from BioChain Institute, Inc. The cells were lysed, and cytosolic and nuclei proteins were isolated according to producer’s recommendations. Protein concentrations in the lysate fractions were measured by Bio-Rad DC Protein Assay. Then, 6 μg of protein were loaded on a 10% Acrylamide/Bis gel. The amounts of p65 in the cytosolic and nuclei fractions were analyzed by Western blotting.
Transfection with siRNA against JNK and p65
A549 cells at a density of 200,000 cells/well were transfected with 10 nM siRNA against JNK2 and p65, using HiPerfect transfection reagent (18 μL/well) according to the procedure by Qiagen NV using the fast-forward protocol.41 The effectiveness of gene silencing was analyzed by Western blotting at 48 hours, 72 hours, or 96 hours by measuring the JNK2 and p65 protein levels in relation to β-actin. A negative siRNA control, SMARTpool (Upstate Biotechnology, Inc.), was used for the JNK experiments and SignalSilence® Control siRNA for the p65 experiments. Cells transfected with siRNA against JNK2 and p65 or negative control siRNA were exposed to NaF and TPA for 20 hours, 48 hours after transfection, and CXCL8 levels were measured by ELISA.
Measurements of CXCL8
CXCL8 protein levels in the cell medium were determined by ELISA assay. The growth medium was harvested and centrifuged at 250× g to remove cells. The final supernatants were stored at −70°C. CXCL8 levels were determined according to the manufacturer’s guidelines. Absorbance was measured and quantified using a plate reader (Sunrise™; Tecan Trading AG, Maennedorf, Switzerland) complete with software (Magellan version 1.10), and color intensity was converted to nanograms of CXCL8 using appropriate standards.
Measurements of CXCL8 mRNA
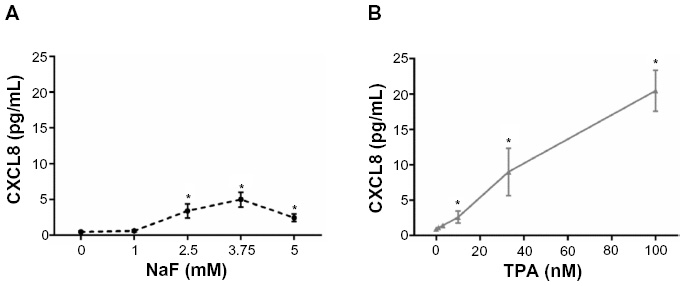
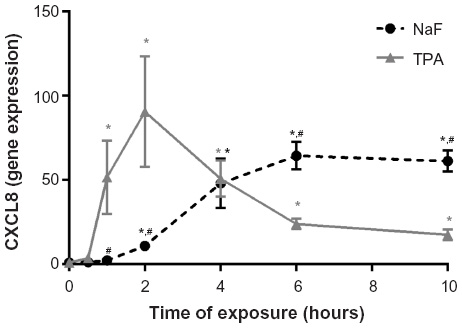
CXCL8 mRNA levels were determined by real-time PCR. Total mRNA was isolated from cells according to the supplier’s recommendations using the Absolutely RNA™RT-PCR Miniprep kit, and reverse transcribed to cDNA on a PCR System 2400 (PerkinElmer) using a High-Capacity cDNA Archive Kit (Applied Biosystems; Thermo Fisher Scientific). Real-time PCR was performed using the Applied Biosystems 7500 Real-Time PCR System, with predesigned TaqMan Gene Expression Assays (18S, Hs99999901_s1 and CXCL8, Hs00174103_m1) and TaqMan Universal PCR Master Mix. For these analyses, 1 μg of total RNA was reverse transcribed to complementary (c)DNA using a High-Capacity cDNA Archive Kit. The cDNAs were diluted 1:20 in a solution of nuclease-free water, TaqMan Universal Master Mix, primers, and probe before performing the real-time PCR. The expression of each gene of interest (GOI) in each sample was normalized against housekeeping genes (HKG), and expressed as the fold change compared to the untreated control, as calculated by the ΔΔCt-method:
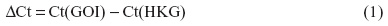
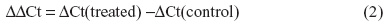

Immunoblotting
Total and phosphorylated protein levels were detected by Western blotting. Cells were resuspended in ice-cold lysis buffer (20 mM Tris-HCL; pH=7.5; 150 mM NaCl; 1 mM EDTA; 1 mM EGTA; 2.4 mM Na-pyrophosphate; 1.0 mM orthovanadate; 1 mM NaF; 21 mM leupeptin; 1.5 mM aprotinin; 15 mM pepstatin A; and 1% Triton™-X), sonicated for 5×1 second, and centrifuged for 8,000× g for 10 minutes. Protein determination was done in the supernatant by the Bio-Rad DC Protein Assay. Proteins (12.5 μg/well) from whole-cell lysates were separated by 10% SDS-PAGE and blotted onto nitrocellulose membranes. To ensure that the protein levels of each well were equal, Ponceau staining was used for loading control. The membranes were then probed with antibodies against the respective phosphorylated MAPKs (p-ERK1/2, p-JNK1/2, or p-p38) prior to incubation with horseradish peroxidase-conjugated secondary antibodies. The blots were developed using the SuperSignal West Dura chemoluminiscence system according to the manufacturer’s instructions. Finally, the membranes were stripped by incubation for 15 minutes at room temperature with Mild Antibody Stripping Solution and reprobed for the total amount of the respective kinases (ERK2, JNK2, p38) and/or β-actin. Using a similar procedure, the membranes were probed with antibodies against different NF-κB components (IκBα, p-IκBα, p65, p-p65) and reprobed with histon H1 and GAPDH.
Statistical analysis
Statistical calculations were performed by Student’s t-test or analysis of variance with post-tests for multiple comparisons, as indicated in the figure legends. Significance was assigned to a P-value ≤0.05.
Results
CXCL8 responses to fluoride and TPA in A549 cells
A549 cells were exposed to NaF (0–5 mM) and TPA (0–100 nM) for 20 hours. NaF significantly increased CXCL8 levels at 2.5 mM, with a maximal response (~6-fold increase) at 3.75 mM, and a subsequent reduction at 5 mM (Figure 1A). In accordance with previous results obtained by propidium iodide and Hoechst 33342 staining,32 NaF appeared to have a marginal effect on cell viability at 3.75 mM, whereas a substantial reduction in viability was observed at 5 mM, as judged visually by microscopy (a decrease in cell density and an increase in rounded/floating cells – not quantified). In comparison, TPA was much more potent, with responses in the nM range, and with a 20-fold increase in CXCL8 release from the cells at 100 nM, as measured after 20 hours’ exposure (Figure 1B). No change in viability was observed (not quantified). The time–course relationships for CXCL8 mRNA expression were also examined. NaF (3.75 mM) only slowly upregulated CXCL8 mRNA levels (~2-fold at 1 hour; ~12-fold at 2 hours), reaching a maximal level (~65-fold) at 6–10 hours. In contrast, TPA triggered a much more rapid and marked mRNA response for CXCL8, with a half-maximal response (45–50-fold) at ~1 hour, a peak at 2 hours (~90-fold), and then a progressive decline up to 10 hours (Figure 2). The cumulative mRNA response for NaF and TPA seemed to be of approximately the same magnitude for up to 10 hours.
 | Figure 1 Concentration-dependent release of CXCL8. |
 | Figure 2 Time-dependent increase in CXCL8 mRNA expression in A549 cells upon exposure to NaF and TPA. |
Importance of time of exposure for the CXCL8 responses
To assess whether short-term or prolonged signaling was required for the CXCL8 responses triggered by NaF and TPA, the culture medium was removed from the A549 cell cultures after incubation for 0.5 hour, 1 hour, 3 hours, 4 hours, 6 hours, 8 hours, and 10 hours. The cells were subsequently washed (three times) and resupplemented with fresh medium up to the time point for CXCL8 analysis at 20 hours. These experiments showed only a slight increase in CXCL8 responses with medium shift after 0.5–3 hours for NaF exposure. Further, after medium shift at 4–6 hours, the response seemed to be approximately half of the response in the cells cultured for 20 hours without medium replacement (Figure 3A), suggesting that prolonged signaling was required for the NaF-induced response. For TPA exposure, on the other hand, the media shift as early as 30 minutes did not reduce the maximal CXCL8 release; it was even higher when TPA was removed from the cells (Figure 3B), suggesting that the early signaling was sufficient.
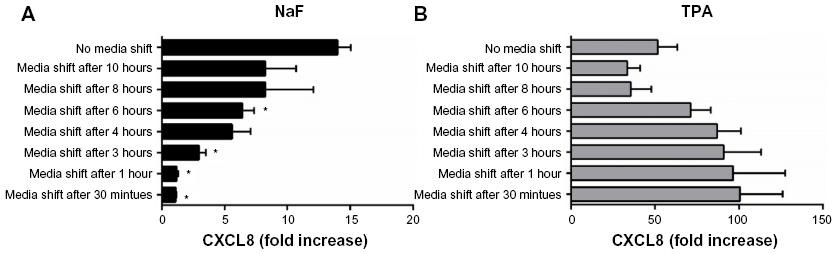 | Figure 3 Effect of short-term exposure on NaF- and TPA-induced CXCL8 responses. |
Involvement of MAPK in NaF- versus TPA-induced CXCL8 release
We have previously shown that NaF induced a time- and concentration-dependent increase in the activation/phosphorylation of the MAPKs ERK1/2, p38, and JNK, with a persistent increase in the period from 1–4 hours in A549 cells.8,9,33 In the present study, we compared the effects of NaF (3.75 mM) and TPA (100 nM) in the same experiments on the duration of MAPK phosphorylation upon exposure up to 4 hours for NaF and TPA, as assessed by Western analysis (Figure 4). TPA strongly enhanced ERK1/2 phosphorylation, with a maximum response already noted after 15–30 minutes. After 2 hours, the TPA-induced ERK1/2 phosphorylation was diminished, but it remained elevated compared to the control at least 4 hours after exposure (Figure 4Ab). NaF induced a much weaker, although sustained, ERK1/2 phosphorylation, with relatively similar levels from 15 minutes to 4 hours (~2.5%–5% of the maximal value for TPA) (Figure 4Aa). Thus, the ERK1/2 phosphorylation after TPA exposure was ~25-fold higher than after NaF exposure. In contrast, NaF exposure led to a stronger induction of p38 phosphorylation, with a distinct and sustained 3-fold increase lasting from 1–4 hours after exposure (Figure 4Ba), whereas TPA only weakly and transiently affected p38 phosphorylation, peaking at 30 minutes (~1.7-fold increase), returning to control levels after 1 hour (Figure 4Bb). Furthermore, NaF induced a very strong JNK1/2 phosphorylation (~20-fold) after 1 hour, which sharply declined at 2 hours (Figure 4Ca). TPA induced a low-to-moderate and transient JNK1/2 response, peaking at 15 minutes, returning to background levels after 1 hour (Figure 4Cb).
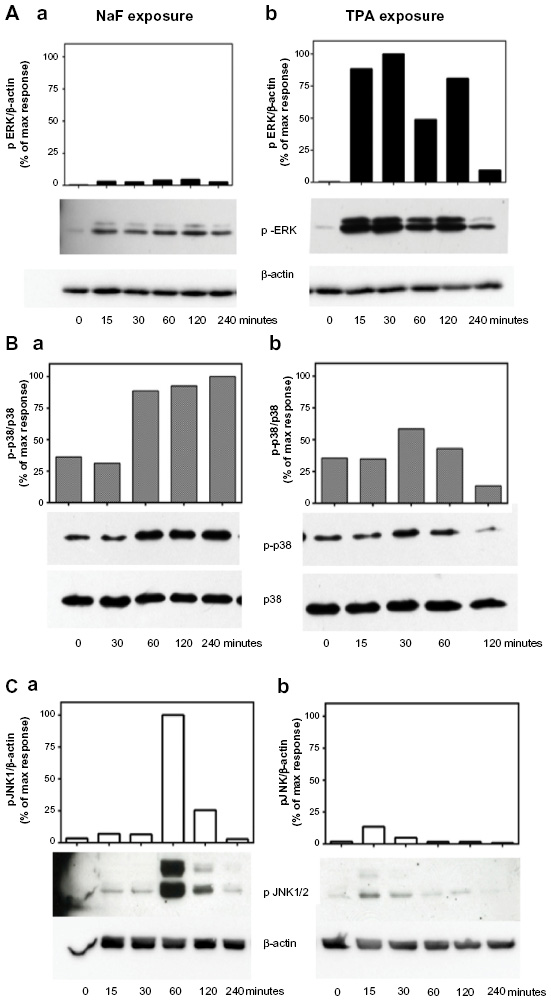 | Figure 4 Time-dependent changes of MAPK ERK1/2, p38, and JNK1/2 phosphorylation upon NaF and TPA exposure. |
Selective pharmacological MAPK inhibitors were then used to assess the role of the three MAPK cascades in TPA-induced CXCL8 responses in A549 cells. Figure 5A shows that pretreatment with the MEK/ERK1/2 inhibitor PD98059 (25 μM) reduced the responses substantially. The p38 inhibitor SB202190 (10 μM) affected CXCL8 release from TPA-exposed cells marginally, whereas the JNK1/2 inhibitor SP600125 (20 μM) seemed to slightly reduce the response, but this was not statistically significant (Figure 5A). Similarly, JNK2 silencing with siRNA only resulted in a slight, statistically nonsignificant reduction in TPA-induced CXCL8 (Figure 5B). The effect of siRNA on the JNK2 levels is shown in Figure 5C. Of note, the level of inhibition of TPA-induced CXCL8 by the three MAPK inhibitors appeared to reflect their activation level by TPA (Figure 4). Thus, the failure of SB202190 and SP600125 (and siJNK2) to elicit a statistically significant suppression of TPA-induced CXCL8 was likely due to a too low activation of p38 and JNK in TPA-exposed cells. In Table 1, the percentage inhibition obtained by PD98059, SB202190, and SP9000125 in TPA-exposed cells are compared to similar experiments in NaF-exposed cells, based upon our previously published study.8
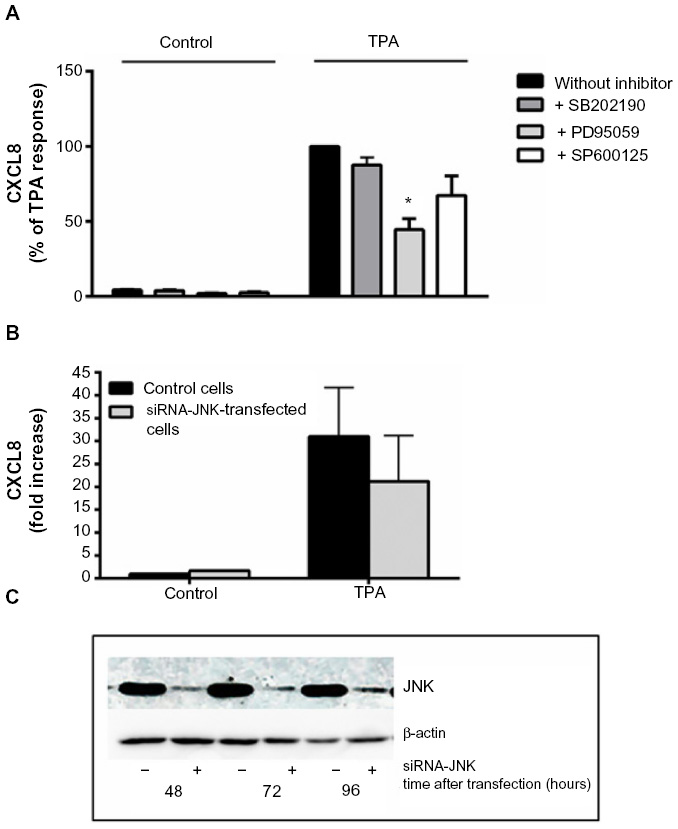 | Figure 5 Involvement of MAPKs in TPA-induced CXCL8 release. |
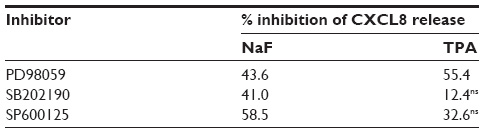 | Table 1 The relative effect of MAPK inhibitors on NaF- and TPA-induced CXCL8 release |