")
Back to Journals » Clinical Ophthalmology » Volume 17
Diagnostic and Management Strategies of Bietti Crystalline Dystrophy: Current Perspectives
Authors Saatci AO, Ataş F, Çetin GO, Kayabaşı M
Received 14 February 2023
Accepted for publication 15 March 2023
Published 24 March 2023 Volume 2023:17 Pages 953—967
DOI https://doi.org/10.2147/OPTH.S388292
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Ali Osman Saatci,1 Ferdane Ataş,2 Gökhan Ozan Çetin,3 Mustafa Kayabaşı1
1Department of Ophthalmology, Dokuz Eylul University, Izmir, Turkey; 2Department of Ophthalmology, Çerkezköy State Hospital, Tekirdağ, Turkey; 3Department of Medical Genetics, Pamukkale University, Denizli, Turkey
Correspondence: Ali Osman Saatci, Department of Ophthalmology, Dokuz Eylul University, Mustafa Kemal Sahil Bulvari, No: 73 A Blok, Daire 9, Narlidere, Izmir, Turkey, Tel +90 5327437071, Email [email protected]
Abstract: Bietti crystalline dystrophy (BCD) is a rare, genetically determined chorioretinal dystrophy presenting with intraretinal crystalline deposits and varying degrees of progressive chorioretinal atrophy commencing at the posterior pole. In some cases, there can be concomitant corneal crystals noted first in the superior or inferior limbus. CYP4V2 gene, a member of the cytochrome P450 family is responsible for the disease and more than 100 mutations have been defined thus far. However, a genotype–phenotype correlation has not been established yet. Visual impairment commonly occurs between the second and third decades of life. By the fifth or sixth decade of life, vision loss can become so severe that the patient may potentially become legally blind. Multitudes of multimodal imaging modalities can be utilized to demonstrate the clinical features, course, and complications of the disease. This present review aims to reiterate the clinical features of BCD, update the clinical perspectives with the help of multimodal imaging techniques, and overview its genetic background with future therapeutic approaches.
Keywords: Bietti crystalline dystrophy, chorioretinal dystrophy, corneal crystals, inherited disorder, vision loss
Introduction
Bietti crystalline dystrophy (BCD) is a rare, genetically determined chorioretinal dystrophy characterized by sparkling crystals in the cornea and retina with progressive chorioretinal atrophy mainly commencing at the posterior pole.1–3 The disease was named after Bietti1 who described the classical clinical features of the disease in three patients in 1937. Marked central vision impairment, nyctalopia, and constriction of visual fields can be noted in conjunction with the disease progression between the 2nd and 4th decades of life.2 The crystalline deposits were also demonstrated in the skin fibroblasts and lymphocytes.4,5
Recent genetic developments and utilization of newer diagnostic techniques including optical coherence tomography (OCT) and optical coherence tomography angiography (OCTA) have added important new information to our current understanding of BCD. Thus, we aim to reiterate the clinical features of BCD, update the clinical perspectives with the help of multimodal imaging techniques, and overview its genetic background with possible future therapeutic approaches.
Genetics and Pathophysiology
BCD is an autosomal recessive disease associated with mutations in the CYP4V2 gene. The BCD locus was first described in 20006 and the CYP4V2 gene was defined later in 2004.7 The protein encoded by CYP4V2 has 525 amino acids and is a member of the cytochrome P450 family. The gene is expressed in the retinal pigment epithelium and other tissues including the heart, brain, lung, liver, kidney, placenta, and lymphocytes7 however clinical findings are limited to the eye. CYP4V2 is involved in converting fatty acid precursors into n-3 polyunsaturated fatty acids and has a preferential activity for hydroxylating the terminal carbon of saturated, medium-chain fatty acids to produce dicarboxylic fatty acids, which are then processed by mitochondrial β-oxidation.8 The disorder is characterized by the reduced conversion of the fatty acid precursors into n-3-polyunsaturated fatty acids. Defect in lipid metabolism causes deficient lipid binding, elongation, or desaturation9 and leads to the accumulation of cholesterol or cholesterol esters in the retina, cornea, conjunctiva, fibroblasts, and lymphocytes.9,10
Over 100 mutations have been defined in the CYP4V2 gene thus far,11 however, a genotype–phenotype correlation has not been established yet, although there are some efforts to correlate the clinical and/or imaging findings with the genotype.12 CYP4V2 consists of 11 exons and the reported mutations spread through all of the exons. Since the disorder is more prevalent in Asian countries and relatively rare in the rest of the world the mutational hot spots lack for the general population, while c.802–8_810delinsGC is the well-known common mutation in Asian populations.13 In addition, the heterogeneity of the BCD phenotype raises the possibility that a variety of lipid metabolism-related factors, including nutrition, inflammation, and more, may have an impact on the phenotype and course of the disease.14
Epidemiology
It is difficult to estimate the prevalence of BCD due to the rarity of the disease and variations of data collection among the ethnic groups and countries stated in the previous papers. Though the disease prevalence is estimated to be 1 in 67.000 subjects, BCD is more prevalent in East Asia, including China, Korea, and Japan.13 However, prevalence of BCD is estimated to be around 1:116,000 according to a recent study.15 In a group of 207 consecutive retinitis pigmentosa (RP) patients, 4 BCD cases were identified, and BCD prevalence was estimated to be 2.5% of all RP patients.16 Matafsi et al16 revealed that BCD was diagnosed in 3% of non-syndromic RP cases and the equivalent frequency increased to 10% when autosomal recessive RP was considered. García-García et al17 suggested that there could be a Mediterranean distribution for the BCD patients in Europe as most of the reported cases were Italian, Lebanese and Spanish patients.17
Clinical Features
Cornea and Lens Features
Crystals are commonly found in the peripheric paralimbal area related to the CYP4V2 expression in the corneal epithelium.18 These corneal crystal deposits can be detected by slit-lamp biomicroscopy or specular microscopy in one-third to one-half of patients with BCD and corneal involvement seems to be more frequent in northern European descent patients.19,20
The crystalline deposits (Figure 1) are occasionally noted first in the superior or inferior limbus and spread into the entire corneal limbus.5 In two BCD cases, Bozkurt et al21 revealed randomly oriented needle- or rod-shaped corneal crystals up to 40 μm in length and 4–8 μm in width in the corneal epithelium and stroma by using the in vivo confocal microscopy.21 García-García et al22 identified a 38-year-old Spanish woman with BCD having central and paracentral keratopathy without limbal involvement with the confocal microscopy. They also mentioned that crystalline deposits were found not only in the proband but also in two elderly heterozygous carriers with a normal slit-lamp examination with the help of in vivo corneal confocal microscopy.22
 |
Figure 1 Corneal crystals. Slit- lamp biomicroscopy photo showing the crystalline deposits located at the limbus. |
Recently, lenticular crystalline deposits have also been described in a few case reports.23–25 Yokoi et al24 reported the presence of crystalline deposits in the corneal stroma, anterior and posterior lens capsule by using slit-lamp biomicroscopy together with the associated intraretinal crystals and advanced chorioretinal atrophy in an Asian man. Chung et al23 observed crystalline deposits underneath the anterior lens capsule by using anterior segment OCT in a 49-year-old man with BCD. Louati et al25 delineated needle-shaped, crystal-like, or rod-shaped deposits in the corneal stroma, anterior lens cortex, posterior lens cortex as well as the anterior and posterior lens capsule in four BCD patients with the slit-lamp biomicroscopy.
Fundus Features
Bietti crystalline dystrophy is a progressive disorder affecting the RPE-choriocapillaris complex.4,26 During the disease course, RPE atrophy and loss of choriocapillaris occur first at the posterior pole and then extend to the peripheral retina as well. In the late stages, bone spicule-like pigmentary changes can also be observed like RP.27 Optic disc maintains its healthy looking while retinal vessels become sclerosed and attenuated in the later stages (Figure 2).26
 |
Figure 2 Features of advanced disease stage. Color fundus (A) and IR-R images. (B) of a left eye showing the marked diffuse chorioretinal atrophy and choroidal sclerosis with some pigment clumping areas. The horizontal SD-OCT section through the fovea (C) revealing the loss of EZ and RPE line with choroidal thinning. |
The origin and biochemical composition of the crystals are still unclear. Kojima et al28 underlined the fact that the crystalline deposits were present mostly at the inner side of the RPE layer and located in the relatively healthy retina as previously reported.28,29 Ancillary imaging techniques could provide additional information about the chorioretinal aspects of the disease. Sharply demarcated hypofluorescent areas on fundus autofluorescence imaging,30 highly reflective crystalline deposits on near-infrared reflectance imaging (NIR),31 outer retinal tubulation,32 outer retinal layer atrophy, hyperreflective spots in or on the RPE-Bruch’s membrane complex, and drusen-like structures,28,33 choroidal neovascularization,34–41 subfoveal neurosensorial detachment42 and macular hole43–46 on OCT have been all reported in BCD patients.
Disease Course and the Staging
Visual impairment commonly occurs between the second and third decades of life.26 The diagnosis sometimes can be challenging since the patients with early disease may not have any visual symptoms. The age at the time of onset, presenting symptoms, and severity of the disease can all vary greatly from case to case, and there can also be an asymmetry between the eyes. In a very recent manuscript, Liu et al47 analyzed the intereye symmetry in a group of 13 BCD patients (26 eyes) by clinical examination and multimodal imaging methods and searched the quantity, size, and distribution of the distinctive retinal crystalline deposits by measuring the crystal number, crystal area and absent autofluorescent area and demonstrated a strong intereye symmetry.47 By the fifth or sixth decade of life, vision loss can become so severe that the patient may potentially become legally blind.3
Disease can be subclassified by Yuzawa et al26 as early, intermediate and advanced disease and later with some minor modifications by Mataftsi et al in the context of clinical features.16
Early Disease
There is retinal crystals scattered throughout the posterior pole with accompanying midperipheric atrophy limited mostly to the macular area that can be assessed with fluorescein and indocyanine angiographies (Figure 3).
 |
Figure 3 Features of early disease stage. Color fundus image of a left eye (A)showing scattered intraretinal crystals throughout the posterior pole. Infrared reflectance (IR-R) image (B) depicting the several bright reflective spots at posterior pole. Spectral domain-optical coherence tomography (SD-OCT) section (C) delineating the hyperreflective deposits at the retina pigment epithelium -Bruch’s membrane complex and ellipsoid zone (EZ) disruption in the left eye (C). |
Intermediate Disease
Number of retinal crystals are reduced in amount or absent at the posterior pole, but can still be observed outside the region of central atrophy that extends beyond the posterior pole (Figure 4).
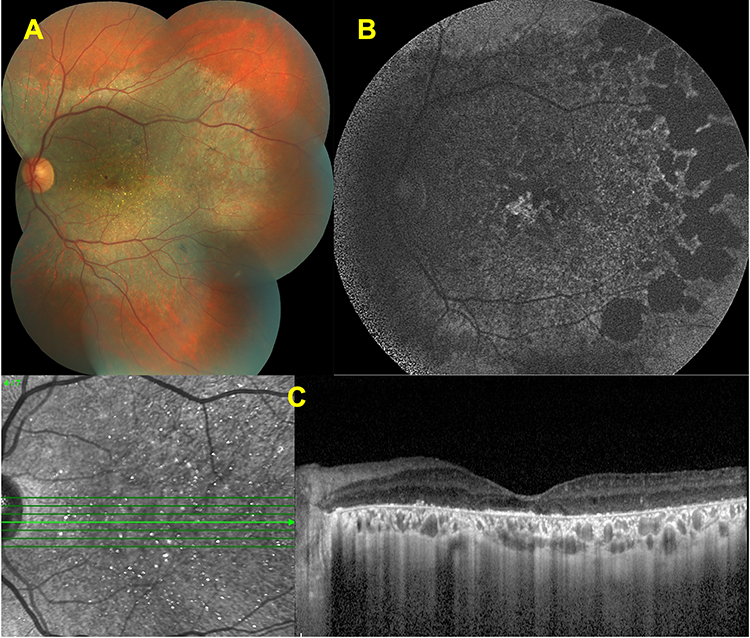 |
Figure 4 Features of intermediate disease stage. Color fundus image of a left eye (A) showing the chorioretinal atrophy extending from the posterior pole and glistening crystals at the macula. Fundus autofluorescence image (B) revealing the well-demarcated hypofluorescent patches corresponding to the atrophy. Spectral domain-optical coherence tomography (SD-OCT) section (C) demonstrating the hyperreflective deposits at the RPE – Bruch’s membrane complex, together with the disruption and loss of the EZ band. |
Advanced Disease
There is nearly total chorioretinal atrophy with very rare crystals seen throughout the fundus (Figure 2).
Clinical Characteristics on Various Ancillary Tests
Fundus autofluorescence imaging provides information about the topographic map of the lipofuscin distribution in the RPE and allows visualization of the areas of RPE loss in eyes with degenerative retinal disorders such as hypoautofluorescent areas.48 It is thus also possible to infer RPE-choriocapillaris atrophy with the help of fundus autofluorescence imaging in BCD patients.30 Hypofluorescent patches were associated with RPE loss in FAF imaging in BCD patients, whereas hyperautofluorescent speckles were interpreted as RPE hyperplasia, as previously documented in a histological investigation.19,30
Near-infrared reflectance (NIR), a noninvasive approach is recently regarded as an useful method for the BCD diagnosis.49 In their retrospective study, Huang et al49 analyzed the multimodal imaging features of 40 eyes of 22 BCD patients. In their group, five legally blind eyes displayed large sclerotic vessels that were visible on NIR and none of these eyes had crystalline deposits. They concluded that NIR imaging could be a helpful diagnostic tool in cases with BCD as it could identify not only the crystalline deposits in the early stages of the disease but also the sclerotic choroidal vessels in more advanced stages where crystalline deposits were undetectable.49
On fluorescein angiography, hyperfluorescent “window defects” are detected related to the areas of RPE atrophy with generally intact choriocapillaris in the earlier disease stages, while lobular hypofluorescent areas also do appear corresponding to the poorly perfused choriocapillaris and chorioretinal atrophy during the disease evolution. These hypofluorescent areas progressively enlarge over time.10,27,50
ICG angiographic results vary depending on the stages of the disease. In the early stages, there were no RPE changes or choriocapillaris loss, but in the advanced stages, there was significant chorioretinal atrophy seen as hypocyanescent areas.51 Most notably, intraretinal crystals had no masking effect or any fluorescence and on fluorescein or ICG angiography. ICG angiography in every case revealed hypocyanescent spots, which most likely corresponded to the RPE changes nearby the atrophic regions.50
Hyperreflective dots present almost in all retinal layers and outer retinal tubulations (ORT) are among the main OCT features.28–30,32,33,52–55 Tomographically, ORT is typically seen in the outer nuclear retinal layers and often observed as circular or ovoid hypo-reflective spaces with hyperreflective borders.32 ORTs are likely the indicator of rearranged photoreceptor layer due to rapid and severe photoreceptor degeneration. Braimah et al56 analyzed the 309 eyes of 157 patients, with retinal dystrophies including retinitis pigmentosa (RP, 183 eyes), Stargardt disease (STGD, 93 eyes) and Best disease (33 eyes) in a retrospective study and found out the presence of ORT in 12 eyes of eight patients (six patients with RP, two with STGD, and four with Best disease). Interestingly, ORTs were more frequently reported in BCD when compared to cone dystrophy and retinitis pigmentosa.28,32 Kojima et al28 showed that ORTs were present in 10 of 292 retinitis pigmentosa patients (3.4%), two of 16 patients with cone dystrophy (12.5%), and all of the 12 patients with BCD (100%) using spectral domain optical coherence tomography (SD-OCT). Iriyama et al32 analyzed 112 patients with RP, 58 patients with cone dystrophy, and seven patients with BCD and ORTs were found in none of the 112 patients with RP, unilaterally in three of 58 patients with cone dystrophy, and bilaterally in five of seven patients with BCD. The association between ORT and disease severity was not further looked for in BCD eyes in the two past manuscripts. Li et al57 suggested that ORTs were commonly observed at intermediate and advanced disease stages. Previously, we58 disclosed that ORTs were present in all intermediate stage eyes and three of seven eyes with advanced disease stage. ORTs might have not been visualized due to already established degeneration related extreme retinal thinning. In a recent Chinese study, only 3 eyes out of 22 eyes (13.6%) with BCD had ORTs by using Cirrus OCT contrary to the reports summarized above but, however, the authors did not mention the disease stage in BCD patients with ORTs.55 Very recently, 42 clinically and genetically diagnosed BCD patients were evaluated in a multicenter, retrospective cohort study for the presence of ORTs and its possible prognostic implications.59 Eighty eyes with good quality of SD OCT sections were included. In 52 eyes, there were evidence of ORT, and the incidence of ORT was significantly higher in stage 2 disease than the other disease stages. The authors considered that ORT might be the sign of the onset of RPE atrophy in early stage BCD and might suggest less risk of rapid progression in late-stage BCD.59
Several bright reflective spots can be observed within different retinal locations on SD-OCT sections of BCD patients.42 Hyperreflective spots are likely corresponded to the crystals located in or on the RPE-Bruch membrane complex.33,52 Pennesi et al mentioned three forms of hyperreflective dots can be distinguished: (1) highly reflective dots in the inner retina, (2) bright reflecting plaques above the Bruch membrane, and (3) partially encapsulated reflective plaques. Interestingly, only some of the hyperreflective spots can be matched to crystalline deposits.30,52 The rest of the hyperreflective spots might be related to glial cell response to retinal degeneration, protein deposits, inflammatory cells, artifacts, or perhaps the protein deposits.33 Li et al57 noted that drusen-like structures seen at early-stage BCD patients resembled to the soft and sawtooth cuticular drusen observed in patients with age-related macular degeneration.60 The presence of hyper-AF even at early disease stages led the authors to hypothesize that this might represent an intermediate alteration that occurred prior to loss of the photoreceptors and RPE.57 In a Cirrus OCT study, Gocho et al29 pointed out that the crystals visible on the infrared images were located adjacent to the healthy looking RPE where outer nuclear layer was preserved. In our previous OCT study,58 we detected inner retinal hyperreflective dots in 20 of 24 eyes, and only in four eyes with the advanced stage there were none. On the other hand, bright reflective plaques were shown in all of the study’s eyes. The presence of choroidal hyperreflective spots presumably corresponding to the crystals is controversial as choroidal hyperreflective dots were described only in some reports.54 Saatci et al58 showed the presence of choroidal hyperreflective spots in 20 of 24 BCD eyes using SD-OCT. There were no choroidal hyperreflective spots in four eyes with advanced disease stage and severe chorioretinal thinning. Zerbib et al61 also mentioned that choroidal crystals could be observed also in the choroid and added that SD-OCT might have allowed the clinicians to detect the hyperreflective foci better due to existing retinal atrophy.61
Longitudinal measurements of central macular thickness and subfoveal choroidal thickness may provide additional information about the disease severity and disease progression during the course as chorioretinal thinning was more pronounced in the later disease stages.58
Swept-source OCT was also utilized in BCD patients and SS-OCT also revealed the tubulations in the outer nuclear layer, destruction of the ellipsoid and interdigitation zone of the retina, hyperreflective retinal deposits in all retinal layers, primarily in the RPE/Bruch membrane complex, and patchy loss of the outer retina and RPE in the advanced stage.31,62,63
Optical coherence tomography angiography provides additional information on the retinal and choroidal perfusion damage in BCD patients.64–67 Hirashima et al65 evaluated 16 eyes of 16 retinitis pigmentosa patients with EYS mutation, and 9 eyes of 9 patients with BCD, 16 eyes of 16 healthy individuals with SS-OCTA. Thinning in the outer choroidal vascular area was associated with the thinning of subfoveal choroid in eyes with BCD and interestingly, they found that the vasculature in the inner choroid, including the choriocapillaris and Sattler’s layers, was absent as the inner choroidal vessels could not be identified in 8 of the 9 BCD eyes. They discovered that the BCD group’s ratio of subfoveal inner choroidal thickness to total choroidal thickness in the BCD group was substantially lower than the RP group.65 Consistently, a recent case report suggested that VD impairment of the retinal plexuses and CC ensued the primary RPE degeneration.68 Miyata et al66 analyzed 13 eyes of 13 patients with BCD using OCTA and showed a flow deficit in the choriocapillaris in 92% of their BCD patients. They also reported that subfoveal choriocapillaris thickness was correlated with visual acuity.66 In a patient with a 10-year of follow-up, Ipek et al69 reported the changes in a 27-year-old female BCD patient and similarly showed that choroidal vessels could be visualized on the outer retinal layer segmentation due to retinal pigment epithelial atrophy and blood flow was reduced on the level of choroidal segmentation with the SS-OCTA. In a retrospective study that included 22 BCD patients (37 eyes) and 22 healthy individuals (37 eyes), Zhang et al67 found that the radial peripapillary capillary density was significantly lower in the BCD patients than in the control group.67 In our recent study, we analyzed the capillary vessel density (VD) and choroidal vascularity index (CVI) in 26 eyes of 13 BCD patients, 26 eyes of RP patients and 26 eyes of 13 healthy subjects.64 The CVI value was lower in BCD and RP patients than in the healthy subjects. We suggested that lower CVI values seemed to be related to the disease severity in BCD patients. We also revealed that VD was also significantly lower in BCD patients when compared to RP patients.64
Different levels of rod and cone dysfunction can be shown on the full field electroretinogram (ffERG), varying from undetectable to normal to reduced amplitudes of scotopic and photopic responses in BCD patients. The ffERG is more likely to be abnormal when the peripheral visual field is severely affected in the mid- to late-stage of the BCD. Though several studies mentioned that ffERG responses seemed to correlate with the disease severity70–72 the ffERG can remain normal or nearly normal even in advanced stages of the BCD.73 Normal ffERG responses can be detected in patients with BCD who have extensive RPE and choroid atrophy, indicating that the neural retina may still be functional despite the disruption of the retinal layers.73
Visual field defects may manifest as peripheral, central, or pericentral scotomas (usually associated with atrophic lesions that extend to the foveal region) in BCD patients.74 Patients with BCD experience substantial visual impairment and peripheral visual field constriction, which typically proceed to legal blindness by the fifth or sixth decade of life.19
Adaptive optics (AO) is a new imaging method that enables the direct observation of the individual rod, cone, and retinal pigment epithelial (RPE) cells.29,75 Gocho et al29 found circular patches and clusters of hyperreflective signals in the AO images that matched the crystals in three individuals with BCD. According to their thinking, the circular spots were the remains of the cone photoreceptors that lay over the crystals. Battu et al75 looked for the ORTs in BCD patients with AO and noted that ORTs appeared as serpentine tubules of varying sizes.
Posterior Segment Complications
Macular Neovascularization
Macular neovascularization (MNV) can complicate the clinical course in almost all of the chorioretinal dystrophies76 as it may be seen with any pathological process disturbing the RPE and Bruch’s membrane.38 Younger age, scant exudation pattern, type 2 MNV, and slow and often limited visual deterioration are common aspects of the MNV secondary to retinal dystrophies.77,78 The efficacy and safety of intravitreal 0.5 mg ranibizumab injection were investigated prospectively in adult patients with MNV due to uncommon causes, such as angioid streak, inflammation, central serous chorioretinopathy, idiopathic, and miscellaneous causes in a randomized 12-month long MINERVA study.79 An individualized PRN ranibizumab treatment regimen depending on the disease activity was showed highly clinically relevant visual improvement for the treatment of MNV irrespective of the underlying etiology and was well tolerated.79 Thus, intravitreal anti-VEGF injection is the generally recommended treatment in eyes with active MNV and BCD nowadays.
MNV in cases with BCD has been reported mostly as anecdotal case reports.34–38,40,41,80 Atmaca et al35 reported the first case of MNV in association with BCD in a 13-year-old girl who developed a unilateral peripapillary MNV and MNV-associated foveal serous retinal detachment that occurred 14 months after the initial diagnosis but no treatment was noted. MNV lesions were commonly located at the foveal and parafoveal region and Fuerst et al36 suggested that regional predilection might be related to the parafoveal disruption of the RPE integrity and photoreceptor outer segment-RPE relationships. A 64-year-old male patient was diagnosed by Gupta et al34 with an inactive juxtafoveal MNV and did not require any treatment. Hua et al38 described a 26-year-old man with BCD and MNV who was treated with a single intravitreal bevacizumab injection, and they argued that reduced activity of the MNV might be related to the already established choriocapillaris destruction. Le Tien et al40 reported a 29-year-old woman with BCD and MNV in her left eye. After three injections of ranibizumab, the vision improved from 20/50 to 20/32, and no recurrence was noted after six months of follow-up.40 Mamatha et al41 reported three BCD patients of whom two had parafoveal MNV. The first one was a 26-year-old male who had a unilateral MNV-related scar thereby no treatment was given. Second, was a 31-year-old male who experienced a subretinal hemorrhage related to an active subfoveal MNV in his left eye together with a disciform scar in his right eye. Scar formation occurred following three intravitreal ranibizumab injections without any recurrence during a six-month follow-up in the left eye.41 Nachiappan et al81 reported a 33-year-old male, BCD patient with a scarred subfoveal MNV in the right eye and subretinal hemorrhage with active subfoveal MNV in the left eye. They administrated three monthly ranibizumab injections into the left eye and the left eye maintained visual acuity with no recurrence 12 months after the final injection.81 Hua et al38 described a 26-year-old male BCD patient with a right subfoveal MNV. Following a single dose of bevacizumab injection a recurrence was noted three months later.38 Han et al37 reported a 30-year-old female with bilateral type 2 macular using OCTA and also OCT and demonstrated the thinning of the RPE and choriocapillaris layer, ellipsoid zone (EZ) band defect, and active type 2 MNV in both eyes. The authors did not provide information about whether the patient received treatment or not.37 We recently reported a 15-year-old Caucasian boy with Bietti crystalline dystrophy who was diagnosed to have a unilateral MNV a year after the initial examination and he was treated with a single intravitreal ranibizumab injection. Visual acuity improved and the macula stayed dry during the 15-month-long follow-up.82 Just very recently, Ll et al83 retrospectively analyzed the presence of MNV in 157 eyes of 79 patients with BCD by the help of multimodal imaging and noticed the presence of MNV in 19 eyes of 12 patients. Median age of these patients was 29.5 years (age range; 15–44 years). Seven cases (58.3%) had bilateral involvement. All MNVs were type 2 membranes together with the subretinal hyperreflective material. While active MNVs were noted in earlier disease stages, MNV activity was seemed to be reduced in advanced cases. Six eyes of five patients received at least one injection of anti-VEGF (maximum 3 times), whereas seven subjects were treatment naive.83 Overall, anti-vascular endothelial growth factor therapy has been reported to be effective for CNV regression and the improvement of decreased vision.34,35,80 SS-OCTA features of a quiescent CNV detected in an otherwise asymptomatic BCD patient are seen in Figure 5.
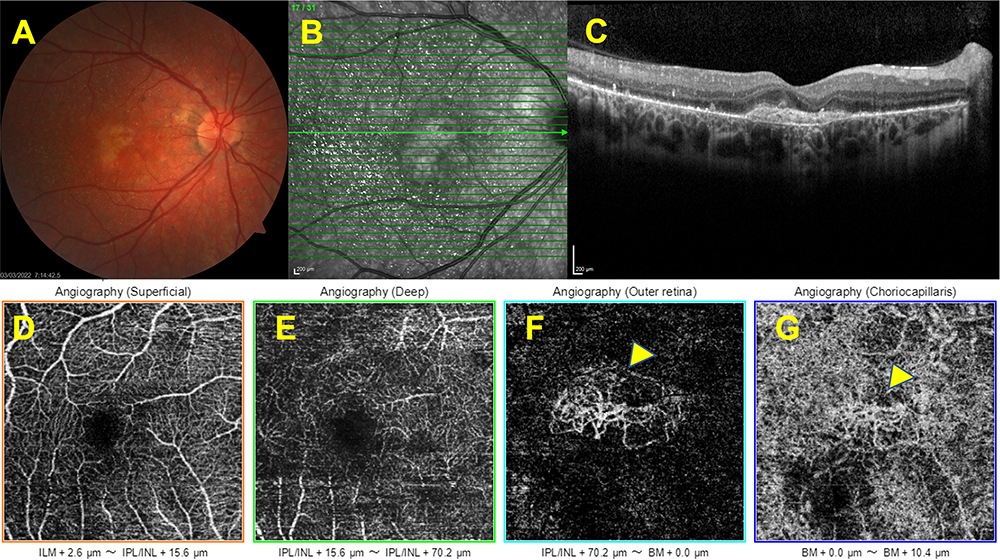 |
Figure 5 Associated macular neovascularization. Right eye, Color fundus image showing the hypopigmented looking well-demarcated lesion at the macula (A). The horizontal SD-OCT section demonstrating the macular neovascularization (B,C). Optical coherence tomography angiographic slabs demonstrating the presence of quiescent choroidal neovascularization (yellow arrows) (D–G). |
Cystoid Macular Edema
Cystoid macular edema (CME) is another rare complication of BCD and can be one of the main causes of central visual deterioration. CME occurrence may be related to poor blood-retinal barrier function, localized intraretinal inflammation, autoimmune processes, tangential vitreous traction, and/or reduced RPE function in the retinal dystrophies and the authors suggested that the occurrence of macular cystoid spaces that was already altered by the atrophic process seemed to be a more degenerative process than due to a blood-retina barrier breakdown in BCD patients.84 Saatci et al85 described 27-year-old male BCD patient with bilateral CME. There was bilateral petaloid-type late hyperfluorescence documented on fluorescein angiography. Though intraretinal crystals reduced in amount during a follow-up of six years CME persisted angiographically and tomographically. No treatment was given to the patient. The treatment of CME in association with BCD is still uncertain but systemic86 and topical87 carbonic anhydrase enzyme inhibitors could be beneficial in CME according to some case reports. Patients with diffuse RPE disease respond better to carbonic anhydrase inhibitors than those with retinal vascular disease, such as diabetes or vascular occlusion.86,88 Figure 6 shows the features of a BCD patient with cystoid macular edema.
 |
Figure 6 Associated macular edema. Left eye, early venous (A) and late composite venous (B) fluorescein angiographic images revealing the increasing macular hyperfluorescence. SD-OCT section showing the cystoid macular changes corresponding to macular edema (C). |
Macular Hole
Macular hole is a rare clinical feature of BCD but its pathogenesis is still somewhat undetermined. Neurosensorial retinal detachment and subsequent central photoreceptor layer defect can be the result of tractional forces transmitted by the Müller cells.89 Abnormal posterior vitreous detachment and epiretinal membrane contraction may also contribute to the formation of the partial or full thickness macular holes. Moreover, macular hole can also develop as the result of high myopia and already existing cystoid macular edema.89 Bagolini and Ioli-Spada4 described the clinical features of eight BCD patients of whom two had been previously followed by Bietti. Among these patients, a 31-year-old highly myopic woman was diagnosed to have an unilateral macular hole together with bilateral retinal detachment. In 1997, we described a 21-year-old man who developed a stage 4 full-thickness unilateral macular hole with a surrounding macular detachment three years after our initial examination.43 More recently, we reported a 39-year-old woman with BCD who developed an unilateral, full-thickness, slightly eccentric, visually asymptomatic macular hole with an intact foveola during our follow-up of 13 years.45 Its full-thickness nature could be disclosed only in two consecutive B-scans 132 µm apart and no surgery was recommended at that time as the visual acuity was relatively good and more frequent visits were advised.45 Nourinia et al44 reported a 42-year-old man with BCD who developed a full-thickness macular hole in the left eye. They performed a left pars plana vitrectomy with the internal limiting membrane (ILM) peeling and gas injection. One month after the surgery, the left macular hole was closed and visual acuity was improved from counting fingers at 2 m to 20/50.80 Figure 7 shows the appearance of a full-thickness macular hole in a BCD patient.
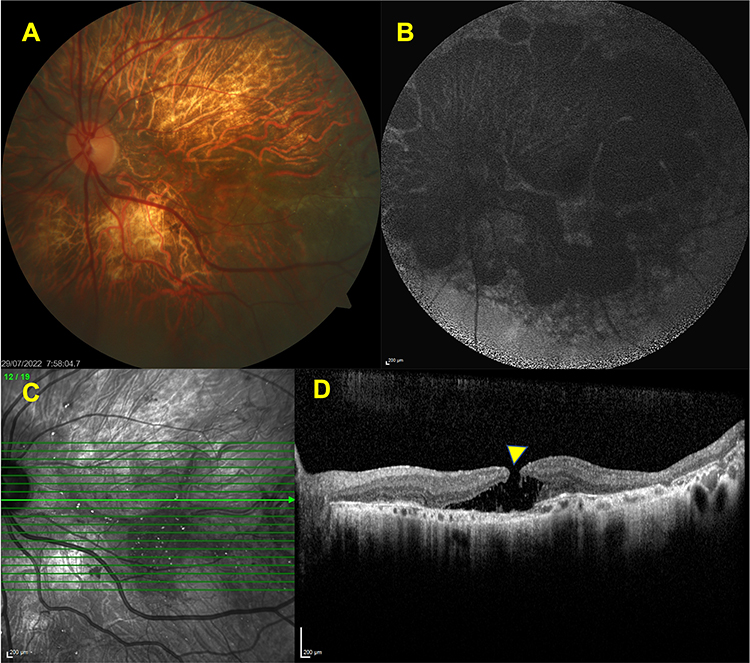 |
Figure 7 Associated macular hole. Left eye, color fundus image (A) delineating the extensive chorioretinal atrophy and choroidal sclerosis. FAF image (B) revealing the multiple hypofluorescent patches corresponding to the posterior pole. SD-OCT section showing the presence of full -thickness hole (yellow arrow) (C,D). |
Incidental Fundus Changes
Bazvand et al reported90 a 39-year-old woman with BCD and bilateral optic drusen that were detected incidentally as they noticed bilateral hyperautofluorescent areas on the optic nerve head consistent with optic nerve head drusen and large hypoautofluorescent areas at the posterior retina consistent with retinal pigment epithelium atrophy on FAF images. We observed unilateral peripapillary myelinated retinal nerve fibers in a patient with BCD (Figure 8A–C). Ten years later, we detected the presence of an asymptomatic retinal vein occlusion with optociliary shunt formation in the same eye (Figure 8D and E).
 |
Figure 8 Incidental findings Right eye, Color fundus image (A) showing the scattered glistening crystals at the posterior pole and peripapillary myelinated nerve fiber (yellow arrow), FAF image (B) demonstrating the patchy hypofluorescent areas with ill-demarcated hyperfluorescent borders at the posterior pole in 2011. SD-OCT section (C) revealing the hyperreflective deposits at the RPE – Bruch’s membrane complex, disruption, and loss of the EZ band. Composite color fundus image (D) demonstrating the marked chorioretinal atrophy and attenuated retinal vessels with the optociliary shunt (green arrows) in 2021. Composite venous fluorescein angiographic image (E) revealing the nonperfused areas related to old central retinal vein occlusion detected in fluorescein angiography image shows extensive ischemic areas throughout posterior pole. |
Future Perspectives
It is difficult to perform studies about the disease pathogenesis and develop new therapeutical approaches because of the clinical and genetic variations present in BCD. Thus, animal models of BCD are in limited number.11 The first murine model was generated in 2014 in Cyp4v3−/− knockout mice, a murine ortholog of human CYP4V2, and successfully mimicked the human BCD.91 This model was used for developing a gene therapy through subretinal delivery of human CYP4V2 carried by an adeno-associated virus (AAV2) vector. This therapeutical approach effectively treated BCD in Cyp4v3−/− knockout mice, which were also on a high-fat diet to induce retinopathy.92 Another animal model was generated recently in zebrafish, which were knock-out for cyp4v7 and cyp4v8, homologous genes of CYP4V2, by CRISPR/Cas9 technology.93 This model showed that the peroxisome proliferator-activated receptor alpha (terim doğru hocam) (PPARα) was impaired in the zebrafish and may be responsible for the abnormal lipid metabolism of the RPE cells. Recently, two more murine models lacking Cyp4v3 were also generated.94,95
There are also some cell models generated using the pluripotent stem cells of CYP4V2 mutated BCD patients to understand the disease pathophysiology and develop treatment options.96,97 In a recent study, Wang et al showed that AAV2 vectors encoding codon optimization of CYP4V2 were superior to AAV2 vectors encoding wild-type CYP4V2 in terms of protein expression and enzyme activity in ARPE19, HEK293, and patient-induced pluripotent stem cells (iPSC) which lack effective CYP4V2.96 In another study which was performed in RPE derived from human CYP4V2 mutant iPSCs, the presence of mitochondrial stress resulting from the excessive accumulation of poly-unsaturated fatty acids was shown to be a key factor of RPE degeneration in BCD.97
Wu et al98 described the generation and characterization of a human-induced pluripotent stem cell line of BCD patients with CYP4V2 mutations. The skin fibroblasts were derived and reprogrammed using a Sendai virus-based approach. They suggested that this iPSC line from BCD patients can be used for patient-specific disease models to develop new treatments.98
There is currently an ongoing clinical study investigating gene replacement therapy in BCD patients that is being conducted at Beijing Tongren Hospital. The study is intended to evaluate the safety and tolerability of a single subretinal administration of VGR-R01, which is a novel AAV vector carrying the human CYP4V2 sequence.99
Conclusion
Bietti crystalline dystrophy is a rare, progressive retinochoroidal dystrophy due to mutations in the CYP4V2 gene. Multimodal imaging modalities including optical coherence tomography and optical coherence tomography angiography provide new insights into its natural course, progression, and related macular complications such as macular neovascularization, macular hole, and cystoid macular edema. Studies on gene therapy hopefully may provide new treatment options in the future.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bietti G. On the familial occurrence of “retinitis punctata albescens” (associated with “dystrophia marginalis cristallinea cornea)”, glistening, vitreous humor and other degenerative ocular changes. Augenvera¨Nderungen Klin Monatsbl Augenheilkd. 1937;99:737–756.
2. Welch RB. Bietti’s tapetoretinal degeneration with marginal corneal dystrophy crystalline retinopathy. Trans Am Ophthalmol Soc. 1977;75:164–179.
3. Osman Saatci A, Can Doruk H. An overview of rare and unusual clinical features of Bietti’s crystalline dystrophy. Med Hypothesis Discov Innov Ophthalmol. 2014;3:51–56.
4. Bagolini B, Ioli-Spada G. Bietti’s tapetoretinal degeneration with marginal corneal dystrophy. Am J Ophthalmol. 1968;65:53–60. doi:10.1016/0002-9394(68)91028-3
5. Furusato E, Cameron JD, Chan CC. Evolution of cellular inclusions in Bietti’s crystalline dystrophy. Ophthalmol Eye Dis. 2010;2010:9–15. doi:10.4137/OED.S2821
6. Jiao X, Munier FL, Iwata F, et al. Genetic linkage of Bietti crystalline corneoretinal dystrophy to chromosome 4q35. Am J Hum Genet. 2000;67(5):1309–1313. doi:10.1016/S0002-9297(07)62960-7
7. Li A, Jiao X, Munier FL, et al. Bietti crystalline corneoretinal dystrophy is caused by mutations in the novel gene CYP4V2. Am J Hum Genet. 2004;74:817–826. doi:10.1086/383228
8. Nakano M, Kelly EJ, Rettie AE. Expression and characterization of CYP4V2 as a fatty acid omega-hydroxylase. Drug Metab Dispos. 2009;37:2119–2122. doi:10.1124/dmd.109.028530
9. Lee J, Jiao X, Hejtmancik JF, et al. The metabolism of fatty acids in human Bietti crystalline dystrophy. Invest Ophthalmol Vis Sci. 2001;42:1707–1714.
10. Wilson DJ, Weleber RG, Klein ML, Welch RB, Green WR. Bietti’s crystalline dystrophy. A clinicopathologic correlative study. Arch Ophthalmol. 1989;107:213–221. doi:10.1001/archopht.1989.01070010219026
11. The human gene mutation database [homepage on the Internet]. Available from: https://www.hgmd.cf.ac.uk/ac/gene.php?gene=CYP4V2Accessed.
12. Chan LW, Sung YC, Wu DC, et al. Predicted protein structure variations indicate the clinical presentation of Cyp4v2-related Bietti crystalline dystrophy. Retina. 2022;42:797–806. doi:10.1097/IAE.0000000000003381
13. Ng DS, Lai TY, Ng TK, Pang CP. Genetics of Bietti crystalline dystrophy. Asia Pac J Ophthalmol. 2016;5:245–252. doi:10.1097/APO.0000000000000209
14. Lockhart CM, Smith TB, Yang P, et al. Longitudinal characterisation of function and structure of Bietti crystalline dystrophy: report on a novel homozygous mutation in CYP4V2. Br J Ophthalmol. 2018;102:187–194. doi:10.1136/bjophthalmol-2016-309696
15. Hanany M, Yang RR, Lam CM, Beryozkin A, Sundaresan Y, Sharon D. An in-depth single-gene worldwide carrier frequency and genetic prevalence analysis of CYP4V2 as the cause of Bietti crystalline dystrophy. Transl Vis Sci Technol. 2023;12:27. doi:10.1167/tvst.12.2.27
16. Mataftsi A, Zografos L, Milla E, Secretan M, Munier FL. Bietti’s crystalline corneoretinal dystrophy: a cross-sectional study. Retina. 2004;24:416–426. doi:10.1097/00006982-200406000-00013
17. Garcia-Garcia GP, Martinez-Rubio M, Moya-Moya MA, Perez-Santonja JJ, Escribano J. Current perspectives in Bietti crystalline dystrophy. Clin Ophthalmol. 2019;13:1379–1399. doi:10.2147/OPTH.S185744
18. Nakano M, Kelly EJ, Wiek C, Hanenberg H, Rettie AE. CYP4V2 in Bietti’s crystalline dystrophy: ocular localization, metabolism of omega-3-polyunsaturated fatty acids, and functional deficit of the p.H331P variant. Mol Pharmacol. 2012;82:679–686. doi:10.1124/mol.112.080085
19. Kaiser-Kupfer MI, Chan CC, Markello TC, et al. Clinical biochemical and pathologic correlations in Bietti’s crystalline dystrophy. Am J Ophthalmol. 1994;118:569–582. doi:10.1016/S0002-9394(14)76572-9
20. Traboulsi EI, Faris BM. Crystalline retinopathy. Ann Ophthalmol. 1987;19:156–158.
21. Bozkurt B, Ozturk BT, Kerimoglu H, Irkec M, Pekel H. In vivo confocal microscopic findings of 2 patients with Bietti crystalline corneoretinal dystrophy. Cornea. 2010;29:590–593. doi:10.1097/ICO.0b013e3181be22ee
22. Garcia-Garcia GP, Lopez-Garrido MP, Martinez-Rubio M, Moya-Moya MA, Belmonte-Martinez J, Escribano J. Genotype-phenotype analysis of Bietti crystalline dystrophy in a family with the CYP4V2 Ile111Thr mutation. Cornea. 2013;32:1002–1008. doi:10.1097/ICO.0b013e31828a27bc
23. Chung JK, Shin JH, Jeon BR, Ki CS, Park TK. Optical coherence tomographic findings of crystal deposits in the lens and cornea in Bietti crystalline corneoretinopathy associated with mutation in the CYP4V2 gene. Jpn J Ophthalmol. 2013;57:447–450. doi:10.1007/s10384-013-0256-6
24. Yokoi Y, Nakazawa M, Mizukoshi S, Sato K, Usui T, Takeuchi K. Crystal deposits on the lens capsules in Bietti crystalline corneoretinal dystrophy associated with a mutation in the CYP4V2 gene. Acta Ophthalmol. 2010;88:607–609. doi:10.1111/j.1755-3768.2009.01529.x
25. Louati Y, Vaclavik V, Moulin A, Schorderet D, Munier FL, Viet Tran H. Crystals deposits in the anterior and posterior lens cortex in Bietti corneo-retinal dystrophy. Ophthalmic Genet. 2021;42:773–779. doi:10.1080/13816810.2021.1952620
26. Yuzawa M, Mae Y, Matsui M. Bietti’s crystalline retinopathy. Ophthalmic Paediatr Genet. 1986;7:9–20. doi:10.3109/13816818609058037
27. Fong AM, Koh A, Lee K, Ang CL. Bietti’s crystalline dystrophy in Asians: clinical, angiographic and electrophysiological characteristics. Int Ophthalmol. 2009;29:459–470. doi:10.1007/s10792-008-9266-7
28. Kojima H, Otani A, Ogino K, et al. Outer retinal circular structures in patients with Bietti crystalline retinopathy. Br J Ophthalmol. 2012;96:390–393. doi:10.1136/bjo.2010.199356
29. Gocho K, Kameya S, Akeo K, et al. High-resolution imaging of patients with Bietti crystalline dystrophy with CYP4V2 mutation. J Ophthalmol. 2014;2014:283603. doi:10.1155/2014/283603
30. Ayata A, Tatlipinar S, Unal M, Ersanli D, Bilge AH. Autofluorescence and OCT features of Bietti’s crystalline dystrophy. Br J Ophthalmol. 2008;92:718–720. doi:10.1136/bjo.2008.138958
31. Oishi A, Oishi M, Miyata M, et al. Multimodal imaging for differential diagnosis of Bietti crystalline dystrophy. Ophthalmol Retina. 2018;2:1071–1077. doi:10.1016/j.oret.2018.02.012
32. Iriyama A, Aihara Y, Yanagi Y. Outer retinal tubulation in inherited retinal degenerative disease. Retina. 2013;33:1462–1465. doi:10.1097/IAE.0b013e31828221ae
33. Halford S, Liew G, Mackay DS, et al. Detailed phenotypic and genotypic characterization of bietti crystalline dystrophy. Ophthalmology. 2014;121:1174–1184. doi:10.1016/j.ophtha.2013.11.042
34. Gupta B, Parvizi S, Mohamed MD. Bietti crystalline dystrophy and choroidal neovascularisation. Int Ophthalmol. 2011;31:59–61. doi:10.1007/s10792-010-9406-8
35. Atmaca LS, Muftuoglu O, Atmaca-Sonmez P. Peripapillary choroidal neovascularization in Bietti crystalline retinopathy. Eye. 2007;21:839–842. doi:10.1038/sj.eye.6702673
36. Fuerst NM, Serrano L, Han G, et al. Detailed functional and structural phenotype of Bietti crystalline dystrophy associated with mutations in CYP4V2 complicated by choroidal neovascularization. Ophthalmic Genet. 2016;37:445–452. doi:10.3109/13816810.2015.1126616
37. Han XY, Zhang LQ, Tang JY, Huang LZ, Tang R, Qu JF. A novel mutation of CYP4V2 gene associated with Bietti crystalline dystrophy complicated by choroidal neovascularization. Int J Ophthalmol. 2022;15:940–946. doi:10.18240/ijo.2022.06.11
38. Hua R, Chen K, Hu Y, Wang X, Chen L. Relapse of choroidal neovascularization in Bietti’s crystalline retinopathy following anti-vascular endothelial growth factor therapy: a case report. Exp Ther Med. 2015;10:1704–1706. doi:10.3892/etm.2015.2716
39. Kobat SG, Gul FC, Yusufoglu E. Bietti crystalline dystrophy and choroidal neovascularization in childhood. Int J Ophthalmol. 2019;12:1514–1516. doi:10.18240/ijo.2019.09.24
40. Le Tien V, Atmani K, Querques G, Massamba N, Souied EH. Ranibizumab for subfoveal choroidal neovascularization in Bietti crystalline retinopathy. Eye. 2010;24:1728–1729. doi:10.1038/eye.2010.116
41. Mamatha G, Umashankar V, Kasinathan N, et al. Molecular screening of the CYP4V2 gene in Bietti crystalline dystrophy that is associated with choroidal neovascularization. Mol Vis. 2011;17:1970–1977.
42. Padhi TR, Kesarwani S, Jalali S. Bietti crystalline retinal dystrophy with subfoveal neurosensory detachment and congenital tortuosity of retinal vessels: case report. Doc Ophthalmol. 2011;122:199–206. doi:10.1007/s10633-011-9274-1
43. Saatci AO, Yaman A, Berk AT, Soylev MF. Macular hole formation in Bietti’s crystalline retinopathy. A case report. Ophthalmic Genet. 1997;18:139–141. doi:10.3109/13816819709057127
44. Nourinia R, Dehghan MH, Fekri S. Outcome of macular hole surgery in Bietti crystalline dystrophy. J Ophthalmic Vis Res. 2017;12:338–341. doi:10.4103/jovr.jovr_154_15
45. Saatci AO, Kayabasi M, Avci R. Asymptomatic unilateral full-thickness macular hole in a patient with Bietti crystalline dystrophy during 13-year follow-up with optical coherence tomography. Turk J Ophthalmol. 2022;52:212–215. doi:10.4274/tjo.galenos.2022.86681
46. Ji SX, Yin XL, He XG, et al. Bietti crystalline dystrophy with bilateral macular holes. Retin Cases Brief Rep. 2009;3:361–363. doi:10.1097/ICB.0b013e3181780832
47. Liu Z, Ayton LN, O’Hare F, et al. Intereye symmetry in Bietti crystalline dystrophy. Am J Ophthalmol. 2022;235:313–325. doi:10.1016/j.ajo.2021.07.009
48. Sparrow JR, Duncker T. Fundus autofluorescence and RPE lipofuscin in age-related macular degeneration. J Clin Med. 2014;3:1302–1321. doi:10.3390/jcm3041302
49. Huang CY, Kang EY, Yeh LK, et al. Predicting visual acuity in Bietti crystalline dystrophy: evaluation of image parameters. BMC Ophthalmol. 2021;21:68. doi:10.1186/s12886-021-01811-y
50. Saatci AO, Yaman A, Oner FH, Ergin MH, Cingil G. Indocyanine green angiography in Bietti’s crystalline retinopathy. Can J Ophthalmol. 2002;37:346–351. doi:10.1016/S0008-4182(02)80005-9
51. Salati C, Lafaut BA, Stevens A, De Laey JJ, Kestelyn P. Indocyanine green angiography in a case of Bietti’s disease. Retina. 1998;18:82–84. doi:10.1097/00006982-199818010-00019
52. Pennesi ME, Weleber RG. High-resolution optical coherence tomography shows new aspects of Bietti crystalline retinopathy. Retina. 2010;30:531–532. doi:10.1097/IAE.0b013e3181c96a15
53. Querques G, Quijano C, Bouzitou-Mfoumou R, Soubrane G, Souied EH. In-vivo visualization of retinal crystals in Bietti’s crystalline dystrophy by spectral domain optical coherence tomography. Ophthalmic Surg Lasers Imaging. 2010;41:1–3.
54. Toto L, Carpineto P, Parodi MB, Di Antonio L, Mastropasqua A, Mastropasqua L. Spectral domain optical coherence tomography and in vivo confocal microscopy imaging of a case of Bietti’s crystalline dystrophy. Clin Exp Optom. 2013;96:39–45. doi:10.1111/j.1444-0938.2012.00784.x
55. Yin H, Jin C, Fang X, et al. Molecular analysis and phenotypic study in 14 Chinese families with Bietti crystalline dystrophy. PLoS One. 2014;9:e94960. doi:10.1371/journal.pone.0094960
56. Braimah IZ, Dumpala S, Chhablani J. Outer retinal tubulation in retinal dystrophies. Retina. 2017;37:578–584. doi:10.1097/IAE.0000000000001220
57. Li Q, Li Y, Zhang X, et al. Utilization of fundus autofluorescence, spectral domain optical coherence tomography, and enhanced depth imaging in the characterization of Bietti crystalline dystrophy in different stages. Retina. 2015;35:2074–2084. doi:10.1097/IAE.0000000000000592
58. Saatci AO, Doruk HC, Yaman A, Oner FH. Spectral domain optical coherence tomographic findings of bietti crystalline dystrophy. J Ophthalmol. 2014;2014:739271. doi:10.1155/2014/739271
59. Wang Y, Chen J, Sun J, et al. Outer retinal tubulation in Bietti crystalline dystrophy associated with the retinal pigment epithelium atrophy. Retina. 2022. doi:10.1097/IAE.0000000000003697
60. Spaide RF, Curcio CA. Drusen characterization with multimodal imaging. Retina. 2010;30(9):1441–1454. doi:10.1097/IAE.0b013e3181ee5ce8
61. Zerbib J, Ores R, Querques G, Bouzitou-Mfoumou R, Souied EH. Choroidal findings in Bietti’s crystalline dystrophy. Retin Cases Brief Rep. 2014;8:130–131. doi:10.1097/ICB.0000000000000022
62. Kovach JL, Isildak H, Sarraf D. Crystalline retinopathy: unifying pathogenic pathways of disease. Surv Ophthalmol. 2019;64:1–29. doi:10.1016/j.survophthal.2018.08.001
63. Kumar V, Gadkar A. Multimodal imaging of Bietti’s crystalline dystrophy. Indian J Ophthalmol. 2018;66:1024–1026. doi:10.4103/ijo.IJO_39_18
64. Atas F, Kayabasi M, Saatci AO. Vessel density and choroidal vascularity index in patients with Bietti crystalline dystrophy and retinitis pigmentosa. Photodiagnosis Photodyn Ther. 2022;40:103181. doi:10.1016/j.pdpdt.2022.103181
65. Hirashima T, Miyata M, Ishihara K, et al. Choroidal vasculature in Bietti crystalline dystrophy with CYP4V2 mutations and in retinitis pigmentosa with EYS mutations. Invest Ophthalmol Vis Sci. 2017;58:3871–3878. doi:10.1167/iovs.17-21515
66. Miyata M, Oishi A, Hasegawa T, et al. Choriocapillaris flow deficit in Bietti crystalline dystrophy detected using optical coherence tomography angiography. Br J Ophthalmol. 2018;102:1208–1212. doi:10.1136/bjophthalmol-2017-311313
67. Zhang SJ, Wang LF, Xiao Z, et al. Analysis of radial peripapillary capillary density in patients with Bietti crystalline dystrophy by optical coherence tomography angiography. Biomed Environ Sci. 2022;35:107–114. doi:10.3967/bes2022.015
68. Montemagni M, Arrigo A, Battaglia Parodi M, et al. Optical coherence tomography angiography in Bietti crystalline dystrophy. Eur J Ophthalmol. 2022:11206721221143156. doi:10.1177/11206721221143156
69. Ipek SC, Ayhan Z, Kadayifcilar S, Saatci AO. Swept-source optical coherence tomography angiography in a patient with Bietti crystalline dystrophy followed for ten years. Turk J Ophthalmol. 2019;49:106–108. doi:10.4274/tjo.galenos.2018.90768
70. Lee KY, Koh AH, Aung T, et al. Characterization of Bietti crystalline dystrophy patients with CYP4V2 mutations. Invest Ophthalmol Vis Sci. 2005;46:3812–3816. doi:10.1167/iovs.05-0378
71. Mansour AM, Uwaydat SH, Chan CC. Long-term follow-up in Bietti crystalline dystrophy. Eur J Ophthalmol. 2007;17:680–682. doi:10.1177/112067210701700434
72. Usui T, Tanimoto N, Takagi M, Hasegawa S, Abe H. Rod and cone a-waves in three cases of Bietti crystalline chorioretinal dystrophy. Am J Ophthalmol. 2001;132:395–402. doi:10.1016/S0002-9394(01)00963-1
73. Rossi S, Testa F, Li A, et al. An atypical form of Bietti crystalline dystrophy. Ophthalmic Genet. 2011;32:118–121. doi:10.3109/13816810.2011.559653
74. Vargas M, Mitchell A, Yang P, et al. Bietti crystalline dystrophy. In: Adam MP, Everman DB, Mirzaa GM, editors. Genereviews((R)). Seattle (WA): Seattle; 1993.
75. Battu R, Akkali MC, Bhanushali D, et al. Adaptive optics imaging of the outer retinal tubules in Bietti’s crystalline dystrophy. Eye. 2016;30:705–712. doi:10.1038/eye.2016.22
76. Iacono P, Da Pozzo S, Papayannis A, Romano F, Arrigo A, Parodi MB. Dystrophy-related choroidal neovascularization. In: Chhablani J, editor. Choroidal Neovascularization. Singapore: Springer Singapore; 2020:139–149.
77. Marano F, Deutman AF, Leys A, Aandekerk AL. Hereditary retinal dystrophies and choroidal neovascularization. Graefes Arch Clin Exp Ophthalmol. 2000;238:760–764. doi:10.1007/s004170000186
78. Battaglia Parodi M, Iacono P, Bandello F. Antivascular endothelial growth factor in hereditary dystrophies. Dev Ophthalmol. 2010;46:107–110.
79. Lai TYY, Staurenghi G, Lanzetta P, et al. Efficacy and safety of ranibizumab for the treatment of choroidal neovascularization due to uncommon cause: twelve-month results of the MINERVA study. Retina. 2018;38:1464–1477. doi:10.1097/IAE.0000000000001744
80. Wang W, Chen W, Bai X, Chen L. Multimodal imaging features and genetic findings in Bietti crystalline dystrophy. BMC Ophthalmol. 2020;20:331. doi:10.1186/s12886-020-01545-3
81. Nachiappan K, Krishnan T, Madhavan J. Ranibizumab for choroidal neovascular membrane in a rare case of Bietti’s crystalline dystrophy: a case report. Indian J Ophthalmol. 2012;60:207–209. doi:10.4103/0301-4738.95873
82. Kayabaşı M, Ataş F, Saatci AO. Unilateral macular neovascularization formation during the follow-up of a 15-year-old boy with Bietti crystalline dystrophy and the successful treatment outcome with a single intravitreal ranibizumab injection. GMS Ophthalmol Cases. 2023;13:Doc06.
83. LI Q, ZHANG S, KANG L, et al. Characterization of macular neovascularization in Bietti’s crystalline dystrophy using multimodal imaging modalities. Retina. 2022. doi:10.1097/IAE.0000000000003695
84. Gaudric A, Audo I, Vignal C, et al. Non-vasogenic cystoid maculopathies. Prog Retin Eye Res. 2022;91:101092. doi:10.1016/j.preteyeres.2022.101092
85. Saatci AO, Doruk HC, Yaman A. Cystoid macular edema in Bietti’s crystalline retinopathy. Case Rep Ophthalmol Med. 2014;2014:964892. doi:10.1155/2014/964892
86. Broadhead GK, Chang AA. Acetazolamide for cystoid macular oedema in Bietti crystalline retinal dystrophy. Korean J Ophthalmol. 2014;28:189–191. doi:10.3341/kjo.2014.28.2.189
87. Khojasteh H, Roohipoor R, Riazi-Esfahani H, Ghasempour M. Topical dorzolamide for cystoid macular edema in Bietti crystalline retinal dystrophy. Retin Cases Brief Rep. 2021;15:306–309. doi:10.1097/ICB.0000000000000792
88. Liew G, Moore AT, Webster AR, Michaelides M. Efficacy and prognostic factors of response to carbonic anhydrase inhibitors in management of cystoid macular edema in retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2015;56:1531–1536. doi:10.1167/iovs.14-15995
89. Bringmann A, Unterlauft JD, Barth T, Wiedemann R, Rehak M, Wiedemann P. Muller cells and astrocytes in tractional macular disorders. Prog Retin Eye Res. 2022;86:100977. doi:10.1016/j.preteyeres.2021.100977
90. Bazvand F, Asadi Khameneh E. Presumed Bietti crystalline dystrophy with optic nerve head drusen: a case report. J Med Case Rep. 2022;16:413. doi:10.1186/s13256-022-03581-7
91. Lockhart CM, Nakano M, Rettie AE, Kelly EJ. Generation and characterization of a murine model of Bietti crystalline dystrophy. Invest Ophthalmol Vis Sci. 2014;55:5572–5581. doi:10.1167/iovs.13-13717
92. Qu B, Wu S, Jiao G, et al. Treating Bietti crystalline dystrophy in a high-fat diet-exacerbated murine model using gene therapy. Gene Ther. 2020;27:370–382. doi:10.1038/s41434-020-0159-3
93. Gao P, Jia D, Li P, et al. Accumulation of lipid droplets in a novel Bietti crystalline dystrophy zebrafish model with impaired PPARalpha pathway. Invest Ophthalmol Vis Sci. 2022;63:32. doi:10.1167/iovs.63.5.32
94. Ma Z, Jiao X, Agbaga MP, et al. A Bietti crystalline dystrophy mouse model shows increased sensitivity to light-induced injury. Int J Mol Sci. 2022;23:13108.
95. Wang Y, Liu Y, Liu S, et al. A novel and efficient murine model of Bietti crystalline dystrophy. Dis Model Mech. 2022;15. doi:10.1242/dmm.049222
96. Wang JH, Lidgerwood GE, Daniszewski M, et al. AAV2-mediated gene therapy for Bietti crystalline dystrophy provides functional CYP4V2 in multiple relevant cell models. Sci Rep. 2022;12:9525. doi:10.1038/s41598-022-12210-8
97. Zhang Z, Yan B, Gao F, et al. PSCs reveal PUFA-provoked mitochondrial stress as a central node potentiating RPE degeneration in Bietti’s crystalline dystrophy. Mol Ther. 2020;28:2642–2661. doi:10.1016/j.ymthe.2020.07.024
98. Wu S, Zhu T, Sun Z, et al. Generation of a human induced pluripotent stem cell line from a Bietti crystalline corneoretinal dystrophy patient with CYP4V2 mutations. Stem Cell Res. 2021;53:102330. doi:10.1016/j.scr.2021.102330
99. Zhao Z, Anselmo AC, Mitragotri S. Viral vector-based gene therapies in the clinic. Bioeng Transl Med. 2022;7:e10258. doi:10.1002/btm2.10258
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.