Back to Journals » Clinical Ophthalmology » Volume 8
Diagnostic challenges in primary orbital fibrosarcoma: a case report
Authors Scruggs B, Ho S, Valenzuela A
Received 9 July 2014
Accepted for publication 14 August 2014
Published 20 November 2014 Volume 2014:8 Pages 2319—2323
DOI https://doi.org/10.2147/OPTH.S70843
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Brittni A Scruggs, Son T Ho, Alejandra A Valenzuela
Department of Ophthalmology, School of Medicine, Tulane University, New Orleans, LA, USA
Abstract: Fibrosarcoma is a rare and malignant spindle cell tumor of mesenchymal origin that infrequently presents in the orbit. Evolving diagnostic criteria confound the identification of these tumors, as well as the interpretation of previous studies on this unusual entity. We report a case of a 66-year-old man with a mass on his left inferior orbit, with associated paresthesia. A computed tomography (CT) scan showed a lesion on the left anteroinferomedial orbit, with bone erosion. An en bloc surgical excision followed by a thorough immunohistologic evaluation allowed diagnosis of an orbital fibrosarcoma. The patient has had no recurrence after 14 months of follow up. Once a commonly identified soft tissue malignancy, fibrosarcoma has become a diagnosis of exclusion as a result of improved diagnostic and classification techniques, such as immunohistochemistry and fluorescence in situ hybridization (FISH). This type of soft tissue tumor is now an uncommon entity, and we report the first case of a primary orbital fibrosarcoma in an adult, using modern diagnostic and classification methods.
Keywords: spindle cell orbital tumor, mesenchymal tumor, soft tissue malignancy, fibroblasts
Introduction
Fibrosarcoma (FS) is a slow-growing, yet locally aggressive, malignant tumor that arises from fibroblasts.1,2 As a result of new diagnostic classifications and changing terminology of soft tissue malignancies, FS is now a diagnosis of exclusion.2,3 This entity, which represents less than 1% of adult soft tissue tumors, most commonly occurs in the deep soft tissue of the extremities.3 Occurrence of this entity in the orbit is exceedingly rare, and these orbital lesions are often associated with recurrence and extension after incomplete removal.1,2 We present a rare case of primary orbital FS in an adult patient, using updated histopathologic, molecular, and cytogenetic studies.
Case report
A 66-year-old man presented with abnormal sensation along the left orbital rim for the prior 6 weeks. Upon palpation, a 2 cm ×1 cm firm tender mass was located anterior to the left medial aspect of the inferior orbital rim. There was no globe displacement or proptosis (Figure 1), but there was paresthesia along the left infraorbital nerve. The patient’s extraocular motility, visual acuity, intraocular pressure, and posterior segment examination were all within normal limits. The patient had no previous history of malignancy, no previous ocular problems, and no significant medical history. Additionally, the family history was noncontributory. A head and orbit computed tomography (CT) scan showed a well-defined lesion in the left anterior aspect of the inferomedial orbit, just behind the rim, with suggestive bone erosion (Figure 2). An inferior fornix approach was used for an excisional biopsy. During surgery, a well-defined, grayish 3.5 cm ×1.5 cm mass was noticed disrupting the inferomedial periorbita in the posterior aspect of the medial left orbital rim, which extended posteriorly toward the medial orbital floor (Figure 3A). The mass did not involve the infraorbital bundle, and after elevating the lesion, the orbital floor was noted to be clearly eroded. Due to these potential malignant features, with the paresthesia and bone erosion, a rectangular en bloc excision was performed including the mass with the surrounding left inferomedial orbital rim and the medial orbital floor (Figure 3B).
 | Figure 1 (A) Clinical photograph showing no globe displacement or ocular abnormalities. (B) Additional clinical photograph demonstrating no mass on visual inspection. |
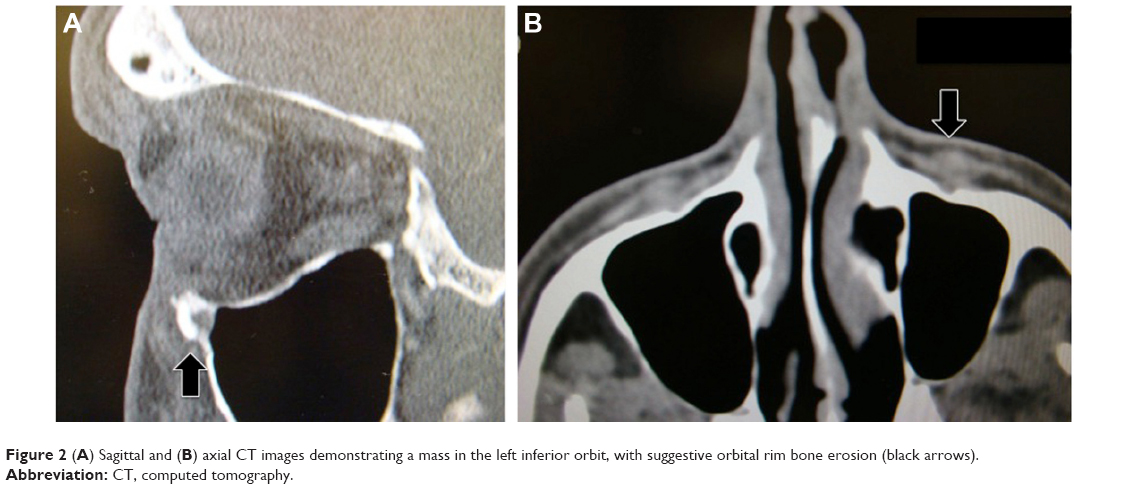 | Figure 2 (A) Sagittal and (B) axial CT images demonstrating a mass in the left inferior orbit, with suggestive orbital rim bone erosion (black arrows). |
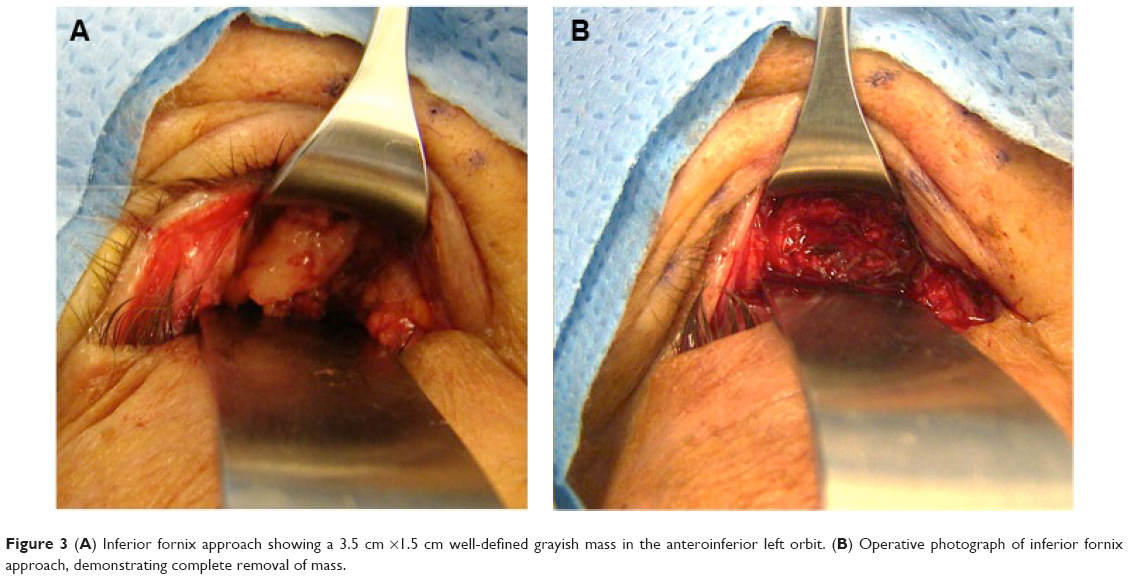 | Figure 3 (A) Inferior fornix approach showing a 3.5 cm ×1.5 cm well-defined grayish mass in the anteroinferior left orbit. (B) Operative photograph of inferior fornix approach, demonstrating complete removal of mass. |
Histopathology demonstrated spindle cells with a herringbone pattern without significant pleomorphism (Figure 4A–C), and the mass demonstrated invasion of bone (Figure 4D). Mitotic activity was low, with a nuclear proliferation index on Ki67 immunostain of 5% or less. The specimen was positive for vimentin and showed cytoplasmic staining for cluster of differentiation (CD)99 with no membranous staining. B-cell lymphoma 2 (Bcl-2) stain showed rare weakly staining, slightly whorled foci of tumor cells. The specimen was negative for smooth muscle actin (SMA), desmin, pancytokeratin, S100, CD34, epithelial membrane antigen (EMA), CAM 5.2, carcinoembryonic antigen (CEA), and CD117. Fluorescence in situ hybridization (FISH) analysis was negative for SYT gene rearrangement. Due to the lack of diagnostically categorizing results, the tumor received the diagnosis of FS. An intermediate grade of 2 was assigned using the French Federation of Cancer Centers (FNCLCC) grading system,4 due to the tumor’s histological characteristics: a low mitotic count (1 point), no necrosis (0 points), and a histological type of poorly differentiated FS (3 points).4 The tumor was considered poorly differentiated, due to a scarcity of intercellular collagen. Staging of the patient using whole body positron emission tomography CT (PET-CT) imaging suggested that this mass was a primary orbital tumor with minimal invasion of the orbital soft tissues, and thus, no further treatment was indicated. The patient has remained alive, well, and disease-free with serial clinical examinations, whole body PET-CTs, and orbital magnetic resonance imaging (MRI) after 14 months of follow up.
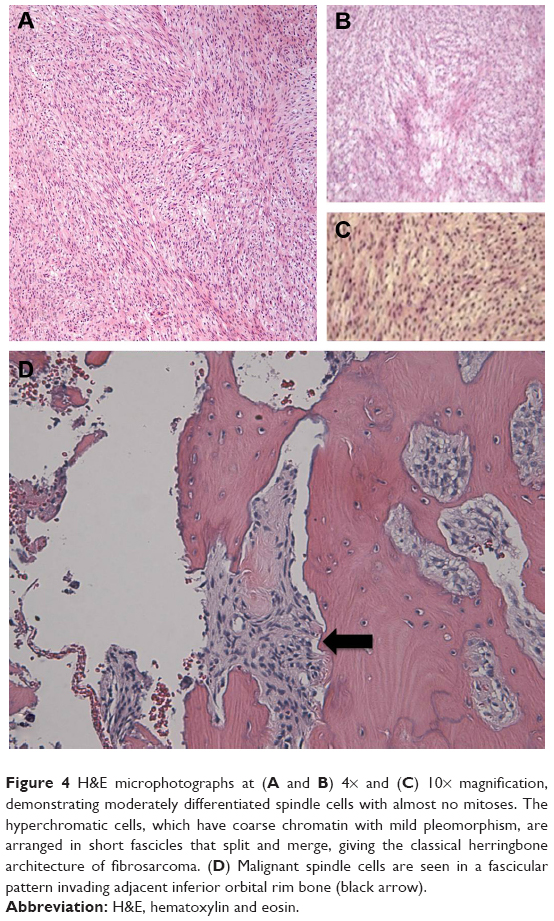 | Figure 4 H&E microphotographs at (A and B) 4× and (C) 10× magnification, demonstrating moderately differentiated spindle cells with almost no mitoses. The hyperchromatic cells, which have coarse chromatin with mild pleomorphism, are arranged in short fascicles that split and merge, giving the classical herringbone architecture of fibrosarcoma. (D) Malignant spindle cells are seen in a fascicular pattern invading adjacent inferior orbital rim bone (black arrow). |
Discussion
In the early 20th century, before new diagnostic classifications and changing terminology of soft tissue malignancies, FS was the most common diagnosis for soft tissue sarcomas in adults.3 Morphologically, FS is composed of relatively monomorphic spindle cells arranged in characteristic long sweeping fascicles or a herringbone pattern, with no more than a moderate degree of pleomorphism; tumors showing a greater degree of pleomorphism should be classified as undifferentiated pleomorphic sarcomas.5,6 Immunohistochemistry shows absent expression of any markers other than vimentin or minimal SMA.3,5 FS typically presents in middle-aged to older adults (median age 50 years), with a preponderance in men.5 These tumors often involve the deep soft tissues of the extremities, trunk, head, and neck; they have been identified in previously irradiated fields and in association with implanted foreign material.3
In recent years, improvements in diagnostic and classification techniques have resulted in FS becoming a diagnosis of exclusion, and these tumors currently represent less than 1% of all adult soft tissue sarcomas.3,5,6 As an example of the increasing rarity of this tumor, a study that reexamined 163 lesions diagnosed as FS between 1960 and 2008 found that 137 (84%) cases would no longer receive this diagnosis.3 This evolution in diagnostic criteria of FS makes interpreting data from previous studies difficult in regards to prognosis or treatment, as these previous studies likely contain tumor populations that would doubtfully receive this diagnosis today.3 Determination of management is further complicated for FS in atypical locations such as the orbit because even the largest and most recent FS studies contain few or no orbital cases.3,5,7–9 Prognosis of FS must be carefully considered when treatment could potentially involve removal of the globe and orbital content. To our knowledge, only one case has been reported of adult-type FS in the orbit using immunohistochemistry and cytogenetic studies; unlike in this case, the tumor occurred in a 3-year-old child and did not affect the orbital rim or floor.10
An extensive differential diagnosis exists for FS and includes other fibroblastic sarcomas, such as low-grade fibromyxoid sarcoma, sclerosing epithelioid FS, and dermatofibrosarcoma protuberans; and non-FS mesenchymal tumors, such as monophasic synovial sarcoma, malignant peripheral nerve sheath tumor, and solitary fibrous tumor.3,5,6 Like FS, these variant fibroblastic sarcomas are negative for lineage-specific markers, but they do not share FS morphological similarities, such as a fascicular “herringbone” growth pattern and low to moderate pleomorphism.3,5,6 In one study, monophasic synovial sarcoma was the most frequent (13%) mimicker of FS; this diagnosis was excluded in our case, due to a lack of expression of cytokeratins or EMA and a negative SYT FISH analysis.3 Malignant peripheral nerve sheath tumors stain for S100, and solitary fibrous tumor express CD34; however, neither marker was present in the specimen.
Previous recommendations for treatment of FS included wide local excision and even orbital exenteration, which is usually reserved for cases of recurrence.2,7,11 For low-grade lesions, some considered surgery alone to be sufficient.8 These recommendations stemmed from an understanding of tumors that had been diagnosed as FS without definitive histopathological evidence. Consequently, these treatment recommendations are not necessarily aligned with the current thinking of orbital FS. Recent investigations of FS do offer some insight in this regard, but few such studies are available due to the increasing rarity of the lesion.3,9 The most comprehensive modern series of FS, which included no orbital cases, evaluated 26 tumors and suggests that modern FS is an aggressive sarcoma with over 80% of the tumors being classified as grade 2 or 3 (FNCLCC grading). That series included multiple local recurrences, along with lymph node and parenchymal metastases. Survival was less than 70% at 2 years and less than 55% at 5 years, but an attempt was not made to correlate grade with outcome, due to the small sample size.3 In another recent study, the mean proliferative index of 20 FSs was reported to be 26.15%±3.30%.12 The aggressive course of the adult-type FS compared with the infantile FS has previously been linked to the higher proliferative index of adult-type tumors,13 but the utility of the proliferative markers has not yet been established for prognosis assessment in FS. In our case, the Ki67 immunostain was less than 5%, indicating a low, and favorable, mitotic index; however, the grade was still determined to be intermediate, based on the poorly differentiated histology.
We decided to proceed with an en bloc excision including the eroded surrounding bone, but we preserved the globe, considering the mass was isolated to the left anteroinferomedial orbit, with minimal disruption of the local periorbita. Lung, bone, and regional lymph node metastases are possible, and postoperative adjuvant radiotherapy and chemotherapy are sometimes considered, although our patient declined in light of the little evidence available in the literature for orbital lesions.7 To our knowledge, no case of primary adult-type orbital FS of an adult patient has been reported using modern diagnostic and classification methods. Due to the rarity of this tumor combined with the continuously evolving diagnostic and classification techniques of FS, case reports such as this could be the most viable source of information on this tumor when it occurs in the orbit.
Acknowledgments
We thank Sharon W Weiss, MD, for her consultation work, and the Department of Pathology at Tulane University Health Sciences Center, for providing excellent histopathology slides for publication.
Disclosure
The authors report no conflicts of interest in this work.
References
Duke-Elder S, MacFaul PA, editors. System of Ophthalmology. Vol XIII: The Ocular Adnexa. Part II: Lacrimal, Orbital, and Para-orbital Diseases. London: Henry Kimpton; 1974. | ||
Rootman J. Diseases of the Orbit. A Multidisciplinary Approach. 2nd ed. Philadelphia, PA: Lippincott Williams and Wilkins; 2003. | ||
Bahrami A, Folpe AL. Adult-type fibrosarcoma: A reevaluation of 163 putative cases diagnosed at a single institution over a 48-year period. Am J Surg Pathol. 2010;34(10):1504–1513. | ||
Coindre JM. Grading of soft tissue sarcomas: review and update. Arch Pathol Lab Med. 2006;130(10):1448–1453. | ||
Folpe AL. Fibrosarcoma: a review and update. Histopathology. 2014;64(1):12–25. | ||
Weiss SW, Goldblum JR. Enzinger and Weiss’s Soft Tissue Tumors. 5th ed. Philadelphia, PA: Mosby; 2007. | ||
Scott SM, Reiman HM, Pritchard DJ, Ilstrup DM. Soft tissue fibrosarcoma. A clinicopathologic study of 132 cases. Cancer. 1989;64(4):925–931. | ||
Mark RJ, Sercarz JA, Tran L, Selch M, Calcaterra TC. Fibrosarcoma of the head and neck. The UCLA experience. Arch Otolaryngol Head Neck Surg. 1991;117(4):396–401. | ||
Hansen T, Katenkamp K, Brodhun M, Katenkamp D. Low-grade fibrosarcoma – report on 39 not otherwise specified cases and comparison with defined low-grade fibrosarcoma types. Histopathology. 2006;49(2):152–160. | ||
Hussain RM, Erickson BP, Rosenberg AE, Dubovy SR, Johnson TE. Recurrent orbital adult-type fibrosarcoma in a 3-year-old girl. Ophthal Plast Reconstr Surg. Epub 2014 May 14. | ||
Lane KA, Bilyk JR, Jakobiec FA. Mesenchymal, fibroosseous, and cartilaginous orbital tumors. In: Albert DM, Miller JW, Azar DT, Blodi BA. Albert and Jakobiec’s Principles and Practice of Ophthalmology. 3rd ed. Edinburgh: Saunders; 2008:3159–3201. | ||
Lin XY, Wang L, Zhang Y, Dai SD, Wang EH. Variable Ki67 proliferative index in 65 cases of nodular fasciitis, compared with fibrosarcoma and fibromatosis. Diagn Pathol. 2013;8:50. | ||
Kihara S, Nehlsen-Cannarella N, Kirsch WM, Chase D, Garvin AJ. A comparative study of apoptosis and cell proliferation in infantile and adult fibrosarcomas. Am J Clin Pathol. 1996;106(4):493–497. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.