")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 18
Diagnosis and Management of Genetic Causes of Middle Aortic Syndrome in Children: A Comprehensive Literature Review
Authors Lazea C , Al-Khzouz C, Sufana C, Miclea D, Asavoaie C , Filimon I, Fufezan O
Received 11 November 2021
Accepted for publication 21 February 2022
Published 16 March 2022 Volume 2022:18 Pages 233—248
DOI https://doi.org/10.2147/TCRM.S348366
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Cecilia Lazea,1,2 Camelia Al-Khzouz,1,3 Crina Sufana,2 Diana Miclea,3,4 Carmen Asavoaie,5 Ioana Filimon,5 Otilia Fufezan5
1Department Mother and Child, University of Medicine and Pharmacy “Iuliu Hatieganu”, Cluj-Napoca, Romania; 2Department of Pediatrics I, Emergency Pediatric Hospital, Cluj-Napoca, Romania; 3Department of Medical Genetics, Emergency Pediatric Hospital, Cluj-Napoca, Romania; 4Department of Molecular Sciences, University of Medicine and Pharmacy “Iuliu Hatieganu”, Cluj-Napoca, Romania; 5Department of Radiology and Medical Imaging, Emergency Pediatric Hospital, Cluj-Napoca, Romania
Correspondence: Cecilia Lazea, Department Mother and Child, University of Medicine and Pharmacy “Iuliu Hatieganu”, 68, Motilor Street, Cluj-Napoca, 400370, Romania, Tel +40 744353764, Email [email protected]; [email protected]
Abstract: Middle aortic syndrome (MAS) is a rare vascular disease representing an important cause of severe hypertension in children. MAS is characterized by segmental or diffuse narrowing of the abdominal and/or distal descending aorta with involvement of the renal and visceral branches. Most cases of MAS are idiopathic, but MAS may occur in genetic and acquired disorders. The most common genetic causes of MAS are neurofibromatosis type I, Williams syndrome, Alagille syndrome, tuberous sclerosis and mucopolysaccharidosis. This review article discusses the pathophysiological aspects, distinctive associated features, and management of genetic forms of MAS in children.
Keywords: middle aortic syndrome, hypertension, neurofibromatosis type 1, Williams syndrome, Alagille syndrome, tuberous sclerosis, mucopolysaccharidoses
Introduction
Definition
Middle aortic syndrome (MAS) is a rare disease characterized by segmental or diffuse narrowing of the abdominal and/or distal descending aorta with involvement of the renal and visceral branches and represents an important cause of severe hypertension in children.1,2 Ostial stenosis of the major branches of the proximal abdominal aorta is a characteristic in MAS.3 MAS was described for the first time in 1963 by Sen et al in 16 patients with inflammatory narrowing of the middle aortic segment.4 It represents 0.5–2% of all cases of aortic narrowing.3,5
Etiology
Most cases of MAS are idiopathic, but MAS may occur in genetic and acquired disorders (Table 1).
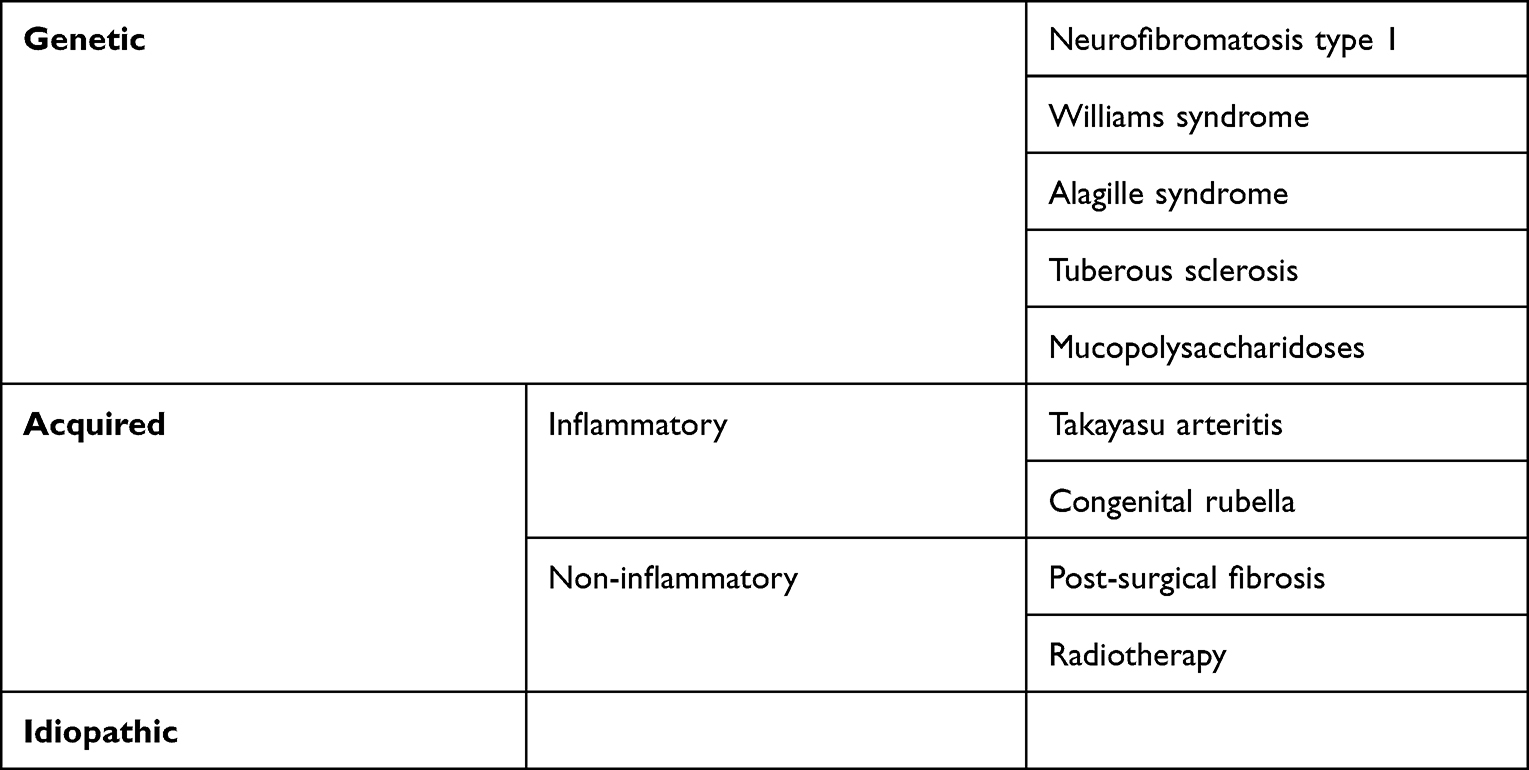 |
Table 1 Causes of Middle Aortic Syndrome in Children |
Genetic Causes of MAS
Genetic forms of MAS are usually described in children and young adults and encountered in 7–36% of the cases.1,6,7 The most common genetic causes of MAS are neurofibromatosis type I, Williams syndrome, Alagille syndrome, tuberous sclerosis and mucopolysaccharidosis.8–15 A recent study has been demonstrated that monogenic cause of MAS was present in 43% of 35 families with MAS and whole-exome sequencing revealed mutations in genes previously associated with vascular disease (NF1, JAG1, ELN, GATA6, RNF213).16 The prevalence of genetic diseases associated with MAS in different studies is presented in Table 2.
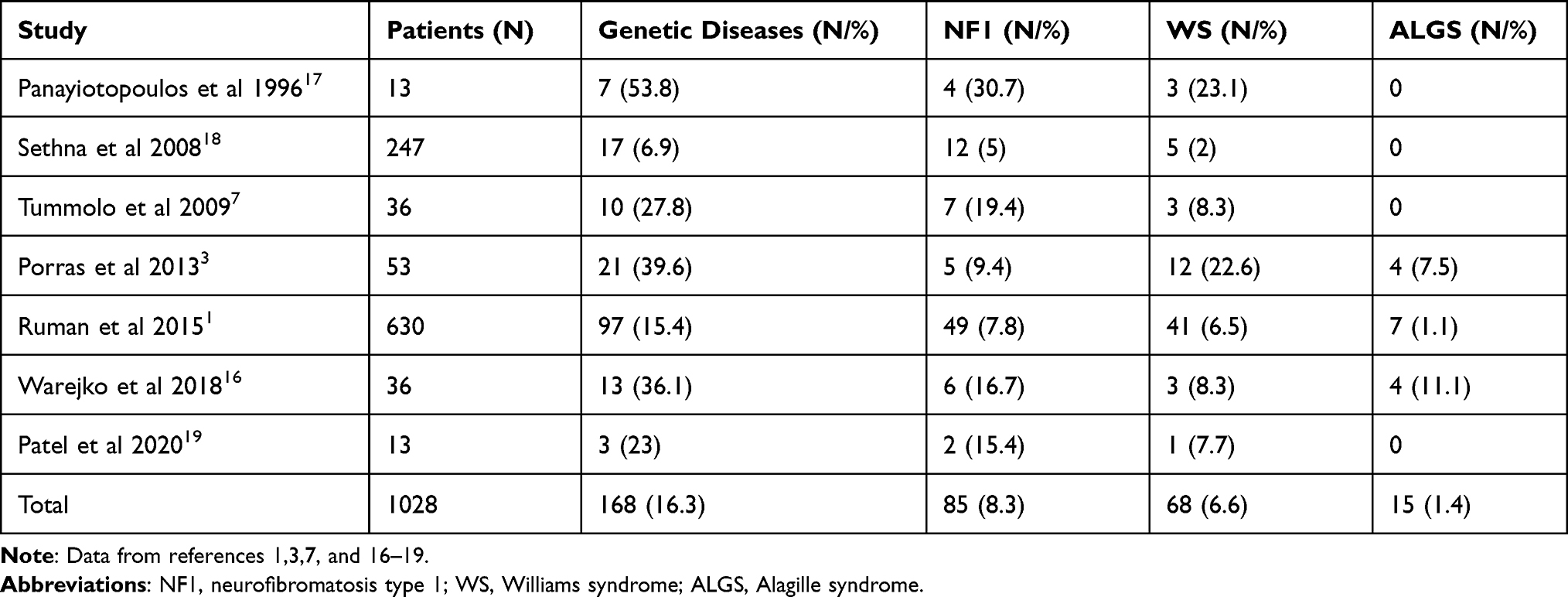 |
Table 2 Prevalence of Genetic Forms of MAS in Children |
Acquired Causes of MAS
Acquired inflammatory diseases which can lead to MAS are Takayasu arteritis and other non-specific arteritis and congenital rubella.3,20–23 Other acquired causes of MAS include post-surgical fibrosis and damage of the vasa-vasorum after surgical resection of an abdominal tumor or impaired growth of the aorta after radiation therapy for neuroblastoma in children.24,25 Takayasu arteritis is a chronic inflammatory disease with unknown precise etiology, with genetic factors’ involvement such as HLA genes and other proposed risk factors as genetic variants in genes encoding immune response regulators and pro-inflammatory cytokines. This form of MAS encountered in 15–18% of the cases.1,26
Idiopathic MAS
Most cases of MAS are idiopathic. Congenital cases have been described due to a developmental anomaly in the fusion and maturation of the embryonic dorsal aortas.5,12
Vessel Involvement
Commonly, idiopathic MAS involves renal and splanchnic branches of the aorta, and the most common anatomic site of the aorta narrowing is infrarenal.1,3 MAS determined by genetic disorders is more often associated with suprarenal stenosis and extra-aortic involvement. Renal arteries are involved in 84% of the cases and in 60% of the cases the stenosis is bilateral, superior mesenteric artery are involved in 44% of the cases, coeliac trunk in 39% of the cases and common iliac artery in 15% of the cases.1,27
The incidence and prevalence of MAS estimation is challenging because of the heterogeneity of aortic branches involvement and etiology.
Presentation
Patients with MAS are usually diagnosed during childhood or adolescence. Presentation in infancy has also been reported and has a poor prognosis, especially in preterm infants.28
Clinical manifestations are severe hypertension, headache, postprandial abdominal migraine, claudication of the inferior limbs, absent femoral pulses, blood pressure discrepancy between upper and lower extremities, abdominal bruit, and failure to thrive.7,12,29 Despite the guidelines which strongly recommend regular measurement of the blood pressure in children, arterial hypertension may be an incidentally finding at a routine clinical examination.30,31
According to associated disease, these patients can have additional physical examination findings as dysmorphic features, skin spots, jaundice, skeletal abnormalities, and organomegaly. The most common complication of MAS is renovascular hypertension, which can lead to heart failure, left ventricle hypertrophy, dilated cardiomyopathy in infants and neonates, cerebrovascular accidents, renal failure, hypertensive retinopathy, and encephalopathy.3,32
Diagnosis
Diagnosis of MAS has increased in the last decades due to improved diagnostic imaging technologies such as angiography, CT angiography, magnetic resonance angiography and ultrasound.1,3,20,33
Treatment
Management of MAS includes medical therapy, endovascular and surgical intervention. The most common antihypertensive agents are calcium-channel blockers, beta-blockers, diuretics, angiotensin-converting enzyme inhibitors and alpha-blockers, and they are effective in mild or moderate aortic or renal stenosis.1,3 Medical treatment is very often insufficient. Endovascular interventions such as balloon angioplasty or stenting represent a palliative procedure because of the increased rate of restenosis, but they are performed to avoid surgical intervention on the developing aorta in children. Repeated balloon dilations with paclitaxel eluting balloons have demonstrated to be an effective and safe therapeutic option in a 15-year-old patient.34 Younger age at the time of intervention could be a risk factor for vascular complication because patients who are diagnosed in the first years of life has a severe form of disease.3 Surgical procedures are indicated when endovascular interventions fail to achieve the long-term blood pressure values control or when the lesions are too complex for percutaneous transluminal angioplasty. The surgical interventions used for MAS are aorto-aortic bypass grafting, graft vascular replacement, patch angioplasty.12,35,36 Prosthetic grafts may necessitate replacement in children who are still growing and can determine mechanical complications as thrombosis and aneurysm formation.12,37,38 A novel technique is represented by tissue expander (TE)-stimulated lengthening of arteries (TESLA), which determines a slow development of the normal distal aorta and the stenotic segment of the aorta can be resected and anastomosis can be performed without a prosthetic graft.39 In this procedure based on stretch-induced growth a tissue-expanding device is placed behind the child’s aorta and it is gradually filled with saline solution for a period of months. During the growth period, the development of the aorta is closely monitored by imaging tests. Another novel technique is represented by Mesenteric Artery Growth Improves Circulation (MAGIC) and consists of an aorta bypass using the mesenteric arteries as a free conduit.40 Both the MAGIC and TESLA procedures provide feasible approaches for aortic bypass and reconstruction using autologous tissues.
Renovascular hypertension may benefit of renal artery reimplantation, arterial reconstruction with autologous or synthetic grafts, nephrectomy, and auto-renal transplantation.41,42 Most studies referring to MAS in children include all causes (acquired, genetic and idiopathic) without a detailed presentation of the genetic ones and those referring to genetic forms of middle aortic syndrome in children are scarce and most of them are case reports. Herein, the most encountered genetic diseases associated with middle aortic syndrome are reviewed. Attention is focused on the genetic and pathophysiological aspects of each disorder with individual description and presentation of distinctive associated features and therapeutic options.
Methods
Search Strategy
A systematic literature search was performed using electronic literature databases (PubMed/Medline and Google Scholar database) and followed Preferred Reporting Items for Systematic Review and Meta-analysis protocol (PRISMA).43 We excluded conference abstracts and limited the search to literature published in English. We also screened all the reference lists of systematic reviews and meta-analysis to find other relevant eligible publications. We excluded all studies in adults and studies referred to idiopathic or acquired MAS.
Study Selection and Data Extraction
Two independent researchers performed the literature search till June 2021. We selected the concept of “middle aortic syndrome”, “mid-aortic syndrome”, “genetic”, “coarctation of the aorta”, “renal artery stenosis”, “coeliac trunk stenosis”, “mesenteric artery stenosis”, “neurofibromatosis”, “Williams syndrome”, “Alagille syndrome”, “tuberous sclerosis”, “mucopolysaccharidoses” and “children aged 0–18 years”. Search term combinations were used.
The criteria to include the patients and studies in the present systematic review were as follows: 1) age under 18 years; 2) studies that reported patients with MAS and genetic diseases; 3) case reports, case series and observational studies; 4) articles in English; and 5) no restriction regarding the year of publication. The exclusion criteria were as follows: studies that did not present complete data about diagnosis and treatment, reviews, conference abstracts, experimental research. After reading the studies in full, the following data were collected: author, year of publication, study design, the country where the study was conducted, number of patients. Data extraction was targeted to patients’ age, clinical findings, vessels involvement, investigations, treatment and outcomes.
Results
The search strategy has identified a total of 475 articles on which only 136 articles met the inclusion criteria. A total of 37 articles of 85 cases, most of them case reports and case series were included in this work. The study selection flowchart is shown in Figure 1.
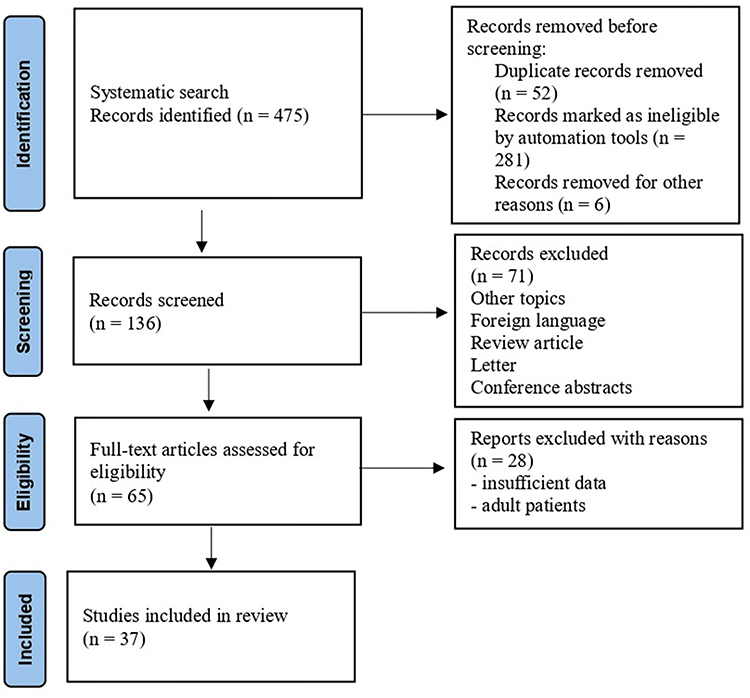 |
Figure 1 PRISMA flow diagram of study selection. Notes: Adapted from Liberati A, Altman DG, Tetzlaff J, et al. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: explanation and elaboration. PLoS Med. 2009;6(7):e1000100. doi: 10.1371/journal.pmed.1000100.43 Copyright: © 2009 Liberati et al. Creative Commons Attribution License. |
The patients age ranged 1–17 years. Five genetic diseases associated with MAS were described in this work: 26 patients with neurofibromatosis, 32 patients with Williams syndrome, 15 patients with Alagille syndrome, 4 patients with tuberous sclerosis and 8 patients with mucopolysaccharidoses. Clinical characteristics, vessel involvement and therapeutic options for patients with MAS and neurofibromatosis, Williams syndrome and Alagille syndrome are depicted in Tables 3–5.
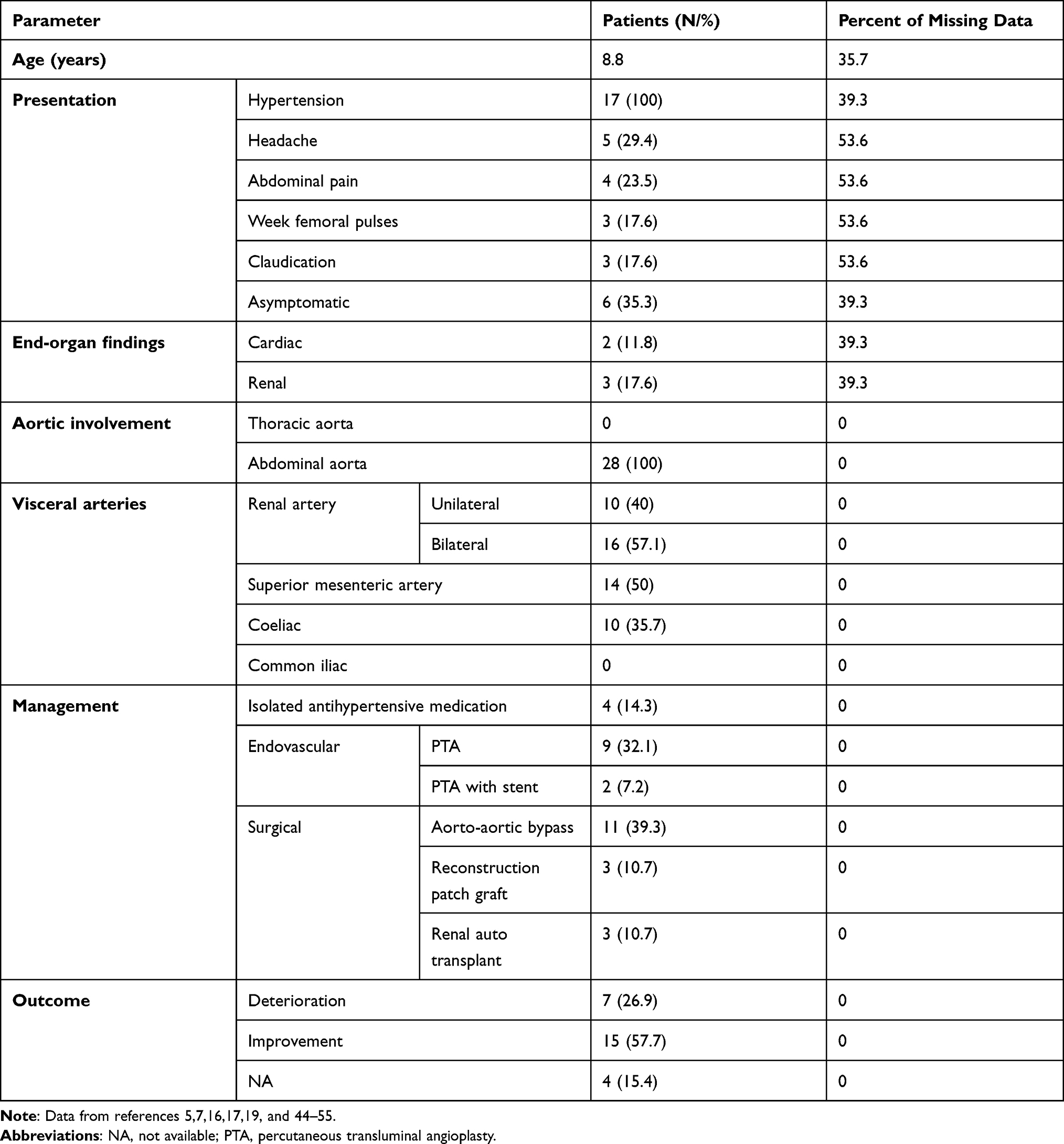 |
Table 3 Characteristics of 26 Children with NF1 and MAS |
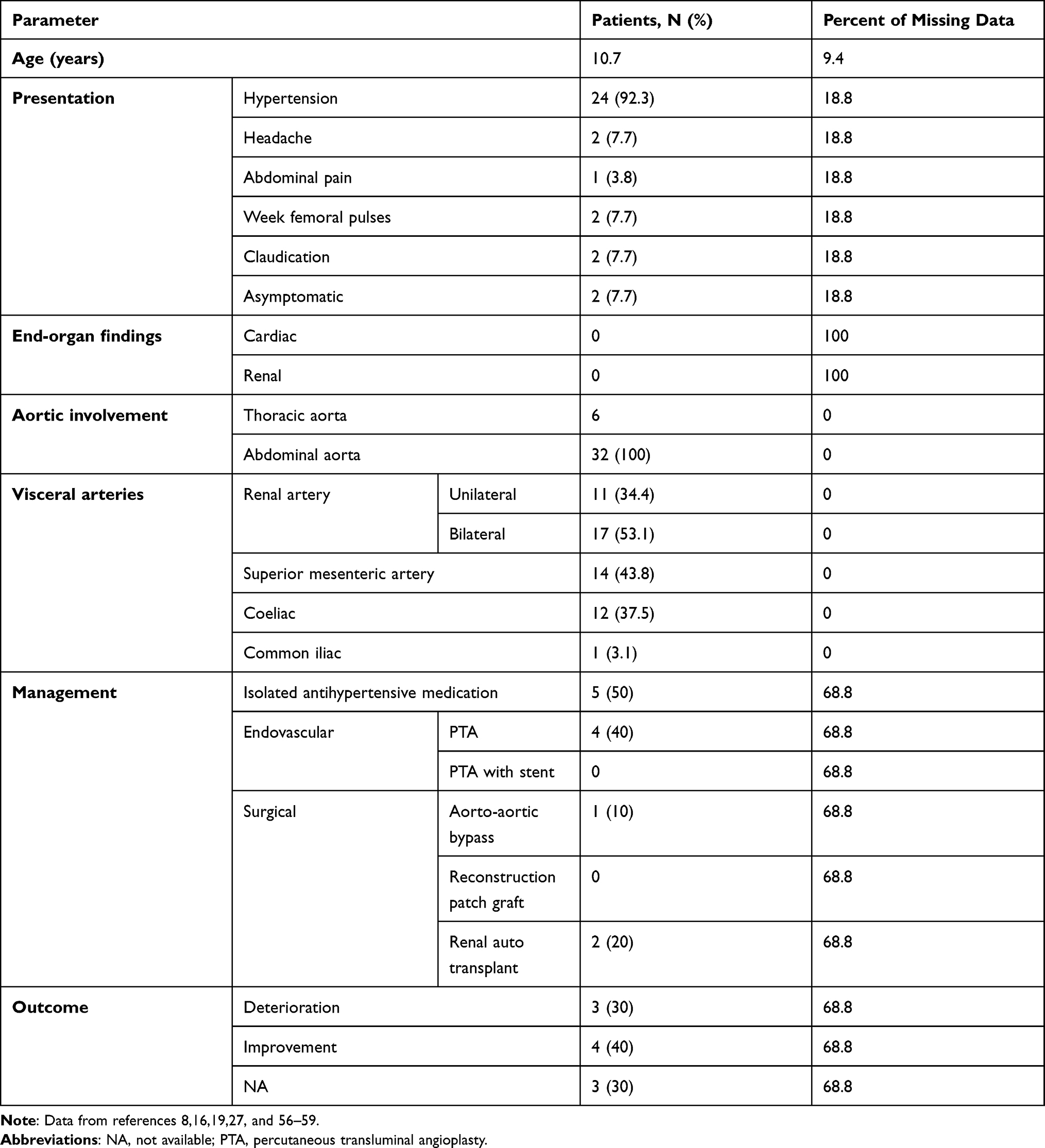 |
Table 4 Characteristics of 32 Children with WS and MAS |
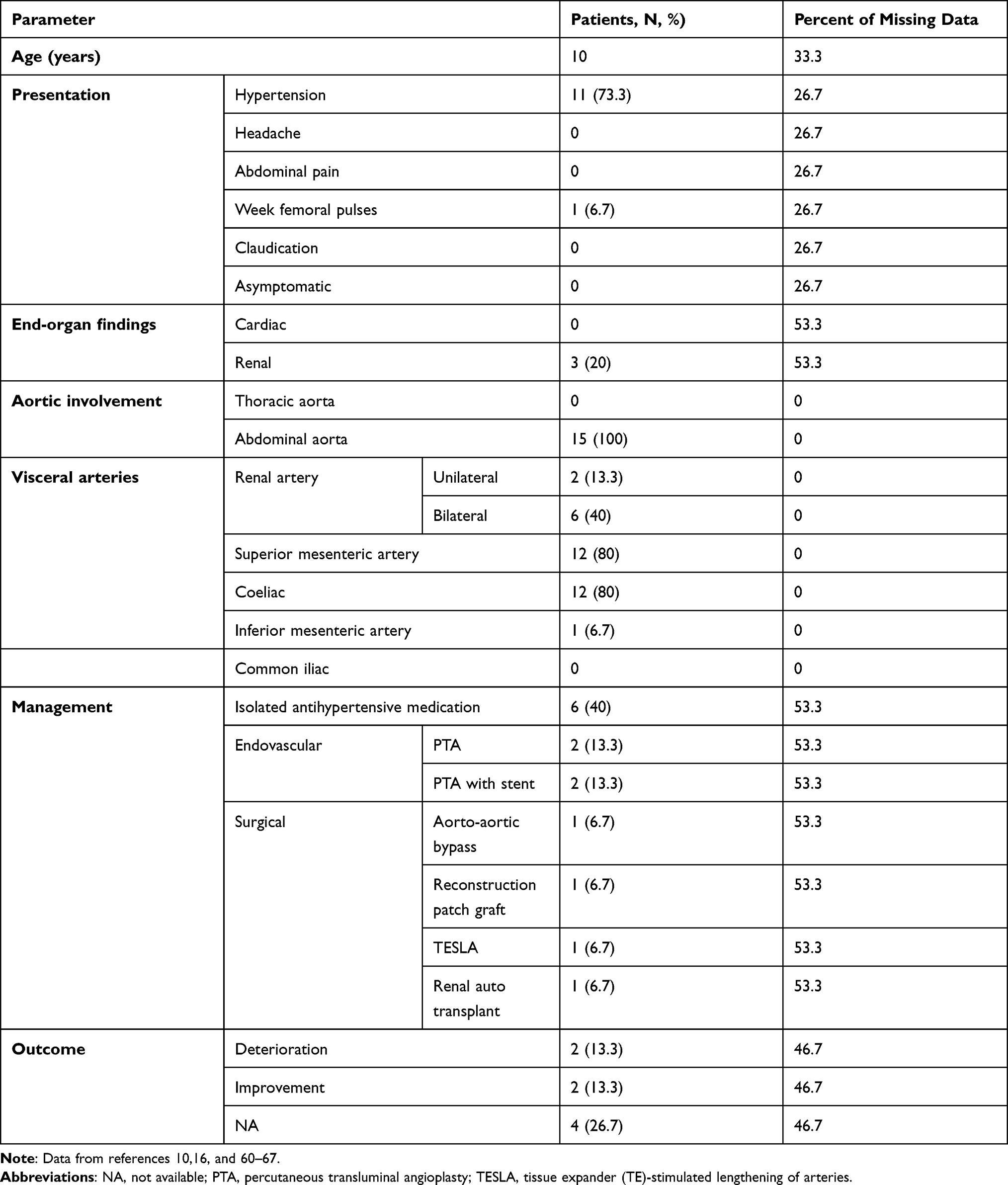 |
Table 5 Characteristics of 15 Children with ALGS and MAS |
Discussion
Neurofibromatosis Type 1
Neurofibromatosis type 1 (NF1) is an autosomal dominant disorder caused by mutations in the tumor suppressor gene NF1 which encodes the neurofibromin, which regulates the cell growth and division. Whole-exome sequencing studies revealed that protein truncating mutations (splice site and frameshift mutations) in NF1 may result in MAS.16
Loss of neurofibromin produces increased mitogenic signaling and leads to increased cellular proliferation or differentiation. Diagnosis criteria for NF1 are as follows: six or more café au lait macules (>0.5 cm at largest diameter in a pre-pubertal child or >1.5 cm in post-pubertal individuals), axillary or groin freckling, two or more neurofibromas or one or more plexiform neurofibromas, two or more Lisch nodules, bony dysplasia, optic pathway glioma and a first-degree relative with neurofibromatosis type 1. Two or more features are required for diagnosis.68
Arterial hypertension is a common finding in neurofibromatosis type 1 and is mainly secondary to vascular disease in children and to pheochromocytoma in older ages. Because many patients are asymptomatic, regular blood pressure assessment and ambulatory blood pressure monitoring enables early diagnosis of hypertension and arterial stenosis.
NF1 is the most common genetic disorder associated with MAS and encountered for 5–22% of the cases.1,3,6,7,16,17,19 Another study found NF1 as the second cause of MAS after Williams syndrome.3
Vascular disease in NF type 1 is determined by loss of neurofibromin expression in the smooth muscle cells and in the vascular endothelium and consists in abnormal proliferative response to arterial injury of the smooth muscle cells and increased neointima formation.69 Vascular abnormalities include aneurysm, stenosis and arteriovenous malformations and affects 18% of the cases.44 Pathologic findings in patients with vascular disease revealed fibromuscular dysplasia with neointimal thickening.45 The true prevalence of vasculopathy in NF-1 is underestimated because most patients are asymptomatic despite multi-vessel involvement and imaging studies are usually reserved for symptomatic patients.
CT angiography and magnetic resonance angiography can diagnose MAS, can determine the location and extent of stenosis of the aorta and its associated visceral branches, as well as the presence of collateral circulation and are also used for postoperative or endovascular intervention follow-up.70 They can also exclude external compression of the aorta by neurofibromas. The typical string-of-pearls involvement of the renal arteries in fibromuscular dysplasia is not present in neurofibromatosis where the vascular involvement is proximal.46,47 A routine abdominal ultrasonography with visualization of the abdominal aorta in a longitudinal view, performed by experienced specialist, could be a useful method of diagnosis in children.48
The most common arteries involved in NF1 children with MAS are renal arteries (97.1% of cases), superior mesenteric artery (50% of cases) and celiac artery (37.5% of cases) – Table 2.
Figure 2 shows thoracic and abdominal magnetic resonance angiogram revealing reduced caliber of the abdominal aorta and narrowed left renal artery at emergence in a 4-year-old boy with NF1.
 |
Figure 2 Thoracic and abdominal magnetic resonance angiogram in a 4-year-old boy with NF1 revealing the reduced caliber of the abdominal aorta (A), narrowed left renal artery (B), superior mesenteric artery (C) and celiac trunk at emergence (D). The diameter of aorta is 6.9 mm (Z score = −2.32) at celiac trunk emergence (arrow 1) and 3.8mm (Z score = −6.7) at bifurcation (arrow 2). |
Treatment of vascular lesions in patients with NF1 includes medical therapy, endovascular and surgical intervention. Very often, hypertension is difficult to control despite multiple antihypertensive agents and may lead to end-organ damage.
The results of balloon angioplasty in children with MAS and NF1 are debated. Srinivasan et al reported similar results to those of children without NF1, with improvement in 84–91% of the cases.71 In other studies, hypertension was more difficult to treat, and the results were disappointing with cured rate in 33% of the patients.9 Other studies found that NF1 represents a risk factor for vascular complications from catheter-based interventions such aneurysm and vascular tears because of predisposition to develop spontaneous aneurysm. In NF1 structure abnormalities of the arterial walls are common, and these abnormalities will lead to increased risk for vascular complication after vascular intervention.3,45,72–74 Further dilatation with repeated angioplasty at a later date could lead to recoil, and a weakened arterial wall may result in later aneurysm formation in 5% to 20% of the cases.74
Williams Syndrome
Williams syndrome (WS) is a rare genetic multisystemic disorder characterized by a distinct facial appearance, cardio-vascular anomalies (most frequently supravalvular aortic stenosis, peripheral pulmonary stenosis, stenosis of medium and large arteries, hypertension), cognitive and developmental delay, growth abnormalities, endocrine abnormalities (hypercalcemia, hypercalciuria, hypothyroidism and early puberty) and connective tissue abnormalities.75–77 Williams syndrome is caused by a microdeletion on chromosome 7q11.23, a region containing 26–28 genes including ELN gene encoding the elastin.78
Hemizygosity of the ELN gene has been demonstrated to be responsible for the vascular pathology in WS because of reduced elasticity of the arterial tree and increased arterial stiffness and these mutations were found in individuals with MAS and without other Williams syndrome phenotypic features.16,79,80 The role of elastin in modulation, proliferation and migration of vascular smooth cells is impaired in WS, and the result of its impaired function is represented by the occlusion of the vascular lumen.81
Vascular abnormalities are present in more than 80% of the patients with WS and involve thoracic and abdominal aorta, renal arteries, mesenteric arteries, coronary arteries and intracranial vessels.79,82,83
Rose et al found a specific morphology of the aorta in patients with WS and MAS with suprarenal narrowing, stenosis of the renal arteries and increased diameter of the infrarenal lumen. The smallest diameter of the aorta is close to the origin of the renal artery, consisted in renal artery stenosis.27
Figure 3 shows thoracic and abdominal CT angiogram revealing reduced calibre of the abdominal aorta and narrowed renal arteries in a 3-month-old infant with WS.
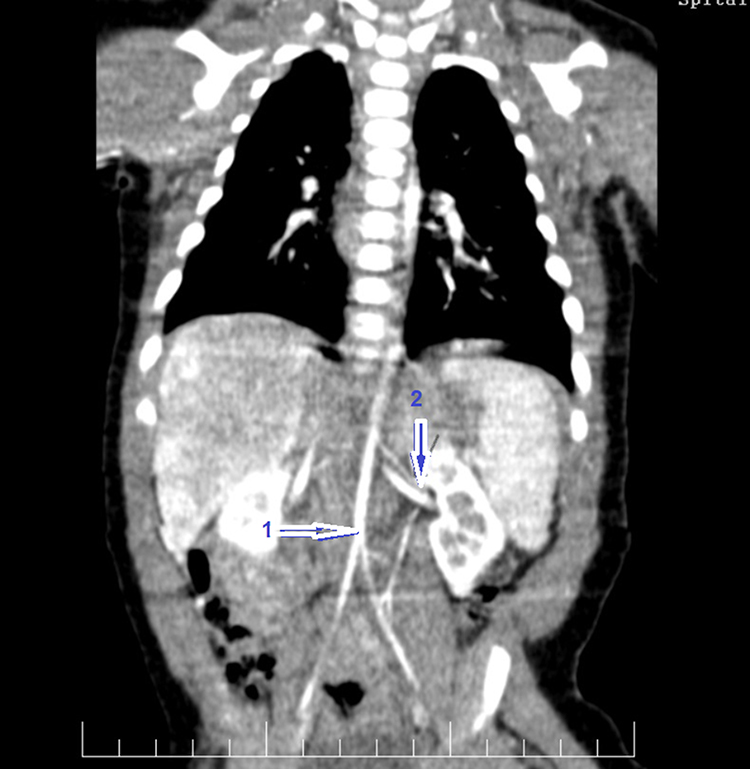 |
Figure 3 Thoracic and abdominal CT angiogram revealing reduced caliber of the abdominal aorta (Z score −3.8) – (arrow 1) and narrowed left renal artery (diameter < 1mm) – (arrow 2) in a 3-month-old boy with WS. |
The frequency of MAS in WS is variable in different studies, ranging 2% to 70%.8,27,56 There is evidence that moderate and severe vascular lesions may have rapid progression over short periods of time.8,57,74,83,84 Arterial hypertension was found in 22–50% of the patients with WS.27,82,83
CT angiography and magnetic resonance angiography are used to diagnose MAS in children with WS and periodic imaging techniques are required in patients with moderate and severe vascular abnormalities characterized by progressive evolution.85,86 Although hypertension in WS patients has been classically attributed to the renal vascular disease, stenosis of other different vessels must be always considered.
Therapeutic options for vascular lesions in patients with WS include medical therapy, endovascular and surgical intervention. The treatment of systemic hypertension in MAS patients with WS includes calcium channel blockers, beta-blockers and angiotensin-converting enzyme inhibitors, and medical therapy is indicated in small children or in patients with unacceptable level of operative risk. There are little data about the interventional treatment and the timing of surgical intervention is controversial in children. Combined antihypertensive treatment alone was used in half of children with MAS and WS. Interventional treatment was used in the other half of the patients and in 20% of the patients vascular angioplasty was associated with renal auto-transplant. Medical treatment, as has been demonstrated in other studies, has better results than interventional procedures.1,56
Alagille Syndrome
Alagille syndrome (ALGS) is a rare autosomal dominant, multisystem disorder resulting from mutations in two genes associated with the Notch signaling pathways: JAGGED1 in most cases and NOTCH2 in a minority of cases determining abnormal development of the intrahepatic bile ducts.87–91 Clinical findings consist in bile duct paucity associated with chronic cholestasis, cardiovascular abnormalities typically peripheral pulmonary artery stenosis, skeletal abnormalities (butterfly vertebrae), ophthalmologic anomalies (posterior embryotoxon), renal anomalies, vascular involvement, and characteristic dysmorphic features.60,92
ALGS is encountered in 1–8% of MAS cases.1,3,16 Vascular abnormalities in ALGS include intracranial vascular abnormalities as aneurysm or moyamoya disease, which may lead to intracranial bleeding in up to 15% of cases and represents a major cause of morbidity and mortality in this disorder.10,90 Other arteries involved are pulmonary arteries, aorta, renal, celiac, mesenteric, subclavian and carotid arteries.91 MAS was found in 1–2.4% of the individuals diagnosed with ALGS in a large study, but the association between ALGS and MAS is underestimated.10,90,93 Superior mesenteric artery and celiac trunk are most often involved (Table 5). Median arcuate ligament syndrome was found to be involved in visceral artery stenoses in patients with ALGS.10
The association between ALGS and MAS is not incidental. Developmental and molecular studies have been demonstrated JAG1 and Notch signalling pathway in vascular development and expression of JAG1 was found in all major arteries during embryogenesis.6,60,94 Mutations of JAG1 and Notch regulation signalling defect will determine defects in angiogenic vascular remodelling and abnormal vessel structure.61,87,95–97 Aortic coarctation and visceral branches stenosis in ALGS are caused by myo-intimal hyperplasia of the vascular wall.62
Whole exome sequencing studies revealed that protein truncating mutations, which usually are associated with other clinical features of ALGS or even missense mutations which may not have additional symptoms of ALGS may result in MAS.16
CT angiography and magnetic resonance angiography are used to diagnose MAS in children with ALGS.63,64,87 Because the vascular disease can be progressive, periodic imaging by ultrasound or angiographic techniques are required.10
Treatment of MAS in patients with ALGS includes medical therapy, endovascular and surgical intervention. Invasive procedure can be difficult because of presence of thick and fibrous media of the vessels.10 Medical treatment was preferred as the only option in 40% of the children with MAS and ALGS and hypertension was controlled in 67% of the cases (Table 5). In 13.3% of the cases, combined interventional therapy was necessary (angioplasty, aortic bypass, TESLA and renal auto-transplant).
Tuberous Sclerosis
Tuberous sclerosis (TS) is a genetic disorder inherited in an autosomal dominant fashion resulting from mutations in the genes TSC1 and TSC2 that affect multiple systems: brain, retina, kidneys, heart, skin and vascular. Diagnosis criteria consist in genetic criteria (identification of either TSC1 or TSC2 pathogenic mutation) and clinical criteria: major features (hypomelanotic macules, angiofibroma, ungual fibromas, Shagreen patch, multiple retinal hamartomas, cortical dysplasia, subependymal nodules, subependymal giant cell astrocytoma, cardiac rhabdomyoma, lymphangioleiomyomatosis, angiomyolipoma) and minor features (“confetti” skin lesions, dental enamel pits, intraoral fibromas, retinal achromic patch, multiple renal cysts, nonrenal hamartoma).98,99 Vascular involvement in tuberous sclerosis consists in aneurysm of the aorta, pulmonary artery, intracranial arteries, subclavian and iliofemoral arteries and stenotic-occlusive lesions of the large and medium size arteries, affecting aorta, common iliac artery, renal artery, mesenteric artery, coronary artery and moya-moya disease.100–104 Abdominal aortic coarctation and renal artery stenosis have been reported most often either in isolation or as components of MAS in four pediatric patients.11,100
The pathogenesis of vascular disease in TS is unclear. Dysplastic and degenerative changes within the arterial wall, including fibrocytic and myofibrocytic intimal proliferation and medial hyperplasia, obliterative dysplasia have been described.101–104
Whole body or targeted vascular screening by duplex ultrasound, magnetic resonance angiography and CT angiography should be performed in TS patients with hypertension or signs or symptoms attributable to stenotic vascular lesions.11,105
Angioplasty represents a useful method of treatment for stenotic lesions in TS.11
Although hypertension in TS patients has been classically attributed to the renal parenchymal disease, vascular occlusive or stenotic disease must be considered.
Mucopolysaccharidoses (MPS)
Mucopolysaccharidoses are rare genetic lysosomal storage disorders, autosomal recessive or X-linked inherited caused by alterations in the functional enzymes, which degrade glycosaminoglycans (GAGs). Progressive pathological accumulation of glycosaminoglycans determines dysfunction of most organ-systems to different degrees, leading to a considerable heterogeneity in clinical presentation, both in age of onset of symptoms and severity.14,106 Somatic involvement includes facial dysmorphism, enlarged liver and spleen, hernia, stiff joints, respiratory infections, recurrent otitis, deafness, cardiac valve disease and neurological impairment.14,106–110 Cardiac involvement is common in MPS, is present at the early stage of the disease because of thickening of the valves’ leaflets and is progressive.107–109 The most affected valves are the mitral and aortic valves. GAGs infiltration of the myocardium can also be present. Great vessels may be affected by increased wall thickness.13,107 Diffuse narrowing of the thoracic and abdominal aorta has been reported in three patients with MPS type 1 (one patient with abdominal aorta stenosis and 2 patients with thoraco-abdominal aorta narrowing), in four patients with MPS type I and in one patient with MPS type VII and was correlated with increased arterial pressure.13–15
Conclusions
Genetic forms of MAS, although rare, represent an important cause of hypertension in children and should be considered in the differential diagnosis of hypertension in paediatric population with genetic disorders. The extent of vascular disease depends on the type of genetic disorder. MAS determined by genetic disorders is more often associated with suprarenal stenosis and extra-aortic involvement. Management of hypertension can be difficult, and each patient needs individualised therapy. Medical treatment is preferred in children with WS and ALGS and MAS. In terms of clinical practice, we recommend evaluating the children with MAS in detail because they may present other specific manifestations for certain genetic diseases, and on the other hand, it is necessary to detect the presence of MAS in children who were already diagnosed with certain genetic diseases.
Ethics Approval and Consent to Participate
All subjects gave their informed consent for inclusion before they participated in the study. The study was conducted in accordance with the Declaration of Helsinki and the patients gave their informed consent for using the results of imaging investigation.
Acknowledgments
We would like to express gratitude to all those who helped during the writing of this manuscript.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research received no external funding.
Disclosure
The authors declare no conflicts of interest for this work.
References
1. Rumman RK, Nickel C, Matsuda-Abedini M, et al. Disease beyond the arch: a systematic review of middle aortic syndrome in childhood. Am J Hypertens. 2015;28(7):833–846. doi:10.1093/ajh/hpu296
2. Forman N, Sinskey J, Shalabi A. A review of middle aortic syndromes in pediatric patients. J Cardiothorac Vasc Anesth. 2020;34(4):1042–1050. doi:10.1053/j.jvca.2019.07.130
3. Porras D, Stein DR, Ferguson MA, et al. Midaortic syndrome: 30 years of experience with medical, endovascular and surgical management. Pediatr Nephrol. 2013;28(10):2023–2033. doi:10.1007/s00467-013-2514-8
4. Sen PK, Kinare SG, Engineer SD, Parulkar GB. The middle aortic syndrome. Br Heart J. 1963;25(5):610–618. doi:10.1136/hrt.25.5.610
5. Connolly JE, Wilson SE, Lawrence PL, Fujitani RM. Middle aortic syndrome: distal thoracic and abdominal coarctation, a disorder with multiple etiologies. J Am Coll Surg. 2002;194(6):774–781. doi:10.1016/s1072-7515(02)01144-4
6. Onat T, Zeren E. Coarctation of the abdominal aorta. Review of 91 cases. Cardiologia. 1969;54(3):140–157. doi:10.1159/000166249
7. Tummolo A, Marks SD, Stadermann M, et al. Mid-aortic syndrome: long-term outcome of 36 children. Pediatr Nephrol. 2009;24(11):2225–2232. doi:10.1007/s00467-009-1242-6
8. Israel G, Krinsky G, Lee V. The “skinny aorta”. Clin Imaging. 2002;26(2):116–121. doi:10.1016/s0899-7071(01)00368-0
9. Booth C, Preston R, Clark G, Reidy J. Management of renal vascular disease in neurofibromatosis type 1 and the role of percutaneous transluminal angioplasty. Nephrol Dial Transplant. 2002;17(7):1235–1240. doi:10.1093/ndt/17.7.1235
10. Salem JE, Bruguiere E, Iserin L, Guiochon-Mantel A, Plouin PF. Hypertension and aortorenal disease in Alagille syndrome. J Hypertens. 2012;30(7):1300–1306. doi:10.1097/HJH.0b013e3283531e1f
11. Salerno AE, Marsenic O, Meyers KE, Kaplan BS, Hellinger JC. Vascular involvement in tuberous sclerosis. Pediatr Nephrol. 2010;25(8):1555–1561. doi:10.1007/s00467-010-1466-5
12. Delis KT, Gloviczki P. Middle aortic syndrome: from presentation to contemporary open surgical and endovascular treatment. Perspect Vasc Surg Endovasc Ther. 2005;17(3):187–203. doi:10.1177/153100350501700302
13. Taylor DB, Blaser SI, Burrows PE, Stringer DA, Clarke JT, Thorner P. Arteriopathy and coarctation of the abdominal aorta in children with mucopolysaccharidosis: imaging findings. AJR Am J Roentgenol. 1991;157(4):819–823. doi:10.2214/ajr.157.4.1909834
14. Wraith JE, Beck M, Giugliani R, Clarke J, Martin R, Muenzer J; HOS Investigators. Initial report from the Hunter Outcome Survey. Genet Med. 2008;10(7):508–516. doi:10.1097/gim.0b013e31817701e6
15. Beaudet AL, DiFerrante NM, Ferry GD, Nichols BL, Mullins CE. Variation in the phenotypic expression of beta-glucuronidase deficiency. J Pediatr. 1975;86(3):388–394. doi:10.1016/s0022-3476(75)80968-1
16. Warejko JK, Schueler M, Vivante A, et al. Whole exome sequencing reveals a monogenic cause of disease in ≈43% of 35 families with midaortic syndrome. Hypertension. 2018;71(4):691–699. doi:10.1161/HYPERTENSIONAHA.117.10296
17. Panayiotopoulos YP, Tyrrell MR, Koffman G, Reidy JF, Haycock GB, Taylor PR. Mid-aortic syndrome presenting in childhood. Br J Surg. 1996;83(2):235–240.
18. Sethna CB, Kaplan BS, Cahill AM, Velazquez OC, Meyers KE. Idiopathic mid-aortic syndrome in children. Pediatr Nephrol. 2008;23(7):1135–1142. doi:10.1007/s00467-008-0767-4
19. Patel RS, Nguyen S, Lee MT, et al. Clinical characteristics and long-term outcomes of midaortic syndrome. Ann Vasc Surg. 2020;66:318–325. doi:10.1016/j.avsg.2019.12.039
20. Patel PA, Cahill AM. Renovascular hypertension in children. CVIR Endovasc. 2021;4(1):10. doi:10.1186/s42155-020-00176-5
21. Kim YS, Cho YH, Sung K, et al. Clinical outcome of extraanatomic bypass for midaortic syndrome caused by takayasu arteritis. Ann Thorac Surg. 2020;109(5):1419–1425. doi:10.1016/j.athoracsur.2019.08.032
22. Daniels SR, Loggie JM, McEnery PT, Towbin RB. Clinical spectrum of intrinsic renovascular hypertension in children. Pediatrics. 1987;80(5):698–704. doi:10.1542/peds.80.5.698
23. Limbacher JP, Hill ME, Janicki PC. Hypoplasia of the abdominal aorta associated with rubella syndrome. South Med J. 1979;72(5):617–619. doi:10.1097/00007611-197905000-00032
24. Chong DST, Constantinou J, Davis M, Hamilton G. Calcification of a synthetic renovascular graft in a child. EJVES Short Rep. 2016;33:13–15. doi:10.1016/j.ejvssr.2016.06.001
25. Sutton EJ, Tong RT, Gillis AM, et al. Decreased aortic growth and middle aortic syndrome in patients with neuroblastoma after radiation therapy. Pediatr Radiol. 2009;39(11):1194–1202. doi:10.1007/s00247-009-1351-1
26. Espinoza JL, Ai S, Matsumura I. New insights on the pathogenesis of takayasu arteritis: revisiting the microbial theory. Pathogens. 2018;7(3):73. doi:10.3390/pathogens7030073
27. Rose C, Wessel A, Pankau R, Partsch CJ, Bürsch J. Anomalies of the abdominal aorta in Williams-Beuren syndrome–another cause of arterial hypertension. Eur J Pediatr. 2001;160(11):655–658. doi:10.1007/s004310100835
28. Izraelit A, Kim M, Ratner V, Levasseur SM, Seigle R, Krishnamurthy G. Mid-aortic syndrome in two preterm infants. J Perinatol. 2012;32(5):390–392. doi:10.1038/jp.2011.130
29. Ten Dam K, van der Palen RL, Tanke RB, Schreuder MF, de Jong H. Clinical recognition of mid-aortic syndrome in children. Eur J Pediatr. 2013;172(3):413–416. doi:10.1007/s00431-012-1800-y
30. Lurbe E, Agabiti-Rosei E, Cruickshank JK, et al. 2016 European Society of Hypertension guidelines for the management of high blood pressure in children and adolescents. J Hypertens. 2016;34(10):1887–1920. doi:10.1097/HJH.0000000000001039
31. Flynn JT, Kaelber DC, Baker-Smith CM, et al.; Subcommittee on Screening and Management of High Blood Pressure in Children. Clinical practice guideline for screening and management of high blood pressure in children and adolescents. Pediatrics. 2017;140(3):e20171904. doi:10.1542/peds.2017-1904
32. Mir A, Stam B, Sperrazza C. A rare cause of cardiomyopathy in an infant: middle aortic syndrome. Cardiol Young. 2017;27(4):794–796. doi:10.1017/S1047951116002018
33. Villegas L, Cahill AM, Meyers K. Pediatric renovascular hypertension: manifestations and management. Indian Pediatr. 2020;57(5):443–451. doi:10.1007/s13312-020-1820-z
34. Zartner P, Hart C, Schneider MBE. Severe midaortic syndrome: a stepwise approach to treatment with drug-eluting balloons: a case report. Eur Heart J Case Rep. 2019;3(1):ytz017. doi:10.1093/ehjcr/ytz017
35. Kim SM, Jung IM, Han A, et al. Surgical treatment of middle aortic syndrome with Takayasu arteritis or midaortic dysplastic syndrome. Eur J Vasc Endovasc Surg. 2015;50(2):206–212. doi:10.1016/j.ejvs.2015.04.032
36. Kim HJ, Choi JW, Hwang HY, Ahn H. Extra-anatomic ascending aorta to abdominal aorta bypass in takayasu arteritis patients with mid-aortic syndrome. Korean J Thorac Cardiovasc Surg. 2017;50(4):270–274. doi:10.5090/kjtcs.2017.50.4.270
37. Barral X, de Latour B, Vola M, Lavocat MP, Fichtner C, Favre JP. Surgery of the abdominal aorta and its branches in children: late follow-up. J Vasc Surg. 2006;43(6):1138–1144. doi:10.1016/j.jvs.2006.01.033
38. Go MR, Bhende S, Smead WL, Vaccaro PS. Long-term complications in two patients after aortoaortic bypass for midaortic syndrome. Ann Vasc Surg. 2013;27(4):
39. Kim HB, Vakili K, Ramos-Gonzalez GJ, et al. Tissue expander-stimulated lengthening of arteries for the treatment of midaortic syndrome in children. J Vasc Surg. 2018;67(6):1664–1672. doi:10.1016/j.jvs.2017.09.052
40. Kim HB, Lee EJ, Vakili K, et al. Mesenteric Artery Growth Improves Circulation (MAGIC) in midaortic syndrome. Ann Surg. 2018;267(6):e109–e111. doi:10.1097/SLA.0000000000002540
41. Stadermann MB, Montini G, Hamilton G, et al. Results of surgical treatment for renovascular hypertension in children: 30 year single centre experience. Nephrol Dial Transplant. 2010;25(3):807–813. doi:10.1093/ndt/gfp537
42. Peker O, Aki FT, Kumbasar U, et al. Surgical management of renovascular hypertension in children and young adults: a 13-year experience. Interact Cardiovasc Thorac Surg. 2019;29(5):746–752. doi:10.1093/icvts/ivz157
43. Liberati A, Altman DG, Tetzlaff J, et al. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: explanation and elaboration. PLoS Med. 2009;6(7):e1000100. doi:10.1371/journal.pmed.1000100
44. Kaas B, Huisman TA, Tekes A, Bergner A, Blakeley JO, Jordan LC. Spectrum and prevalence of vasculopathy in pediatric neurofibromatosis type 1. J Child Neurol. 2013;28(5):561–569. doi:10.1177/0883073812448531
45. Oderich GS, Sullivan TM, Bower TC, et al. Vascular abnormalities in patients with neurofibromatosis syndrome type I: clinical spectrum, management, and results. J Vasc Surg. 2007;46(3):475–484. doi:10.1016/j.jvs.2007.03.055
46. Kurien A, John PR, Milford DV. Hypertension secondary to progressive vascular neurofibromatosis. Arch Dis Child. 1997;76(5):454–455. doi:10.1136/adc.76.5.454
47. Petrak B, Bendova S, Seeman T, et al. Mid-aortic syndrome with renovascular hypertension and multisystem involvement in a girl with familiar neurofibromatosis von Recklinghausen type 1. Neuro Endocrinol Lett. 2007;28(6):734–738.
48. Saif I, Seriki D, Moore R, Woywodt A. Midaortic syndrome in neurofibromatosis type 1 resulting in bilateral renal artery stenosis. Am J Kidney Dis. 2010;56(6):1197–1201. doi:10.1053/j.ajkd.2010.04.023
49. Gogou M, Keivanidou A, Giannopoulos A. Aortic dysfunction along with subaortic ridge in a patient with neurofibromatosis type 1 and a history of midaortic syndrome. Hellenic J Cardiol. 2018;59(6):362–364. doi:10.1016/j.hjc.2018.01.003
50. Humbert J, Roussey-Kesler G, Guerin P, et al. Diagnostic and medical strategy for renovascular hypertension: report from a monocentric pediatric cohort. Eur J Pediatr. 2015;174(1):23–32. doi:10.1007/s00431-014-2355-x
51. Jóźwik-Plebanek K, Prejbisz A, Januszewicz A, Madej K, Litwin M, Januszewicz M. A 21-year-old woman with neurofibromatosis type 1, mid-aortic syndrome, and hypertension. Pol Arch Intern Med. 2019;129(2):131–132. doi:10.20452/pamw.4393
52. Lazea C, Asavoaie C, Al-Khzouz C, Popa L. Rare complications of neurofibromatosis 1 diagnosed incidentally in two children. Ther Clin Risk Manag. 2018;14:1547–1552. doi:10.2147/TCRM.S173237
53. Stabouli S, Vargiami E, Maliachova O, et al. Arterial stiffness in a toddler with neurofibromatosis type 1 and refractory hypertension. Case Rep Pediatr. 2018;2018:5957987. doi:10.1155/2018/5957987
54. Veean S, Thakkar N, Gupta S, Keshavamurthy J. A case of coarctation of the abdominal aorta and renal artery stenosis due to neurofibromatosis type 1. Postgrad Med J. 2017;93(1098):235–236. doi:10.1136/postgradmedj-2016-134460
55. West CA, Delis KT, Service GJ, Driscoll DJ, McPhail IR, Gloviczki P. Middle aortic syndrome: surgical treatment in a child with neurofibromatosis. J Vasc Surg. 2005;42(6):1236. doi:10.1016/j.jvs.2004.07.031
56. Sumboonnanonda A, Robinson BL, Gedroyc WM, Saxton HM, Reidy JF, Haycock GB. Middle aortic syndrome: clinical and radiological findings. Arch Dis Child. 1992;67(4):501–505. doi:10.1136/adc.67.4.501
57. Hall EK, Glatz J, Kaplan P, et al. A case report of rapid progressive coarctation and severe middle aortic syndrome in an infant with Williams syndrome. Congenit Heart Dis. 2009;4(5):373–377. doi:10.1111/j.1747-0803.2009.00287.x
58. Byoun JT, Cho JY, Yun KH, Rhee SJ, Yu ST, Oh SJ. Mid-aortic syndrome in Williams-Beuren syndrome with an atypical small-sized deletion of chromosome 7q11.23 misdiagnosed as takayasu arteritis. Int Heart J. 2021;62(1):207–210. doi:10.1536/ihj.20-495
59. Opoka-Winiarska V, Tomaszek MB, Sobiesiak A, et al. The importance of FDG PET/CT in the diagnostic process of the middle aortic syndrome in a 15-year-old boy patient with suspected systemic vasculitis and final diagnosis of Williams-Beuren syndrome. Rheumatol Int. 2020;40(8):1309–1316. doi:10.1007/s00296-020-04550-3
60. Quiros-Tejeira RE, Ament ME, Heyman MB, et al. Variable morbidity in Alagille syndrome: a review of 43 cases. J Pediatr Gastroenterol Nutr. 1999;29(4):431–437. doi:10.1097/00005176-199910000-00011
61. Raas-Rothschild A, Shteyer E, Lerer I, Nir A, Granot E, Rein AJ. Jagged1 gene mutation for abdominal coarctation of the aorta in Alagille syndrome. Am J Med Genet. 2002;112(1):75–78. doi:10.1002/ajmg.10652
62. Bérard E, Sarles J, Triolo V, et al. Renovascular hypertension and vascular anomalies in Alagille syndrome. Pediatr Nephrol. 1998;12(2):121–124. doi:10.1007/s004670050418
63. Shefler AG, Chan MK, Ostman-Smith I. Middle aortic syndrome in a boy with arteriohepatic dysplasia (Alagille syndrome). Pediatr Cardiol. 1997;18(3):232–234. doi:10.1007/s002469900160
64. Yokoyama K, Minami T, Seki M, Okada Y, Kumagai H, Yamagata T. A boy with Alagille syndrome coexisting with mid-aortic syndrome and renovascular hypertension. J Cardiol Cases. 2019;21(1):28–31. doi:10.1016/j.jccase.2019.09.010
65. Kayhan A, Ilkhchoui Y, Venu N, Jensen DM, Oto A. Multiple abdominal vascular anomalies in a patient with Alagille syndrome. J Vasc Interv Radiol. 2010;21(6):937–940. doi:10.1016/j.jvir.2010.02.007
66. Quek SC, Tan L, Quek ST, Yip W, Aw M, Quak SH. Abdominal coarctation and Alagille syndrome. Pediatrics. 2000;106(1):E9. doi:10.1542/peds.106.1.e9
67. Yucel H, Hoorntje SJ, Bravenboer B. Renal abnormalities in a family with Alagille syndrome. Neth J Med. 2010;68(1):38–39.
68. Ferner RE, Huson SM, Thomas N, et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet. 2007;44(2):81–88. doi:10.1136/jmg.2006.045906
69. Xu J, Ismat FA, Wang T, Yang J, Epstein JA. NF1 regulates a Ras-dependent vascular smooth muscle proliferative injury response. Circulation. 2007;116(19):2148–2156. doi:10.1161/CIRCULATIONAHA.107.707752
70. Sebastià C, Quiroga S, Boyé R, Perez-Lafuente M, Castellà E, Alvarez-Castells A. Aortic stenosis: spectrum of diseases depicted at multisection CT. Radiographics. 2003;23:S79–91. doi:10.1148/rg.23si035506
71. Srinivasan A, Krishnamurthy G, Fontalvo-Herazo L, et al. Angioplasty for renal artery stenosis in pediatric patients: an 11-year retrospective experience. J Vasc Interv Radiol. 2010;21(11):1672–1680. doi:10.1016/j.jvir.2010.07.012
72. Hamilton SJ, Friedman JM. Insights into the pathogenesis of neurofibromatosis 1 vasculopathy. Clin Genet. 2000;58(5):341–344. doi:10.1034/j.1399-0004.2000.580501.x
73. Friedman JM, Arbiser J, Epstein JA, et al. Cardiovascular disease in neurofibromatosis 1: report of the NF1 Cardiovascular Task Force. Genet Med. 2002;4(3):105–111. doi:10.1097/00125817-200205000-00002
74. Hetzer R, Absi D, Miera O, et al. Extraanatomic bypass technique for the treatment of midaortic syndrome in children. Ann Thorac Surg. 2013;96(1):183–189. doi:10.1016/j.athoracsur.2013.03.025
75. Kaplan P, Wang PP, Francke U. Williams (Williams Beuren) syndrome: a distinct neurobehavioral disorder. J Child Neurol. 2001;16(3):177–190. doi:10.1177/088307380101600305
76. Eronen M, Peippo M, Hiippala A, et al. Cardiovascular manifestations in 75 patients with Williams syndrome. J Med Genet. 2002;39(8):554–558. doi:10.1136/jmg.39.8.554
77. Pober BR. Williams-Beuren syndrome. N Engl J Med. 2010;362(3):239–252. doi:10.1056/NEJMra0903074
78. Nickerson E, Greenberg F, Keating MT, McCaskill C, Shaffer LG. Deletions of the elastin gene at 7q11.23 occur in approximately 90% of patients with Williams syndrome. Am J Hum Genet. 1995;56(5):1156–1161.
79. Keating MT. Genetic approaches to cardiovascular disease. Supravalvular aortic stenosis, Williams syndrome, and long-QT syndrome. Circulation. 1995;92(1):142–147. doi:10.1161/01.cir.92.1.142
80. Salaymeh KJ, Banerjee A. Evaluation of arterial stiffness in children with Williams syndrome: does it play a role in evolving hypertension? Am Heart J. 2001;142(3):549–555. doi:10.1067/mhj.2001.116763
81. Karnik SK, Brooke BS, Bayes-Genis A, et al. A critical role for elastin signaling in vascular morphogenesis and disease. Development. 2003;130(2):411–423. doi:10.1242/dev.00223
82. Del Pasqua A, Rinelli G, Toscano A, et al. New findings concerning cardiovascular manifestations emerging from long-term follow-up of 150 patients with the Williams-Beuren-Beuren syndrome. Cardiol Young. 2009;19(6):563–567. doi:10.1017/S1047951109990837
83. Collins RT. Cardiovascular disease in Williams syndrome. Circulation. 2013;127(21):2125–2134. doi:10.1161/CIRCULATIONAHA.112.000064
84. Arrington C, Tristani-Firouzi M, Puchalski M. Rapid progression of long-segment coarctation in a patient with Williams’ syndrome. Cardiol Young. 2005;15(3):312–314. doi:10.1017/S104795110500065X
85. Gray JC, Krazinski AW, Schoepf UJ, et al. Cardiovascular manifestations of Williams syndrome: imaging findings. J Cardiovasc Comput Tomogr. 2013;7(6):400–407. doi:10.1016/j.jcct.2013.11.007
86. Das KM, Momenah TS, Larsson SG, Jadoon S, Aldosary AS, Lee EY. Williams-Beuren syndrome: computed tomography imaging review. Pediatr Cardiol. 2014;35(8):1309–1320. doi:10.1007/s00246-014-0998-z
87. Turnpenny PD, Ellard S. Alagille syndrome: pathogenesis, diagnosis and management. Eur J Hum Genet. 2012;20(3):251–257. doi:10.1038/ejhg.2011.181
88. McDaniell R, Warthen DM, Sanchez-Lara PA, et al. NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch signaling pathway. Am J Hum Genet. 2006;79(1):169–173. doi:10.1086/505332
89. Li L, Krantz ID, Deng Y, et al. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet. 1997;16(3):243–251. doi:10.1038/ng0797-243
90. Kamath BM, Baker A, Houwen R, Todorova L, Kerkar N. Systematic review: the epidemiology, natural history, and burden of Alagille syndrome. J Pediatr Gastroenterol Nutr. 2018;67(2):148–156. doi:10.1097/MPG.0000000000001958
91. Kamath BM, Spinner NB, Emerick KM, et al. Vascular anomalies in Alagille syndrome: a significant cause of morbidity and mortality. Circulation. 2004;109(11):1354–1358. doi:10.1161/01.CIR.0000121361.01862.A4
92. Ayoub MD, Kamath BM. Alagille syndrome: diagnostic challenges and advances in management. Diagnostics (Basel). 2020;10(11):907. doi:10.3390/diagnostics10110907
93. Kamath BM, Podkameni G, Hutchinson AL, et al. Renal anomalies in Alagille syndrome: a disease-defining feature. Am J Med Genet A. 2012;158A(1):85–89. doi:10.1002/ajmg.a.34369
94. Shutter JR, Scully S, Fan W, et al. Dll4, a novel Notch ligand expressed in arterial endothelium. Genes Dev. 2000;14(11):1313–1318. doi:10.1101/gad.14.11.1313
95. Villa N, Walker L, Lindsell CE, Gasson J, Iruela-Arispe ML, Weinmaster G. Vascular expression of Notch pathway receptors and ligands is restricted to arterial vessels. Mech Dev. 2001;108(1–2):161–164. doi:10.1016/s0925-4773(01)00469-5
96. Jones EA, Clement-Jones M, Wilson DI. JAGGED1 expression in human embryos: correlation with the Alagille syndrome phenotype. J Med Genet. 2000;37(9):658–662. doi:10.1136/jmg.37.9.658
97. Leimeister C, Schumacher N, Steidl C, Gessler M. Analysis of HeyL expression in wild-type and Notch pathway mutant mouse embryos. Mech Dev. 2000;98(1–2):175–178. doi:10.1016/s0925-4773(00)00459-7
98. Hinton RB, Prakash A, Romp RL, Krueger DA, Knilans TK; International Tuberous Sclerosis Consensus Group. Cardiovascular manifestations of tuberous sclerosis complex and summary of the revised diagnostic criteria and surveillance and management recommendations from the International Tuberous Sclerosis Consensus Group. J Am Heart Assoc. 2014;3(6):e001493. doi:10.1161/JAHA.114.001493
99. Uysal SP, Şahin M. Tuberous sclerosis: a review of the past, present, and future. Turk J Med Sci. 2020;50(SI–2):1665–1676. doi:10.3906/sag-2002-133
100. Flynn PM, Robinson MB, Stapleton FB, Roy S, Koh G, Tonkin IL. Coarctation of the aorta and renal artery stenosis in tuberous sclerosis. Pediatr Radiol. 1984;14(5):337–339. doi:10.1007/BF01601889
101. Jost CJ, Gloviczki P, Edwards WD, Stanson AW, Joyce JW, Pairolero PC. Aortic aneurysms in children and young adults with tuberous sclerosis: report of two cases and review of the literature. J Vasc Surg. 2001;33(3):639–642. doi:10.1067/mva.2001.111976
102. Moon SB, Shin WY, Park YJ, Kim SJ. An abdominal aortic aneurysm in an 8-month-old girl with tuberous sclerosis. Eur J Vasc Endovasc Surg. 2009;37(5):569–571. doi:10.1016/j.ejvs.2009.01.002
103. Carette MF, Antoine M, Bazelly B, Cadranel J, Khalil A. Primary pulmonary artery aneurysm in tuberous sclerosis: CT, angiography and pathological study. Eur Radiol. 2006;16(10):2369–2370. doi:10.1007/s00330-006-0173-x
104. Rolfes DB, Towbin R, Bove KE. Vascular dysplasia in a child with tuberous sclerosis. Pediatr Pathol. 1985;3(2–4):359–373. doi:10.3109/15513818509078795
105. Morin CE, Morin NP, Franz DN, Krueger DA, Trout AT, Towbin AJ. Thoracoabdominal imaging of tuberous sclerosis. Pediatr Radiol. 2018;48(9):1307–1323. doi:10.1007/s00247-018-4123-y
106. Wraith JE. The mucopolysaccharidoses: a clinical review and guide to management. Arch Dis Child. 1995;72(3):263–267. doi:10.1136/adc.72.3.263
107. Muenzer J, Beck M, Eng CM, et al. Multidisciplinary management of Hunter syndrome. Pediatrics. 2009;124(6):e1228–39. doi:10.1542/peds.2008-0999
108. Lachman R, Martin KW, Castro S, Basto MA, Adams A, Teles EL. Radiologic and neuroradiologic findings in the mucopolysaccharidoses. J Pediatr Rehabil Med. 2010;3(2):109–118. doi:10.3233/PRM-2010-0115
109. Braunlin EA, Harmatz PR, Scarpa M, et al. Cardiac disease in patients with mucopolysaccharidosis: presentation, diagnosis and management. J Inherit Metab Dis. 2011;34(6):1183–1197. doi:10.1007/s10545-011-9359-8
110. Alkhzouz C, Lazea C, Bucerzan S, et al. Clinical and genetic characteristics of Romanian patients with mucopolysaccharidosis type II. JIMD Rep. 2017;33:19–25. doi:10.1007/8904_2016_535
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.