")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 16
Diagnosis and Screening of Patients with Hereditary Transthyretin Amyloidosis (hATTR): Current Strategies and Guidelines
Authors Benson MD, Dasgupta NR, Rao R
Received 7 May 2020
Accepted for publication 12 July 2020
Published 14 August 2020 Volume 2020:16 Pages 749—758
DOI https://doi.org/10.2147/TCRM.S185677
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
MD Benson,1 NR Dasgupta,1,2 R Rao2
1Department of Pathology & Laboratory Medicine, Indiana University School of Medicine, Indianapolis, IN, USA; 2Division of Cardiology, Indiana University School of Medicine, Indianapolis, IN 46040, USA
Correspondence: MD Benson 635 Barnhill Drive, A-128, Indianapolis, IN 46202, USA
Tel +1 317-278-3428
Fax +1 317-274-4304
Email [email protected]
Abstract: The outlook for transthyretin amyloidosis (ATTR) is changing with the availability of new and emerging treatments. ATTR now appears to be more common than previously thought and is no longer viewed as an obscure diagnosis with a grim prognosis. Now more than ever, there is growing emphasis on the need for early diagnosis because the treatments appear to be most effective if started in earlier stages of the disease. Diagnosing ATTR is a challenge as it may initially present with nonspecific symptoms and it is often thought of as a diagnosis of exclusion. Increased awareness is imperative as new treatments offer hope and have the potential to change the disease trajectory. ATTR commonly presents with neurological and cardiac features. Transthyretin (TTR) is a protein produced in the liver which misfolds either due to genetic mutations or due to aging and results in deposition of amyloid fibrils in organs and tissues. Apart from the traditional imaging modalities, newer techniques including echocardiographic strain imaging, magnetic resonance imaging (MRI), and nuclear scintigraphy, as well as the increased availability of genetic testing are aiding in making a timely diagnosis. In this review, we present the current understanding of the ATTR disease process, diagnostic and surveillance approaches, newer treatment modalities, and the future directions.
Keywords: transthyretin, amyloidosis, neuropathy, cardiomyopathy
Introduction
Transthyretin amyloidosis (ATTR) is a systemic disease with the potential to disrupt the function of practically every organ of the body. Traditionally, it has been characterized as affecting peripheral nerves, the gastrointestinal tract, and the heart but even the central nervous system can be affected as the result of vascular and leptomeningeal amyloid deposition. Timely and appropriate diagnosis can be relatively difficult because the initial presenting symptoms may be vague and initially attributed to other more common conditions. Delays in the diagnosis of ATTR can have serious consequences for patients who may develop severe end-organ impairment. The development of new treatments that appear to be most effective if started early makes it imperative for clinicians to recognize the constellation of signs and symptoms that may be clues to the diagnosis of transthyretin amyloidosis.
Amyloidosis is a disease that is confusing to many clinicians, because it does not indicate a single condition. Rather, amyloidosis is a generic term used to describe the deposition of misfolded proteins extracellularly in a beta-pleated sheet formation with resultant impairment of organ and tissue function. Over 30 different proteins have been demonstrated to form amyloid fibrils.1 The two most common forms of amyloidosis are immunoglobulin light chain amyloidosis (AL) and transthyretin amyloidosis (ATTR).2 The accepted nomenclature for the different forms of amyloidosis is “A” for amyloid followed by the abbreviation of the precursor protein that forms the amyloid fibrils.1 For example, in ATTR amyloidosis, transthyretin (TTR) is the precursor protein that forms amyloid fibrils and in AL the precursor protein is a monoclonal immunoglobulin light chain. The clinical course of each specific type of amyloidosis is determined by the amyloid-forming protein and the organ predilection. This review will focus on transthyretin amyloidosis, a form of amyloidosis that commonly attacks the heart and peripheral nerves.
Transthyretin is predominately made by the liver and serves as a transport protein for thyroid hormone and retinol/vitamin A. It is also synthesized in small amounts locally in the central nervous system choroid plexus and ocular retinal pigment epithelium.2 TTR in blood predominately exists as a tetramer; however, hereditary destabilizing mutations or aging can cause disassociation of the tetramer into monomers which may misfold and deposit as amyloid fibrils.2
Transthyretin amyloidosis can occur either due to a mutation in the transthyretin gene that is inherited in an autosomal dominant fashion with variable penetrance or due to age-related protein misfolding. The current nomenclature guidelines recommend that familial forms of ATTR, previously known as familial amyloid polyneuropathy (FAP) or familial amyloid cardiomyopathy (FAC), now be called hereditary ATTR (hATTR) and the description of the predominant features and mutation should also be included if known. For example, a hATTR patient with a mutation at position 30 could be described as having ATTRVal30Met with cardiomyopathy (CM), neuropathy, or mixed phenotype.2 The age-related form of amyloidosis primarily affects the heart and is called wild-type amyloidosis (ATTRwt, formally known as senile cardiac amyloidosis) to indicate that wild-type or normal transthyretin protein can form amyloid fibrils.
Over 100 different mutations in transthyretin have been described.2 Clinical manifestations often correlate with the specific point mutation.2 Certain mutations are traditionally thought to present clinically with a neurologic phenotype with progressive peripheral sensory-motor polyneuropathy while others present with restrictive heart failure.2 However, there may be substantial overlap of organ involvement and many mutations present with a mixed phenotype with features of both neuropathy and cardiomyopathy (Figure 1). Certain mutations are endemic in different areas of the world and ethnicities so it is important to understand a patient’s ancestral heritage. The most prevalent TTR mutation worldwide is the Val30Met mutation and ATTR classically presents with peripheral neuropathy.2 In the United States, the most common hereditary mutation is Val122Ile and organ involvement may be limited to the heart. The Val122Ile mutation in transthyretin is present in 3–4% of African Americans in the United States, but ATTR may have variable penetrance.2
 |
Figure 1 In hereditary transthyretin amyloidosis point mutations in the transthyretin gene can lead to a predominately neurologic or cardiac phenotype, however, there is often substantial overlap and a mixed phenotype with both neurologic and cardiac features is common. |
The age-related form of amyloidosis primarily affects the heart and is called wild-type amyloidosis and abbreviated ATTRwt (formally known as senile cardiac amyloidosis) to indicate that wild-type or normal transthyretin protein can form amyloid fibrils.2 ATTRwt is more common in men and individuals over age 60. The exact cause of ATTRwt and reasons for male predominance are unknown.
Diagnostic Approach
It is not unusual that patients have been evaluated by several physicians with no satisfactory explanation of their symptoms or concerns. The two most common presenting signs and symptoms of ATTR amyloidosis are peripheral neuropathy and/or congestive heart failure and therefore should be included in the differential diagnosis of both conditions. It is important for the clinician to look for the underlying cause of peripheral neuropathy or congestive heart failure to make a specific diagnosis before labeling the conditions as “idiopathic.” It is important to remember that CHF, atrial fibrillation, and peripheral neuropathy are not diagnoses, they are symptom complexes. They do not clinically “get to the heart” of the matter. Common misdiagnoses for ATTR polyneuropathy include chronic demyelinating polyneuropathy (CIDP), Charcot Marie Tooth, Guillain Barré, idiopathic axonal neuropathy, lumbar spinal stenosis, motor neuron disease, diabetic neuropathy, and alcoholic polyneuropathy.5 ATTR cardiomyopathy is often misdiagnosed as hypertensive heart disease, hypertrophic cardiomyopathy, or undifferentiated heart failure with preserved ejection fraction.3 A comprehensive history and physical, family history, laboratory studies, cardiac imaging, neurologic testing, genetic testing, and biopsies of affected organs are often needed to differentiate between these other conditions and confirm a diagnosis of ATTR amyloidosis.
Although the nerves and heart are most commonly affected in ATTR amyloidosis, other organ systems such as the central nervous system and gastrointestinal tract may also be impacted. Therefore, leptomeningeal ATTR amyloidosis should be included in the differential diagnosis of strokes, intracranial hemorrhage, and episodes of loss of consciousness. Additionally, gastrointestinal ATTR amyloidosis should be included in the differential diagnosis of early satiety, alternating constipation and diarrhea, unintentional weight loss which might be ascribed to Crohn’s disease, irritable bowel syndrome, and progressive systemic sclerosis.
Integrating the History, Physical, and Test Results: Red Flags for Transthyretin Amyloidosis
One sign or symptom alone is often not sufficient to make the diagnosis of ATTR amyloidosis unless there is a known hereditary mutation. It is often necessary to integrate many signs and symptoms that may raise the suspicion for ATTR amyloidosis. Red flags for transthyretin amyloidosis may include multisystem organ involvement, bilateral carpal tunnel syndrome, spinal stenosis, a discordance between the electrocardiogram (ECG) voltage and wall thickness seen on cardiac imaging, orthostatic hypotension, intolerance to standard heart failure medications such as angiotensin-receptor enzyme inhibitors (ACE inhibitors) and beta-blockers, paradoxical low flow/low gradient aortic stenosis, alternating diarrhea and constipation, and unexplained peripheral neuropathy (Figure 2).3,4
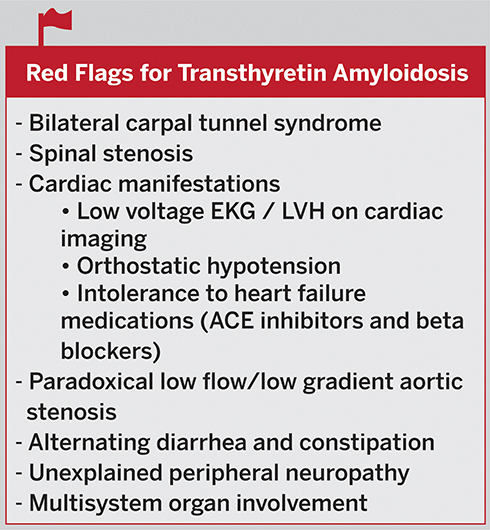 |
Figure 2 Red Flags for Transthyretin Amyloidosis. |
Imaging clues for cardiac involvement may include an apical sparing pattern on global longitudinal strain on echocardiography with an apical-to-basal strain ratio >2.1, diffuse subendocardial late gadolinium enhancement or increased extracellular volume on MRI, and increased uptake of the bone avid tracers technetium 99m technetium pyrophosphate (99mTc-PYP), 99mTc 3,3-disphonphone-1,2-propanodicarboxylic acid (99mTc-DPD), or 99m technetium hydroxymethylene diphosphate (99mTc99-HMDP) on nuclear bone scintigraphy.3,4
Neurologic clues may include carpal tunnel syndrome, progressive axonal polyneuropathy or dysautonomia. Amyloidosis should be included in the differential diagnosis of peripheral neuropathy associated with autonomic involvement, especially in patients without diabetes.5 Additionally, cardiomyopathy or neuropathy associated with symptoms of other organ involvement should raise suspicion for transthyretin amyloidosis.5
Transthyretin Amyloid Cardiomyopathy
Pathophysiology of Cardiac ATTR
Deposition of amyloid fibrils in the heart leads to an increase in global wall thickness which makes the heart less compliant and leads to diastolic heart dysfunction. Initially, the left ventricular ejection fraction (LVEF) is preserved despite a reduction in the LV cavity volume and effective stroke volume because the ratio of the left ventricular stroke volume to left ventricular end-diastolic volume (LVEF) remains normal. In more advanced disease the LVEF may become reduced. An LVEF < 50% portends an ingravescent clinical course. Amyloid fibrils are inert and do not conduct electricity very well so there may be a discordance between the left ventricular wall thickness on cardiac imaging and the ECG voltage. Amyloid fibrils deposit in the atria, ventricular myocardium, conduction system, and valves leading to atrial and ventricular arrhythmias, heart block, electromechanical dissociation, atrial thrombi, and valvular thickening.
Diagnosis of Cardiac ATTR
Cardiac amyloidosis is more common than previously thought and is likely underdiagnosed. A prospective study performed in 2015 that included 120 patients admitted to the hospital over age 60, with heart failure with preserved ejection fraction (EF > 50%) and a left ventricular wall thickness ≥12 mm found that 13% of patients had moderate to severe uptake of 99mTc-DPD.6 TTR genetic testing was performed in all patients with positive scans and was found to be negative in all cases suggesting a diagnosis of wild-type transthyretin amyloidosis. Additionally, in older patients with calcific aortic stenosis up to 15% of patients have wild-type ATTR amyloidosis and up to 30% of patients with low flow low gradient aortic stenosis may have ATTR amyloidosis.7
Left ventricular hypertrophy without a long history of hypertension may be a clue to an infiltrative cardiomyopathy. In patients with unexplained left ventricular hypertrophy (wall thickness >1.4 cm) on cardiac imaging, first see if there is a discordance between the ECG voltage and wall thickness on cardiac imaging.4 Then obtain a detailed history that focuses on any past history of bilateral carpal tunnel syndrome, vitreous opacities, spinal stenosis, neuropathy, diarrhea and constipation, erectile dysfunction, and a family history of heart failure. Do not dismiss the fact that a personal or family history of heart attack or sudden death may have been CHF or cardiac arrhythmias due to amyloidosis. The physical exam should focus on evaluation for vitreous opacities, clinical evidence of congestive heart failure, and sensory and motor neuropathy (deep tendon reflexes, vibration sense, light touch, muscle strength). You also want to evaluate for signs and symptoms of Immunoglobulin Light Chain amyloidosis (AL) which may present with periorbital purpura, macroglossia, congestive heart failure, hepatomegaly, and peripheral neuropathy.
It can sometimes be difficult to differentiate between the different types of amyloidosis based on the history and physical examination and imaging studies. The two most common forms of amyloidosis that can affect the heart are ATTR and AL (95% of cases), but there are less common forms that can affect the heart as well (ex. ApoA1, immunoglobulin heavy chain, fibrinogen alpha chain, and gelsolin). AL amyloidosis with cardiac involvement is considered a medical emergency because death may occur within 6 months if untreated and should be ruled out in all patients with suspected cardiac amyloidosis. The following laboratory tests should be obtained to evaluate for AL amyloidosis: serum free light chains, serum protein electrophoresis (SPEP) and urine protein electrophoresis (UPEP) with immunofixation, and a urinalysis to look for proteinuria. If the lab work reveals an abnormal ratio of serum kappa and lambda free light chains (less than 0.26 or greater than 1.65), a hematology consult and/or bone marrow biopsy may be required.3
Once AL amyloidosis has been ruled out in patients with findings concerning for cardiac amyloidosis, either a biopsy or nonbiopsy diagnosis of ATTR can be made. Diagnosis without biopsy can be made by obtaining a nuclear scintigraphy scan using 99mTc-PYP, 99mTc-DPD, or 99mTc-HMDP.3 These bone tracers are known to have high uptake in the heart with ATTR amyloidosis. 99mTC-PYP is the bone tracer that is most commonly used in the United States.3
If the nuclear scan is positive and shows more uptake of tracer in the heart than the bone, this would be suggestive of a diagnosis of ATTR amyloidosis as long as AL amyloidosis has been ruled out. Genetic testing should then be performed to evaluate for a genetic mutation in TTR. If a known pathologic mutation in TTR is found the presumptive diagnosis is hereditary ATTR. Alternatively, if the DNA analysis is normal and the patient has a positive nuclear scintigraphy scan, the diagnosis would be wild-type ATTR. Alternatively, cardiac biopsy can be performed to make the diagnosis of amyloidosis.3 The benefit of a biopsy diagnosis is that the amyloid can be specifically typed by mass spectrometry or immunohistochemistry to determine the specific amyloid-forming protein. If ATTR amyloidosis is confirmed on biopsy this should again be followed by genetic testing (Figure 3).
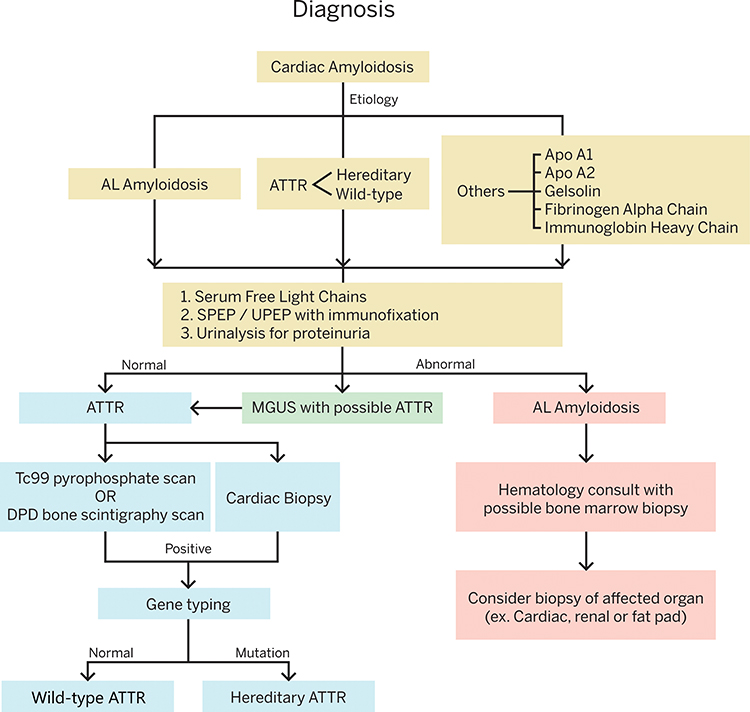 |
Figure 3 Diagnosis of Transthyretin Amyloid Cardiomyopathy. It is essential to first consider and rule out Immunoglobulin Light Chain Cardiac Amyloidosis, before doing more specific testing for transthyretin amyloidosis. Abbreviations: AL, immunoglobulin light chain amyloidosis; ATTR, transthyretin amyloidosis; Apo, apolipoprotein; SPEP, serum protein electrophoresis; UPEP, urine protein electrophoresis; Tc, technetium; MGUS, monoclonal gammopathy of unknown significance. |
Treatment of ATTR CM
Nonspecific treatment for ATTR-CM should focus on volume management with diuretics and control of arrhythmias. Standard therapy for heart failure with reduced ejection fraction such as angiotensin-converting enzymes, angiotensin-receptor blockers, beta-blockers, and angiotensin-receptor blockers-neprilysin inhibitors may not be well tolerated due to hypotension.2 Specific treatments for ATTR cardiomyopathy fall under the general categories of synthesis inhibitors of TTR protein by the liver either by liver transplantation or gene silencers, TTR tetramer stabilizers, and fibril disrupters (Figure 4).2
 |
Figure 4 Treatment for Transthyretin Amyloidosis. Abbreviations: siRNA, small interfering RNA; TUDCA, tauroursodeoxycholic acid. |
Since the majority of TTR is made by the liver, liver transplant can prevent the production of mutant TTR in patients with hereditary ATTR. Liver transplantation has been most effective in patients with the Val30Met mutation with early onset of disease that is limited to peripheral neuropathy. The FAPWTR registry reports there have been 2236 liver transplants for ATTR performed as of December 31, 2017. Important prognostic factors are modified body mass index, duration of the symptoms (<7 years), age (<50 years), absence of amyloid cardiomyopathy, and degree of autonomic involvement.8 Although liver transplantation can prevent the production of mutant TTR, once organs have been “seeded” with mutant TTR, normal or wild-type TTR may continue to form amyloid fibrils. Emerging new medical therapies may soon replace liver transplant and as such in the last few years there has been a decline in the reported number of transplant patients in the registry.
In ATTR cardiomyopathy the left ventricular cavity is small with thick walls causing restrictive cardiomyopathy with low cardiac output. As a result, mechanical circulatory support is not beneficial in end-stage congestive heart failure.9 Heart transplant may be considered in severe ATTR cardiomyopathy with limited extracardiac involvement but will not slow the progression of the disease in other organs. Patients with the Val122Ile mutation often have disease primarily limited to the heart so isolated heart transplantation may be appropriate.2 In certain mutations, combined heart and liver transplant is an option. More than 50 cases of combined heart and liver transplantation have been reported so far, however, due to the advances in medical therapy, the numbers of liver transplantation will likely decrease. AL cardiac amyloidosis typically has a poor prognosis with a median survival of about 7.1 months. In carefully selected patients, with recent advances in chemotherapy, initial high dose chemotherapy followed by heart transplant and subsequent autologous bone marrow transplant has resulted in better outcomes.9
The two drugs approved for hereditary ATTR polyneuropathy that have been shown to slow or improve neuropathy are inotersen (Tegsedi,™ Akcea Therapeutics, Boston, MA, USA) and patisiran (Onpattro,™ Alnylam Pharmaceuticals, Cambridge, MA, USA).10,11 Both drugs decrease the synthesis of mutant and wild-type TTR by the liver. Many patients with hereditary ATTR have a mixed phenotype and have both neuropathy and cardiomyopathy. While not a primary end point of these trials, imaging and functional data suggest that these drugs may also stabilize or reverse hereditary ATTR cardiomyopathy.12,13
Patisiran is administered intravenously every 3 weeks with pretreatment with steroids and Benadryl to offset drug reactions.10 Second generation formulations of inotersen and patisiran are currently being studied prospectively in hereditary and wild-type ATTR cardiomyopathy. The second generation of patisiran will be administered subcutaneously every 3 months and is being studied in the Helios B trial (Alnylam Pharmaceuticals, Cambridge, MA, US) in ATTR hereditary and wild-type cardiomyopathy. The CARDIO-TTRansform trial (Ionis Pharmaceuticals, Carlsbad, CA, US and Akcea Therapeutics, Boston, MA, US) will study a modified form of inotersen which is anticipated to have less side effects and is administered subcutaneously once per month in hereditary and wild-type ATTR cardiomyopathy.
Revusiran (Alnylam Pharmaceuticals, Cambridge, MA, US) is a subcutaneous small interfering RNA that was studied prospectively in patients with hereditary ATTR cardiomyopathy in the ENDEAVOR trial, but the trial was stopped prematurely at 6.7 months by the sponsor due to increased deaths in the treatment arm. Eighteen patients (12.9%) died in the revusiran arm and 2 patients (3%) died in the placebo arm.14 The sponsor was unable to identify a drug-related mechanism to explain the increased mortality in the revusiran group.14,15
Tafamidis (Vynaqel,™ Pfizer, New York, NY, US), Diflunisal, and AG10 are stabilizers of the TTR tetramer that inhibit dissociation into monomers and fibril formation. Tafamidis is the first FDA approved therapy for wild-type and hereditary ATTR cardiomyopathy. The ATTR-ACT study showed that daily administration of tafamidis 20 mg or 80 mg decreased all-cause mortality, cardiovascular hospitalizations, and decreased the decline in 6-minute walk distance and quality of life compared to placebo at 30 months.16 NYHA class I and II patients received the greatest benefit, suggesting that early identification and treatment are essential. Tafamidis, however, is priced at $225,000/year and is the most expensive cardiovascular drug ever marketed in the United States.17 A recent cost-effective analysis of tafamidis suggested that the price would have to be reduced by 92.6% to be considered cost-effective and more accessible to patients.17 Diflunisal is a nonsteroidal anti-inflammatory drug that has been shown to reduce the progression of ATTR polyneuropathy compared to placebo in a randomized, double-blind trial with 130 patients.18 Diflunisal has been used off label to treat ATTR cardiomyopathy, but must be used with caution in heart failure patients as it may exacerbate heart failure or negatively affect renal function. A retrospective analysis of 81 patients with ATTR cardiomyopathy treated with diflunisal for a median of 1 year showed a statistically significant increase in serum TTR concentration, a decrease in left atrial volume index, and serum troponin I. More long-term data are needed, but diflunisal may represent a cost-effective tetramer stabilizer in ATTR cardiomyopathy.19 The efficacy of the tetramer stabilizer AG10 (Eidos Therapeutics, San Francisco, CA, USA) is currently being studied prospectively in the randomized double-blind ATTRibute-CM trial.
Alternative therapies that may disrupt fibril formation, but for which more data are needed include doxycycline, epigallocatechin gallate found in green tea, and taurooursodeoxycholic acid (TUDCA).
Transthyretin Amyloid Polyneuropathy
Pathophysiology of ATTR Polyneuropathy
Amyloid deposits first in the endoneurial blood vessels leading to degeneration of Schwann cells as well as the blood-nerve barrier. It is thought that small unmyelinated nerve cells are impacted first so in early disease myelin fiber density and nerve conduction velocity studies (NCV)/electromyogram (EMG) remain normal, despite the patient experiencing symptoms of loss of pain and temperature sensation.20 Patients may also present with autonomic symptoms early in the disease course due to amyloid deposition in autonomic ganglia. Later in the disease course, there is additional disruption of Schwann cells and amyloid deposition in the dorsal root ganglia with resultant demyelination and distal axon deterioration.20 Amyloid deposition may also occur within the blood vessel walls and lumens. As the disease progresses patients develop length-dependent loss of sensory and later motor weakness that can be interpreted on NCV and EMG as a large fiber peripheral neuropathy.20
Diagnosis of Hereditary ATTR Polyneuropathy
Initial symptoms of hereditary ATTR polyneuropathy may include distal sensory symptoms such as numbness, pain and paresthesias in the feet, autonomic dysfunction, early satiety, diarrhea and constipation, erectile dysfunction, and weight loss. As the disease advances the neuropathy may ascend to include not only the lower limbs but also the upper extremities, and result in loss of motor function, deep tendon reflexes, muscular weakness, and gait disorders.5,20 Hereditary ATTR polyneuropathy is endemic in Brazil, Japan, Sweden, and Portugal. The prevalence in endemic countries is approximately 1 in 1000.5 In endemic countries, ATTR polyneuropathy should be considered in patients with length-dependent small-fiber polyneuropathy with autonomic dysfunction, family history of ATTR amyloidosis, renal dysfunction, vitreous opacities, heart arrhythmias, and/or unexplained weight loss. ATTR amyloidosis can also occur in nonendemic countries due to sporadic mutations. The worldwide prevalence of hereditary ATTR polyneuropathy is estimated to be 1 in 1,000,000.5
Nerve conduction studies and EMGs are performed to detect the presence of peripheral neuropathy. The diagnosis of hereditary ATTR polyneuropathy can be confirmed in patients with polyneuropathy with genetic testing of the TTR gene and a biopsy demonstrating amyloid deposits. Biopsies may be obtained from salivary glands, abdominal fat pad, nerve tissue, skin, kidney, or gastrointestinal tract.5 Positive Congo red staining of the biopsy with apple-green birefringence under polarized light can confirm a diagnosis of amyloidosis. Mass spectroscopy or immunohistochemistry can then be used to type the amyloid-forming protein (Figure 5). Many patients with hereditary ATTR polyneuropathy also have cardiomyopathy so bone scintigraphy with 99m technetium pyrophosphate, 99mtechnetium 3,3-disphonphone-1,2-propanodicarboxylic acid, or 99m technetium hydroxymethylene diphosphate scan or an endomyocardial cardiac biopsy can be performed to confirm cardiac involvement.
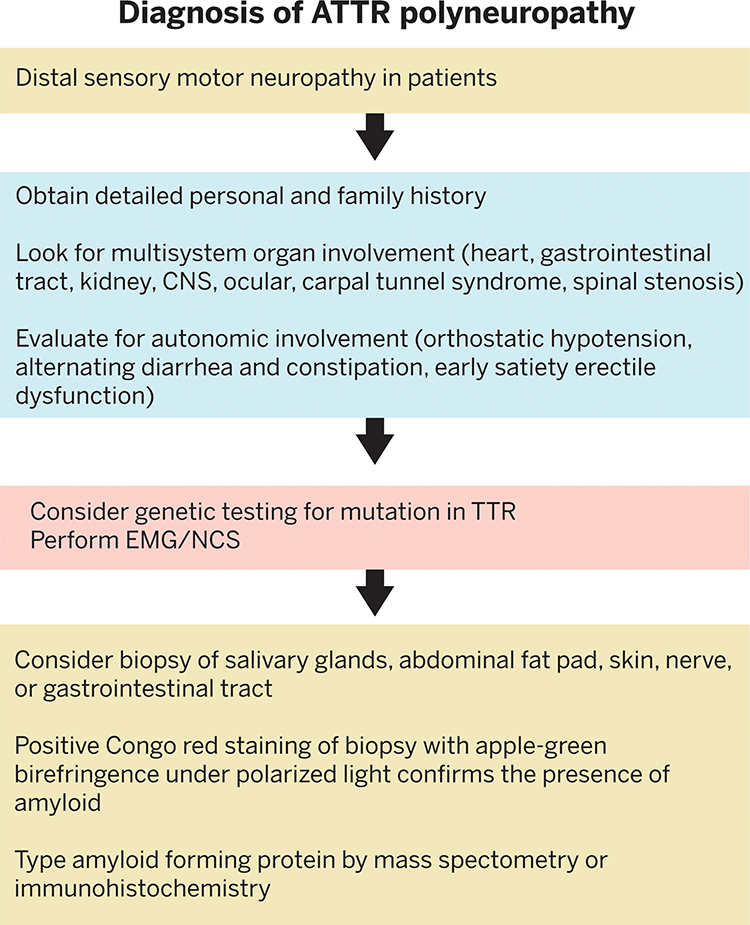 |
Figure 5 Diagnosis of Transthyretin Amyloid Polyneuropathy. Abbreviations: CNS, central nervous system; EMG, electromyography; NCS, nerve conduction study. |
Medical Treatment of Hereditary ATTR Polyneuropathy
There are two FDA approved drugs for the treatment of hereditary ATTR polyneuropathy, inotersen (Tegsedi™, Akcea Therapeutics, Boston, MA, US) and patisiran (Onpattro™, Alnylam Pharmaceuticals, Cambridge, MA, US).10,11 Both drugs inhibit the production of TTR by the liver and have been shown to slow progression and/or improve polyneuropathy. Inotersen is a 2′-O-methoxyethyl–modified antisense oligonucleotide that is administered via subcutaneous injection. The modified neurologic impairment score +7 and Norfolk Quality of Life Scale favored therapy with inotersen compared to placebo in preventing the progression of neuropathy and maintaining quality of life.11 Treatment-emergent side effects included severe thrombocytopenia and glomerulonephritis and rejection in patients with liver transplantation. These side effects are manageable with routine monitoring. Patients receiving inotersen require weekly monitoring of platelet counts, biweekly monitoring of serum creatinine, estimated glomerular filtration rate, urine protein to creatinine ratio and urinalysis plus periodic monitoring of liver function. Patients are also required to take vitamin A supplementation during treatment. Treatment should not be initiated in patients with a platelet count less than 100,000/mm3 or a history of glomerulonephritis related to inotersen. A trial is currently underway to evaluate the safety and tolerability of a second-generation antisense oligonucleotide therapeutic administered subcutaneously every 4 weeks (AKCEA-TTR-LRx) that is anticipated to have less side effects.
Patisiran (Onpattro,™Alnylam Pharmaceuticals, Boston, MA, USA) is a small interfering RNA that is administered every 3 weeks intravenously with pretreatment with steroids and Benadryl.10 Patisiran was studied in the APOLLO trial which showed stabilization or improvement in the modified Neurologic Impairment Score and Norfolk Quality of Life Scale. Adverse reactions in the patisiran group that were more common than placebo included peripheral edema (30% vs 22%) and infusion-related reactions (19% vs 9%). Vitamin A supplementation is recommended. No routine safety monitoring is required. The Helios A study is currently in progress to evaluate the safety and efficacy of a second-generation subcutaneous formulation (vutrisiran) administered every 12 weeks.
Tafamidis (Vynadqel,™ Pfizer, New York, NY, US) is a TTR tetramer stabilizer that was approved for hereditary stage I neuropathy in Europe but did not receive FDA approval in the United States for treatment of peripheral neuropathy.21,22 Diflunisal is a nonsteroidal anti-inflammatory drug that has been shown to slow neuropathy in a randomized double-blind study involving 130 patients but approval from the FDA for this condition has not been pursued.18
Conclusion
Transthyretin amyloidosis is thought to be underdiagnosed. Because of the multi-system involvement, clinicians must often integrate clues from the patient’s personal and family history, physical exam, cardiac imaging, neurologic testing, genetic testing, and ophthalmologic and gastrointestinal findings to make a diagnosis. Screening should be considered in patients with unexplained left ventricular hypertrophy or peripheral neuropathy. Additionally, transthyretin amyloidosis should be considered in older patients with heart failure with preserved ejection fraction or aortic stenosis. The currently available treatments appear to be most effective if started early, so making a timely diagnosis is essential.
Future Directions
There is currently a significant amount of research in the field of ATTR amyloidosis that will undoubtedly affect diagnosis and treatment. Earlier diagnosis of this fatal disease will hopefully improve survival and quality of life. New noninvasive techniques are being developed to diagnose ATTR amyloidosis at earlier stages. Additionally, new therapeutic approaches may focus on gene editing and methods to remove amyloid deposition from affect organs and reverse end-organ damage.
Acknowledgments
The authors would like to thank Fredrik H. Skarstedt for creating the graphics and Alisa L. Gumm, RN for amyloid research and clinical support.
Disclosure
Noel R Dasgupta has consulted for Akcea Therapeutics, Ionis Pharmaceuticals, Pfizer Pharmaceuticals, and Alnylam Pharmaceuticals and been on the speaker’s bureau for Akcea Therapeutics and Pfizer Pharmaceuticals. Merrill D. Benson and Roopa Rao have no disclosures.
References
1. Benson MD, Buxbaum JN, Esienberg DS, et al. Amyloid nomenclature 2018: recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid. 2018;25(4):215–219. doi:10.1080/13506129.2018.1549825
2. Thomas VE, Smith J, Benson MD. Amyloidosis: diagnosis and new therapies for a misunderstood and underdiagnosed disease. Neurodegener Dis Manag. 2019;9(6). doi:10.2217/nmt-2019-0020
3. Maurer MS, Bokhari S, Dorbala S, et al. Expert consensus recommendations for the suspicion and diagnosis of transthyretin cardiac amyloidosis. Circ Heart Fail. 2019;12(9). doi:10.1161/CIRCHEARTFAILURE.119.006075.
4. Wittles RM, Bokhari S, Thibaud D, et al. Screening for transthyretin amyloid cardiomyopathy in everyday practice. J Am Coll Cardiol HF. 2019;7:709–716.
5. Adams D, Ando Y, Melo Beirao J, et al. Expert consensus recommendations to improve diagnosis of ATTR amyloidosis with polyneuropathy. J Neurol. 2020. doi:10.1007/s00415-019-09688-0
6. Gonzalez-Lopez E, Gallego-Delgado M, Guzzo-Merello G. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J. 2015;36(38):2585–2594. doi:10.1093/eurheartj/ehv338
7. Ternacle J, Krapf L, Mohty D, et al. Aortic stenosis and cardiac amyloidosis. JACC. 2019;74(21):2638–2651. doi:10.1016/j.jacc.2019.09.056
8. Carvalho A, Rocha A, Lobato L. Liver Transplantation in transthyretin amyloidosis: issues and challenges. Liver Transpl. 2015;21(3):282–292. doi:10.1002/lt.24058
9. Sousa M, Mohan G, Guglin M, et al. Heart transplantation in cardiac amyloidosis. Heart Fail Rev. 2017;22:317–327. doi:10.1007/s10741-017-9601-z
10. Adams D, Gonzalez-Duarte A, O’Riordan WD, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. NEJM. 2018;379:11–21. doi:10.1056/NEJMoa1716153
11. Benson MD, Waddington-Cruz M, Berk JL, et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. NEJM. 2018;379:22–31. doi:10.1056/NEJMoa1716793
12. Solomon SD, Adams D, Kristen A, et al. Effects of patisiran, an RNA interference therapeutic, on cardiac parameters in patients with hereditary transthyretin-mediated amyloidosis. Circulation. 2019;139(4):431–443. doi:10.1161/CIRCULATIONAHA.118.035831
13. Dasgupta NR, Rissing SM, Smith J, et al. Inotersen therapy of transthyretin amyloid cardiomyopathy. Amyloid. 2020;27(1):52–58. doi:10.1080/13506129.2019.1685487
14. Judge DP, Kristin AV, Grogen M, et al. Phase 3 multicenter study of revusiran in patients with hereditary transthyretin-mediated (hATTR) amyloidosis with cardiomyopathy. Cardiovasc Drugs Ther. 2020;1–14.
15. Janas MM, Zlatev L, Liu J, et al. Safety of 2ʹ-deoxy-2ʹ-flouro nucleotides in GalNAc-siRNA conjugates. Nucleic Acids Res. 2019;47(7):3306–3320. doi:10.1093/nar/gkz140
16. Maurer MS, Schwartz JH, Gundapaneni MS, et al. Tafamidis treatment for patients with Transthyretin Amyloid Cardiomyopathy. NEJM. 2018;379(11):1007–1016. doi:10.1056/NEJMoa1805689
17. Kazi DS, Bellows BK, Baron SJ, et al. Cost-effectiveness of tafamidis for transthyretin amyloid cardiomyopathy. Circulation. 2020;141(15):1214–1224. doi:10.1161/CIRCULATIONAHA.119.045093
18. Berk JL, Suhr OB, Obici L, et al. Repurposing diflunisal for familial amyloid polyneuropathy. JAMA. 2013;310(24):2658–2667. doi:10.1001/jama.2013.283815
19. Graham L, Pipilas A, Mussinelli R, et al. Stabilization of cardiac function with diflunisal in transthyretin (ATTR) cardiac amyloidosis. J Card Fail. 2019.
20. Schwarzlow C, Kazamel M. Hereditary transthyretin amyloidosis: clinical presentation and management updates. J Clin Neuromuscul Dis. 2020;21:144–156. doi:10.1097/CND.0000000000000270
21. Coelho T, Maia LF, da Silva A. Tafamidis for transthyretin familial polyneuropathy: a randomized controlled trial. Neurology. 2012;79(8):785–792. doi:10.1212/WNL.0b013e3182661eb1
22. Waddington Cruz M, Benson MD. A review of tafamidis for the treatment of transthyretin- related amyloidosis. Neurol Ther. 2015;4(2):61–79. doi:10.1007/s40120-015-0031-3
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.