Back to Journals » Clinical Optometry » Volume 8
Diagnosis and treatment of anterior uveitis: optometric management
Authors Harthan J ![]() , Opitz D
, Opitz D ![]() , Fromstein S, Morettin C
, Fromstein S, Morettin C
Received 21 July 2015
Accepted for publication 30 September 2015
Published 31 March 2016 Volume 2016:8 Pages 23—35
DOI https://doi.org/10.2147/OPTO.S72079
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Mr Simon Berry
Jennifer S Harthan,1 Dominick L Opitz,2 Stephanie R Fromstein,1 Christina E Morettin3
1Cornea Center for Clinical Experience, 2Ophthalmology Services and Practice Development, 3Urgent Eye Care Service, Illinois College of Optometry, Chicago, IL, USA
Abstract: Anterior uveitis encompasses inflammation of the iris and/or ciliary body and is one of the most common types of ocular inflammation that primary eye care practitioners will encounter. Anterior uveitis may be caused by a variety of etiologies, including infectious, noninfectious, and masquerade diseases. The short-term and long-term treatment of uveitis should include the evaluation of location, duration, pathology, and laterality, in addition to presenting signs and symptoms of the disease. A complete review of systems, thorough examination, and laboratory testing, may assist the practitioner in narrowing the list of possible causes for the uveitis. This is imperative as once a list of diagnoses has been made, a targeted approach to treatment can be pursued.
Keywords: anterior uveitis, iritis, inflammation
Introduction
Uveitis, or inflammation of the uvea (which consists of the iris, ciliary body, and choroid), may be caused by a number of different etiologies.1,2 Anterior uveitis is defined by the presence of cells or cellular aggregates that are visible in the anterior chamber during examination.3 Anterior uveitis is one of the most common types of ocular inflammation that eye care practitioners will encounter.1–4 It may present as acute, subacute, or chronic.4 An estimated 14–17 cases per 100,000 and 38,000 new cases are confirmed each year in the USA.5 Uveitis accounts for ~10% (30,000 new cases annually) of legal blindness in the USA, and it may affect all ages, although it is most common in individuals in the third and fourth decades.3,5–8 Under certain circumstances, the body produces a normal inflammatory response as a protective mechanism to heal itself. In chronic or uncontrolled cases, excessive infiltration of neutrophils, macrophages, and lymphocytes may cause permanent damage to the ocular tissue.3,4 Anterior uveitis may be caused by infectious, noninfectious, and masquerade diseases. Diagnosis and treatment of the underlying disease are imperative not only to treat the disease but also to preserve vision and potentially uncover underlying systemic diseases. If untreated or not appropriately treated, acute inflammation can develop into chronic, sight-threatening inflammation,9 emphasizing the role of the primary eye care practitioner in appropriately and effectively managing these patients.
The primary eye care practitioner is often the first health care provider to see and treat cases of anterior uveitis.4 In order to effectively manage and treat ocular inflammation, an understanding of the immunoinflammatory cascade is necessary. There are two main components to this pathway: the innate immune system and the adaptive immune system. The innate immune system uses macrophages, mast cells, and neutrophils to destroy invading microbes,3 and it is able to immediately recognize certain bacterial, viral, and foreign antigens and becomes engaged without a prior history of exposure to an antigen. The adaptive immune system improves its immunological memory of previous encounters to foreign antigens that is dependent on antigen presentation, cell activation, cell differentiation, and cell memory.3 Repeated exposure to this same antigen leads to an increased immune response and then provides long-lasting immunologic protection to foreign antigens. Antigen-presenting cells, B- and T-lymphocytes, and macrophages are the cells responsible for the adaptive immune system.3
The ability of the immune system to use both the innate and adaptive immune systems in response to an antigen in a sufficient manner is one way in which the body regulates itself. When unregulated immune responses take place, tissue injury and disease occur, as we will demonstrate in our discussion of anterior uveitis.4
Diagnosis
The diagnosis of uveitis is multifaceted, and important diagnostic clues lie in both the clinical course and the clinical presentation. There are a number of important distinctions that must be made in order to fully characterize the condition; the Name Meshing System for diagnosis of uveitis provides a useful framework for organizing these divisions. Using this systematic approach, uveitis management should involve evaluation of location, duration, pathology, and laterality, in addition to presenting signs and symptoms.9–11
Evaluation of location is an important first step in the diagnosis of uveitis. Anterior uveitis is an umbrella term encompassing inflammation of the iris (iritis), anterior ciliary body (cyclitis), or both (iridocyclitis).12 This constitutes an important distinction from intermediate or posterior segment uveitis in terms of severity, threat to vision, and long-term sequelae, all of which are poorer for the more posterior iterations.13 Microscopically, aqueous cells are diagnostic of an iritis, often accompanied by aqueous flare composed of albumin.13 Although anterior vitreous cells serve as an evidence of cyclitis, they are rarely seen in isolation and are limited in clinical significance.14,15
Another important distinction in anterior uveitis is to understand the acute course versus the chronic course of the disease. According to the Standardization of Uveitis Nomenclature criteria, acute or limited cases refer to an episode of <3 months duration, while chronic or persistent cases extend beyond that time frame.12 This division is important clinically, as the etiology and management strategy differ between the two.11 The symptoms of chronic anterior uveitis are generally less severe, more commonly granulomatous in nature, and more likely to be accompanied by signs of chronicity, including band keratopathy and iris changes such as atrophy and lenticular changes.11 Recurrent anterior uveitis refers to relapsing inflammation separated by >3 months without treatment.12
The pathology of an anterior uveitis can be either granulomatous or nongranulomatous. Granulomatous inflammation is associated with large, mutton-fat keratic precipitates (KPs) composed mainly of epitheloid cells on the corneal endothelium.13 Granulomatous uveitis tends to be chronic and is often associated with systemic conditions and autoimmune reactions.9 It may also be associated with infectious etiologies such as syphilis, Lyme disease, tuberculosis (TB), and herpetic viral infections.9 Herpetic infections usually cause nongranulomatous uveitis in acute cases and granulomatous uveitis in chronic cases.9 Nongranulomatous inflammation, by contrast, tends to be associated with smaller lymphocytic cells in the anterior chamber.9 It is most often acute and idiopathic or associated with human leukocyte antigen B27 (HLA-B27) conditions.16 While this distinction is useful in directing treatment and targeting a systemic workup, a granulomatous uveitis may initially present as nongranulomatous, as may the reverse.17
Laterality is another element that should be considered when diagnosing and – ultimately – when considering a systemic workup for anterior uveitis patients. Following a similar pattern to granulomatous versus nongranulomatous inflammation, bilateral presentations tend to be associated with chronic, systemic conditions, whereas unilateral conditions tend to be acute and idiopathic or infectious.9
The final diagnostic element involves the clinical features of the inflammation. Presenting signs and symptoms can give you important diagnostic clues as to the underlying etiology. The most common patient symptom is blurry vision, secondary to cells and flare in the aqueous.13 Pain and photophobia are also common, owing mainly to ciliary muscle spasm, though anterior chamber infiltration, corneal epithelial edema, and pupillary muscle involvement can also contribute to light sensitivity.13 Pain is generally described as dull, aching, or throbbing and referred to the temple or periorbital region.13 Pain may be more localized and severe if associated with elevated intraocular pressure (IOP).18 There is a notable absence of mucopurulent discharge, which may help to differentiate this condition from other forms of anterior segment inflammation.9 In cases of chronic uveitis, the patient may be completely free of symptoms, and the inflammation may be noted on routine evaluation.
Common presenting signs of acute anterior uveitis include circumlimbal injection and anterior chamber cells and flare. Circumlimbal injection is secondary to the enlargement of episcleral vessels adjacent to the inflamed ciliary body.13 The patient may also present with diffuse injection or a mixed pattern.17 Cells and flare in the aqueous are due to inflammatory cells and protein in the anterior chamber. Anterior chamber cells are diagnostic of anterior uveitis and may or may not be accompanied by flare.13 Flare does not necessarily indicate active inflammation, as chronic uveitis can cause damage to the vasculature of the iris and ciliary body thus increasing protein permeability.14 If cells are adequately dense, a hyopyon may form, which is a collection of inflammatory cells collecting inferiorly in the anterior chamber.17 To visualize the cells and flare, the anterior chamber must be observed with a small beam (1 mm) with high illumination and magnification. The Standardization of Uveitis Nomenclature (SUN) Working Group developed a grading scheme for both anterior chamber cells and flare. The SUN grading scheme for anterior chamber cells is clinically evaluated with a 1 mm beam and includes: Grade 0, <1 cells in the field; Grade 0.5+, 1–5 cells in the field; Grade 1+, 6–15 cells in the field; Grade 2+, 16–25 cells in the field; Grade 3+, 25–50 cells in the field; and Grade 4+, >50+ cells in the field. The SUN grading scheme for anterior chamber flare includes: Grade 0, none; Grade 1+, faint; Grade 2+, moderate (iris and lens detail clear); Grade 3+, marked (iris and lens detail hazy); and Grade 4+, intense (fibrin/plastic aqueous).12,13,17 Grading the level of inflammation is critical in initiating and monitoring the response to treatment. Flare may additionally be visualized by laser flare photometry, which may play a role moving forward in providing a qualitative method to measure and follow anterior chamber inflammation.17 In addition to cellular entities in the anterior chamber, there are often also cellular deposits on the endothelium known as KPs.17 Their appearance can be of great diagnostic importance in differentiating the etiology of the inflammation. Fine KPs are generally associated with nongranulomatous inflammation, whereas larger, greasy, mutton-fat KPs are associated with granulomatous inflammation.13 Pigmented KPs on the endothelium are indicative of a prior episode or chronic inflammation.13 There is currently research underway using confocal biomicroscopy to further differentiate subtypes of KPs that will hopefully help us better differentiate between infectious or noninfectious subtypes. KPs should be classified based on appearance: globular, infiltrating, infectious, smooth-rounded, granulomatous, stippled, dendritiform, infectious, and crusciform.
Another important structure in the evaluation of anterior uveitis is the iris. Miosis can occur secondary to spasm of the iris sphincter or distention of iris vessels.18 Iris nodules are indicative of granulomatous inflammation and are seen in two forms – Busacca nodules are seen on the anterior stroma while Koeppe’s nodules are seen at the pupillary margin. Both are composed of leukocytes and should be differentiated from infected nodules.13 Busacca nodules are more commonly seen in granulomatous forms of anterior uveitis, whereas Koeppe’s nodules are seen in both granulomatous and nongranulomatous anterior uveitis.13 With chronic inflammation and ischemia, iris atrophy can develop.13 Posterior synechiae – adhesions between the lens and the iris – can ultimately extend 360°, preventing the flow of aqueous via this route. Finally, neovascularization of the iris stroma can occur with prolonged inflammation.13
Patients with anterior uveitis can have altered IOP. There are several mechanisms that can contribute to these changes. The first and most commonly encountered is IOP reduction. This occurs when the ciliary body becomes inflamed, resulting in low production of aqueous humor at the ciliary body.13,17 IOP can become elevated if aqueous humor outflow is hindered through the trabecular meshwork (TM) as with trabeculitis or when inflammatory cells and pigment clog the TM. A more severe complication is when the IOP elevates due to peripheral anterior synechiae obstructing the TM or if posterior synechiae creates pupillary block.13,17 Other mechanisms of elevated IOP include long-term steroid treatment or neovascularization, which can also occur in the angle and may cause a rise in IOP due to secondary angle closure.17 Fortunately, iris rubeosis in uveitis tends to be less severe and more reversible than ischemic neovascularization and usually resolves after therapy.13,17
Finally, any evaluation of a patient with anterior uveitis should include a dilated fundus examination. Not only can uveitis cause secondary effects on the lens or spillover inflammation, but also posterior segment findings indicate a panuveitis, which is a distinct – and significantly more serious – condition requiring immediate referral and a different treatment/management approach. A dilated fundus examination will help determine if there are signs of true intermediate or posterior uveitis with spillover, such as snowballs or snowbanking, inflammation of the optic nerve or along blood vessels, or active lesions of the retina and/or choroid.
By characterizing uveitis as anterior or posterior, acute or chronic, granulomatous or nongranulomatous, unilateral or bilateral, and by noting important clinical signs and symptoms, the primary care practitioner is able to formulate an appropriate diagnosis that is suggestive of an infectious or noninfectious underlying etiology, which will help the clinician devise a targeted work up. Furthermore, they can devise a targeted systemic workup to minimize cost and inconvenience to the patient. Finally, it allows them to initiate a thoughtful treatment tailored to specific aspects of the patient’s clinical presentation. Accurate – and complete – diagnosis is a critical first step in the management of the patient with anterior uveitis.
Systemic associations
Although the majority of uveitic cases are idiopathic (48%–70%),11,19 there is a diverse set of etiologies that span a wide spectrum of infectious and noninfectious causes. A complete review of systems helps guide the systemic evaluation of a patient with anterior uveitis (Table 1). Systemic consideration for an underlying etiology is suspected when the uveitis is bilateral, granulomatous, recurrent, intermediate and/or posterior, or presents with minimal signs and symptoms.11,20–22 The following section reviews some of the more common underlying etiologies for anterior uveitis.
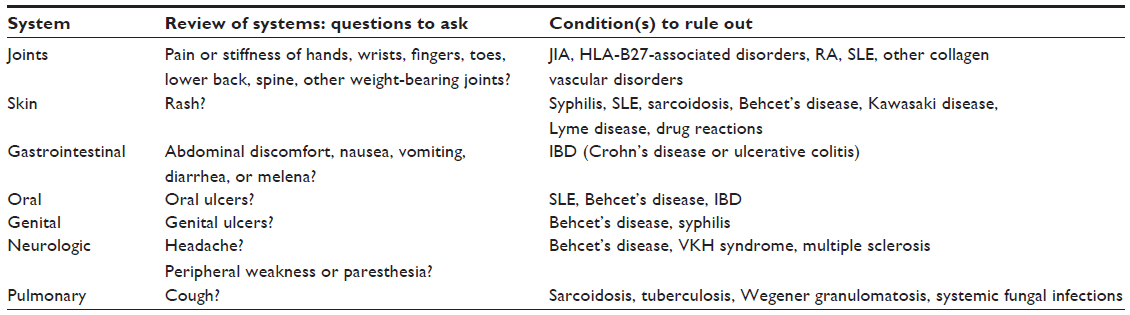 | Table 1 Review of systems to be included with any patient that presents with uveitis |
Immune-mediated (noninfectious) causes
HLA-B27 seronegative spondyloarthropathies are a group of inflammatory disorders with negative rheumatoid factor and are associated with HLA-B27, a class I major histocompatibility complex.23–27 It is the most common noninfectious etiology of anterior uveitis accounting for up to 50% of acute and recurrent anterior uveitis cases.27 Patients will usually present with a recurrent unilateral, bilateral, or alternating nongranulomatous anterior uveitis and may have fine endothelial KPs.23–27 The HLA-B27 seronegative spondyloarthropathies include ankylosing spondylitis, reactive arthritis syndrome, psoriatic arthritis, inflammatory bowel disease (IBD).
Ankylosing spondylitis is a chronic systemic disease of unknown cause. It is characterized by inflammation of both sacroiliac joints and the spine. The most common extra-articular manifestation is anterior uveitis, occurring in 25% of patients with ankylosing spondylitis.28 Reactive arthritis syndrome, previously known as Reiter syndrome, presents as the classic triad of arthritis, urethritis, and conjunctivitis. In total, 3%–12% of these patients present with uveitis.29 Psoriatic arthritis has classically been defined as the triad of psoriasis (skin and/or nail), chronic recurrent erosive polyarthritis, and a negative test for rheumatoid factor.30–32 IBD encompasses a variety of different conditions, the two most common being Crohn’s disease and ulcerative colitis, both characterized by chronic inflammation of the gastrointestinal tract, with unknown etiology. In Crohn’s disease, the entire intestinal tract (mouth to anus) is affected, predominantly the ileum and cecum of the small intestine. Ulcerative colitis affects predominantly the rectosigmoid region of the large intestine.33 In patients with IBD, the uveitis may have an insidious onset.34,35
Juvenile idiopathic arthritis (JIA) is the most common form of arthritis in children and adolescents with unknown etiology. It is an umbrella term wherein there are seven categories of JIA. The majority of JIA-associated uveitis patients have oligoarticular onset JIA (78%–90%), while 7%–14% have the polyarticular variety.36 As uveitis in JIA can present in a quiet eye, recommended screening guidelines as per Cassidy et al37 is 3 months for high-risk JIA patients, 6 months for moderate-risk JIA patients, and 12 months for low-risk JIA patients.36 In total, 37.3% of JIA-associated uveitis patients experience complications from the disease or associated steroid treatment, such as cataract, posterior synechiae, band keratopathy, glaucoma, or macular edema (listed in decreasing order of incidence).38 Patients with JIA typically present with an acute recurrent unilateral or bilateral nongranulomatous anterior uveitis, with or without symptoms.39
Sarcoidosis is a multisystem disease of unknown origin, where the hallmark sign is noncaseating granulomatous inflammation. It predominantly affects the lungs, and ocular involvement is present in up to 50% of patients.40–43 Anterior uveitis is the most common ocular sequelae, which is typically bilateral, chronic, and granulomatous. The granulomatous nature of the uveitis is characterized by mutton-fat KPs, TM inflammation, and iris nodules.40–43 Up to 25% of patients with ocular sarcoidosis develop posterior involvement, including vitritis, intermediate uveitis snowballs, periphlebitis, panuveitis, optic nerve involvement, granulomas along vessels “candle wax drippings”, and cystoid macular edema.40–43
Systemic lupus erythematous (SLE) is a multisystem autoimmune disease characterized by the production of autoantibodies targeted against cell nuclei (antinuclear antibodies), a type III hypersensitivity reaction. SLE is most commonly associated with scleritis, episcleritis, secondary Sjogren’s syndrome, lupus retinopathy, and choroidopathy.44,45 Anterior uveitis seldom occurs in isolation and is more commonly associated with scleritis or posterior uveitis.46
Behcet’s disease is a chronic, relapsing inflammatory disorder of unknown etiology, characterized by the classic triad of oral and genital ulcers, ocular inflammation, and skin lesions. It is associated with HLA-B51.47,48 Patients with Behcet’s disease typically present with a nongranulomatous anterior and/or posterior uveitis, KPs, and posterior synechiae. In total, 19%–31% of these cases present with a hypopyon.49,50
Vogt–Koyanagi–Harada disease is a multisystem autoimmune disorder involving the ocular, auditory, central nervous, and integumentary organ systems. It affects the pigmented tissues within these structures, due to a T-cell-mediated immune response to self-antigens on melanocytes. It presents as a chronic, bilateral, diffuse, granulomatous uveitis.51–53
Fuch’s heterochronic iridocyclitis (FHI) is characterized by a low-grade chronic, unilateral, nongranulomatous uveitis of unknown etiology. Recent studies suggest that rubella may be a possible cause of FHI. The uveitis in patients with FHI usually presents with fine stellate endothelial KPs, iris atrophy with or without heterochromia, abnormal angle vessels, and without posterior synechiae, and patients are usually asymptomatic. The most severe sequelae include glaucoma and cataracts.54,55
Multiple sclerosis (MS) is an autoimmune demyelinating disease of the central nervous system. Although the most notable ocular manifestation of this disease is optic neuritis (40%–73% of patients with MS will develop optic neuritis), it has been reported that 0.4%–26.9% of patients with MS will develop uveitis.56–58 It is most common for patients with MS to present with an intermediate uveitis, pars planitis with or without granulomatous anterior uveitis with mutton-fat KPs (although rare to have anterior uveitis is this condition).
Infectious causes
Bacterial/spirochete
Cat-scratch disease (Bartonella) occurs in immunocompetent individuals of all ages worldwide. It is the leading cause of regional lymphadenopathy in children and young adults. Patients will present with tender, swollen lymph nodes at or near the site of the bite or scratch. It is the most common cause of neuroretinitis. Other forms of ocular inflammation include intermediate uveitis, anterior uveitis, conjunctivitis, retinal vasculitis, and orbital abscess.59–61
Lyme disease (Borrelia) is a multisystem disorder caused by the spirochete Borrelia bergdorferi infection and its sequelae and is transmitted via tick bites. In the USA, cases are highly concentrated in the northeast, mid-Atlantic, and upper midwest regions.62,63 There are three stages of Lyme disease: the early stage, disseminated stage, and persistent stage. In the early stage of the disease, 60%–80% of patients present with the classic “Bull’s eye” red macular rash at the site of the tick bite 2–28 days after the bite.64 Fever, malaise, fatigue, arthralgias, and myalgias often accompany the rash. The disseminated stage occurs several weeks after the initial exposure, and the patients may develop skin, nervous system, joint, heart, and eye problems. The skin, nervous system, and joints are affected at the persistent stage of this disease. Late neurologic involvement manifests as subacute or chronic encephalopathy, with subtle memory and cognitive dysfunction, progressive encephalomyelitis with white matter lesions, and peripheral neuropathy. Uveitis may be present at the disseminated and persistent stages of the disease; anterior uveitis, intermediate uveitis, posterior uveitis, neuroretinitis, retinal vasculitis, choroiditis, and panuveitis have all been reported.65–69
Syphilis is a sexually transmitted infection of the spirochete bacterium, Treponema pallidum. It has the potential to cause morbidity to any of the major organs of the body and can reveal itself at any time, lending to its name as the “Great Masquerader”. If untreated, it will progress through four stages.70–72 With primary syphilis, a chancre at the inoculation site will be present from 2 to 6 weeks after infection. Uveitis does not typically present at this stage. Secondary syphilis occurs 4–10 weeks after infection. A disseminated maculopapular rash on the palms of hands and soles of feet and lymphadenopathy are the most common clinical manifestations; however, fever, malaise, headache, nausea, anorexia, hepatitis, and meningitis can occur. Approximately 10% of cases with secondary syphilis present with uveitis.70–72 Patients with latent syphilis have no evidence of systemic disease. The patient is not contagious, and the disease is not detectable. One-third of patients will progress to tertiary syphilis, which is the most common stage in syphilis to present with uveitis.73–75 Once a patient has tertiary syphilis, he/she may have cardiovascular syphilis, neurosyphilis, or benign tertiary syphilis. Uveitis is often present in patients with tertiary syphilis. Anterior uveitis in patients with syphilis may present as unilateral or bilateral, granulomatous, or nongranulomatous with or without iris nodules, dilated iris vessels, and iris atrophy. Uveitis may manifest during any stage of syphilis. Patients with syphilis may present with posterior uveitis as diffuse or focal chorioretinitis, neuroretinitis, necrotizing retinitis, retinal vasculitis, intermediate uveitis, or panuveitis.73–75
TB is an airborne infectious disease caused by Mycobacterium tuberculosis. It commonly affects the lungs. The classic presentation of an active TB infection includes chronic cough, fever, night sweats, and weight loss. Granulomas may be noted on the iris, angle, or choroid. Uveitis may present in active TB and in patients with or without systemic TB symptoms.76 Uveitis is the most common ocular manifestation of TB. The most common uveitis seen in TB is disseminated chorioretinitis, but it can also present as acute anterior uveitis, chronic granulomatous anterior uveitis, intermediate uveitis, vitritis, or endophthalmitis. Broad-based posterior synechiae and retinal vasculitis are common with the uveitis presentation.77–81
Viral
Viral infections are the most common infectious underlying etiology of anterior uveitis. Increased IOP, iris atrophy, and unilateral presentations are common with viral etiologies. Herpes simplex 1 and 2 are ubiquitous in humans and acquired via direct contact with active infection. In most patients, this virus remains latent in neural ganglia, most notably the trigeminal ganglia. Virus reactivation can be induced by stress, UV exposure, and illness or found in immunocompromised individuals. Virus reactivation usually results in fever and malaise with eruption of skin vesicles along the neural dermatome. Anterior uveitis that occurs with HSV is usually unilateral, granulomatous, or nongranulomatous, with elevated IOP due to concomitant trabeculitis. It may also present with intermediate uveitis, posterior uveitis, or panuveitis.82–84 With intermediate and posterior involvement, uveitis treatment may include oral corticosteroids, oral or intravenous antivirals, and/or glaucoma medications.
Varicella zoster virus (VZV), human herpesvirus 3, primary infection is termed “chicken pox”, although with the Varivax vaccine (Merck & Co, Inc., Whitehouse Station, NJ, USA) the incidence of chicken pox in children has significantly decreased since the mid-1990s. VZV remains dormant in neural ganglia and, when reactivated, causes herpes zoster (HZ). HZ typically manifests as unilateral pain in a dermatomal distribution accompanied by a maculopapular vesicular rash. When reactivated along the trigeminal nerve, it is termed herpes zoster ophthalmicus.84–88 Uveitis occurs in ~20% of herpes zoster ophthalmicus patients and usually occurs 1–3 weeks after the initial presentation.25,28 Less common findings of HSV and VZV include retinitis, multifocal choroiditis, acute retinal necrosis, and progressive outer retinal necrosis.88 Epstein–Barr virus, human herpesvirus 4, is ubiquitous in humans and is the cause of infectious mononucleosis (IM). It is also associated with certain cancers, including Hodgkin’s lymphoma, Burkitt’s lymphoma, and nasopharyngeal carcinoma, and some that are associated with human immunodeficiency virus (HIV), including hairy leukoplakia and central nervous system lymphomas. It has also been linked to a higher risk of autoimmune diseases. Uveitis is generally found during primary infection during IM.89,90 Cytomegalovirus (CMV), human herpesvirus 5, is ubiquitous in humans and can cause major morbidity and mortality in immunocompromised patients. The most common manifestation is CMV retinitis, now rarely seen in patients with HIV/AIDS with the now frequent use of highly active antiretroviral therapy. In rare instances, immunocompetent individuals may present with unilateral, hypertensive, chronic, or recurrent uveitis due to CMV. The uveitis that presents with CMV is clinically identical to that which is seen with HSV and VZV. Anterior uveitis can present as unilateral, hypertensive, granulomatous, or nongranulomatous and that can be chronic or recurrent.91–95
HIV is a retrovirus that infects CD4+ T-lymphocytes, a part of cell-mediated immunity. Most opportunistic infections occur when the CD4+ count is <200 cells/μL, with the most severe infections occurring when <50 cells/μL, including but not limited to CMV retinitis, VZV retinitis, presumed ocular histoplasmosis syndrome, syphilis, toxoplasmosis, and intraocular lymphoma.96,97 Along with these opportunistic infections that have previously been covered, patients can develop immune recovery uveitis and iatrogenic uveitis. Immune recovery uveitis typically occurs in patients who have previously had CMV retinitis 2–16 weeks after bringing CD4+ counts >100 cells/μL with highly active antiretroviral therapy. The uveitis presents as increased anterior chamber reaction with vitreous haze, causing a reduction in visual acuity.78,79 Few of these patients develop a panuveitis and posterior segment sequelae. Iatrogenic uveitis was first described with the use of rifabutin, which typically manifests as a panuveitis, occasionally with hypopyon, cystoid macular edema, and retinal vasculitis. With topical steroids and discontinuation of the drug, this uveitis typically resolves within days. Intravitreal or intravenous cidofovir can also induce anterior uveitis, occasionally with hypotony and occasionally with an associated vitritis.75,93,98–100
Fungal
Presumed ocular histoplasmosis syndrome is classically defined by the triad of peripapillary atrophy, retinal chorioretinal atrophy “punched out histo spots”, and maculopathy without vitritis. There is a high incidence along the Ohio–Mississippi river valley within the USA.101–103
Parasitic
Toxoplasmosis is caused by the parasite Toxoplasma gondii and is the leading cause of posterior uveitis worldwide.104 It is spread through eating undercooked meat or from exposure to cat feces. Classically, toxoplasmosis presents as a yellow-white or gray exudative chorioretinal lesion with ill-defined borders in the retina with a marked vitritis. This is known as “headlight-in-the-fog”. Anterior uveitis may also be present, usually with a granulomatous response, including mutton-fat KPs, posterior synechiae, iris nodules, and fibrin deposition.105–107
Toxocariasis is an infectious disease caused by the larvae of the parasites Toxocara canis or Toxocara cati that are present in the small intestine of dogs and cats, respectively. The larvae reach the eye via the bloodstream and deposit, most commonly, in the retina. This results in a posterior granulomatous response. The posterior granuloma is a hazy, white elevated lesion with vitritis. Chronic endophthalmitis with associated retinal detachment, low-grade anterior uveitis, posterior synechiae, and hypopyon may also be evident. Papillitis, macular edema, and vitreous exudates are also associated findings. If present in a child, the chief complaint may not be reduced visual acuity, but leukocoria.108–111
Masquerade and drug-induced uveitis
Lymphoma is an underlying etiology of anterior uveitis in the masquerade category, which is common in elderly patients with chronic anterior uveitis cases. The list is exhaustive of medications that can cause uveitis. Some of the more common medications are discussed. Drug-induced uveitis is almost always reversible within weeks when the medication is discontinued and topical treatment is initiated. Rifabutin has been shown to cause panuveitis. Cidofovir can also induce anterior uveitis, occasionally with hypotony and with an associated vitritis.94,112 Bisphosphonates (ie, zoledronate and pamidronate) inhibit bone resorption that are used in the management of osteoporosis, multiple myeloma, osteogenesis imperfecta, Paget’s disease of the bone, and other disorders of fragility of bones. The drugs in this class can cause nongranulomatous anterior uveitis, usually occurring in the first 24–48 hours.113 Tumor necrosis factor-alpha inhibitors (ie, etanercept and adalimumab) are used in autoimmune and immune-mediated disorders such as rheumatoid arthritis, ankylosing spondylitis, IBD, and psoriasis. Our patients who are at risk of developing immune-mediated uveitis can be on medications that can induce uveitis. The uveitis is generally nongranulomatous anterior uveitis.114
Laboratory testing
Ordering the appropriate laboratory testing is imperative not only to treat uveitis itself but also to ensure that the patient is receiving the appropriate systemic care for an underlying disorder that they may, or may not, know they have as well as addressing public health concerns. It is now a standard of care to order/refer for the appropriate laboratory testing when there is suspicion for an underlying systemic condition. Most practitioners discourage a “shotgun” approach to laboratory testing, as there can be a struggle to find meaning in a mix of results that can be misleading. Table 2 shows the complete laboratory reference.
 | Table 2 Review of systems and appropriate laboratory testing |
As up to 50% of acute unilateral, anterior nongranulomatous uveitis is idiopathic, the majority of cases do not require an extensive workup. Laboratory testing should be considered when the uveitis is recurrent, chronic, bilateral, granulomatous, nonresponsive to treatment, intermediate, posterior, or with children younger than 15 years old.3,4,10,11 A basic blood workup for an anterior uveitis includes an HLA-B27, rapid plasma regain, fluorescent treponemal antibody absorption, Lyme Ab, and chest X-ray depending on geographic location. For intermediate uveitis, a magnetic resonance imaging may be necessary if the patient has neurologic signs or symptoms to rule out MS. For posterior or panuveitis, a HLA-B27 is not necessary.3,4,10,11 If the uveitis appears on examination to be infectious (toxoplasmosis or toxocariasis), the appropriate antibody should be obtained. Table 3 outlines the appropriate testing to be ordered once a thorough review of systems and a thorough ophthalmologic examination is complete. Obtaining a rheumatoid factor in anyone or an antinuclear antibody in adult is usually unnecessary. The exception might be an antinuclear antibody in a child with anterior uveitis and concern for juvenile rheumatoid arthritis. Note that rheumatoid arthritis and SLE are typically associated with scleritis, not uveitis.3,4,10,11
 | Table 3 Blood work |
Treatment
When determining the management approach for a patient with anterior uveitis, the practitioner should attempt to determine the etiology of the inflammation to establish both short-term and long-term control of the inflammation. The first step at uncovering an etiology is to discuss with the patient a thorough review of systems. The second step is to determine whether the anterior uveitis is acute, recurrent, or chronic, which is often established during case history.3,4,9–14 There are several examination findings that may suggest that a patient had a prior episode of iritis, including remnants of KPs, pigment clumps on the anterior lens capsule, posterior synechiae, and anterior synechiae.3,4 The presence of band keratopathy and loss of contour to the iris face may suggest that the presenting uveitis is more chronic in nature.3,4 The next step is to determine whether the uveitis is unilateral or bilateral, which can usually be determined based on history and through examination. However, this may be difficult when a patient presents with an acute uveitis in one eye and has a history and/or examination findings consistent with a previous episode in the fellow eye. The fourth step is to determine whether the anterior uveitis is granulomatous or nongranulomatous, which is determined through examination.3,4 The presence of KPs or iris nodules would indicate that the uveitis is granulomatous.3,4,9–14 Once an etiology has been established, appropriate treatment may commence.
Initial treatment of anterior uveitis involves topical corticosteroids. The most common topical corticosteroid prescribed for the treatment of anterior uveitis is prednisolone acetate 1%, followed by dexamethasone 0.1% and prednisolone sodium phosphate 1%.3,4,116–118 When a patient presents with an acute anterior uveitis, the eye care practitioner should prescribe corticosteroids hourly while the patient is awake for at least 1 week in duration.3,4,115–118 Difluprednate 0.05% emulsion is a difluorinated prednisolone derivative that may be dosed four times daily and has been found to be similar in efficacy as compared to prednisolone acetate 1% when dosed eight times daily.115–118 (Table 4 shows commercially available topical corticosteroids).
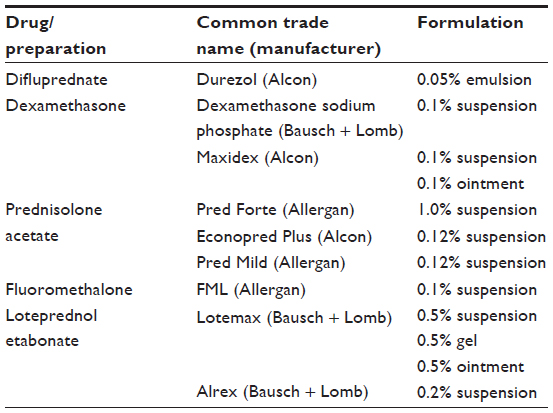 | Table 4 Most commonly prescribed topical ophthalmic corticosteroids |
Patients are typically scheduled for a follow-up after 1 week of treatment with corticosteroids. During this visit and each subsequent visit, the number of anterior chamber cells/high-power field (HPF) should be half of what it was at the initial examination.3,4,116 If the number of cells is not decreasing, then the primary eye care practitioner should suspect one of two things: the patient is either not using the drops correctly or an infectious etiology should be suspected, such as herpes. It is also important that the patient properly shake the steroid suspension as failure to do so may result in a subtherapuetic response.3,4,64–66 The patient should continue hourly instillation of topical corticosteroids with weekly follow-up visits until there are five or fewer cells/HPF. Once five or fewer cells/HPF are present in the anterior chamber, the frequency of drop instillation should be reduced to every 2 hours.3,4,116–118 At each subsequent visit, the inflammation should continue to improve. Once the inflammation is reduced and is showing signs of continual improvement, the follow-up examination schedule can be lengthened to every 2 weeks. The proper tapering schedule of topical corticosteroids is one drop every 2 hours for 2 weeks, one drop four times per day for 2 weeks, one drop three times per day for 2 weeks, one drop two times per day for 2 weeks, one drop one time per day for 2 weeks, and then topical therapy should be discontinued.3,4 If the disease flares at any time during the follow-up process or the inflammation begins to increase, the primary eye care practitioner should consider either a slower taper or a referral for periocular corticosteroid injections.116–118 Cycloplegia, typically homatropine 5%, is also commonly prescribed one drop twice daily until there are fewer than five cells/HPF in the anterior chamber and then it is discontinued.3,4,116–118 Cycloplegia helps to lessen the pain a patient may experience that results from increased ciliary spasm. Cycloplegia also reduces the risk of posterior synechia development.3,4,116–118
In certain cases, additional therapies may be needed in conjunction with topical corticosteroid and cycloplegic therapies. In all cases of anterior uveitis, it is imperative to determine whether the inflammation also includes posterior involvement, and for this reason, all patients who present with an anterior uveitis should have a dilated fundus examination. Patients who are HLA-B27 positive may have a hypopyon present along with their inflammation and often need many weeks of hourly corticosteroids before a taper is started. In addition to hourly topical corticosteroids, these patients may also be prescribed oral corticosteroids, which is equivalent to prednisone 1 mg/kg/d for the first 7 days or periocular injections (dexamethasone 2 mg, 0.5 mL).3,4,116–118 Patients who present with a hypopyon need to have the steroids tapered by one increment each month as it can recur rapidly without the slow taper.3,4,115–118 Patients with an iritis secondary to herpes (often have transillumination defects) should have an oral antiviral added to their therapy. Suspicion of herpetic etiology – especially elevated IOP in the affected eye warrants prophylactic treatment with oral antivirals. Acyclovir 400 mg (800 mg for zoster) five times per day, valacyclovir 500 mg (1,000 mg for zoster) three times per day, or famciclovir 500 mg three times per day are all acceptable dosages to effectively treat patients with active herpes infections and ocular inflammation.82–88 Antiviral coverage can be reduced to two times per day for acyclovir or one time per day for valacyclovir or famciclovir once the ocular inflammation is showing signs of reduction and once the patient’s corticosteroid therapy has been tapered to one drop, three times per day.82–88 Long-term control can be extremely difficult in patients with anterior uveitis. The goal of long-term therapy is to keep the eye free of inflammation, which is the most challenging aspect in managing uveitis. In many cases of anterior uveitis, long-term therapy is not necessary, and the inflammatory process will not recur. However, in some instances, the iritis may recur during the initial corticosteroid taper or within 3 months of stopping it.3,4,115–118 Corticosteroids are an acceptable option of maintaining long-term control of the inflammation if the dose required is low (three times per day or lower) and likely to be well tolerated, and the patient’s iritis is not believed to be associated with a systemic disease.3,4,116–118 IOP in corticosteroid responders should be identified and closely monitored.3,4,116–118 Immunomodulatory therapy may be used for the long-term control of uveitis when patients cannot tolerate corticosteroids either because of increased IOP or early cataract formation or in the setting of systemic disease.3,4,116–118
Conclusion
When a patient presents clinically with an acute or chronic presentation of anterior uveitis, the primary eye care practitioner is responsible for completing a thorough examination to determine an appropriate etiology for the diagnosis. By understanding the diagnosis and underlying etiology to the best of one’s ability allows for the practitioner to effectively manage the disease in a targeted manner versus solely treating the presenting clinical signs and symptoms. Making an appropriate diagnosis of anterior uveitis is based on a complete review of systems, an understanding of systemic diseases, and laboratory testing. Laboratory testing should be tailored to a specific diagnosis or diagnoses rather than ordering a general battery of testing. Once a list of diagnoses has been made, a targeted approach to treatment can be pursued, or the decision may be made to refer to our ophthalmology colleagues. Treatment should be customized to each patient as suboptimal treatment may lead to complications and loss of vision.
Disclosure
The authors report no conflicts of interest in this work.
References
Snell RS, Lemp MA. The eyeball. In: Snell RS, Lemp MA, editors. Clinical Anatomy of the Eye. 2nd ed. Malden, MA: Blackwell Science; 1998:140–156. | |
Rao NA, Forster DJ. Basic principles. In: Podos SM, Yanoff M, editors. The Uvea, Uveitis, and Intraocular Neoplasms. Vol 2. New York: Gower Medical Publications; 1992:1–17. | |
Huang JJ, Gau PA. Ocular Inflammatory Disease and Uveitis Manual. Diagnosis and Treatment. Philadelphia: Lippincott Williams & Wilkins; 2010:1–9. | |
Foster CS, Vitale AT. Diagnosis and Treatment of Uveitis. 2nd ed. New Delhi: Jaypee Brothers Medical Publishers Ltd; 2013:20–32. | |
Smit RLMJ, Baarsman GS, DeVries J. Classification of 750 consecutive uveitis patients in the Rotterdam Eye Hospital. Int Ophthalmol. 1993;17:71–75. | |
Silverstein A. Changing trends in the etiological diagnosis of uveitis. Documenta Ophthalmologic. 1997;94:25–37. | |
Roday MJH, Stilma JS, Rothova A. Blindness from uveitis in a hospital population in Sierra Leone. Br J Ophthalmol. 1994;9:690–693. | |
Rothova A, Suttorp-van Schulten MSA, Treffers WF, et al. Causes and frequency of blindness in patients with intraocular inflammatory diseases. Br J Ophthalmol. 1992;70:137–141. | |
Whitley W, Sheppard J. The basics of uveitis. Rev Optom. 2011. Available at: http://www.reviewofoptometry.com/continuing_education/tabviewtest/lessonid/107773/. | |
Smith RE, Nozik RM. Uveitis: A Clinical Approach to Diagnosis and Management. Baltimore: Williams & Wilkins; 1983. | |
Gutteridge IF, Hall AJ. Acute anterior uveitis in primary care. Clin Exp Optom. 2007;90(2):70–82. | |
Jabs DA, Nussenblatt RB, Rosenbaum JT. Standardization of uveitis nomenclature (SUN) for reporting clinical data – results of the first international workshop. Am J Opthalmol. 2005;150:509–516. | |
Agrawal RV, Murthy S, Sangwan V, Biswas J. Current approach in diagnosis and management of anterior uveitis. Indian J Ophthalmol. 2010;58(1):11–19. | |
Yanoff M, Duker JS. Opthalmology. 2nd ed. St Louis: Mosby; 2004. | |
Dohm K. Practical pearls for managing anterior uveitis. Rev Optom. 2015. Available at: http://www.reviewofoptometry.com/content/d/systemic_disease/c/52474/. | |
Kabat AG. Uveitis. In: Bartlett J, Jannus S, editors. Clinical Ocular Pharmacology. 3rd ed. St Louis: Elsevier; 2008:587. | |
Herbort CP. Appraisal, workup and diagnosis of anterior uveitis: a practical approach. Middle East Afr J Ophthalmol. 2009;16(4):159–167. | |
Hogan MJ, Kimura SJ, Thygeson P. Signs and symptoms of uveitis. Am J Ophthalmol. 1959;47:155–170. | |
Gritz DC, Wong IG. Incidence and prevalence of uveitis in Northern California; the Northern California Epidemiology of Uveitis Study. Ophthalmology. 2004;111:491–500. | |
Rosenbaum JT. Acute anterior uveitis and spondyloarthropathies. Rheum Dis Clin North Am. 1992;18:143–151. | |
Rosenbaum JT. Nibbling away at the diagnosis of idiopathic uveitis. JAMA Ophthalmol. 2015;133:146–147. | |
Rosenbaum JT. Systemic associations of anterior uveitis. Int Ophthalmol Clin. 1991;31:131–142. | |
Rudwaleit M, van der Heijde D, Landewe R, et al. The Assessment of SpondyloArthritis international Society classification criteria for peripheral spondyloarthritis and for spondyloarthritis in general. Ann Rheum Dis. 2011;70:25–31. | |
Rudwaleit M, Landewé R, van der Heijde D, et al. The development of Assessment of SpondyloArthritis international Society classification criteria for axial spondyloarthritis (part I): classification of paper patients by expert opinion including uncertainty appraisal. Ann Rheum Dis. 2009;68:770–776. | |
Rudwaleit M, van der Heijde D, Landewé R, et al. The development of Assessment of SpondyloArthritis international Society classification criteria for axial spondyloarthritis (part II): validation and final selection. Ann Rheum Dis. 2009;68:777–783. | |
Rosenbaum JT. Characterization of uveitis associated with spondyloarthritis. J Rheumatol. 1989;16:792–796. | |
Rosenbaum JT. Uveitis in spondyloarthritis including psoriatic arthritis, ankylosing spondylitis, and inflammatory bowel disease. Clin Rheumatol. 2015;34:999–1002. | |
Arnett FC. The seronegative spondyloarthropathies. Curr Opin Rheumatol. 1992;4:460–462. | |
Lee DA, Barker SM, Su WP, Allen GL, Liesegang TJ, Ilstrup DM. The clinical diagnosis of Reiter’s syndrome. Ophthalmic and nonophthalmic aspects. Ophthalmology. 1986;93:350–356. | |
Gerber LH, Murray CL, Perlman SG, et al. Human lymphocyte antigens characterizing psoriatic arthritis and its subtypes. J Rheumatol. 1982;9:703–707. | |
Taylor W, Gladman D, Helliwell P, Marchesoni A, Mease P, Mielants H. Classification criteria for psoriatic arthritis: development of new criteria from a large international study. Arthritis Rheum. 2006;54:2665–2673. | |
Taylor WJ, Robinson PC. Classification criteria: peripheral spondyloarthropathy and psoriatic arthritis. Curr Rheumatol Rep. 2013;15:317. | |
Palumbo PJ, Ward LE, Sauer WG, Scudamore HH. Musculoskeletal manifestations of inflammatory bowel disease. Ulcerative and granulomatous colitis and ulcerative proctitis. Mayo Clin Proc. 1973;48:411–416. | |
Lyons JL, Rosenbaum JT. Uveitis associated with inflammatory bowel disease compared with uveitis associated with spondyloarthropathy. Arch Ophthalmol. 1997;115:61–64. | |
Hopkins DJ, Horan E, Burton IL, Clamp SE, de Dombal FT, Goligher JC. Ocular disorders in a series of 332 patients with Crohn’s disease. Br J Ophthalmol. 1974;58:732–737. | |
Hofer M, Southwood TR. Classification of childhood arthritis. Best Pract Res Clin Rheumatol. 2002;16:379–396. | |
Cassidy J, Kivlin J, Lindsley C, James Nocton. Ophthalmic examinations in children with juvenile rheumatoid arthritis. Pediatrics. 2006;117(5):1843–1845. | |
Sabri K, Saurenmann RK, Silverman ED, Levin AV. Course, complications, and outcome of juvenile arthritis-related uveitis. J AAPOS. 2008;12:539–545. | |
Burgos-Vargas R, Pacheco-Tena C, Vazquez-Mellado J. The juvenile-onset spondyloarthritides: rationale for clinical evaluation. Best Pract Res Clin Rheumatol. 2002;16:551–572. | |
Birnbaum AD, Oh FS, Chakrabarti A, Tessler HH, Goldstein DA. Clinical features and diagnostic evaluation of biopsy-proven ocular sarcoidosis. Arch Ophthalmol. 2011;129:409–413. | |
Jabs DA, Johns CJ. Ocular involvement in chronic sarcoidosis. Am J Ophthalmol. 1986;102:297–301. | |
Karma A, Huhti E, Poukkula A. Course and outcome of ocular sarcoidosis. Am J Ophthalmol. 1988;106:467–472. | |
Crick RP, Hoyle C, Smellie H. The eyes in sarcoidosis. Br J Ophthalmol. 1961;45:461–481. | |
Gold DH, Morris DA, Henkind P. Ocular findings in systemic lupus erythematosus. Br J Ophthalmol. 1972;56:800–804. | |
Silpa-Archa S, Lee JJ, Foster CS. Ocular manifestations in systemic lupus erythematosus. Br J Ophthalmol. 2015;46:1757–1762. | |
Sivaraj RR, Durrani OM, Denniston AK, Murray PI, Gordon C. Ocular manifestations of systemic lupus erythematosus. Rheumatology (Oxford). 2007;46:1757–1762. | |
Ando K, Fujino Y, Hijikata K, Izawa Y, Masuda K. Epidemiological features and visual prognosis of Behcet’s disease. Jpn J Ophthalmol. 1999;43:312–317. | |
Nussenblatt RB. Uveitis in Behcet’s disease. Int Rev Immunol. 1997;14:67–79. | |
Mishima S, Masuda K, Izawa Y, Mochizuki M, Namba K. The eighth Frederick H. Verhoeff Lecture. presented by Saiichi Mishima, MD Behcet’s disease in Japan: ophthalmologic aspects. Trans Am Ophthalmol Soc. 1979;77:225–279. | |
Baer JC, Raizman MB, Foster CS. Ocular Behçet’s disease in the United States. Clinical presentation and visual outcome in 29 patients. In: Masahiko U, Shigeaki O, Koki A, editors. Proceedings of the Fifth International Symposium on the Immunology and Immunopathology of the Eye. Tokyo, New York: Elsevier Science; 1990:383. | |
Beniz J, Forster DJ, Lean JS, Smith RE, Rao NA. Variations in clinical features of the Vogt-Koyanagi-Harada syndrome. Retina. 1991;11:275–280. | |
Kimura R, Sakai M, Okabe H. Transient shallow anterior chamber as initial symptom in Harada’s syndrome. Arch Ophthalmol. 1981;99:1604–1606. | |
Kimura R, Kasai M, Shoji K, Kanno C. Swollen ciliary processes as an initial symptom in Vogt-Koyanagi-Harada syndrome. Am J Ophthalmol. 1983;95:402–403. | |
Jones NP. Fuchs’ heterochromic uveitis: a reappraisal of the clinical spectrum. Eye (Lond). 1991;5(pt 6):649–661. | |
Fearnley IR, Rosenthal AR. Fuchs’ heterochromic iridocyclitis revisited. Acta Ophthalmol Scand. 1995;73:166–170. | |
Nussenblatt RB, Whitcup SM, Palestine AG. Intermediate Uveitis. Uveitis: Fundamentals and Clinical Practice. 2nd ed. St Louis: Mosby; 1996:58–68. | |
Bamford CR, Ganley JP, Sibley WA, Laguna JF. Uveitis, perivenous sheathing and multiple sclerosis. Neurology. 1978;28:119–124. | |
Lim JI, Tessler HH, Goodwin JA. Anterior granulomatous uveitis in patients with multiple sclerosis. Ophthalmology. 1991;98:142–145. | |
Solley WA, Martin DF, Newman NJ, et al. Cat scratch disease: posterior segment manifestations. Ophthalmology. 1999;106:1546–1553. | |
Ormerod LD, Dailey JP. Ocular manifestations of cat-scratch disease. Curr Opin Ophthalmol. 1999;10:209–216. | |
Soheilian M, Markomichelakis N, Foster CS. Intermediate uveitis and retinal vasculitis as manifestations of cat scratch disease. Am J Ophthalmol. 1996;122:582–584. | |
Centers for Disease Control and Prevention. Reported Lyme Disease cases by State. Atlanta, GA: CDC; 2013. | |
Campbell GL, Paul WS, Schriefer ME, Craven RB, Robbins KE, Dennis DT. Epidemiologic and diagnostic studies of patients with suspected early Lyme disease, Missouri, 1990–1993. J Infect Dis. 1995;172:470–480. | |
Guidelines for laboratory evaluation in the diagnosis of Lyme disease. American College of Physicians. Ann Intern Med. 1997;127:1106–1108. | |
Isogai E, Isogai H, Kotake S, et al. Detection of antibodies against Borrelia burgdorferi in patients with uveitis. Am J Ophthalmol. 1991;112:23–30. | |
Karma A, Viljanen M, Pirttila T, Vesti E, Stenborg T, Tervo T. [Ocular Lyme borreliosis]. Duodecim. 1993;109:35–42. Finnish. | |
Karma A, Seppala I, Mikkila H, Kaakkola S, Viljanen M, Tarkkanen A. Diagnosis and clinical characteristics of ocular Lyme borreliosis. Am J Ophthalmol. 1995;119:127–135. | |
Mikkila H, Karma A, Viljanen M, Seppala I. The laboratory diagnosis of ocular Lyme borreliosis. Graefes Arch Clin Exp Ophthalmol. 1999;237:225–230. | |
Flach AJ, Lavoie PE. Episcleritis, conjunctivitis, and keratitis as ocular manifestations of Lyme disease. Ophthalmology. 1990;97:973–975. | |
Barile GR, Flynn TE. Syphilis exposure in patients with uveitis. Ophthalmology. 1997;104:1605–1609. | |
Margo CE, Hamed LM. Ocular syphilis. Surv Ophthalmol. 1992;37:203–220. | |
Deschenes J, Seamone CD, Baines MG. Acquired ocular syphilis: diagnosis and treatment. Ann Ophthalmol. 1992;24:134–138. | |
Tamesis RR, Foster CS. Ocular syphilis. Ophthalmology. 1990;97:1281–1287. | |
Mendelsohn AD, Jampol LM. Syphilitic retinitis. A cause of necrotizing retinitis. Retina. 1984;4:221–224. | |
McLeish WM, Pulido JS, Holland S, Culbertson WW, Winward K. The ocular manifestations of syphilis in the human immunodeficiency virus type 1-infected host. Ophthalmology. 1990;97:196–203. | |
Suzuki J, Oh I, Kezuka T, Sakai J, Goto H. Comparison of patients with ocular tuberculosis in the 1990s and the 2000s. Jpn J Ophthalmol. 2010;54:19–23. | |
Cutrufello NJ, Karakousis PC, Fishler J, Albini TA. Intraocular tuberculosis. Ocul Immunol Inflamm. 2010;18:281–291. | |
Gupta A, Bansal R, Gupta V, Sharma A, Bambery P. Ocular signs predictive of tubercular uveitis. Am J Ophthalmol. 2010;149:562–570. | |
Gupta A, Sharma A, Bansal R, Sharma K. Classification of intraocular tuberculosis. Ocul Immunol Inflamm. 2015;23:7–13. | |
Madge SN, Prabhakaran VC, Shome D, Kim U, Honavar S, Selva D. Orbital tuberculosis: a review of the literature. Orbit. 2008;27:267–277. | |
Rosen PH, Spalton DJ, Graham EM. Intraocular tuberculosis. Eye (Lond). 1990;4(pt 3):486–492. | |
North RD, Pavan-Langston D, Geary P. Herpes simplex virus types 1 and 2: therapeutic response to antiviral drugs. Arch Ophthalmol. 1976;94:1019–1021. | |
Pavan-Langston D, Grene B. Herpes simplex ocular disease. Compr Ther. 1984;10:30–36. | |
Pavan-Langston D. Herpes simplex and herpes zoster keratouveitis: diagnosis and management. Bull N Y Acad Med. 1977;53:731–748. | |
Pavan-Langston D. Varicella-zoster ophthalmicus. Int Ophthalmol Clin. 1975;15:171–185. | |
Pavan-Langston D. Ocular viral infections. Med Clin North Am. 1983;67:973–990. | |
Pavan-Langston D. Herpes zoster ophthalmicus. Neurology. 1995;45:S50–S51. | |
Womack LW, Liesegang TJ. Complications of herpes zoster ophthalmicus. Arch Ophthalmol. 1983;101:42–45. | |
Henle W, Henle G. Epstein-Barr virus and infectious mononucleosis. N Engl J Med. 1973;288:263–264. | |
Morishima N, Miyakawa S, Akazawa Y, Takagi S. A case of uveitis associated with chronic active Epstein-Barr virus infection. Ophthalmologica. 1996;210:186–188. | |
Atherton SS, Newell CK, Kanter MY, Cousins SW. T cell depletion increases susceptibility to murine cytomegalovirus retinitis. Invest Ophthalmol Vis Sci. 1992;33:3353–3360. | |
Dunn JP, Jabs DA. Cytomegalovirus retinitis in AIDS: natural history, diagnosis, and treatment. AIDS Clin Rev. 1995–1996:99–129. | |
Jabs DA, Enger C, Bartlett JG. Cytomegalovirus retinitis and acquired immunodeficiency syndrome. Arch Ophthalmol. 1989;107:75–80. | |
Kirsch LS, Arevalo JF, Chavez de la P, Munguia D, de Clercq E, Freeman WR. Intravitreal cidofovir (HPMPC) treatment of cytomegalovirus retinitis in patients with acquired immune deficiency syndrome. Ophthalmology. 1995;102:533–542. | |
Klemola E, Stenstrom R, von ER. Pneumonia as a clinical manifestation of cytomegalovirus infection in previously healthy adults. Scand J Infect Dis. 1972;4:7–10. | |
Karavellas MP, Azen SP, MacDonald JC, et al. Immune recovery vitritis and uveitis in AIDS: clinical predictors, sequelae, and treatment outcomes. Retina. 2001;21:1–9. | |
Schrier RD, Song MK, Smith IL, et al. Intraocular viral and immune pathogenesis of immune recovery uveitis in patients with healed cytomegalovirus retinitis. Retina. 2006;26:165–169. | |
Rosberger DF, Heinemann MH, Friedberg DN, Holland GN. Uveitis associated with human immunodeficiency virus infection. Am J Ophthalmol. 1998;125:301–305. | |
Rose-Nussbaumer J, Goldstein DA, Thorne JE, et al. Uveitis in human immunodeficiency virus-infected persons with CD4+ T-lymphocyte count over 200 cells/mL. Clin Experiment Ophthalmol. 2014;42:118–125. | |
Holland GN, Pepose JS, Pettit TH, Gottlieb MS, Yee RD, Foos RY. Acquired immune deficiency syndrome. Ocular manifestations. Ophthalmology. 1983;90:859–873. | |
Wong VG. Focal choroidopathy in experimental ocular histoplasmosis. Trans Am Ophthalmol Soc. 1972;70:615–630. | |
Walma D Jr, Schlaegel TF Jr. Presumed histoplasmic choroiditis. A clinical analysis of 43 cases. Am J Ophthalmol. 1964;57:107–110. | |
Watzke RC, Claussen RW. The long-term course of multifocal choroiditis (presumed ocular histoplasmosis). Am J Ophthalmol. 1981;91:750–760. | |
Holland GN. Ocular toxoplasmosis: a global reassessment. Part I: epidemiology and course of disease. Am J Ophthalmol. 2003;136:973–988. | |
Holland GN. Ocular toxoplasmosis: new directions for clinical investigation. Ocul Immunol Inflamm. 2000;8:1–7. | |
Commodaro AG, Belfort RN, Rizzo LV, et al. Ocular toxoplasmosis: an update and review of the literature. Mem Inst Oswaldo Cruz. 2009;104:345–350. | |
Novais EA, Commodaro AG, Santos F, et al. Patients with diffuse uveitis and inactive toxoplasmic retinitis lesions test PCR positive for Toxoplasma gondii in their vitreous and blood. Br J Ophthalmol. 2014;98:937–940. | |
Ashton N. Larval granulomatosis of the retina due to Toxocara. Br J Ophthalmol. 1960;44:129–148. | |
Wilkinson CP, Welch RB. Intraocular toxocara. Am J Ophthalmol. 1971;71:921–930. | |
Gillespie SH. Human toxocariasis. Commun Dis Rep CDR Rev. 1993;3:R140–R143. | |
Gillespie SH, Dinning WJ, Voller A, Crowcroft NS. The spectrum of ocular toxocariasis. Eye (Lond). 1993;7(pt 3):415–418. | |
Kirsch LS, Arevalo JF, De Clercq E, et al. Phase I/II study of intravitreal cidofovir for the treatment of cytomegalovirus retinitis in patients with the acquired immunodeficiency syndrome. Am J Ophthalmol. 1995;119:466–476. | |
Macarol V, Fraunfelder FT. Pamidronate disodium and possible ocular adverse drug reactions. Am J Ophthalmol. 1994;118:220–224. | |
Lim LL, Fraunfelder FW, Rosenbaum JT. Do tumor necrosis factor inhibitors cause uveitis? A registry-based study. Arthritis Rheum. 2007;56:3248–3252. | |
Korenfeld M. Difluprednate: changing the landscape of ocular pharmacology. Exp Rev Ophthalmol. 2008;3(6):619–625. | |
Foster CS, Davanzo R, Flynn TE, et al; for the Durezol Study Group. Durezol (difluprednate ophthalmic emulsion 0.05%) compared with PredForte 1% ophthalmic suspension in the treatment of endogenous anterior uveitis. J Ocul Pharmocol Ther. 2010;26(5):475–483. | |
Freeman WR, Green RL, Smith RE. Echographic localization of corticossteroids after periocular injection. Am J Ophthalmol. 1987;103:281–288. | |
Smith RE, Nozik RA. Uveitis: A Clinical Approach to Diagnosis and Management. Baltimore: Williams & Wilkins; 1989:51–76. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.