")
Back to Journals » Journal of Blood Medicine » Volume 6
Diagnostic and therapeutic considerations in idiopathic hypereosinophilia with warm autoimmune hemolytic anemia
Authors Sweidan A, Brys A, Sohn D, Sheth M
Received 8 June 2015
Accepted for publication 28 July 2015
Published 7 September 2015 Volume 2015:6 Pages 257—260
DOI https://doi.org/10.2147/JBM.S90078
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Martin Bluth
Alexander J Sweidan,1,2 Adam K Brys,3 David D Sohn,1,2 Milan R Sheth4
1University of California Los Angeles, Los Angeles, CA, USA; 2Department of Internal Medicine, St Mary Medical Center, Long Beach, CA, USA; 3School of Medicine, Duke University, Durham, NC, USA; 4Long Beach Memorial Hospital, Long Beach, CA, USA
Abstract: Hypereosinophilic syndrome (HES) encompasses numerous diverse conditions resulting in peripheral hypereosinophilia that cannot be explained by hypersensitivity, infection, or atopy and that is not associated with known systemic diseases with specific organ involvement. HES is often attributed to neoplastic or reactive causes, such as chronic eosinophilic leukemia, although a majority of cases remains unexplained and are considered idiopathic. Here, we review the current diagnosis and management of HES and present a unique case of profound hypereosinophilia associated with warm autoimmune hemolytic anemia requiring intensive management. This case clearly illustrates the limitations of current knowledge with respect to hypereosinophilia syndrome as well as the challenges associated with its classification and management.
Keywords: hypereosinophilia, eosinophils, myeloproliferative disorder, autoimmune hemolytic anemia, idiopathic autoimmune hemolytic anemia, leukemia
Introduction
Peripheral hypereosinophilia is most commonly associated with allergic causes, including drug hypersensitivity, parasitic infection, or atopic disease. Hypereosinophilic syndrome (HES) encompasses a group of disorders that cannot be explained by these common causes.1 True HES is rare, with an estimated incidence of 0.36 cases per 100,000 person-years and affects predominantly men between the ages of 20 and 50 years.2 Following exclusion of common known causes of hypereosinophilia, HES is diagnosed by confirming the presence of ≥1.5×109 cells/L on two occasions separated by at least 1 month and by determining the evidence of organ involvement.3 HES can be subclassified broadly as 1) a primary disorder of myeloid cells resulting in monoclonal proliferation (eg, neoplasia), 2) a secondary expansion of polyclonal, reactive eosinophils (eg, cytokine driven proliferation), or 3) an otherwise unexplained increase in eosinophils (idiopathic HES).4
Warm autoimmune hemolytic anemia (WAIHA) is a potentially life-threatening condition characterized by the presence of intravascular hemolysis, spherocytes in peripheral smear, and anti-red blood cell (RBC) antibodies, known as warm agglutinins that bind to RBC antigens at room temperature.5 Common etiologies of WAIHA include drug reaction, viral infection, autoimmune or connective tissue disease, and hematological malignancy, particularly chronic lymphocytic leukemia.6 Despite potential underlying lymphoproliferative and immune dysregulation, to our knowledge, there have been only two reported cases of idiopathic HES associated with WAIHA.7,8 Here, we present the diagnosis and management of an unusual case of profound hypereosinophilia (49.9×109 cells/L) associated with WAIHA, but without identifiable neoplastic etiology.
Case report
A 25-year-old male with a past medical history, significant only for frequent use of ibuprofen for musculoskeletal pain, presented to the emergency department with acute onset fever and malaise. On arrival, the patient was quickly found to be hypotensive and tachypneic, with scleral icterus evident on physical exam. Otherwise, no cardiac, pulmonary, gastrointestinal, neurological, or dermatological irregularities were reported. Preliminary laboratory evaluation demonstrated profound anemia (hemoglobin: 5.4, hematocrit: 13.0), and the patient was admitted to the intensive care unit for further management, with a working diagnosis of systemic inflammatory response syndrome with severe anemia.
A direct antiglobulin test was positive for anti-erythrocyte IgG, supporting the diagnosis of WAIHA.9 Peripheral blood smear showed significant eosinophilia (49.9×109 cells/L) with no blasts (Figure 1). A subsequent bone marrow biopsy was negative for the presence of a leukemic or lymphoproliferative process and found no evidence of PDGFR-A mutation or BCR/ABL translocation (Figure 2). Flow cytometry was performed and similarly showed no evidence of a leukemic or lymphoproliferative process. Further workup with transthoracic echocardiography and CT scan of the neck and torso found no neoplastic disease. A survey for infectious etiologies associated with eosinophilia revealed a negative Entamoeba histolytica, Toxocara, Trichinella, Strongyloides stercoralis, Hepatitis B and C, and HIV serologies. A QuantiFERON Gold® (Cellestis Ltd, Carnegie, VIC, Australia) as well as blood cultures, urine cultures, stool cultures, stool white blood cell count, and stool ova and parasites were unremarkable. To rule out other causes of eosinophilia, a cortisol stimulation test (adrenal insufficiency), anti-neutrophil cytoplasmic antibodies (eosinophilic granulomatosis with polyangiitis), and vitamin B12 (myeloproliferative disease) were examined, all of which were found to be within normal limits. Of note, an abdominal ultrasound revealed mild hepatosplenomegaly.
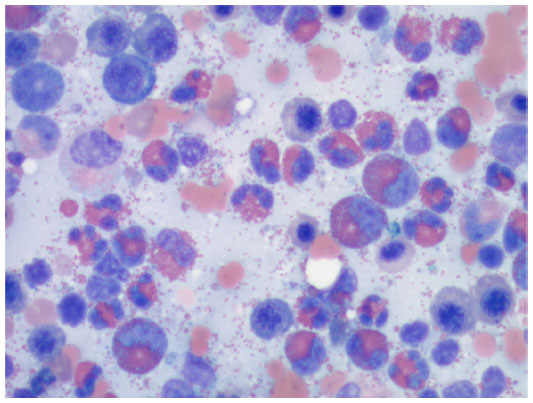 | Figure 1 Examination of peripheral blood smear. |
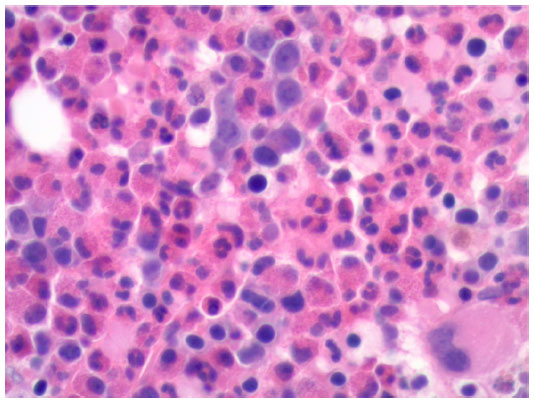 | Figure 2 Bone marrow biopsy. |
On admission to the intensive care unit, the patient received transfusion of 4 units of packed RBCs and was treated with high-dose intravenous glucocorticoids (methylprednisone 120 mg intravenous every 6 hours for 1 day, followed by prednisone 60 mg oral administration daily). His hemoglobin and eosinophil count stabilized (hemoglobin: 9.2, hematocrit: 26.5, eosinophil count: 0.8×109 cells/L) and the patient was discharged in 5 days, with a prednisone taper to complete his glucocorticoid therapy, and instructed to avoid nonsteroidal anti-inflammatory drugs (NSAIDs) as possible precipitants of hemolytic anemia. The treatment plan for this patient consisted of short-interval follow-up, with hematology and oncology to reassess blood count. The patient was instructed to return to the hospital for any concerning symptoms such as fatigue, fevers, chills, and night sweats. Unfortunately, the patient was lost to follow-up.
Discussion
A general diagnostic approach for a patient with hypereosinophilia is displayed in Figure 3. Following exclusion of infectious and atopic etiologies as causes of hypereosinophilia, the differential diagnosis in our patient consisted of adverse drug hypersensitivity to ibuprofen, chronic eosinophilic leukemia with AIHA, or idiopathic HES with AIHA.
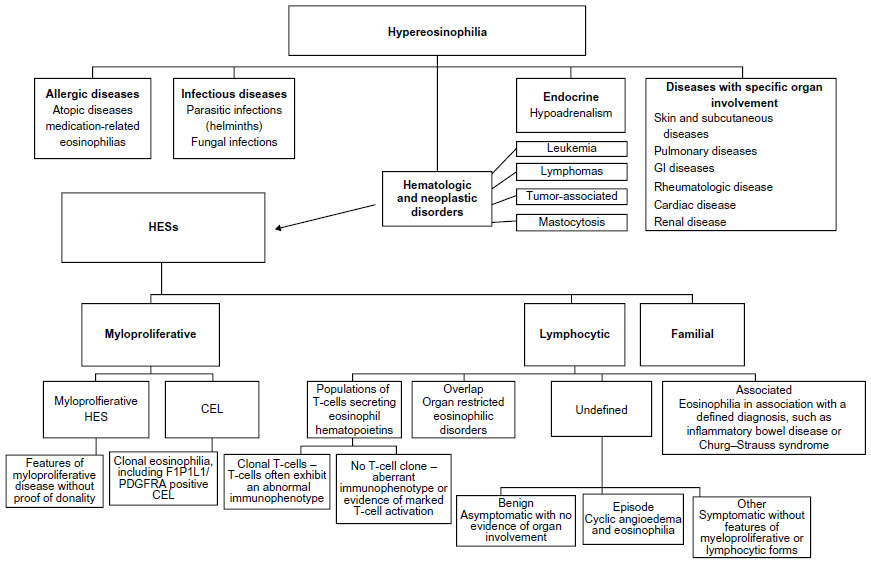 | Figure 3 A general diagnostic approach for patients with hypereosinophilia. |
Despite being considered as generally benign agents, NSAIDs such as ibuprofen are known precipitants of adverse drug reactions associated with hypereosinophilia. Drug reaction with eosinophilia and systemic symptoms (DRESS) is a rare, potentially life-threatening condition that infrequently may result from use of NSAIDs and, more rarely, may be associated with autoimmune phenomena, including hemolytic anemia.10,11 Alternatively, simultaneous drug-induced immune hemolytic anemia resulting from NSAID use may explain this patient’s presentation.12 Although the likelihood of an adverse immune reaction to ibuprofen may have been elucidated through an elution test, such a test was not available at the time of this patient’s presentation and may be of questionable value.13 However, DRESS or other drug hypersensitivities would not be expected to result in an eosinophil count of nearly 50×109 cells/L, and no rash was present in this patient, suggesting an underlying neoplastic process may be more likely.14
Neoplastic etiologies of hypereosinophilia syndrome may be myeloproliferative or lymphoproliferative. Lymphoproliferative syndromes including B-cell acute lymphoblastic leukemia, Hodgkin lymphoma, adult T-cell leukemia/lymphoma, and cutaneous T-cell lymphoma (Sézary syndrome) may produce a reactive eosinophilia in response to dysregulated cytokine secretion, particularly IL-5.15 However, blood smear and bone marrow biopsy demonstrated no evidence of underlying lymphoproliferative disease. Therefore, this patient’s hypereosinophilia most likely had an underlying myeloproliferative origin.
Myeloproliferative etiologies of hypereosinophilia include primary HES, acute eosinophilic leukemia, chronic eosinophilic leukemia, and chronic myeloid leukemia. In this patient, the only abnormality observed was the presence of increased mature eosinophils on peripheral blood smear, making chronic myeloid leukemia and acute eosinophilic leukemia unlikely. While PDGFR-A, the most common mutation associated with myeloproliferative HES, was not found in our workup, other underlying mutations, such as in PDGFR-B or FGFR1, may have been present and resulted in HES. Interestingly, one case of chronic eosinophilic leukemia associated with warm hemolytic anemia has previously been reported in the literature.16
Although a presumptive diagnosis of myeloproliferative hypereosinophilia syndrome could be made, like the majority of cases of HES, a definitive diagnosis was not possible. Nevertheless, our case effectively demonstrates the diagnostic process to be followed in patients presenting with HES, as well as various treatment considerations. Fortunately, our patient did not suffer significant complications from severe hypereosinophilia that may include cutaneous manifestations, pulmonary symptoms, cardiac symptoms (eg, myocarditis), and neurological abnormalities, although the prognosis remains guarded as there is a known possibility for such patients to develop acute myeloid leukemia, sometimes years later.17 In this particular case, the use of high-dose glucocorticoids alone was sufficient to elicit response, and, in combination with transfusion, effectively suppress symptomatic hemolytic anemia. In other cases of HES associated with mutations in PDGFR-A or -B, the tyrosine kinase imatinib may be associated with an effective clinical response.18
Conclusion
Hypereosinophilia syndrome represents a poorly characterized cohort of heterogeneous diseases that may have incredibly diverse presentations. Despite advances in molecular diagnostics and cancer research, the majority of cases of HES lack definitive diagnosis. Here, we have presented a rare case of HES associated with AIHA and reviewed the relevant diagnostic and therapeutic considerations. Further work is needed to better characterize the underlying molecular and genetic factors underlying the development of HES, create improved diagnostic tools, and improve treatment options for affected patients.
Disclosure
The authors report no conflicts of interest in this work.
References
Sheikh J, Weller PF. Clinical overview of hypereosinophilic syndromes. Immunol Allergy Clin North Am. 2007;27:333–355. | |
Crane MM, Chang CM, Kobayashi MG, Weller PF. Incidence of myeloproliferative hypereosinophilic syndrome in the United States and an estimate of all hypereosinophilic syndrome incidence. J Allergy Clin Immunol. 2010;126:179–181. | |
Chusid MJ, Dale DC, West BC, Wolff SM. The hypereosinophilic syndrome: analysis of fourteen cases with review of the literature. Medicine. 1975;54:1–27. | |
Klion AD. How I treat hypereosinophilic syndromes. Blood. 2009;114:3736–3741. | |
Roumier M, Loustau V, Guillaud C, et al. Characteristics and outcome of warm autoimmune hemolytic anemia in adults: new insights based on a single-center experience with 60 patients. Am J Hematol. 2014;89: E150–E155. | |
Dacie SJ. The immune haemolytic anaemias: a century of exciting progress in understanding. Br J Haematol. 2001;114:770–785. | |
Arquati M, Amodei V, Foa P, Tedeschi A. Autoimmune hemolytic anemia in a patient with idiopathic hypereosinophilia. Am J Hematol. 1995;49:356. | |
Hirszel P, Cashell AW, Whelan TV, Dolan R, Yoshihashi A. Urinary Charcot-Leyden crystals in the hypereosinophilic syndrome with acute renal failure. Am J Kidney Dis. 1988;12:319–322. | |
Lechner K, Jager U. How I treat autoimmune hemolytic anemias in adults. Blood. 2010;116:1831–1838. | |
Jeung YJ, Lee JY, Oh MJ, Choi DC, Lee BJ. Comparison of the causes and clinical features of drug rash with eosinophilia and systemic symptoms and Stevens-Johnson syndrome. Allergy Asthma Immunol Res. 2010;2:123–126. | |
Chen YC, Chang CY, Cho YT, Chiu HC, Chu CY. Long-term sequelae of drug reaction with eosinophilia and systemic symptoms: a retrospective cohort study from Taiwan. J Am Acad Dermatol. 2013;68:459–465. | |
Garratty G. Immune hemolytic anemia associated with drug therapy. Blood Rev. 2010;24:143–150. | |
Yazer MH, Triulzi DJ. The role of the elution in antibody investigations. Transfusion. 2009;49:2395–2399. | |
Kardaun SH, Sekula P, Valeyrie-Allanore L, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): an original multisystem adverse drug reaction. Results from the prospective RegiSCAR study. Br J Dermatol. 2013;169:1071–1080. | |
Wilkins HJ, Crane MM, Copeland K, Williams WV. Hypereosinophilic syndrome: an update. Am J Hematol. 2005;80:148–157. | |
Kuk JS, Maceachern JA, Soamboonsrup P, et al. Chronic eosinophilic leukemia presenting with autoimmune hemolytic anemia and erythrophagocytosis by eosinophils. Am J Hematol. 2006;81:458–461. | |
Bain BJ, Fletcher SH. Chronic eosinophilic leukemias and the myeloproliferative variant of the hypereosinophilic syndrome. Immunol Allergy Clin North Am. 2007;27:377–388. | |
Cools J, DeAngelo DJ, Gotlib J, et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med. 2003;348:1201–1214. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.