Back to Journals » Pediatric Health, Medicine and Therapeutics » Volume 10
Diagnostic and Management Challenges in Congenital Nephrotic Syndrome
Authors Reynolds BC ![]() , Oswald RJA
, Oswald RJA ![]()
Received 24 September 2019
Accepted for publication 5 December 2019
Published 17 December 2019 Volume 2019:10 Pages 157—167
DOI https://doi.org/10.2147/PHMT.S193684
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Roosy Aulakh
Ben Christopher Reynolds,1 Robert James Alan Oswald2
1Department of Paediatric Nephrology, Royal Hospital for Children, Glasgow G51 4TF, UK; 2Tayside Children’s Hospital, Ninewells Hospital & Medical School, Dundee DD1 9SY, UK
Correspondence: Ben Christopher Reynolds
Department of Paediatric Nephrology, Royal Hospital for Children, 1345 Govan Road, Glasgow G51 4TF, UK
Email [email protected]
Abstract: Congenital Nephrotic Syndrome (CNS) is defined as nephrotic range proteinuria, hypoalbuminaemia and edema in the first three months of life. CNS is most commonly genetic in cause, with international variance in the incidence of causative mutations. Initially defined by the histopathological appearance, increasingly sophisticated and accessible genetic analyses now provide a body of evidence to suggest that there is a disparity between the histological appearance, the genotype of individuals and the severity of the clinical disease. Through the evolution of management approaches CNS has changed from being an invariably fatal condition to one with appreciable ongoing morbidity and mortality but comparably good outcomes to other causes of paediatric end-stage renal disease, especially following transplantation. This review briefly summarises the more commonly recognised genetic mutations leading to CNS, addresses common management decisions, and concludes with potential therapies for the future.
Keywords: nephrectomy, genetics, infantile nephrotic syndrome
Introduction
First described by Gautier and Miville in 1942, congenital nephrotic syndrome (CNS) is defined as the triad of nephrotic range proteinuria (>200mg/mmol creatinine), hypoalbuminaemia and clinically detectable edema, occurring in the first three months of life.1 It is a separate entity from idiopathic childhood nephrotic syndrome. Congenital nephrotic syndrome is most frequently genetic in aetiology, with a minority being secondary to congenital infections such as syphilis or toxoplasmosis. Inheritance is autosomal recessive, with an incidence of 1–3 per 100,000 live births. Mutations in NPHS1 are the commonest cause and are particularly prevalent in Finland (“Finnish-type” CNS) where the incidence of CNS rises to 1 in 10,000.
CNS was historically defined by histopathological appearance, with five discrete patterns described; Finnish type, Diffuse mesangial sclerosis (DMS), Focal Segmental Glomerulosclerosis (FSGS), Membranous glomerulopathy and minimal change disease.2–7 Emerging diagnostic and mechanistic tools challenge these distinctions.8 The increasing range and accessibility of genetic analyses have demonstrated that the genotype-phenotype correlation is not as strict as once believed.
This review will discuss how the diagnostic pathway for children with CNS has changed and summarise some of the more frequently recognised genes in which mutations may cause a CNS phenotype. There remains a dichotomy in management between bilateral versus unilateral nephrectomy; the arguments for both are compared, with consideration of other management approaches. Common challenges in the management of these children are also briefly summarised, ending with novel approaches currently under investigation in the pre-clinical environment.
Basic Pathophysiology
Filtration by the glomerulus is performed by a structural unit, the glomerular filtration unit (GFU), which constitutes the architectural arrangement of the capillary endothelium, the glomerular basement membrane (GBM), and the podocyte. Podocytes are highly differentiated cells comprising a cell body, major processes and foot processes. These foot processes are vital to the integrity of the slit diaphragm (SD), a highly specialised intercellular junction between podocytes. Disruption of these slit diaphragms is highly associated with proteinuria and glomerular disease. Almost exclusively, monogenic causes of congenital nephrotic syndrome are related to mutations within genes relevant to the podocyte and structural integrity of the GFU.
Though foot process effacement is typically associated with significant proteinuria, there are clinical situations where this association does not hold true. Effacement has been ascribed to the development of lamellipodia, thick protrusions from the foot process component of the podocyte.9,10 Suvanto et al demonstrated reduced expression of slit diaphragm proteins in CNS kidneys compared to controls, though expression of cytosolic proteins was preserved.11
Clinical Presentation
The diagnosis of CNS may be suspected antenatally, with placentomegaly being a commonly reported feature.12–14 However, presentation is more typically in the neonatal or infant period. Infants may present with clinically evident edema, or more subtle features such as poor feeding and lethargy. There may be associated dysmorphic features or co-morbidities such as ocular abnormalities which may suggest the diagnosis. Common physical abnormalities associated with CNS are summarised in Table 1.
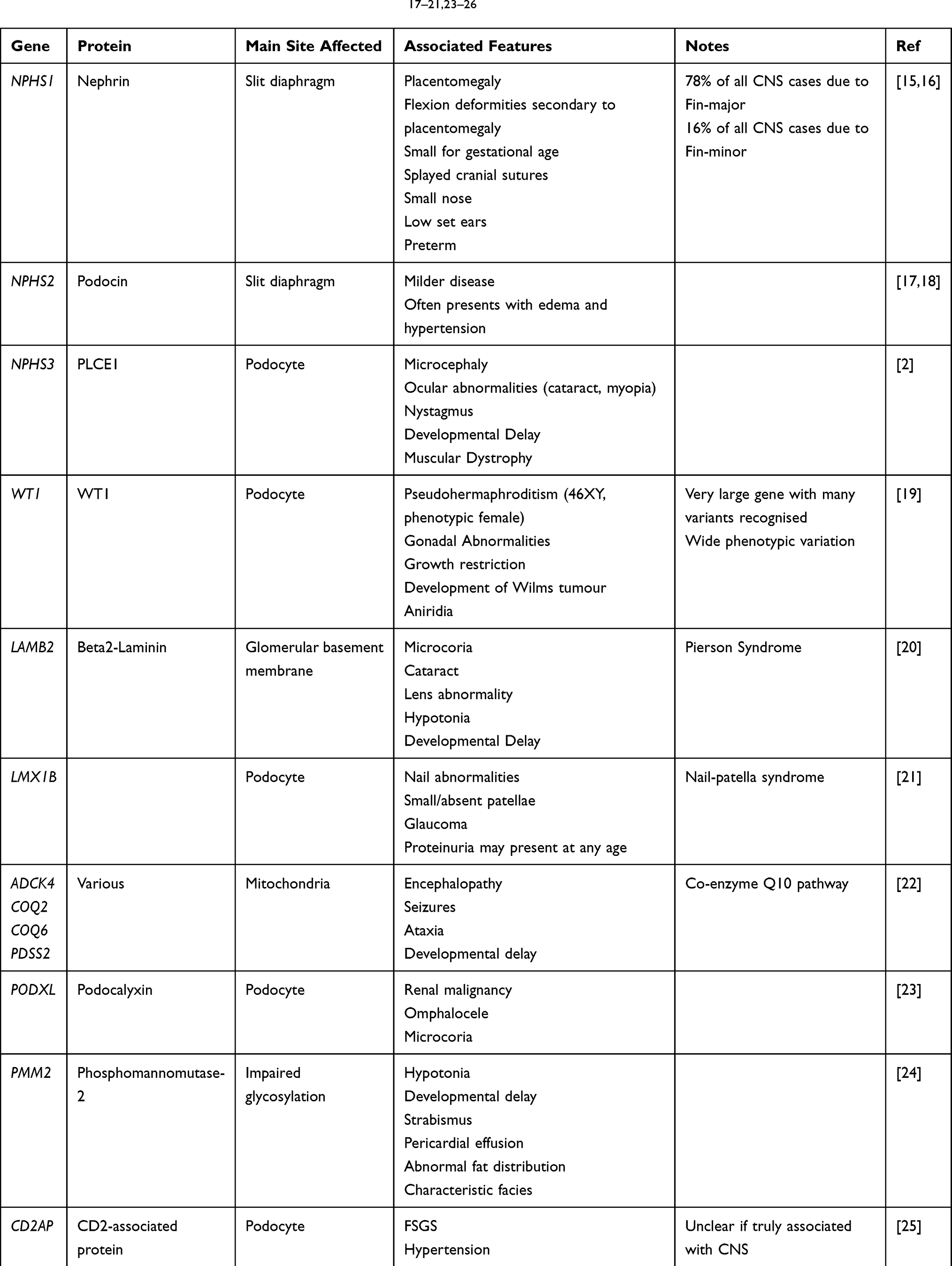 |
Table 1 CNS Genotypes and Their Associated Features17–21,23–26 |
Initial investigations aim to establish the likely diagnosis, exclude important secondary cause, and identify any complications that may require immediate management (Table 2).
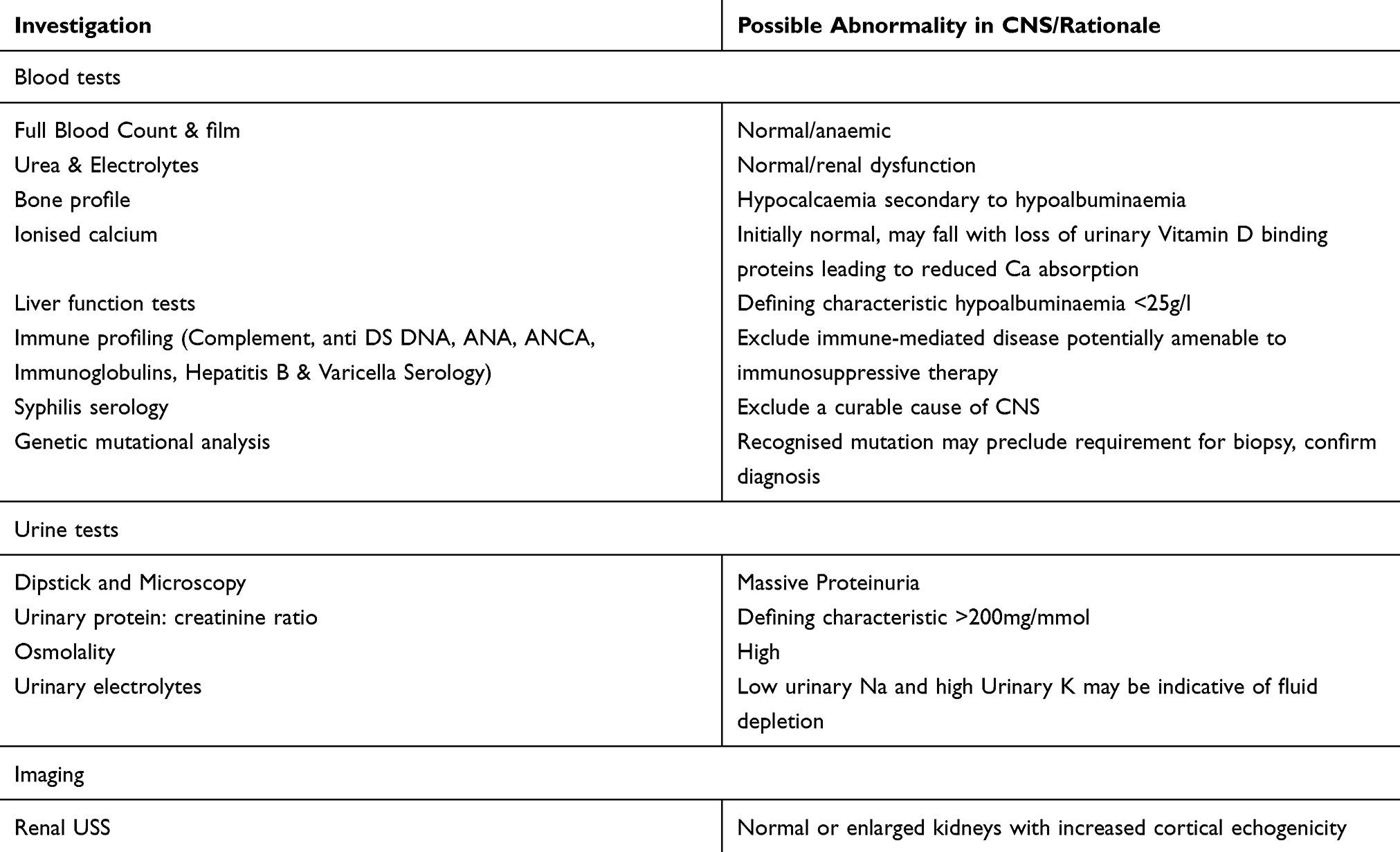 |
Table 2 Preliminary Investigations Where CNS Suspected |
The diagnosis of CNS is strongly suggested by the detection of massive nephrotic range proteinuria. Though classically nephrotic range is considered as >200mg/mmol, infants with CNS will often have proteinuria ten to a hundred-fold greater. Thus, the measurement of a urinary protein: creatinine ratio on a single urine specimen can be indicative of the diagnosis.
Traditionally, the diagnosis was clinical. Persisting nephrotic range proteinuria presenting aged less than 3 months, and in the absence of an apparent secondary cause (i.e. congenital infection) would suggest the diagnosis. Histological confirmation either by percutaneous renal biopsy or by examination of nephrectomy material would then confer a confirmed diagnosis.
Histologically, Finnish-type NS is associated with expansion of the mesangial matrix and hypercellularity leading to progressive glomerulosclerosis. Microcystic dilatation of the proximal tubules is a classical feature. There is an absence of immune complex deposition on immunofluorescence.27
Conversely, diffuse mesangial sclerosis more typically demonstrates small condensed glomeruli with collagen deposition in the mesangium, a feature absent in the Finnish-type.28 Immunofluorescence may demonstrate mesangial IgM and C3 deposition.
In membranous nephropathy, localised inflammation causes an increase in the thickness of the capillary membrane. Deposition of immune complexes with the associated inflammation leads to podocyte effacement.
FSGS shows sclerosis of segments of the glomerular tuft. These lesions are best demonstrated with PAS and silver-methenamine staining highlighting the increased collagen deposition. They will often abut normal glomeruli highlighting its focal nature.
However, the undertaking of percutaneous renal biopsy is not without risk. Though it can confirm the presence of histological features consistent with a diagnosis of CNS, the correlation between mutational analysis and histology is less definitive than originally believed. Unless there is significant diagnostic uncertainty, awaiting genetic analysis is often more prudent. Furthermore, nephrectomy often forms a part of the subsequent management, providing a wealth of histological tissue to support the diagnosis later on.
Genetic Analyses
The advent of genetic mutational analysis has led to more specific diagnoses of CNS. It is increasingly recognised that the histological appearance may not correlate as readily with the genetic findings.5,15,16,19
The majority of cases of CNS are caused by mutations in four notable genes; NPHS1, NPHS2, WT1 and LAMB2.2,29 However, as genetic assessment becomes more readily accessible and expansive, causative mutations in other genes are increasingly recognised e.g. PMM2, PODXL.23,24 For example, a large UK paediatric cohort of steroid-resistant nephrotic syndrome, including CNS patients, had whole-exome sequencing undertaken, focussing on 53 genes recognised to associate with NS. Identified several novel likely pathogenic variants in NPHS1 and NPHS2.17
NPHS1
NPHS1 encodes for the production of nephrin.16,30–32 Nephrin is the major structural component of the slit diaphragm, being key to its integrity.31,33 Mutation in NPHS1 leads to a loss of nephrin expression within the SD. This loss of interaction between the intracellular components of nephrin with podocin and other intra-podocyte processes leads to a reduction in actin polymerisation and corresponding change in foot process morphology and GBM functionality.16,34 Fin-major (c.121delCT; p.L41fs) and Fin-minor (c.3325C>T; p.R1109X) mutations are the cause of 98% of the NPHS1 mutations seen in the Finnish population; however other mutations are more prevalent in other populations with multiple pathogenic mutations in the nephrin gene now identified.30,35 Similar to other genetic diseases whereby mutation can affect protein expression, the severity of the CNS may be associated with the impact on nephrin expression. Absence of nephrin leads to CNS, whilst altered (but not absent) expression is associated with a severe nephrosis presenting beyond infancy. Disruption of the nephrin signalling pathway, due to disease or injury, leads to changes in the actin cytoskeleton morphology with migration of healthy podocytes to cover areas where podocyte loss has occurred.36 Deficiency in nephrin and neph1 leads to an upregulation of other intra-podocyte factors (podocin, NCK1/2 & CD2AP). Though the mechanism for this upregulation is not fully understood, it is felt to be in response to the failing SD and the resultant damage to the podocyte cytoskeleton.11
Typically, infants with NPHS1 mutation will be born in late prematurity (35–38 weeks’ gestation).37 Through their antenatal course, ultrasound scans may highlight a relatively large placenta and there may be a suggestion of crowding, restricting movement in utero. This can lead to flexion deformities of the elbows, hips and knees, and may also have an impact on respiratory development.13,14 Infants can also present with a small nose and low set ears, and there is evidence of delayed ossification with gaping anterior and posterior fontanelles.
NPHS2
NPHS2 encodes for podocin, a membranous protein which forms an anchoring hairpin structure between the plasma membrane and the internal actin cytoskeleton of the podocyte.38 Podocin promotes the integration of nephrin in the lipid rafts of the SD (24), with loss of this signalling and integration leading to nephrotic syndrome. The phenotype tends to be less aggressive than those arising from NPHS1 mutations. In the main, these children present in early childhood but may avoid detection until early adulthood.
WT1
The WT1 gene was initially identified in conjunction with WAGR syndrome (Wilm’s tumour, Aniridia, Genitourinary malformation and mental Retardation).39,40 Located on chromosome 11p13, WT1 is a tumour suppressor gene essential for normal gonadal and kidney development, and in later life promotes podocyte stabilisation. WT1 mutation may present with early onset NS in the context of Deny-Drash Syndrome (DDS) (with underlying diffuse mesangial sclerosis (DMS)) or later in life, e.g. Frasier syndrome. DDS associated with genotypical males (46XY) present with Wilm’s tumour, pseudohermaphroditism (i.e. phenotypically female) and progressive glomerulopathy. Those of the female genotype (46XX) present with Wilm’s tumour and glomerulopathy without the pseudohermaphroditism. There are other incomplete forms of the condition which present with DMS. In Frasier syndrome, the underlying pathological findings are more typically of FSGS. Though initially considered to contribute only 3% of CNS cases worldwide, finer tools for genetic diagnosis are suggesting a greater contribution. A recent UK registry review identified WT1 as the second commonest pathogenic genetic variant in children aged under 2 years.15 Patients with WT1 mutation may have less symptomatic nephrosis in terms of oedema and hypoalbuminemia but are more likely to have progressive and rapid reduction in renal function. WT1 is a very large gene, partly explaining the very broad range of clinical findings associated with mutation.
LAMB2
Unlike the aforementioned mutations which all impact the podocyte directly, the glomerular basement membrane is affected by mutations in LAMB2. This gene encodes for laminin-β2 a major structural protein providing anchoring within the GBM to podocytes. Being expressed in several systems, mutation in LAMB2 leads to Pierson syndrome, comprising CNS, ocular manifestations and neurological defects in children.20 The renal phenotype of this condition is variable from mild glomerulosclerosis to diffuse mesangial disease.41
Secondary CNS
The development of nephrotic range proteinuria in infancy is rare, with most cases in the developed world resulting from genetic mutation leading to CNS. However, secondary NS with an antecedent infection should be considered and excluded.7,18,42 Syphilis has seen a global resurgence43 and is implicated in the development of CNS, with histological evidence of glomerular immune deposition which may demonstrate an antigen-dose effect.44 Appropriate antibiotic therapy for syphilis may lead to resolution of the CNS (35).
Congenital toxoplasmosis infection has been identified as a cause of nephrotic syndrome.45 Similarly, appropriate treatment should lead to resolution of the nephrosis.
Neonatal cytomegalovirus (CMV) infection has also been implicated in nephrotic syndrome, though whether this is causative remains controversial given that evidence of CMV infection is relatively common and CNS is very rare.18,46,47 It is plausible that CMV infections represent a further “hit” in a complex multi-factorial condition.
The increasing availability of mutational analysis and technology that permits interrogation of multiple relevant genes on a single sample has altered the diagnostic pathway for infants with significant proteinuria. The ongoing identification of new mutations has progressed our understanding of the pathophysiology of podocytopathies, including CNS. Though individualised patient therapy based on mutational analysis is still outside the scope of most services, this may become an achievable goal in the future.8
Management
The primary goal in the management of CNS in infancy and early life is optimisation of nutrition and minimisation of complications, to achieve an adequate weight and height for transplantation to proceed as soon as possible. Nutritional management in these children is extremely challenging, and mandates specialist dietetic input early on. These patients are at risk from several co-morbidities that may impact on their long-term functioning and quality of life. The low incidence of CNS has essentially precluded randomised controlled trials of management strategies though long-term registry data, particularly from Finland, has provided some reassuring outcome data that these children can have an excellent outlook.48–50 The management of complications can be broadly divided into those associated with the nephrotic state, those arising from impaired glomerular filtration rate (GFR), and those associated with co-morbidities, e.g. retinal detachment in Pierson syndrome. This review will focus on the former – the challenges of managing the nephrotic state and its complications in young infants.
Managing Edema
The hallmark of CNS is significant protein loss. This leads to problematic edema, protein malnutrition, and complications relating to the wastage of specific proteins, e.g. immunoglobulins. What is the optimal strategy to reduce protein loss? The two predominant strategies in the last 25 years have been:
- Early bilateral nephrectomy to minimise protein loss, optimisation of growth and renal replacement therapy
- Medical management with non-steroidal anti-inflammatory (indomethacin) and angiotensin-converting enzyme inhibitor (ACEi) medication, sometimes with unilateral nephrectomy.
Bilateral Nephrectomy
Due to the far higher incidence in Finland, many aspects of management have been heavily influenced by the strategies and outcomes reported there. The standard approach has been early bilateral nephrectomy to optimise growth and permit earlier transplantation. This achieves a rapid correction of protein deficiency, avoiding the requirement for long-term intravenous albumin administration, and with improvements in quality of life measures.3 Unilateral nephrectomy may not be as impactful on protein loss leading to poorer growth, and ongoing intravenous albumin requirements. Uraemia and complications such as dyslipidaemia may also not be addressed by this less aggressive approach. There is a suggested distinction between children with “severe” protein loss, who should undergo bilateral nephrectomy, and less severe forms of the disease.3,51 Outcomes showed good growth following nephrectomy and progression to transplantation. Finnish registry data on outcomes with end-stage renal disease (ESRD) demonstrate that these patients are not disadvantaged in terms of graft survival, overall mortality, or developmental attainment compared to other infants with ESRD of other causes.18,48,52
Subsequent work by Holmberg et al proposed bilateral nephrectomy as the best means of controlling protein deficit and attaining a stable nutritional platform from which to progress to renal transplantation.3 Native nephrectomy prior to transplantation in a nephrotic patient is recommended to enable recovery of serum proteins which leads to a reduced complication rate including graft thrombosis and intravascular depletion. A recent cohort of nephrotic patients was described in this circumstance, of whom 3 had CNS, all undergoing bilateral nephrectomy; 2 synchronous and 1 staged prior to transplant. Within that cohort, unilateral nephrectomy was reported to effect a 40% reduction in proteinuria; this was a mixed cohort and therefore may not be generalisable to CNS.53
Unilateral Nephrectomy
Unilateral nephrectomy is proposed as a less aggressive alternative to bilateral nephrectomy. Multiple case series have reported reductions in the burden of albumin infusion and a positive impact on growth. Additional short-term growth benefits seen after bilateral nephrectomy appear to diminish beyond a year postintervention.54,55 Reported cohorts are heterogeneous and individual outcome data is often not presented. It would therefore be prudent in any future evaluation of management in nephrotic syndrome to include genetic variant data to enable distinctions to be made.56 This has been replicated in more recent work.56–58
Dufek reported on a heterogenous cohort of 80 patients, in whom just under half the patient cohort had nephrectomies performed to manage the gross proteinuria caused in this condition.57 Initially, when reviewed at 12 months there was a weight advantage to those receiving nephrectomies consistent with the Finnish cohorts leading to beneficial growth and optimised conditions for transplantation. However, this advantage was lost over the subsequent 6 months, which adds weight to the argument for a more conservative approach. They also report no increase in the complications of CNS between conservative or nephrectomised patients. The cohort suggests poor correlation between genotype and phenotype in CNS, a familiar finding.4
It is accepted that transplantation in those under 2 yrs carries with it a significant increase in morbidity.49,59 Dialysis carries with it its own independent morbidity.60,61 It would appear pragmatic where there is not the increased risk of Wilm’s (WT1) that a management strategy of staged nephrectomy gives CNS patients the opportunity to be managed, potentially in their home environment with domestic administration of albumin, allowing them to grow and avoid dialysis, and the possibility of pre-emptive transplantation.
As an increasing number of genes are implicated in the development of CNS it seems prudent to consider moderation of the Finnish approach and whether it is applicable to all CNS genotypes.
Albumin Replacement and Medical Management
The development of edema is a core feature of the nephrotic syndrome. Whether the increased interstitial fluid is secondary to reduced oncotic pressure, hypotension, and renin-angiotensin-aldosterone activation – the “underfill” hypothesis, or sodium and water retention with intravascular expansion and overflow into the interstitium – the “overflow” hypothesis, the mainstay of early management is the infusion of human albumin solution with the co-administration of loop diuretics.42,57,58
The frequency of these infusions places a heavy burden on the family of the affected child, more so if delivery is required in an in-patient setting. Delivery of IV albumin by the family in the home setting can mitigate some of these quality of life issues, without an increase in adverse events.62–64
Deliberate reduction in renal blood flow using angiotensin-converting enzyme inhibitors (ACEi) and prostaglandin inhibitors (PGi) such as indomethacin leads to a reduced protein loss through reducing intraglomerular pressure, and may reduce the number of albumin infusions required.29,56,57,59,65–67 There is some evidence to suggest the use of ACEi alone is ineffective at reducing the level of proteinuria.57,68 Although the use of amiloride to limit sodium retention through ENaC activation seems similarly physiologically sound, it is not used routinely. Amiloride has been studied in conjunction with loop diuretics and has shown to have an additive effect.69,70
Medical management may also include trials of immunosuppressive therapy. The standard management of idiopathic nephrotic syndrome includes corticosteroids and often subsequently calcineurin inhibitors, e.g. cyclosporine.71 Pre-clinical models investigating these medications demonstrate either observable beneficial effects on the podocyte or, in the case of rituximab, direct binding to the podocyte which may or may not be related to its therapeutic effect in other forms of nephrotic syndrome. Literature exists of infants with CNS trialled on corticosteroids, cyclosporin, or rituximab, in some circumstances with apparent benefit.72–75 Limited patient numbers render it impossible to definitively confirm a clinical effect, and all immunomodulatory therapy carries an adverse event profile. Pragmatically, if the diagnosis is unclear utilisation of immunotherapy until a genetic diagnosis is made is potentially justifiable but should be stopped if no clinical benefit is demonstrated.76
Overall, two main management strategies persist. Bilateral nephrectomy, especially in those with “severe” disease, or those with significant malignant risk, i.e. WT1 mutations, with early consideration of transplantation. Unilateral nephrectomy combined with medical anti-proteinuric strategies including ACEi and indomethacin, with optimised supportive renal care.
Other Complications Arising from Proteinuria
Nutrition and Growth
The development of protein malnutrition is intrinsic to CNS. Involvement of a specialist paediatric renal dietitian in the management of these patients is mandatory and should occur as soon as the diagnosis is suspected. A high calorific and protein content diet in combination with fluid and salt restriction are the cornerstones of the nutritional management.3,14,18,29,58 Regular dietetic review as renal function declines and the degree of protein loss lessens is a cornerstone of management. Early consideration of supplemental feeding, i.e. nasogastric tube, gastrostomy to provide optimal nutrition from the earliest stages is vital. The primary endpoint for these patients is to achieve a height and weight at which they can be transplanted, minimising their morbidity and mortality.
Vascular Access
Long-term central venous access is frequently essential for the secure regular delivery of IV albumin during the markedly proteinuric stage. The presence of a central venous catheter has associated risks including infection, bleeding, displacement and the need for anaesthesia for insertion and removal in children.77–79 The most significant risk in this particular patient population is thrombosis – the combination of being relatively hypercoagulable secondary to proteinuria, and the presence of foreign material within the vessels provide a strong impetus to thrombus formation; an “aggravated” clot.80,81 Anticoagulation therapy should be considered, either oral warfarin or subcutaneous low molecular weight heparin. The evidence base in preference for either therapy does not exist and may be guided by patient/family preference. Due to loss of anti-thrombin III in the urine, the dose required to achieve satisfactory prophylactic anti-coagulant concentrations may be several-fold higher than in other patient populations.82
Infective Risk
Several disease factors predispose these children to a higher risk of infective complications, including urinary loss of immunoglobulins, requirement for central venous catheter, and suboptimal nutrition. Administration of intravenous immunoglobulin in CNS is not recommended as intravascular persistence of the extraneous immunoglobulin is a matter of hours, providing no significant benefit.83 Prophylaxis against encapsulated organisms is sometimes provided in the form of regular phenoxymethylpenicillin (“Penicillin V”) though the clinical benefit is uncertain.58,84
Loss of Other Albumin-Bound Proteins
Urinary loss of thyroid-binding globulin leads to a functionally hypothyroid state requiring thyroxine replacement therapy.85 Infantile hyperbilirubinaemia carries an increased risk of kernicterus due to a fractionally greater free bilirubin – such infants should be aggressively managed with phototherapy where the diagnosis is recognised early enough. Hyperlipidemia and hypercholesterolemia are common (22) – use of medication to lower such as statins do not yet have an established role in CNS.
Transplantation
Transplantation is predominantly curative for the majority of cases of congenital nephrotic syndrome.3,29,48,49,51,57,58,86,87 Long-term graft survival is however dependent upon a number of pre and peri-transplant factors including adequate growth to accommodate a graft and very close monitoring and management in the early post-transplant phase, particularly graft vascular thrombus.
Living-related donation from a parent entails receiving a graft with heterozygosity for the causative mutation (if identified). Though there are theoretical concerns regarding this, in practice there is no evidence of poorer graft outcome, or the development of disease “recurrence” in patients receiving a living-related rather than deceased donor kidney.88 More detailed donor genetic analysis is suggested in NPHS2 mutation, as the pR229Q variant is relatively common (~4% population) and is associated with adult onset NS in compound heterozygotes. In that circumstance, donation from an alternative donor should be considered.
Disease “recurrence” following transplant has been reported, though in truth it is the development of a different pathophysiology. A subset of patients with Fin-major mutation who lack expression of normal nephrin develop an anti-nephrin antibody which may present with recurrence of proteinuria in the transplanted patient.89 This process can be rapid post-transplant.90 These patients typically respond to clearance of antibody with plasma exchange and anti-CD20 antibody (e.g. rituximab).
Areas for Future Development
Previous podocyte models have been limited by the absence of a truly representative in vitro slit diaphragm. Novel techniques in three-dimensional cell cultures and “organoid” development hold promise for ongoing mechanistic investigation.91,92
As our understanding of the genetic permutations associated with CNS develops, translation into tailored treatments mirroring the developments in other genetic conditions like cystic fibrosis and juvenile idiopathic arthritis may occur. As an example, murine models of Pierson Syndrome showing disruption of the GBM have been shown to be amenable to treatment with intravenous human LM-521 leading to a reduction in GBM damage and subsequent protein loss.93
Summary
The work carried out primarily by the Finnish group revolutionised the management of CNS in the 1980s and 1990s, changing it from a disease with little chance of survival beyond the first few weeks and months of life, to a chronic condition potentially curable through transplantation.
The evolution of next-generation genetic sequencing has led to clinically more meaningful stratification of CNS and opened the door to more moderate clinical approaches in patients with less severe nephrosis, reducing or halting the progression to ESRD.
Our understanding of the genetic basis of CNS alongside the creation of realistic facsimiles of the SD in vitro provide a research environment where targeted gene-specific therapies may transform management of this condition.
Management of CNS should be individualised. Both unilateral and bilateral nephrectomy offer benefits. Management of complications arising from proteinuria has very little evidence base to support and continues to be based on “best practice”.
Acknowledgments
The authors would like to thank the patients and families who have supported this work.
Special thanks go to Dr. Paul French, Consultant Pathologist, Royal Hospital for Children, for providing the images for this manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Gautier P, Miville D. Syndrome de nephrose lipoidique congenitale. Rev Med Suisse Rom. 1942;62:740.
2. Hinkes BG, Mucha B, Vlangos CN, et al. Nephrotic syndrome in the first year of life: two thirds of cases are caused by mutations in 4 genes (NPHS1, NPHS2, WT1, and LAMB2). Pediatrics. 2007;119(4):e907–e919. doi:10.1542/peds.2006-2164
3. Holmberg C, Antikainen M, Ronnholm K, Ala Houhala M, Jalanko H. Management of congenital nephrotic syndrome of the Finnish type. Pediatr Nephrol. 1995;9(1):87–93. doi:10.1007/BF00858984
4. Kari JA, Montini G, Bockenhauer D, et al. Clinico-pathological correlations of congenital and infantile nephrotic syndrome over twenty years. Pediatr Nephrol. 2014;29(11):2173–2180. doi:10.1007/s00467-014-2856-x
5. Machuca E, Benoit G, Nevo F, et al. Genotype–phenotype correlations in non-Finnish congenital nephrotic syndrome. J Am Soc Nephrol. 2010;21(7):1209. doi:10.1681/ASN.2009121309
6. Sharief SN, Hefni NA, Alzahrani WA, et al. Genetics of congenital and infantile nephrotic syndrome. World J Pediatr. 2019;15(2):198–203. doi:10.1007/s12519-018-00224-0
7. Wang -J-J, Mao J-H. The etiology of congenital nephrotic syndrome: current status and challenges. World J Pediatr. 2016;12(2):149–158. doi:10.1007/s12519-016-0009-y
8. Wiggins RC. The spectrum of podocytopathies: a unifying view of glomerular diseases. Kidney Int. 2007;71(12):1205–1214. doi:10.1038/sj.ki.5002222
9. Garg P. A review of podocyte biology. Am J Nephrol. 2018;47(suppl1)):3–13. doi:10.1159/000481633
10. Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nature Rev Mol Cell Biol. 2005;6:56. doi:10.1038/nrm1549
11. Suvanto M, Jahnukainen T, Kestilä M, Jalanko H. Podocyte proteins in congenital and minimal change nephrotic syndrome. Clin Exp Nephrol. 2015;19(3):481–488. doi:10.1007/s10157-014-1020-z
12. Huttunen NP. Congenital nephrotic syndrome of Finnish type. Study of 75 patients. Arch Dis Child. 1976;51(5):344–348. doi:10.1136/adc.51.5.344
13. Morgan G, Postlethwaite RJ, Lendon M, Houston IB, Savage JM. Postural deformities in congenital nephrotic syndrome. Arch Dis Child. 1981;56(12):959. doi:10.1136/adc.56.12.959
14. Savage JM, Jefferson JA, Maxwell AP, Hughes AE, Shanks JH, Gill D. Improved prognosis for congenital nephrotic syndrome of the Finnish type in Irish families. Arch Dis Child. 1999;80(5):466. doi:10.1136/adc.80.5.466
15. Bierzynska A, McCarthy HJ, Soderquest K. Genomic and clinical profiling of a national nephrotic syndrome cohort advocates a precision medicine approach to disease management. Kidney Int. 2017;91(4):937–947. doi:10.1016/j.kint.2016.10.013
16. Patrakka J, Kestilä M, Wartiovaara J, et al. Congenital nephrotic syndrome (NPHS1): features resulting from different mutations in Finnish patients. Kidney Int. 2000;58(3):972–980. doi:10.1046/j.1523-1755.2000.00254.x
17. Bierzynska A, McCarthy HJ, Soderquest K, et al. Genomic and clinical profiling of a national nephrotic syndrome cohort advocates a precision medicine approach to disease management. Kidney Int. 2017;91(4):937–947. doi:10.1016/j.kint.2016.10.013
18. Jalanko H. Congenital nephrotic syndrome. Pediatr Nephrol. 2009;24(11):2121–2128. doi:10.1007/s00467-007-0633-9
19. Chernin G, Vega-Warner V, Schoeb DS, et al. Genotype/phenotype correlation in nephrotic syndrome caused by WT1 mutations. Clin J Am Soc Nephrol. 2010;5(9):1655–1662. doi:10.2215/CJN.09351209
20. Aydin B, İpek MŞ, Ozaltin F, et al. A Novel Mutation of Laminin Β-2 Gene in Pierson Syndrome Manifested with Nephrotic syndrome in the Early Neonatal Period. Genet Couns. 2013;24:141–147.
21. Dreyer SD, Morello R, German MS, et al. LMX1B transactivation and expression in nail-patella syndrome. Hum Mol Genet. 2000;9(7):1067–1074. doi:10.1093/hmg/9.7.1067
22. Heeringa SF, Chernin G, Chaki M, et al. COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J Clin Invest. 2011;121(5):2013–2024. doi:10.1172/JCI45693
23. Kang HG, Lee M, Lee KB, et al. Loss of podocalyxin causes a novel syndromic type of congenital nephrotic syndrome. Exp Mol Med. 2017;49(12):e414–e414. doi:10.1038/emm.2017.227
24. Altassan R, Witters P, Saifudeen Z, et al. Renal involvement in PMM2-CDG, a mini-review. Mol Genet Metab. 2018;123(3):292–296. doi:10.1016/j.ymgme.2017.11.012
25. Lowik MM, Groenen PJ, Pronk I, et al. Focal segmental glomerulosclerosis in a patient homozygous for a CD2AP mutation. Kidney Int. 2007;72(10):1198–1203. doi:10.1038/sj.ki.5002469
26. Hinkes BG. NPHS3: new clues for understanding idiopathic nephrotic syndrome. Pediatr Nephrol. 2008;23(6):847–850. doi:10.1007/s00467-008-0747-8
27. Fogo AB, Lusco MA, Najafian B, Alpers CE. AJKD atlas of renal pathology: congenital nephrotic syndrome of Finnish type. Am J Kidney Dis. 2015;66(3):e11–e12. doi:10.1053/j.ajkd.2015.07.008
28. Fogo AB, Lusco MA, Najafian B, Alpers CE. AJKD atlas of renal pathology: diffuse mesangial sclerosis. Am J Kidney Dis. 2015;66(4):e23–e24. doi:10.1053/j.ajkd.2015.08.007
29. Niaudet P Congenital and infantile nephrotic syndrome [Review]. UpToDate. 2019. Available from: https://www.uptodate.com/contents/congenital-and-infantile-nephrotic-syndrome. Published March 8, 2019.
30. Kestila M, Lenkkeri U, Mannikko M, et al. Positionally cloned gene for a novel glomerular protein–nephrin–is mutated in congenital nephrotic syndrome. Mol Cell. 1998;1(4):575–582. doi:10.1016/S1097-2765(00)80057-X
31. Ruotsalainen V, Ljungberg P, Wartiovaara J, et al. Nephrin is specifically located at the slit diaphragm of glomerular podocytes. Proc National Acad Sci. 1999;96(14):7962. doi:10.1073/pnas.96.14.7962
32. Zenker M, Machuca E, Antignac C. Genetics of nephrotic syndrome: new insights into molecules acting at the glomerular filtration barrier. J Mol Med. 2009;87(9):849. doi:10.1007/s00109-009-0505-9
33. Wartiovaara J, Ofverstedt L-G, Khoshnoodi J, et al. Nephrin strands contribute to a porous slit diaphragm scaffold as revealed by electron tomography. J Clin Invest. 2004;114(10):1475–1483. doi:10.1172/JCI22562
34. Menon MC, Chuang PY, He CJ. The glomerular filtration barrier: components and crosstalk. Int J Nephrol. 2012;2012:9. doi:10.1155/2012/749010
35. Lenkkeri U, Männikkö M, McCready P, et al. Structure of the gene for congenital Nephrotic Syndrome of the Finnish Type (NPHS1) and characterization of mutations. Am J Human Genet. 1999;64(1):51–61. doi:10.1086/302182
36. Wharram BL, Goyal M, Wiggins JE, et al. Podocyte depletion causes glomerulosclerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J Am Soc Nephrol. 2005;16(10):2941. doi:10.1681/ASN.2005010055
37. Hallman N, Hjelt L. Congenital nephrotic syndrome. J Pediatr. 1959;55(2):152–162. doi:10.1016/S0022-3476(59)80083-4
38. Boute N, Gribouval O, Roselli S, et al. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet. 2000;24:349. doi:10.1038/74166
39. Call KM, Glaser T, Ito CY, et al. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms’ tumor locus. Cell. 1990;60(3):509–520. doi:10.1016/0092-8674(90)90601-A
40. Gessler M, Poustka A, Cavenee W, Neve RL, Orkin SH, Bruns GAP. Homozygous deletion in Wilms tumours of a zinc-finger gene identified by chromosome jumping. Nature. 1990;343(6260):774–778. doi:10.1038/343774a0
41. Choi HJ, Lee BH, Kang JH, et al. Variable phenotype of Pierson syndrome. Pediatr Nephrol. 2008;23(6):995–1000. doi:10.1007/s00467-008-0748-7
42. Rheault MN. Nephrotic and nephritic syndrome in the newborn. Clin Perinatol. 2014;41(3):605–618. doi:10.1016/j.clp.2014.05.009
43. Public Health England. Sexually Transmitted Infections and Chlamydia Screening in England, 2016. Public Health England; 2016.
44. Suskind R, Winkelstein JA, Spear GA. Nephrotic syndrome in congenital syphilis. Arch Dis Child. 1973;48(3):237–239. doi:10.1136/adc.48.3.237
45. Shahin B, Papadopoulou ZL, Jenis EH. Congenital nephrotic syndrome associated with congenital toxoplasmosis. J Pediatr. 1974;85(3):366–370. doi:10.1016/S0022-3476(74)80117-4
46. Batisky DL, Roy S
47. Besbas N, Bayrakci US, Kale G, et al. Cytomegalovirus-related congenital nephrotic syndrome with diffuse mesangial sclerosis. Pediatr Nephrol. 2006;21(5):740–742. doi:10.1007/s00467-006-0051-4
48. Holtta T, Bonthuis M, Van Stralen KJ, et al. Timing of renal replacement therapy does not influence survival and growth in children with congenital nephrotic syndrome caused by mutations in NPHS1: data from the ESPN/ERA-EDTA registry. Pediatr Nephrol. 2016;31(12):2317–2325. doi:10.1007/s00467-016-3517-z
49. Kim MS, Stablein D, Harmon WE. Renal transplantation in children with congenital nephrotic syndrome: a report of the North American Pediatric Renal Transplant Cooperative Study (NAPRTCS). Pediatr Transplant. 1998;2(4):305–308.
50. Trautmann A, Bodria M, Ozaltin F, et al. Spectrum of steroid-resistant and congenital nephrotic syndrome in children: the PodoNet registry cohort. Clin J Am Soc Nephrol. 2015;10(4):592–600. doi:10.2215/CJN.06260614
51. Holmberg C, Jalanko H, Koskimies O, et al. Renal transplantation in children with congenital nephrotic syndrome of the Finnish type. Transplant Proc. 1990;22(1):158–159.
52. Dufek S, Ylinen E, Trautmann A, et al. Infants with congenital nephrotic syndrome have comparable outcomes to infants with other renal diseases. Pediatr Nephrol. 2019;34(4):649–655. doi:10.1007/s00467-018-4122-0
53. Ghane Sharbaf F, Bitzan M, Szymanski KM, et al. Native nephrectomy prior to pediatric kidney transplantation: biological and clinical aspects. Pediatr Nephrol. 2012;27(7):1179–1188. doi:10.1007/s00467-012-2115-y
54. Coulthard MG. Management of Finnish congenital nephrotic syndrome by unilateral nephrectomy. Pediatr Nephrol. 1989;3(4):451–453. doi:10.1007/BF00850226
55. Mattoo TK, al-Sowailem AM, al-Harbi MS, Mahmood MA, Katawee Y, Hassab MH. Nephrotic syndrome in 1st year of life and the role of unilateral nephrectomy. Pediatr Nephrol. 1992;6(1):16–18. doi:10.1007/BF00856821
56. Licht C, Eifinger F, Gharib M, Offner G, Michalk DV, Querfeld U. A stepwise approach to the treatment of early onset nephrotic syndrome. Pediatr Nephrol. 2000;14(12):1077–1082. doi:10.1007/s004670000386
57. Dufek S, Holtta T, Trautmann A, et al. Management of children with congenital nephrotic syndrome: challenging treatment paradigms. Nephrol Dial Transplant. 2018.
58. Berody S, Heidet L, Gribouval O, et al. Treatment and outcome of congenital nephrotic syndrome. Nephrol Dial Transplant. 2019;34(3):458–467. doi:10.1093/ndt/gfy015
59. Kovacevic L, Reid CJ, Rigden SP. Management of congenital nephrotic syndrome. Pediatr Nephrol. 2003;18(5):426–430. doi:10.1007/s00467-003-1131-3
60. Alexander RT, Foster BJ, Tonelli MA, et al. Survival and transplantation outcomes of children less than 2 years of age with end-stage renal disease. Pediatr Nephrol. 2012;27(10):1975–1983. doi:10.1007/s00467-012-2195-8
61. Shroff R, Ledermann S. Long-term outcome of chronic dialysis in children. Pediatr Nephrol. 2009;24(3):463–474. doi:10.1007/s00467-007-0700-2
62. Reynolds BC, Pickles CW, Lambert HJ, et al. Domiciliary administration of intravenous albumin in congenital nephrotic syndrome. Pediatr Nephrol. 2015;30(11):2045–2050. doi:10.1007/s00467-015-3177-4
63. Balaguer A, González de Dios J. Home versus hospital intravenous antibiotic therapy for cystic fibrosis. Cochrane Database Syst Rev. 2015;2015(12):CD001917–CD001917.
64. Pederiva F, Khalil B, Morabito A, Wood SJ. Impact of short bowel syndrome on quality of life and family: the patient’s perspective. Eur J Pediatr Surg. 2019;29(02):196–202. doi:10.1055/s-0037-1621737
65. Guez S, Giani M, Melzi ML, Antignac C, Assael BM. Adequate clinical control of congenital nephrotic syndrome by enalapril. Pediatr Nephrol. 1998;12(2):130–132. doi:10.1007/s004670050420
66. Pomeranz A, Wolach B, Bernheim J, Korzets Z, Bernheim J. Successful treatment of Finnish congenital nephrotic syndrome with captopril and indomethacin. J Pediatr. 1995;126(1):140–142. doi:10.1016/S0022-3476(95)70518-X
67. Wong W, Morris MC, Kara T. Congenital nephrotic syndrome with prolonged renal survival without renal replacement therapy. Pediatr Nephrol. 2013;28(12):2313–2321. doi:10.1007/s00467-013-2584-7
68. Birnbacher R, Forster E, Aufricht C. Angiotensin converting enzyme inhibitor does not reduce proteinuria in an infant with congenital nephrotic syndrome of the Finnish type. Pediatr Nephrol. 1995;9(3):400. doi:10.1007/BF02254232
69. Andersen RF, Buhl KB, Jensen BL, et al. Remission of nephrotic syndrome diminishes urinary plasmin content and abolishes activation of ENaC. Pediatr Nephrol. 2013;28(8):1227–1234. doi:10.1007/s00467-013-2439-2
70. Deschênes G, Guigonis V, Doucet A. Mécanismes physiologiques et moléculaires de la constitution des œdèmes au cours du syndrome néphrotique. Arch De Pédiatrie. 2004;11(9):1084–1094. doi:10.1016/j.arcped.2004.03.029
71. Noone DG, Iijima K, Parekh R. Idiopathic nephrotic syndrome in children. Lancet. 2018;392(10141):61–74. doi:10.1016/S0140-6736(18)30536-1
72. Büscher AK, Beck BB, Melk A, et al. Rapid response to cyclosporin a and favorable renal outcome in nongenetic versus genetic steroid-resistant nephrotic syndrome. Clin j Am Soc Nephrol. 2016;11(2):245–253. doi:10.2215/CJN.07370715
73. Giglio S, Provenzano A, Mazzinghi B, et al. Heterogeneous genetic alterations in sporadic nephrotic syndrome associate with resistance to immunosuppression. J Am Soc Nephrol. 2015;26(1):230–236. doi:10.1681/ASN.2013111155
74. Ransom RF, Lam NG, Hallett MA, Atkinson SJ, Smoyer WE. Glucocorticoids protect and enhance recovery of cultured murine podocytes via actin filament stabilization. Kidney Int. 2005;68(6):2473–2483. doi:10.1111/j.1523-1755.2005.00723.x
75. Büscher AK, Kranz B, Büscher R, et al. Immunosuppression and renal outcome in congenital and pediatric steroid-resistant nephrotic syndrome. Clin j Am Soc Nephrol. 2010;5(11):2075–2084. doi:10.2215/CJN.01190210
76. Preston R, Stuart HM, Lennon R. Genetic testing in steroid-resistant nephrotic syndrome: why, who, when and how? Pediatr Nephrol. 2019;34(2):195–210. doi:10.1007/s00467-017-3838-6
77. Ares G, Hunter CJ. Central venous access in children: indications, devices, and risks. Curr Opin Pediatr. 2017;29(3):340–346. doi:10.1097/MOP.0000000000000485
78. Duesing LA, Fawley JA, Wagner AJ. Central venous access in the pediatric population with emphasis on complications and prevention strategies. Nutr Clin Pract. 2016;31(4):490–501. doi:10.1177/0884533616640454
79. Wolf J, Curtis N, Worth LJ, Flynn PM. Central line-associated bloodstream infection in children: an update on treatment. Pediatr Infect Dis J. 2013;32(8):905–910. doi:10.1097/INF.0b013e3182996b6e
80. Kanfer A. Coagulation factors in nephrotic syndrome. Am J Nephrol. 1990;10(suppl1):63–68. doi:10.1159/000168196
81. Lau KK, Chan HH, Massicotte P, Chan AK. Thrombotic complications of neonates and children with congenital nephrotic syndrome. Curr Pediatr Rev. 2014;10(3):169–176.
82. Chander A, Nagel K, Wiernikowski J, et al. Evaluation of the use of low-molecular-weight heparin in neonates: a retrospective, single-center study. Clin Appl Thromb Hemost. 2013;19(5):488–493. doi:10.1177/1076029613480557
83. Harris HW
84. Ljungberg P, Holmberg C, Jalanko H. Infections in infants with congenital nephrosis of the Finnish type. Pediatr Nephrol. 1997;11(2):148–152. doi:10.1007/s004670050246
85. Kacer M, Whyte DA, Boydstun I, Wilson TA. Congenital nephrotic syndrome and persistent hypothyroidism after bilateral nephrectomy. J Pediatr Endocrinol Metab. 2008;21(6):597–601.
86. Downie ML, Gallibois C, Parekh RS, Noone DG. Nephrotic syndrome in infants and children: pathophysiology and management. Paediatr Int Child Health. 2017;37(4):248–258. doi:10.1080/20469047.2017.1374003
87. Mahan JD, Mauer SM, Sibley RK, Vernier RL. Congenital nephrotic syndrome: evolution of medical management and results of renal transplantation. J Pediatr. 1984;105(4):549–557. doi:10.1016/S0022-3476(84)80418-7
88. Niaudet P. Living donor kidney transplantation in patients with hereditary nephropathies. Nat Rev Nephrol. 2010;6(12):736–743. doi:10.1038/nrneph.2010.122
89. Holmberg C, Jalanko H. Congenital nephrotic syndrome and recurrence of proteinuria after renal transplantation. Pediatr Nephrol. 2014;29(12):2309–2317. doi:10.1007/s00467-014-2781-z
90. Grenda R, Jarmużek W, Rubik J, Piątosa B, Prokurat S. Rituximab is not a “magic drug” in post-transplant recurrence of nephrotic syndrome. Eur J Pediatr. 2016;175(9):1133–1137. doi:10.1007/s00431-016-2747-1
91. Hall G, Gbadegesin RA. Translating genetic findings in hereditary nephrotic syndrome: the missing loops. Am j Physiol Renal physiol. 2015;309(1):F24–F28. doi:10.1152/ajprenal.00683.2014
92. Kim YK, Refaeli I, Brooks CR, et al. Gene-edited human kidney organoids reveal mechanisms of disease in podocyte development. Stem Cells. 2017;35(12):2366–2378. doi:10.1002/stem.2707
93. Lin M-H, Miller JB, Kikkawa Y, et al. Laminin-521 protein therapy for glomerular basement membrane and podocyte abnormalities in a model of pierson syndrome. J Am Soc Nephrol. 2018;29(5):1426. doi:10.1681/ASN.2017060690
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.