Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 13
Differential expression of RNA-binding proteins in bronchial epithelium of stable COPD patients
Authors Ricciardi L ![]() , Dal Col J
, Dal Col J ![]() , Casolari P, Memoli D
, Casolari P, Memoli D ![]() , Conti V
, Conti V ![]() , Vatrella A
, Vatrella A ![]() , Vonakis BM
, Vonakis BM ![]() , Papi A
, Papi A ![]() , Caramori G
, Caramori G ![]() , Stellato C
, Stellato C ![]()
Received 22 February 2018
Accepted for publication 1 August 2018
Published 12 October 2018 Volume 2018:13 Pages 3173—3190
DOI https://doi.org/10.2147/COPD.S166284
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Luca Ricciardi,1,* Jessica Dal Col,1,* Paolo Casolari,2 Domenico Memoli,1 Valeria Conti,1 Alessandro Vatrella,1 Becky M Vonakis,3 Alberto Papi,2 Gaetano Caramori,4 Cristiana Stellato1,3
1Department of Medicine, Surgery and Dentistry “Scuola Medica Salernitana”, University of Salerno, Salerno, Italy; 2Interdipartimental Study Center for Inflammatory and Smoke-related Airway Diseases (CEMICEF), Cardiorespiratory and Internal Medicine Section, University of Ferrara, Ferrara, Italy; 3Division of Allergy and Clinical Immunology, Johns Hopkins University School of Medicine, Baltimore, MD, USA; 4Department of Biomedical Sciences, Dentistry and Morphological and Functional Imaging (BIOMORF), University of Messina, Messina, Italy
*These authors contributed equally to this work
Purpose: Inflammatory gene expression is modulated by posttranscriptional regulation via RNA-binding proteins (RBPs), which regulate mRNA turnover and translation by binding to conserved mRNA sequences. Their role in COPD is only partially defined. This study evaluated RBPs tristetraprolin (TTP), human antigen R (HuR), and AU-rich element-binding factor 1 (AUF-1) expression using lung tissue from COPD patients and control subjects and probed their function in epithelial responses in vitro.
Patients and methods: RBPs were detected by immunohistochemistry in bronchial and peripheral lung samples from mild-to-moderate stable COPD patients and age/smoking history-matched controls; RBPs and RBP-regulated genes were evaluated by Western blot, ELISA, protein array, and real-time PCR in human airway epithelial BEAS-2B cell line stimulated with hydrogen peroxide, cytokine combination (cytomix), cigarette smoke extract (CSE), and following siRNA-mediated silencing. Results were verified in a microarray database from bronchial brushings of COPD patients and controls. RBP transcripts were measured in peripheral blood mononuclear cell samples from additional stable COPD patients and controls.
Results: Specific, primarily nuclear immunostaining for the RBPs was detected in structural and inflammatory cells in bronchial and lung tissues. Immunostaining for AUF-1, but not TTP or HuR, was significantly decreased in bronchial epithelium of COPD samples vs controls. In BEAS-2B cells, cytomix and CSE stimulation reproduced the RBP pattern while increasing expression of AUF-1-regulated genes, interleukin-6, CCL2, CXCL1, and CXCL8. Silencing expression of AUF-1 reproduced, but not enhanced, target upregulation induced by cytomix compared to controls. Analysis of bronchial brushing-derived transcriptomic confirmed the selective decrease of AUF-1 in COPD vs controls and revealed significant changes in AUF-1-regulated genes by genome ontology.
Conclusion: Downregulated AUF-1 may be pathogenic in stable COPD by altering posttranscriptional control of epithelial gene expression.
Keywords: airway epithelium, AUF-1, COPD, inflammation, posttranscriptional gene regulation
Introduction
Posttranscriptional gene regulation (PTR) critically controls immune and inflammatory responses through coordinated changes in mRNA turnover and translation rates, adapting the amplitude and timing of protein expression to cell environment changes.1 Regulated mRNA degradation of effector genes – cytokines, chemokines, and enzymes – as well as transcription and signaling factors contributes to the physiological cessation of acute inflammatory reactions; conversely, aberrant mRNA stabilization and sustained translation can support inflammatory gene overexpression and failed resolution of inflammatory responses, leading to chronic disease.1,2
During inflammatory responses, triggered signaling pathways coordinate transcriptional regulation with posttranscriptional events by targeting downstream factors – RNA-binding proteins (RBPs), microRNA (miRNA), and other small noncoding RNAs (sncRNA) – that associate with mRNA through conserved sequences mainly present in their untranslated regions to form dynamic ribonucleoprotein (RNP) complexes.2 Competitive or cooperative transcript binding of the different factors, as well as stimulus-driven remodeling of RBP composition, regulates mRNA stability and translation and ultimately conveys stimulus-specific PTR.3,4
Regulated responses to oxidative stress and aging are fundamental biological processes that are critically influenced by PTR regulation;5 importantly, alterations of these processes are also key pathogenic determinants of COPD.6 This disease affects over 350 million individuals globally (www.goldcopd.org), with 44 million cases in Europe (http://www.europeanlung.org/en/). It is the only chronic noncommunicable disease showing increasing morbidity and mortality, projected to become the third cause of death worldwide by 2020, and it is one of the strongest independent risk factors for the development of lung cancer among long-term smokers.7 However, while investigation of RBP-driven regulation and even its therapeutic targeting is well under way in human lung cancer,8–10 the role of RBPs in COPD pathogenesis is, by comparison, less explored.11,12
The etiology of COPD features a complex interplay of genetic and environmental factors, such as atmospheric pollution and cigarette smoking: the latter is involved in >90% of COPD cases in Westernized countries.13 The progressive chronic airflow limitation in COPD is due to two major pathological processes: remodeling and narrowing of small airways and destruction of the lung parenchyma with consequent loss of the airways’ alveolar attachments as a result of pulmonary emphysema. These changes determine diminished lung recoil, higher resistance to flow, and closure of small airways at higher lung volumes during expiration, with consequent air trapping in the lung. Hyperinflation of the lungs develops subsequently, which gives rise to the sensation of dyspnea and decrease exercise tolerance.14
Both the small-airway remodeling and the pulmonary emphysema are likely the results of chronic inflammation. Chronic lung inflammation in stable COPD is characterized by infiltration of neutrophils, monocytes, CD8+ cytotoxic, and CD4+ Th1 and Th17 T lymphocytes. Cell recruitment is initially triggered by chemotactic signals elicited in macrophages and epithelial cells by smoke and air pollutants, through oxidative stress- and toll-like receptor-mediated signaling.15 Driven by a globally defective response to the increased oxidative burden upon the respiratory system,16 epigenetic changes in immune and epithelial cells maintain overexpression of cytokines, chemokines, and enzymes leading to tissue destruction and accelerated lung aging process, defined as inflammaging.15,17,18
Many of the key genes orchestrating this process, deregulated in COPD and in large part expressed in the epithelium – such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), IL-6, interferon-γ (IFN-γ), granulocyte-monocyte colony stimulating factor, TGF-β, and VEGF, and chemokines such as CXCL1, CXCL5, CXCL8, CCL2, CCL1119,20 – are subjected to PTR in which the RBPs human antigen R (HuR), tristetraprolin (TTP), and AU-rich element binding factor 1 (AUF-1, also denominated heteronuclear ribonucleoprotein D [HRNPD]) take part;21–24 yet, the involvement of these factors has not been evaluated to date in COPD pathophysiology.
Genome-wide profiling of RBP-associated mRNAs indicated that functionally related mRNAs bearing shared conserved sequences (such as adenylate/uridylate-rich elements [ARE]) can be coordinately regulated by one or more RBPs.25 In particular, studies consistently identified HuR, TTP, and AUF-1 as the three ARE-binding proteins mainly regulating genes involved in proliferation/apoptosis, oxidative stress responsiveness, angiogenesis, immune response skewing in immune cells, and their targets.26–28 This points at the relevance of these RBPs as master regulators of homeostatic and pathologic immune responses and lends a strong rationale for investigating their expression and role in COPD pathogenesis.
HuR is the ubiquitous member of the Hu RBP family and acts mainly as a positive regulator of mRNA stability, partially by competing with ARE-binding RBPs, such as TTP and AUF-1, that limit gene expression by increasing the rate of mRNA decay of their targets.26 TTP, encoded by the ZFP-36 gene, is an immediate/early response gene inducible by inflammatory signaling that promotes rapid decay of TNF-α and many other inflammatory and immune genes.29 Function of AUF-1 is carried out by four isoforms – p37, p40, p42, p45 – generated from alternative splicing of the AUF/HRNPD gene, which mediate mRNA stabilization or decay according to the isoforms involved, their expression levels, and nucleocytoplasmic distribution.30 Animal models indicate that TTP and AUF-1 are critically involved in the resolution of inflammation by accelerating the decay of overexpressed inflammatory genes: mouse knockout for TTP show early onset of severe inflammatory arthritis, myeloid hyperplasia, autoimmune dysfunction, and cachexia through overexpression of TNF-α and GM-CSF, due to their aberrant transcript stabilization;31 similarly, aberrantly stable TNFα mRNA is found in AUF-1−/− mice in which endotoxin challenge provokes high mortality rates,32 along with spontaneous onset of chronic pruritic eczema resembling atopic dermatitis, coupled with a Th2-skewed response with hypereosinophilia and increased immunoglobulin E (IgE) levels.33
This study investigates the expression of HuR, TTP, and AUF-1 in COPD with ex vivo, in vitro, and in silico approaches. The RBPs were evaluated by immunohistochemistry (IHC) in the lower airways of stable COPD patients and control subjects; regulation of RBPs and RBP-regulated genes by inflammatory stimuli modeling COPD milieu was evaluated in the human bronchial epithelial cell line BEAS-2B and, based on the results, further probed with selective silencing of the RBP AUF-1. Transcripts of RBPs were also measured in peripheral blood mononuclear cell (PBMC) samples from additional stable COPD and control smokers to probe whether changes in RBPs were specific to lung inflammation or traceable as a potential marker of systemic inflammation in COPD.34,35 Lastly, expression profiling of RBPs and RBP-dependent genes in primary bronchial epithelial cells was investigated using a published microarray database obtained from cells isolated by bronchial brushings of stable COPD patients and control subjects.36
Patients and methods
Study population
Bronchial rings and peripheral lung samples were obtained from subjects recruited from the Respiratory Unit of the University Hospital of Ferrara, Italy, among patients undergoing lung resection for peripheral lung carcinoma (Table 1). Smokers with mild-to-moderate stable COPD (n=12) were compared with age- and smoke history-matched smokers with normal lung function (NLF) (n=12). Diagnosis of COPD was defined according to international guidelines as the presence of post-bronchodilator FEV1/FVC ratio <70% or the presence of cough and sputum production for at least 3 months in each of 2 consecutive years.6 All patients were in stable condition at the time of the surgery and had not suffered acute exacerbations or upper respiratory tract infections in the preceding 2 months. None had received glucocorticoids or antibiotics within the month preceding surgery, or inhaled bronchodilators within the previous 48 hours. Patients had no history of asthma or other allergic diseases. All former smokers had stopped smoking for >1 year. Each patient was subjected to medical history, physical examination, chest radiography, electrocardiogram, routine blood tests, and pulmonary function tests during the week prior to surgery. Pulmonary function tests (Biomedin Spirometer, Padova, Italy) were performed as previously described37 according to published guidelines.
 | Table 1 Study population providing bronchial and lung samples for immunohistochemical study |
PBMC samples were obtained at the pulmonary outpatient clinic of the University Hospital in Salerno, Italy, from stable COPD and control smokers with NLF (Table S1). The study was approved by the local ethics committees of the University Hospitals of Ferrara and Salerno, and the participating patients and control subjects signed the approved informed consent forms.
Lung sample preparation and IHC
Collection, processing, and immunohistochemical analysis of bronchial rings and lung tissue samples as well as data analysis were performed as published.16 The primary Abs (anti-human) used were rabbit polyclonal anti-AUF-1 (HPA004911; Atlas Antibodies, Bromma, Sweden); mouse monoclonal anti-HuR (sc-5261; Santa Cruz Biotechnology Inc., Dallas, TX, USA); and rabbit polyclonal anti-TTP (LS-B1572; LSBio, Seattle, WA). Negative Ab controls were carried using nonspecific isotype-matched Ig at their respective primary Ab concentrations. Image analysis was performed16 using an integrated microscope (Olympus, Albertslund, Denmark), video camera (JVC Digital color, Tatstrup, Denmark), automated microscope stage (Olympus), and PC running Image-Pro Plus software (Media Cybernetics) to quantify the RBP staining areas. Immunostaining counting and interpretation were done blinded without prior knowledge of clinicopathologic parameters. The scoring system for IHC is described in the Supplementary materials.
Cell culture and experimental protocols
The SV40-immortalized human tracheal epithelial cell line BEAS-2B (ATCC) was cultured in F12/DMEM (EuroClone) containing 5% heat-inactivated FBS (EuroClone), 2 mM L-glutamine (Lonza), penicillin (100 U/mL)–streptomycin (100 mg/mL) (Lonza), and 0.2% fungizone (EuroClone).38 For cell challenge protocols, cells were kept in medium only or stimulated using 200 μM hydrogen peroxide (H2O2) or cytomix (10 nM each rHuIL-1β, TNFα, IFN-γ, GoldBio) (n=3 each) for increasing time points (1, 3, 6, 24, and 48 hours). Cigarette smoke extract (CSE) was prepared as described.39 Briefly, smoke from one full-strength Marlboro cigarette (Phillip Morris, London, UK), with filter removed, was extracted in 1.5 minutes by controlled vacuum and bubbled into 10 mL of F12/DMEM. The solution was passed through a 0.2 μm filter, and the optical density (OD) was measured at 320 λ wavelength; the OD was then adjusted with culture medium to a reading of 0.85 considered as the 100% stock, which was then diluted with culture medium to a range of CSE solution percentages (see “Results” section) that were used within 30 minutes from preparation. Cells were serum-starved overnight prior to challenge with CSE, carried out for 24 hours. As a negative control, medium was obtained by the same procedure but with unlit cigarette, in which OD was unchanged from that of unprocessed medium.
For AUF-1 gene silencing, cells were seeded in six-well plates and transfected at 50%–60% confluency with 100 nM AUF1 siRNA (5′-AAGAUC CUAUCACAGGGCGATdTdT-3′)40 or a scrambled control siRNA (5′-GAGUCAACCUUAUGAUACUdTdT-3′) using the nonliposomal cationic vehicle FuGENE HD (Promega). After 48 hours, cells were exposed to cytomix or medium for additional 48 hours.
All cell monolayers were harvested using trypsin/EDTA (Lonza). The cells were counted, and viability was assessed by trypan blue exclusion in all conditions and found to be consistently >95% of total cell count. Supernatants of all experiments were collected, centrifuged, and stored at −80°C for subsequent analysis.
Isolation of PBMC was performed by standard Ficoll gradient separation (Sigma).
RNA extraction, cDNA synthesis, and qPCR
Total RNA was extracted using TriFast reagent (EuroClone) and reverse transcription was prepared using the Moloney murine leukemia virus reverse transcriptase (Applied Biological Materials) following the manufacturer’s protocol. Template cDNA was subjected to quantitative real-time PCR (qRT-PCR) with the FluoCycle II SYBR Master Mix (Euroclone) according to the manufacturer’s protocol. Primer sequences were: GAPDH forward: 5′-GAAGGTGAAGGTCGGAGTC-3′, reverse: 5′-GAAGATGGTGATGGGATTTC-3′; AUF-1 5′-GATCCTAAAAGGGCCAAAGC-3′, reverse: 5′-CCACTGTTGCTGTTGCTGAT-3; HuR 5′-CGCAGAGATTCAGGTTCTCC-3′, reverse: 5′-CCAAACCCTTTGCACTTGTT-3′; TTP 5′-CGCTACAAGACTGAGCTATG-3′, reverse: 5′-CCTGGAGGTAGAACTTGTG-3. Primers were published38,41 or designed with Primer-BLAST software (https://www.ncbi.nlm.nih.gov/tools/primer-blast/). Reactions were run in duplicate on a LightCycler 480 II (Roche), using the following setup: 5 minutes, 95°C; 15 seconds at 95°C, 45 cycles; 15 seconds, 60°C. Target expression was normalized to GAPDH by the cycle threshold (Ct) method and expressed using the 2−ΔΔCt calculation as fold over control and in selected cases, 2−ΔΔCt as fold over housekeeping gene levels.
Protein extraction and Western blot
Nuclear and cytoplasmic proteins were separated, quantified, and subjected to Western blot analysis as described.42,43 Briefly, proteins were fractionated using SDS-PAGE and transferred onto nitrocellulose membranes. After blocking with 5% milk for 1 hour, membranes were stained with primary antibodies (anti-RBP Abs as in IHC; mouse monoclonals anti-Lamin A/C [SAB420023; Sigma-Aldrich Co., St Louis, MO, USA], anti-tubulin [2128; Cell Signaling Technology], anti-PARP-1 [F-2, Sc-8007; Santa Cruz Biotechnology Inc.], anti-Cleaved Caspase-3 [Asp175, #9661; Cell Signaling Technology]) at 4°C overnight, then labeled with horseradish peroxidase-conjugated secondary antibodies. Immunoblotting analysis and densitometry were performed via digital image system (ChemiDoc MP; Bio-Rad Laboratories Inc., Hercules, CA, USA) using the enhanced chemiluminescent substrate ECL (#32106; Thermo Fisher Scientific, Waltham, MA, USA).
Analysis of secreted proteins
BEAS-2B supernatants were screened for secreted proteins using specific IL-6 and CCL2 ELISA kits (Elabscience; detection threshold 0.122 pg/mL, and Cloud-Clone Corp., Kathy, TX, USA; detection threshold 15 pg/mL, respectively) and using the Proteome Profiler Human Cytokine Array Kit (R&D Systems, Inc., Minneapolis, MN, USA), following the manufacturer’s protocol. Absorbance values were measured using an Infinite M200 PRO plate reader (Tecan).
Bioinformatics analysis
We interrogated a microarray database originated from epithelial cells obtained by bronchial brushings of stable COPD patients (n=6), and smokers and nonsmokers (n=12 each) as NLF controls,36 deposited in the Gene Expression Omnibus (GEO) repository (GEO ID: GSE5058). Fluorescence intensity data from individual data sets were extracted and normalized on the medians for fold change (FC) comparison of RBP expression among groups. For RBPs, FCs were set at ≥2.0 with a false discovery rate (FDR) ≤0.05. Data sets were subsequently analyzed for expression of AUF-1-associated transcripts and annotated according to genome-wide RNP-IP and PAR-CLIP analysis.40,44 For this analysis, FC was set at ≥2.0 with FDR ≤0.05. Heatmaps were generated using tMEV tools v4_9_0.45
Genome ontology analysis
Characterization of the AUF-1-dependent gene clustering according to the database comparison was performed using NCBI PANTHER version 11 (www.pantherdb.org) with Bonferroni correction for multiple testing. The system clusters genes based on functional properties, using published studies and evolutionary relationships as sources.46
Statistical analysis
To determine differences for IHC data between groups, analyses of variance were used for clinical data and unpaired t-test with post hoc Mann–Whitney U-test. Categorical values were compared by chi-squared test.16 Data from qRT-PCR, Western blot densitometry, and ELISA were analyzed using Student’s paired t-test; time- and concentration-dependent responses were analyzed using ANOVA test with Fisher’s post hoc multiple comparison analysis. P<0.05 was considered significant. Statistical analysis was performed using GraphPad Prism 5 (GraphPad Software, Inc. La Jolla, CA, USA).
Results
Characterization of RBP expression in human lung tissue and PBMC in stable COPD patients and control subjects
Expression of HuR, TTP, and AUF-1 was first evaluated by IHC in bronchial rings and peripheral lung tissue samples obtained from patients with mild-to-moderate stable COPD and control smokers with NLF. The clinical characteristics of the study population are described in Table 1 and in the “Methods” section. As expected, smokers with COPD had significantly lower FEV1 (percent of predicted) and FEV1/FVC ratio compared to controls (P<0.01).
Specific immunostaining was detected for all three RBPs in bronchial tissue (Figure 1) and peripheral lung (Figure 2), with mainly nuclear localization in both structural and inflammatory cells. In bronchial tissue, the number of bronchial epithelial cells with nuclei positively stained for AUF-1 was significantly lower in patients with COPD compared to control smokers (Figure 1), whereas no statistically significant difference in other AUF-1-positive cells was found between the two groups in bronchial tissue and peripheral lung samples (Figures 1 and 2). In contrast, the localization and global expression of HuR and TTP were not significantly different between stable COPD patients and control subjects in both bronchial and peripheral lung tissues (Figures 1 and 2). No statistical difference was found in RBP expression patterns between current and former smokers.
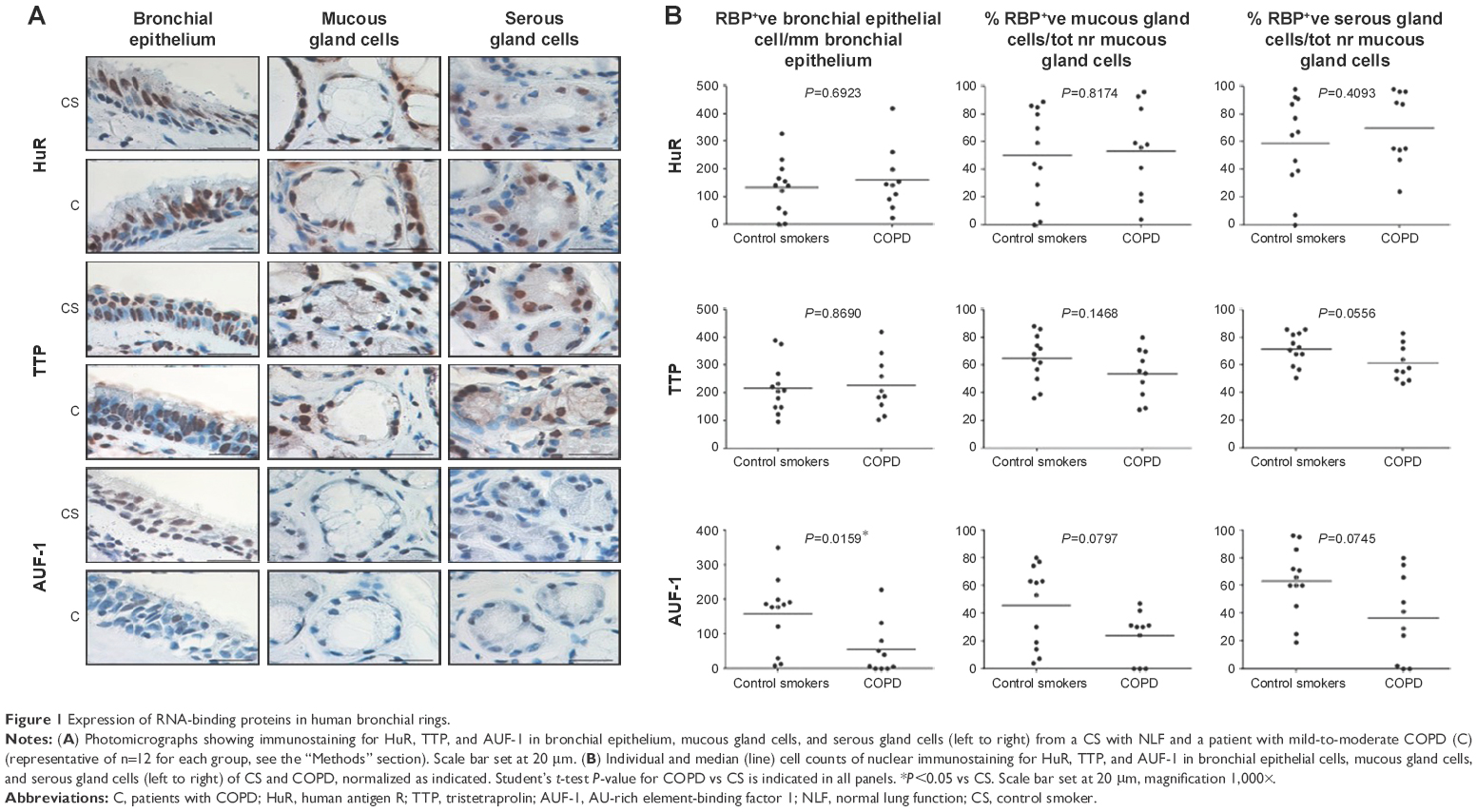 | Figure 1 Expression of RNA-binding proteins in human bronchial rings. |
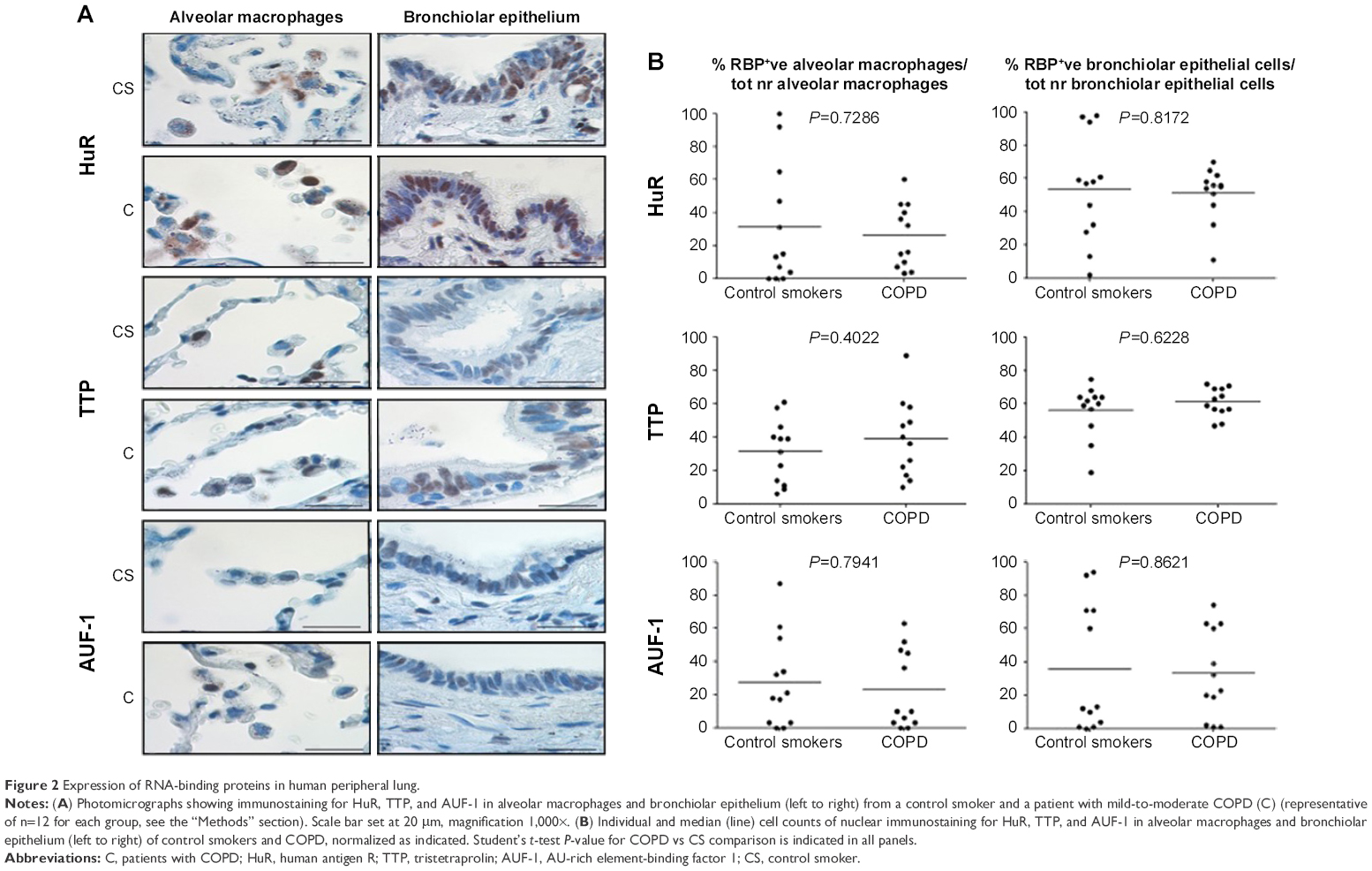 | Figure 2 Expression of RNA-binding proteins in human peripheral lung. |
Evaluation of RBP mRNA levels in PBMCs obtained from additional stable COPD patients and control smokers with NLF (n=5 and 4, respectively; Table S1) by RT-PCR showed no differences between the two groups (Figure S1).
In vitro modulation of RBP expression in BEAS-2B cells by proinflammatory and oxidant stimuli
To investigate whether epithelial RBP expression could be modulated by a Th1-skewed cytokine milieu and oxidative stress, we evaluated HuR, TTP, and AUF-1 levels by Western blot analysis in BEAS-2B cells following challenge with cytomix or hydrogen peroxide (200 μM) (Figure 3). Cytomix induced a time-dependent, statistically significant decrease of the expression of main AUF-1 isoforms compared to unstimulated control (to a maximum of 60% decrease at 48 hours for p42 isoform in nuclear fraction, shown in bar graph (Figure 3) and 49% decrease in cytoplasmic fraction, P<0.05 in both cases). Such effect was stimulus-specific, as AUF-1 levels were unchanged by hydrogen peroxide stimulation. By contrast, HuR and TTP expressions were not affected by cell treatments beyond consistent but small variations. Cleavage of PARP-1 and caspase 3 as markers of apoptosis was not detected upon any cell treatment (Figure S2). RT-PCR analysis (Figure S3A) confirmed at mRNA level the lack of stimulus-induced changes in HuR and TTP expression; interestingly, AUF-1 mRNA was unchanged by cytomix despite clear protein downregulation. Instead, in line with protein results, hydrogen peroxide treatment did not change RBP transcript levels (Figure S3B).
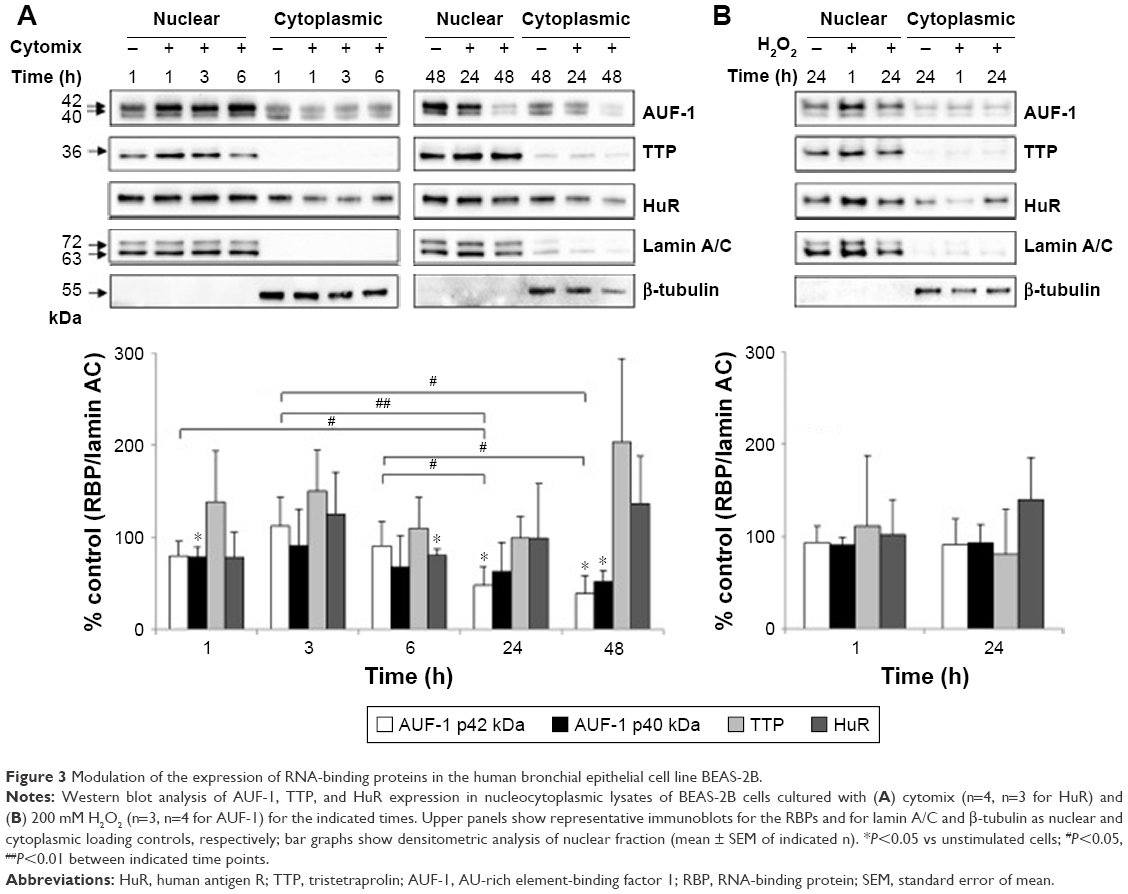 | Figure 3 Modulation of the expression of RNA-binding proteins in the human bronchial epithelial cell line BEAS-2B. |
To test whether the RBP pattern induced by cytomix could be recapitulated by a broader COPD inflammatory modeling encompassing both inflammatory and oxidant-driven triggers, BEAS-2B cells were exposed to CSE for 24 hours, as described39,47 (Figure 4). Expression of AUF-1 was downregulated in a concentration-dependent fashion, but a marked decrease in cell viability at the highest CSE concentration tested (20% and 10%) (Figure 4A) has led to exclusion of these two points from densitometric analysis; however, expression of AUF-1 main isoforms remained significantly decreased also by exposure to 3% CSE, a concentration displaying close to 90% cell viability (mean ± SEM, 88±2), to a maximum decrease of 54% in cytoplasmic extracts (P<0.05, Figure 4B and C). Lack of cleavage of PARP-1 in all samples indicated absence of apoptosis (not shown).
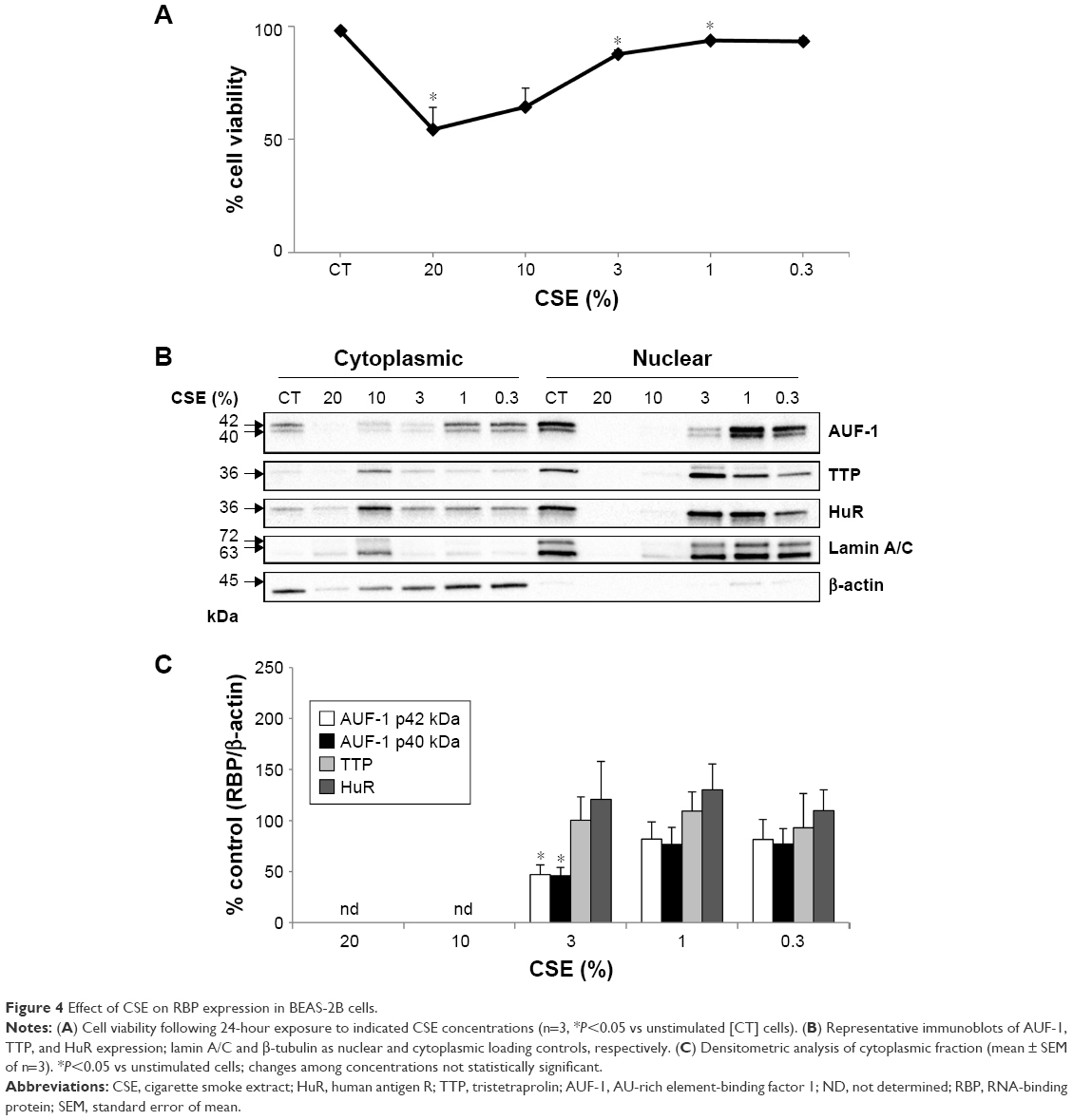 | Figure 4 Effect of CSE on RBP expression in BEAS-2B cells. |
Effect of cytomix-induced and siRNA-mediated loss of AUF-1 on RBP and inflammatory gene expression in BEAS-2B cells
We then implemented silencing of AUF-1 expression in BEAS-2B cells (Figure 5) to investigate its potential effects on RBPs and the expression of their downstream targets. Following transfection with a specific AUF-1 siRNA,40 levels of AUF-1 protein in unstimulated cells were significantly, although partially, diminished compared to both mock (Fugene only)- and scrambled siRNA-transfected cells (Figure 5A) up to 49% and 58%, respectively, according to densitometric analysis (P>0.05 for both comparisons). Furthermore, cytomix treatment further lowered AUF-1 levels to a maximum average of 27% compared to unstimulated, AUF-1 silenced cells (Figure 5B, P<0.05). Levels of TTP protein in AUF-1-silenced cells displayed a small (close to twofold) yet significant increase in conditions of maximal AUF-1 loss induced by cytomix in these cells, while HuR levels were not affected by AUF-1 silencing (Figure 5B). In these experiments, AUF-1 mRNA, detected by RT-PCR, remained unchanged in cytomix-stimulated untransfected cells (Figure S3C) as in the previous experiment set (Figure S3A), while a consistent but small decrease in AUF-1 mRNA (25% of control, P<0.05) was detected in scrambled-transfected cells and, predictably, in cells transfected with AUF-1 siRNA (48% of control, P<0.05). Regarding the other RBPs, while HuR mRNA levels were unchanged among conditions, cytomix induced a consistent reduction in TTP mRNA (58% and 51% of control in untransfected and scrambled-transfected cells, respectively; P<0.05 in both cases) that was lost in AUF-1-silenced cells (Figure S3C). Interestingly, a small but significant increase in TTP protein was indeed detectable in these cells (Figure 5B). Furthermore, silencing of AUF-1 did not induce cell apoptosis, as verified by lack of cleavage of PARP-1 (Figure S4) or of caspase 3 (data not shown).
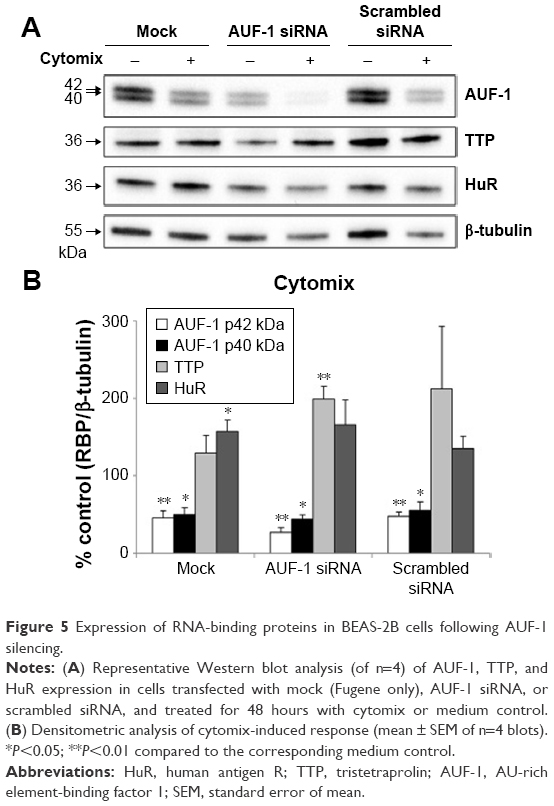 | Figure 5 Expression of RNA-binding proteins in BEAS-2B cells following AUF-1 silencing. |
We then evaluated changes in AUF-1-regulated genes in conditions of relative loss of AUF-1 (Figure 6), first by assessing the expression of IL-6 and of the chemokine CCL2, chosen as readouts as they are overexpressed in airway epithelium in COPD and significantly increased as well in the AUF-1-deficient animal model.19,33 RT-PCR analysis indicated that both transcripts were upregulated upon treatment in untransfected cells (Figure 6A) although not in a statistically significant fashion compared to unstimulated cells, likely due to variability in small sample size (n=4). Following AUF-1 silencing, basal mRNA levels did not change significantly compared to scrambled siRNA-transfected cells and upon cytomix stimulation, their increase was comparable to that caused by treatment in scrambled- and mock-transfected cells (Figure 6A), in which a similar decrease in AUF-1 protein was present (Figure 5). Levels of IL-6 and CCL2 proteins, detected by ELISA in the supernatants (Figure 6B), were instead significantly upregulated by cytomix, but also in this case without additional potentiation of cytokine secretion in AUF-1-silenced cells. Levels of both proteins following cytomix were comparable in scrambled and AUF1-silenced cells also when normalized on cell number/condition (data not shown).
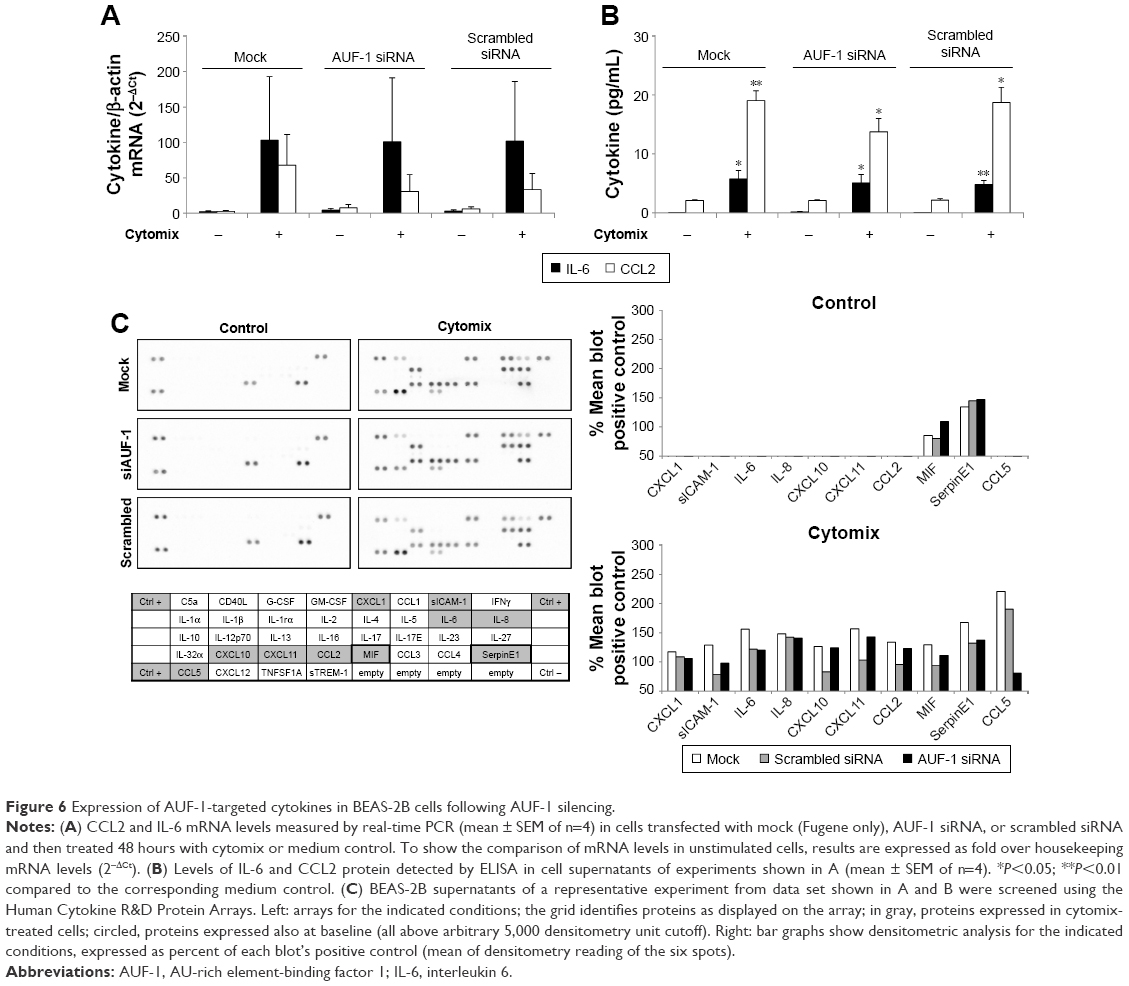 | Figure 6 Expression of AUF-1-targeted cytokines in BEAS-2B cells following AUF-1 silencing. |
To gain a broader view of potential differences in cytomix-induced epithelial responses upon loss of AUF-1, supernatants of a representative experiment were further screened with an inflammatory cytokine protein array carrying additional RBP targets, as well as cytokines and chemokines involved in COPD pathogenesis (Figure 6C). As for IL-6 and CCL2, stimulus-induced upregulation of COPD-relevant chemokines such as CXCL1, CXCL8, and CXCL10, as well as other proteins was densitometrically comparable among conditions with the exception of CCL5, which was markedly reduced in AUF1 siRNA-transfected cells.
Validation of decreased AUF-1 levels and changes in AUF-1-regulated genes in COPD patients vs control subjects in primary airway epithelial cell transcriptome database
In order to verify the overall RBP profile in primary airway epithelial cells, including the decreased AUF-1 expression and the potential downstream effects on target expression, we interrogated a public microarray database originated from primary airway epithelium obtained by bronchial brushings of COPD patients, and smokers and nonsmokers with NLF (GEO ID: GSE5058)36 (Figure 7). Bronchial epithelial AUF-1 expression was found to be significantly lower in COPD vs both controls (FC =−2.7 and −3.5 vs nonsmokers and smokers, respectively), while FC levels for HuR and TTP did not change significantly among groups, showing however a trend toward reciprocal changes in COPD. We then evaluated in the same database a compiled list of 152 validated AUF-1 target genes.40,44 Thirty of them were significantly upregulated and 22 were downregulated in COPD patients compared to both control groups, with greatest changes vs control smokers (Figure 8). Genome ontology analysis indicates that AUF-1 regulation has a potentially significant impact on several pathogenic pathways of stable COPD.
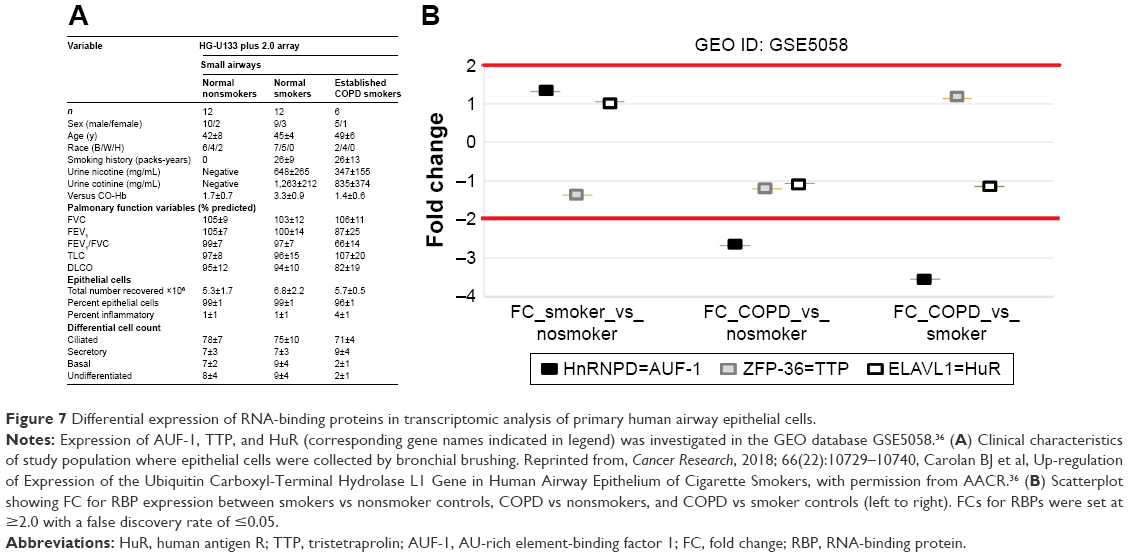 | Figure 7 Differential expression of RNA-binding proteins in transcriptomic analysis of primary human airway epithelial cells. |
 | Figure 8 Expression of AUF-1-dependent genes in airway epithelial gene array database GSE5058. |
Discussion
This study provides for the first time the expression profile of RBPs chiefly regulating inflammatory responses – HuR, TTP, and AUF-1 – in the lower airways of patients with COPD, a chronic lung inflammatory disease. In particular, immunohistochemical analysis showed that the expression of AUF-1 was selectively decreased in the bronchial, but not in the bronchiolar, epithelium from patients with stable COPD compared to control smokers, while HuR and TTP expression levels were comparable between groups in both bronchial and lung tissues. Importantly, the selective loss of AUF-1 in epithelium was confirmed by unbiased search in a primary bronchial epithelial gene array database derived from bronchial brushings of COPD patients and controls with similar clinical characteristics. In contrast, expression of RBP transcripts did not differ in PBMCs of stable COPD compared to control smokers, suggesting that AUF-1 downregulation, rather than systemic,34 may be related to airway-specific conditions.
Bronchial epithelium is an early and key contributor to the pathogenesis of COPD48 although at variance with asthma, its functional activity in stable COPD patients is less characterized, with most studies focused on the role of mucus-secreting epithelial cells and on epithelial stem cells.13 Inhaled cigarette smoke, pollutants, and irritants activate epithelial innate immune responses via TLR and DAMPS, producing both barrier alteration and triggering recruitment of immune cells and their activation through the release of chemotactic and proinflammatory mediators. This gene expression program is largely mediated at transcriptional level by activation of NF-κB, documented in the bronchial epithelium of mild/moderate stable COPD patients and, to a lesser extent, in control smokers in comparison with control nonsmokers.49 In the same disease context, our study uncovers the potential contribution of altered posttranscriptional gene regulatory mechanisms in mediating defective response to oxidative stress and inflammation in COPD, suggested by decreased levels of the RBP AUF-1 in ex vivo and in vitro experiments. Very few studies so far investigated AUF-1 in models of human lung inflammation. In vitro, cytosolic levels of AUF-1 increased significantly in primary airway epithelial cells infected with human rhinovirus, concomitant with a decreased expression of CXCL10,50 while cigarette smoke-induced upregulation of CXCL8 was not AUF-1-dependent.51 Recently, decreased levels of AUF-1 mRNA were found in bronchoalveolar lavage cells and PBMC of patients with sarcoidosis, another chronic inflammatory lung disease.52 The epithelial-specific loss of AUF-1 observed in COPD subjects in our study suggests that lower AUF-1 levels may become determinants of non-resolving inflammation, by altering the RNP complexes necessary for coordinate degradation of epithelial transcripts involved in the inflammatory response.
Importantly, the selective loss of AUF-1 in epithelium was reproducible in vitro by exposure of BEAS-2B cells to established COPD/oxidative stress models such as CSE, which provides both a proinflammatory trigger and an oxidant overload stimulation;39 stimulation with H2O2 did not change AUF-1 levels, suggesting selectivity in this response. Cytomix, a cytokine association representing the Th1-skewed airway milieu in COPD,53 instead reproduced CSE effect and was associated with a concomitant upregulation of known AUF-1 targets such as IL-6, CCL2, CXCL1, and CXCL8.3,33,54 The pleiotropic cytokine IL-6 is also expressed in airway epithelium and is found in increased amounts in the sputum, exhaled breath, BAL fluid of patients with COPD,55 in particular following exacerbations as well as in plasma, as important biomarkers of systemic inflammation.35,55,56 Importantly, AUF1 binds to AU-rich elements in IL-6 mRNA and promotes its degradation.57 The chemokine CCL2 is a potent monocyte, T-cell, and mast cell chemoattractant and a basophil activator expressed by alveolar macrophages and T cells, and is overexpressed in airway epithelial cells in COPD.58,59 Levels of the chemokine CCL2 are increased in the sputum and in the BAL fluid of patients with COPD.19,20 Involvement of CCL2 in macrophage and mast cell recruitment in the lung is supported by several experimental mouse models of inflammation and emphysema, constituting an attractive therapeutic target.20 Although well known to be regulated posttranscriptionally in airway epithelium by TTP and HuR,60 association of CCL2 mRNA with AUF-1 has not been yet fully characterized; similarly, direct association of AUF-1 with other epithelial genes relevant to COPD pathogenesis overexpressed upon cytomix treatment, such as CCL5, CXCL10, and CXCL11, remains to be studied.
In our study, upregulation of IL-6, CCL2, and the entire chemokine/cytokine profile induced by cytomix in BEAS-2B cells was recapitulated, but without further modification, following siRNA-mediated silencing of AUF-1. This result does not yet clarify to what extent, and how, AUF-1 could be deemed necessary for cytokine upregulation in this model. First, limitations of the experimental setting – such as achieving only partial knockdown of AUF-1 protein and/or near-maximal stimulation of epithelial response by cytomix – may have hindered the uncovering of AUF1 function in this model. Furthermore, the majority of the transcripts coding for the cytokine/chemokine profile induced by cytomix – including CCL2 and IL-6 mRNA – are dynamically regulated by multiple ARE-binding proteins, including TTP and HuR,23,29,40,60 as well as by miRNA working either in cooperation or in competition, contextually to signaling-driven environmental changes.3,4 Proteomic analysis of transcript-associated RNPs, models of AUF-1 overexpression, and binding site mutational studies will be necessary to further probe AUF-1 role in epithelial responses and in COPD. Moreover, stimulus-induced posttranslational mechanisms, such as promotion of ubiquitination,61,62 may be regulating AUF-1 protein levels and require definition, also as potential means of modulating AUF-1-mediated gene regulation.
Further support to a role for AUF-1 in shaping epithelial gene expression in COPD is provided by the significant changes found, in the bronchial epithelial GEO database, in the global expression of a compiled list of bona fide AUF-1 mRNA targets21,40,44 – including IL6 and CCL2 – within the COPD array samples compared to controls, concomitant with the loss of AUF-1 expression detected in the same data set. Genome ontology analysis of AUF-1-regulated gene expression profile in COPD samples indicates a potentially significant impact of this RBP in the pathogenesis of stable COPD through coordinate regulation of genes involved in inflammation and angiogenesis, alteration of DNA repair, defective response to hypoxia and oxidative stress, and altered metabolism of serotonin – which is increased in COPD patients and linked to exposure to cigarette smoke.13,63,64 Taken together, these findings suggest the occurrence of AUF-1-driven PTR in COPD according to a ribonomic paradigm, whereby transcripts structurally related by sharing RBP recognition motifs are co-regulated by a core RBP – likely shaping a specific RNP configuration – according to their participation to a specific function.65
The marginal yet consistent changes in HuR and TTP levels in our in vitro systems holds further investigation, rather than rule out their participation to epithelial gene regulation in COPD. In primary and transformed airway epithelial cultures, upon cytokine challenge, HuR functions as a positive regulator of mRNA stability for multiple chemokines, while TTP limits the half-life of inflammatory transcripts and mediates glucocorticoid-induced gene regulation – indicating their potential role in integrated posttranscriptional gene control, yet to be fully characterized in lung inflammatory diseases where airway epithelial responses are pathogenic. Furthermore, similarities in binding sites and partially overlapping mRNA target pools indicate major complexity of the interplay among TTP, HuR, and AUF-1, as well as with other ncRNA species participating to RNP complexes,4,40,66 warranting further studies to understand how pathogenic immune responses in the lung ultimately rely on altered RBP-mediated control of mRNA decay and translation.
Increasing knowledge on how immune responses rely on PTR is likely to yield a strong rationale to evaluate PTR mechanisms for therapeutic intervention. Indeed, a major limitation in the current treatment of stable COPD patients is the reduced anti-inflammatory efficacy of glucocorticoids,14 indicating a major unmet need for therapeutic approaches able to override this limitation. The development of high-throughput screening methods for RNP interactions and generation of small-molecule inhibitors for lung cancer67–70 indicates that uncovering PTR mechanisms in COPD may also sustain the development of new anti-inflammatory strategies, potentially transferable to other chronic lung inflammatory diseases.
Acknowledgments
This study was funded by POR Campania FESR 2007–2013–RETE DELLE BIOTECNOLOGIE IN CAMPANIA – Project “Terapie Innovative di Malattie Infiammatorie croniche, metaboliche, Neoplastiche e Geriatriche” – TIMING (University of Salerno, CS), FARB 2014/2015 (University of Salerno, CS, VA), and FAR 2015 (University of Ferrara, GC).
Author contributions
LR and JDC are primary authors and conducted all the in vitro studies (cell cultures, Western blot, ELISA, and RT-PCR); they conducted data collection, analysis, and interpretation, and drafted the figures and manuscript parts. PC performed and analyzed all the IHC studies; DM performed all in silico analyses; AV and VC provided clinical recruitment of COPD and smoker control patients (Salerno) and isolation and processing of PBMC samples; BMV made contributions to data interpretation and advice on RBP protein detection; AP provided clinical samples from COPD and control smokers for IHC (Ferrara); GC and CS made significant contribution to study conception, design, data interpretation, manuscript writing, and revision. All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Anderson P. Post-transcriptional regulons coordinate the initiation and resolution of inflammation. Nat Rev Immunol. 2010;10(1):24–35. | ||
Gerstberger S, Hafner M, Tuschl T. A census of human RNA-binding proteins. Nat Rev Genet. 2014;15(12):829–845. | ||
Chowdhury S, Dijkhuis A, Steiert S, Lutter R. IL-17 attenuates degradation of ARE-mRNAs by changing the cooperation between AU-binding proteins and microRNA16. PLoS Genet. 2013;9(9):e1003747. | ||
Tiedje C, Ronkina N, Tehrani M, et al. The p38/MK2-driven exchange between tristetraprolin and HuR regulates AU-rich element-dependent translation. PLoS Genet. 2012;8(9):e1002977. | ||
Abdelmohsen K, Kuwano Y, Kim HH, Gorospe M. Posttranscriptional gene regulation by RNA-binding proteins during oxidative stress: implications for cellular senescence. Biol Chem. 2008;389(3):243–255. | ||
Kirkham PA, Caramori G, Casolari P, et al. Oxidative stress-induced antibodies to carbonyl-modified protein correlate with severity of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2011;184(7):796–802. | ||
Caramori G, Casolari P, Cavallesco GN, Giuffrè S, Adcock I, Papi A. Mechanisms involved in lung cancer development in COPD. Int J Biochem Cell Biol. 2011;43(7):1030–1044. | ||
Fallahi M, Amelio AL, Cleveland JL, Rounbehler RJ. CREB targets define the gene expression signature of malignancies having reduced levels of the tumor suppressor tristetraprolin. PLoS One. 2014;9(12):e115517. | ||
Giaginis C, Alexandrou P, Tsoukalas N, et al. Hu-antigen receptor (HuR) and cyclooxygenase-2 (COX-2) expression in human non-small-cell lung carcinoma: associations with clinicopathological parameters, tumor proliferative capacity and patients’ survival. Tumour Biol. 2015;36(1):315–327. | ||
Muralidharan R, Panneerselvam J, Chen A, Zhao YD, Munshi A, Ramesh R. HuR-targeted nanotherapy in combination with AMD3100 suppresses CXCR4 expression, cell growth, migration and invasion in lung cancer. Cancer Gene Ther. 2015;22(12):581–590. | ||
Juan Y, Haiqiao W, Xie W, et al. Cold-inducible RNA-binding protein mediates airway inflammation and mucus hypersecretion through a post-transcriptional regulatory mechanism under cold stress. Int J Biochem Cell Biol. 2016;78:335–348. | ||
Baker JR, Vuppusetty C, Ito K, Barnes P, Yasuo K. RNA-binding protein HuR inhibits the expression of sirtuin-1 in patients with COPD. Paper presented at: European Respiraory Journal. 2017. Epub 2017 Dec 6. | ||
Caramori G, Kirkham P, Barczyk A, di Stefano A, Adcock I. Molecular pathogenesis of cigarette smoking-induced stable COPD. Ann N Y Acad Sci. 2015;1340:55–64. | ||
Barnes PJ, Burney PG, Silverman EK, et al. Chronic obstructive pulmonary disease. Nat Rev Dis Primers. 2015;1:15076. | ||
Barnes PJ. Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol. 2008;8(3):183–192. | ||
Caramori G, Adcock IM, Casolari P, et al. Unbalanced oxidant-induced DNA damage and repair in COPD: a link towards lung cancer. Thorax. 2011;66(6):521–527. | ||
Cosio MG, Saetta M, Agusti A. Immunologic aspects of chronic obstructive pulmonary disease. N Engl J Med. 2009;360(23):2445–2454. | ||
Ito K, Barnes PJ. COPD as a disease of accelerated lung aging. Chest. 2009;135(1):173–180. | ||
Barnes PJ. The cytokine network in asthma and chronic obstructive pulmonary disease. J Clin Invest. 2008;118(11):3546–3556. | ||
Caramori G, di Stefano A, Casolari P, et al. Chemokines and chemokine receptors blockers as new drugs for the treatment of chronic obstructive pulmonary disease. Curr Med Chem. 2013;20(35):4317–4349. | ||
Moore AE, Chenette DM, Larkin LC, Schneider RJ. Physiological networks and disease functions of RNA-binding protein AUF1. Wiley Interdiscip Rev RNA. 2014;5(4):549–564. | ||
Anderson P. Post-transcriptional control of cytokine production. Nat Immunol. 2008;9(4):353–359. | ||
Stoecklin G, Tenenbaum SA, Mayo T, et al. Genome-wide analysis identifies interleukin-10 mRNA as target of tristetraprolin. J Biol Chem. 2008;283(17):11689–11699. | ||
Hamilton T, Novotny M, Pavicic PJ, et al. Diversity in post-transcriptional control of neutrophil chemoattractant cytokine gene expression. Cytokine. 2010;52(1–2):116–122. | ||
Keene JD. RNA regulons: coordination of post-transcriptional events. Nat Rev Genet. 2007;8(7):533–543. | ||
Srikantan S, Gorospe M. HuR function in disease. Front Biosci. 2012;17(1):189–205. | ||
Sanduja S, Blanco FF, Young LE, Kaza V, Dixon DA. The role of tristetraprolin in cancer and inflammation. Front Biosci. 2012;17:174–188. | ||
White EJ, Brewer G, Wilson GM. Post-transcriptional control of gene expression by AUF1: mechanisms, physiological targets, and regulation. Biochim Biophys Acta. 2013;1829(6–7):680–688. | ||
Tiedje C, Diaz-Muñoz MD, Trulley P, et al. The RNA-binding protein TTP is a global post-transcriptional regulator of feedback control in inflammation. Nucleic Acids Res. 2016;44(15):7418–7440. | ||
Raineri I, Wegmueller D, Gross B, Certa U, Moroni C. Roles of AUF1 isoforms, HuR and BRF1 in ARE-dependent mRNA turnover studied by RNA interference. Nucleic Acids Res. 2004;32(4):1279–1288. | ||
Taylor GA, Carballo E, Lee DM, et al. A pathogenetic role for TNF alpha in the syndrome of cachexia, arthritis, and autoimmunity resulting from tristetraprolin (TTP) deficiency. Immunity. 1996;4(5):445–454. | ||
Lu JY, Sadri N, Schneider RJ. Endotoxic shock in AUF1 knockout mice mediated by failure to degrade proinflammatory cytokine mRNAs. Genes Dev. 2006;20(22):3174–3184. | ||
Sadri N, Schneider RJ. Auf1/Hnrnpd-deficient mice develop pruritic inflammatory skin disease. J Invest Dermatol. 2009;129(3):657–670. | ||
Agustí A, Faner R. Systemic inflammation and comorbidities in chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2012;9(2):43–46. | ||
Bradford E, Jacobson S, Varasteh J, et al. The value of blood cytokines and chemokines in assessing COPD. Respir Res. 2017;18(1):180. | ||
Carolan BJ, Heguy A, Harvey BG, Leopold PL, Ferris B, Crystal RG. Up-regulation of expression of the ubiquitin carboxyl-terminal hydrolase L1 gene in human airway epithelium of cigarette smokers. Cancer Res. 2006;66(22):10729–10740. | ||
Marwick JA, Caramori G, Casolari P, et al. A role for phosphoinositol 3-kinase delta in the impairment of glucocorticoid responsiveness in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2010;125(5):1146–1153. | ||
Fan J, Ishmael FT, Fang X, et al. Chemokine transcripts as targets of the RNA-binding protein HuR in human airway epithelium. J Immunol. 2011;186(4):2482–2494. | ||
Yanagisawa S, Baker JR, Vuppusetty C, et al. The dynamic shuttling of SIRT1 between cytoplasm and nuclei in bronchial epithelial cells by single and repeated cigarette smoke exposure. PLoS One. 2018;13(3):e0193921. | ||
Yoon JH, de S, Srikantan S, et al. PAR-CLIP analysis uncovers AUF1 impact on target RNA fate and genome integrity. Nat Commun. 2014;5:5248. | ||
Tominaga K, Srikantan S, Lee EK, et al. Competitive regulation of nucleolin expression by HuR and miR-494. Mol Cell Biol. 2011;31(20):4219–4231. | ||
dal Col J, Dolcetti R. GSK-3beta inhibition: at the crossroad between Akt and mTOR constitutive activation to enhance cyclin D1 protein stability in mantle cell lymphoma. Cell Cycle. 2008;7(18):2813–2816. | ||
Casolaro V, Fang X, Tancowny B, et al. Posttranscriptional regulation of IL-13 in T cells: role of the RNA-binding protein HuR. J Allergy Clin Immunol. 2008;121859(4):853–859. | ||
Mazan-Mamczarz K, Kuwano Y, Zhan M, et al. Identification of a signature motif in target mRNAs of RNA-binding protein AUF1. Nucleic Acids Res. 2009;37(1):204–214. | ||
Howe EA, Sinha R, Schlauch D, Quackenbush J. RNA-Seq analysis in MeV. Bioinformatics. 2011;27(22):3209–3210. | ||
Mi H, Huang X, Muruganujan A, et al. PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017;45(D1):D183–D189. | ||
Bourgeois JS, Jacob J, Garewal A, Ndahayo R, Paxson J. The Bioavailability of Soluble Cigarette Smoke Extract Is Reduced through Interactions with Cells and Affects the Cellular Response to CSE Exposure. PLoS One. 2016;11(9):e0163182. | ||
Gao W, Li L, Wang Y, et al. Bronchial epithelial cells: the key effector cells in the pathogenesis of chronic obstructive pulmonary disease? Respirology. 2015;20(5):722–729. | ||
di Stefano A, Caramori G, Oates T, et al. Increased expression of nuclear factor-kappaB in bronchial biopsies from smokers and patients with COPD. Eur Respir J. 2002;20(3):556–563. | ||
Spurrell JC, Wiehler S, Zaheer RS, Sanders SP, Proud D. Human airway epithelial cells produce IP-10 (CXCL10) in vitro and in vivo upon rhinovirus infection. Am J Physiol Lung Cell Mol Physiol. 2005;289(1):L85–L95. | ||
Hudy MH, Proud D. Cigarette smoke enhances human rhinovirus-induced CXCL8 production via HuR-mediated mRNA stabilization in human airway epithelial cells. Respir Res. 2013;14:88. | ||
Navratilova Z, Novosadova E, Hagemann-Jensen M, et al. Expression Profile of Six RNA-Binding Proteins in Pulmonary Sarcoidosis. PLoS One. 2016;11(8):e0161669. | ||
Ricciardolo FL, Caramori G, Ito K, et al. Nitrosative stress in the bronchial mucosa of severe chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2005;116(5):1028–1035. | ||
Sirenko OI, Lofquist AK, Demaria CT, Morris JS, Brewer G, Haskill JS. Adhesion-dependent regulation of an A+U-rich element-binding activity associated with AUF1. Mol Cell Biol. 1997;17(7):3898–3906. | ||
di Stefano A, Caramori G, Barczyk A, et al. Innate immunity but not NLRP3 inflammasome activation correlates with severity of stable COPD. Thorax. 2014;69(6):516–524. | ||
Bhowmik A, Seemungal TA, Sapsford RJ, Wedzicha JA. Relation of sputum inflammatory markers to symptoms and lung function changes in COPD exacerbations. Thorax. 2000;55(2):114–120. | ||
Paschoud S, Dogar AM, Kuntz C, Grisoni-Neupert B, Richman L, Kühn LC. Destabilization of interleukin-6 mRNA requires a putative RNA stem-loop structure, an AU-rich element, and the RNA-binding protein AUF1. Mol Cell Biol. 2006;26(22):8228–8241. | ||
de Boer WI, Sont JK, van Schadewijk A, Stolk J, van Krieken JH, Hiemstra PS. Monocyte chemoattractant protein 1, interleukin 8, and chronic airways inflammation in COPD. J Pathol. 2000;190(5):619–626. | ||
Traves SL, Culpitt SV, Russell RE, Barnes PJ, Donnelly LE. Increased levels of the chemokines GROalpha and MCP-1 in sputum samples from patients with COPD. Thorax. 2002;57(7):590–595. | ||
Panganiban RP, Vonakis BM, Ishmael FT, Stellato C. Coordinated post-transcriptional regulation of the chemokine system: messages from CCL2. J Interferon Cytokine Res. 2014;34(4):255–266. | ||
Laroia G, Sarkar B, Schneider RJ. Ubiquitin-dependent mechanism regulates rapid turnover of AU-rich cytokine mRNAs. Proc Natl Acad Sci U S A. 2002;99(4):1842–1846. | ||
Laroia G, Schneider RJ. Alternate exon insertion controls selective ubiquitination and degradation of different AUF1 protein isoforms. Nucleic Acids Res. 2002;30(14):3052–3058. | ||
Lau WK, Li X, Yeung DS, Chan KH, Ip MS, Mak JC. The involvement of serotonin metabolism in cigarette smoke-induced oxidative stress in rat lung in vivo. Free Radic Res. 2012;46(11):1413–1419. | ||
To M, Yamamura S, Akashi K, et al. Defect of adaptation to hypoxia in patients with COPD due to reduction of histone deacetylase 7. Chest. 2012;141(5):1233–1242. | ||
Keene JD. The globalization of messenger RNA regulation. Natl Sci Rev. 2014;1(2):184–186. | ||
Sedlyarov V, Fallmann J, Ebner F, et al. Tristetraprolin binding site atlas in the macrophage transcriptome reveals a switch for inflammationresolution. Mol Syst Biol. 2016;12(5):868. | ||
Wu X, Lan L, Wilson DM, et al. Identification and validation of novel small molecule disruptors of HuR-mRNA interaction. ACS Chem Biol. 2015;10(6):1476–1484. | ||
Yang WY, Gao R, Southern M, Sarkar PS, Disney MD. Design of a bioactive small molecule that targets r(AUUCU) repeats in spinocerebellar ataxia 10. Nat Commun. 2016;7:11647. | ||
Alonso N, Guillen R, Chambers JW, Leng F. A rapid and sensitive high-throughput screening method to identify compounds targeting protein-nucleic acids interactions. Nucleic Acids Res. 2015;43(8):e52. | ||
Wang Z, Bhattacharya A, Ivanov DN. Identification of Small-Molecule Inhibitors of the HuR/RNA Interaction Using a Fluorescence Polarization Screening Assay Followed by NMR Validation. PLoS One. 2015;10(9):e0138780. |
Supplementary materials
Scoring system for immunohistochemistry in the bronchial rings
The immunostaining for all the RBPs studied was scored in the bronchial epithelium, as previously described.1 Briefly, immunostained cells in the bronchial epithelium lined over the epithelial basement membrane were counted in several nonoverlapping high-power fields until the whole specimen was examined. As previous studies have shown that lung carcinogenesis may be associated per se with changes in RBP expression,2,4–6 we deliberately avoided measuring the area of bronchial surface epithelium involved with squamous metaplasia and preneoplastic lesions of the bronchial epithelium. Results of the single immunohistochemistry are expressed as the area of stained bronchial surface epithelium to total bronchial epithelium area and as the area of reactive glands to total bronchial submucosal glands area, measured by computerized image analysis. Group data were expressed as mean and standard error of the mean (SEM).
Scoring system for immunohistochemistry in the peripheral lung
Staining analysis was performed as previously published.3 A bronchiole was taken to be an airway with no cartilage and glands in its wall. According to a validated method,3 the number of bronchiolar epithelial cells with positive staining (nuclear and/or cytosolic) was expressed as a percentage of the total number of epithelial cells counted in each bronchiolar section and the number of positively stained endoalveolar macrophages was expressed as a percentage of the total cells with the morphological appearance of alveolar macrophages counted inside of the alveoli. Group data were expressed as mean and standard error of the mean (SEM).
 | Table S1 Study population providing peripheral blood mononuclear cells (PBMC) |
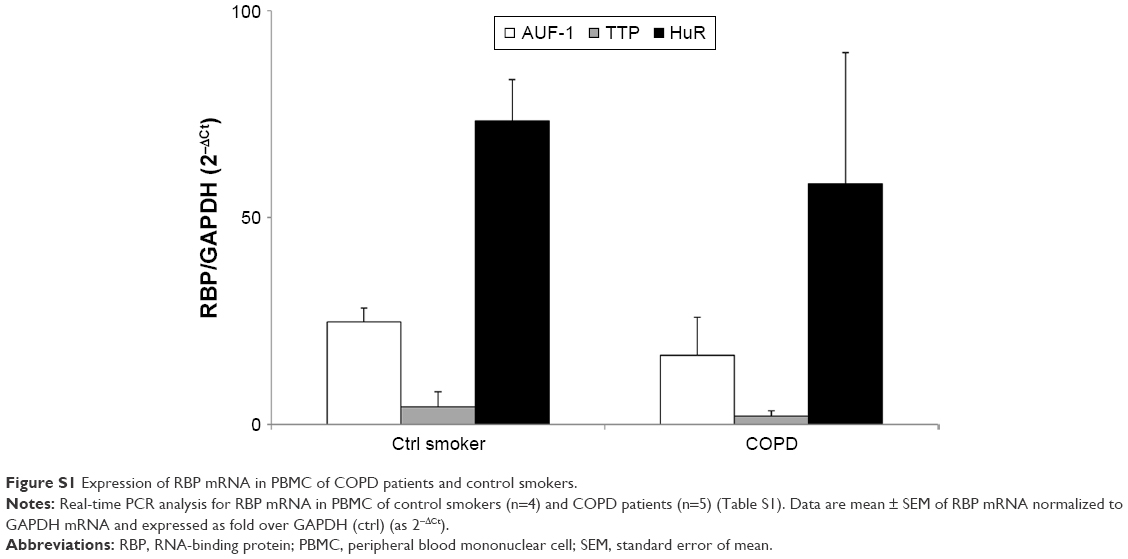 | Figure S1 Expression of RBP mRNA in PBMC of COPD patients and control smokers. |
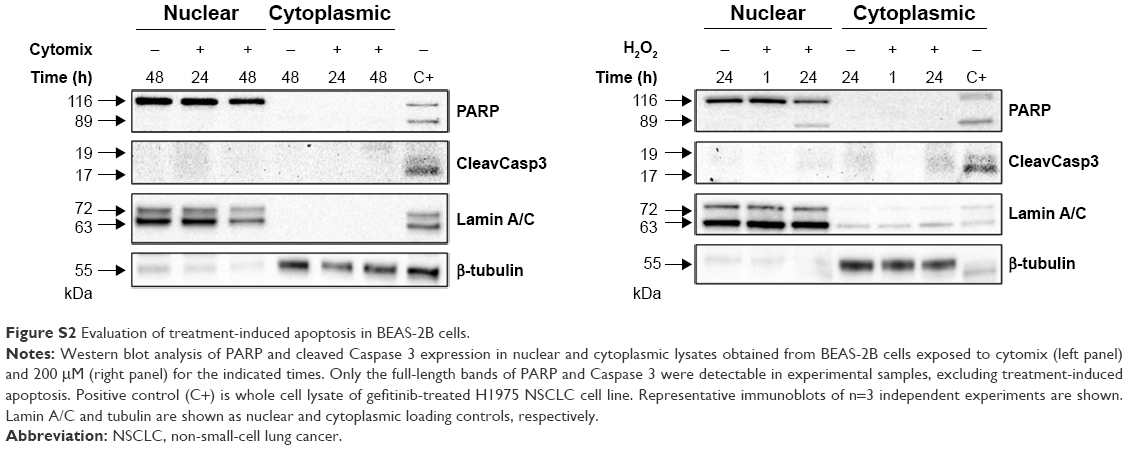 | Figure S2 Evaluation of treatment-induced apoptosis in BEAS-2B cells. |
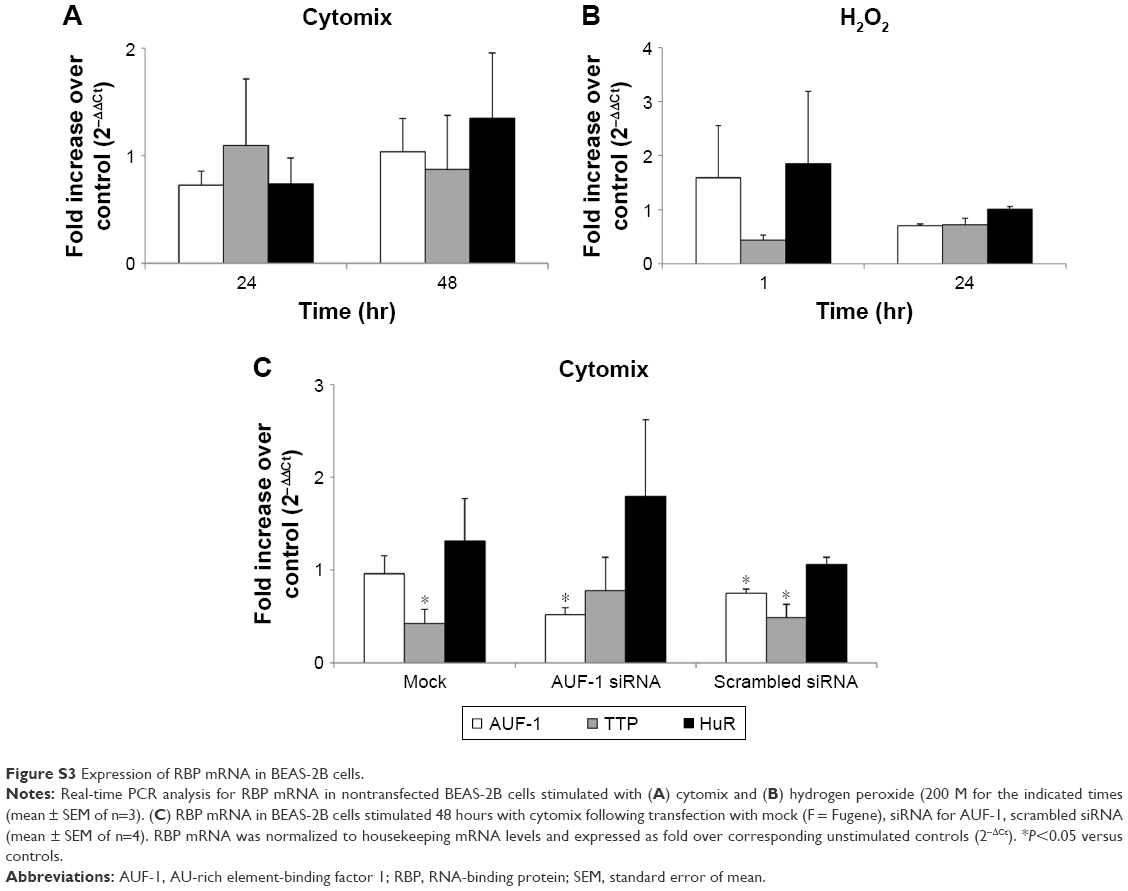 | Figure S3 Expression of RBP mRNA in BEAS-2B cells. |
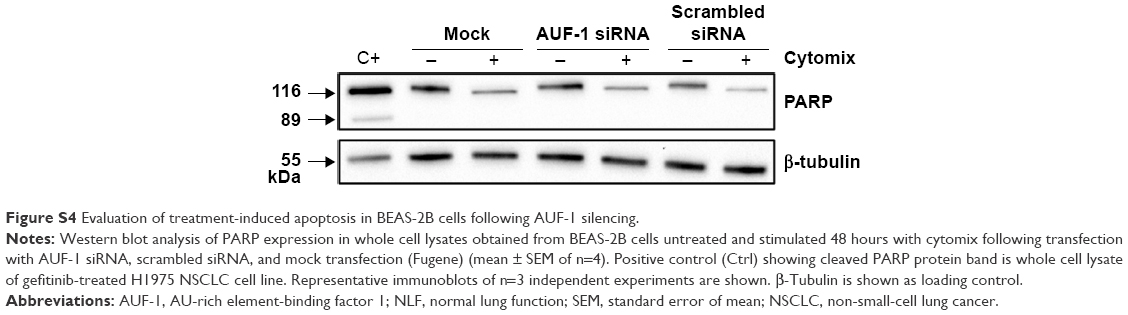 | Figure S4 Evaluation of treatment-induced apoptosis in BEAS-2B cells following AUF-1 silencing. |
References
Caramori G, Casolari P, Di Gregorio C, et al. MUC5AC expression is increased in bronchial submucosal glands of stable COPD patients. Histopathology. 2009;55(3):321–331. | ||
Lafzi A, Kazan H. Inferring RBP-Mediated Regulation in Lung Squamous Cell Carcinoma. PLoS One. 2016;11(5):e0155354. | ||
Caramori G, Adcock IM, Casolari P, et al. Unbalanced oxidant-induced DNA damage and repair in COPD: a link towards lung cancer. Thorax. 2011;66(6):521–527. | ||
Fallahi M, Amelio AL, Cleveland JL, Rounbehler RJ. CREB targets define the gene expression signature of malignancies having reduced levels of the tumor suppressor tristetraprolin. PLoS One. 2014;9(12):e115517. | ||
Giaginis C, Alexandrou P, Tsoukalas N, et al. Hu-antigen receptor (HuR) and cyclooxygenase-2 (COX-2) expression in human non-small-cell lung carcinoma: associations with clinicopathological parameters, tumor proliferative capacity and patients’ survival. Tumour Biol. 2015;36(1):315–327. | ||
Muralidharan R, Panneerselvam J, Chen A, Zhao YD, Munshi A, Ramesh R. HuR-targeted nanotherapy in combination with AMD3100 suppresses CXCR4 expression, cell growth, migration and invasion in lung cancer. Cancer Gene Ther. 2015;22(12):581–590. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.