Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 14
Diagnosing Alpha-1-Antitrypsin Deficiency Using A PCR/Luminescence-Based Technology
Authors Veith M, Klemmer A, Anton I ![]() , El Hamss R
, El Hamss R ![]() , Rapun N, Janciauskiene S
, Rapun N, Janciauskiene S ![]() , Kotke V, Herr C
, Kotke V, Herr C ![]() , Bals R, Vogelmeier CF, Greulich T
, Bals R, Vogelmeier CF, Greulich T ![]()
Received 22 July 2019
Accepted for publication 21 October 2019
Published 18 November 2019 Volume 2019:14 Pages 2535—2542
DOI https://doi.org/10.2147/COPD.S224221
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Richard Russell
Martina Veith,1 Andreas Klemmer,1 Iker Anton,2 Rachid El Hamss,2 Noelia Rapun,2 Sabina Janciauskiene,3 Viktor Kotke,1 Christian Herr,4 Robert Bals,4 Claus Franz Vogelmeier,1 Timm Greulich1
1Department of Medicine, Pulmonary and Critical Care Medicine, Member of the German Center for Lung Research Marburg, University Medical Center Giessen And Marburg, Germany; 2Progenika Biopharma, S.A. A Grifols Company, Derio, Bizkaia, Spain; 3Department of Respiratory Medicine, Member of the German Center for Lung Research (DZL), Hannover Medical School, Biomedical Research in End Stage and Obstructive Lung Disease Hannover (BREATH), Hannover 30625, Germany; 4Department of Internal Medicine V, Pulmonology, Allergology, Respiratory and Intensive Care Medicine, Saarland Hospital, Homburg/Saar, Germany
Correspondence: Martina Veith
Department of Medicine, Pulmonary and Critical Care Medicine, Member of the German Center for Lung Research Marburg, University Medical Center Giessen and Marburg, Germany Tel +49 642 1586 4723
Fax +49 642 158 6370
Email [email protected]
Purpose: Alpha-1-antitrypsin deficiency (AATD) is a rare hereditary condition resulting from the mutations in the SERPINA1 (serine protease inhibitor) gene and is characterized by low circulating levels of the alpha-1 antitrypsin (AAT) protein. The traditional algorithm for laboratory testing of AATD involves the analysis of AAT concentrations (nephelometry), phenotyping (isoelectric focusing, IEF), and genotyping (polymerase chain reaction, PCR); in selected cases, full sequencing of the SERPINA1 gene can be undertaken. New technologies arise that may make diagnosis easier and faster.
Methods: We developed and evaluated a new diagnostic algorithm based on Luminex xMAP (multi-analyte profiling) technology using Progenika A1AT Genotyping Test. In an initial learning phase, 1979 samples from individuals suspected of having AATD were examined by both, a traditional and a “new” algorithm. In a second phase, 1133 samples were analyzed with the Luminex xMAP only.
Results: By introducing a Luminex xMAP based algorithm, we were able to simultaneously identify 14 mutations in SERPINA1 gene (instead of two- S and Z-by using our old algorithm). Although the quantity of IEF assays remained unchanged, the nephelometric measurements and sequencing were reduced by 79% and 63.4%, respectively.
Conclusion: The new method is convenient, fast and user-friendly. The application of the Luminex xMAP technology can simplify and shorten the diagnostic workup of patients with suspected AATD.
Keywords: SERPINA1, diagnosis, Luminex xMAP technology, mutations
Introduction
Alpha-1-antitrypsin deficiency (AATD) is caused by mutations of the SERPINA1 gene. Up to date, more than 100 mutations within the SERPINA1 have been identified that induce a reduced level of AAT protein.1 The most common mutations are PI*Z (Glu342Lys) and PI*S (Glu264Val), each caused by a single nucleotide polymorphism. The carriers of AATD are at an increased risk of developing early-onset emphysema and consequently chronic obstructive pulmonary disease. Another common clinical manifestation of AATD is hepatic disease, and less frequently panniculitis and other skin diseases.
The mutations causing AATD are distributed worldwide and remain significantly under-diagnosed.2,3 Diagnosis is often delayed for several years and many patients remain yet to be identified.2–4 Early diagnosis is key for AATD-related disease control and treatment. The World Health Organisation recommends testing for AATD all COPD patients5 and the recent European Respiratory Society statement supports this recommendation.6 However, implementation of this proposal is limited.7 One of the reasons may be a time-consuming multi-step diagnostic workup.8 Traditionally, AATD is diagnosed by measuring the plasma concentration of AAT through nephelometry, the presence of the S- or Z-allele is determined by PCR and IEF is used to analyze samples with suspected mutations or to identify rare mutations (conventional workflow).9
The Luminex xMAP-technology is a combination of flow cytometry, encoding microspheres, lasers and digital signal processing which allows simultaneous, high-throughput and multiplex detection of up to 100 targets in protein or nucleic acid study.10 This technology is based on very small (5.6 micron) polystyrene beads which are color-coded and which are coupled to specific oligonucleotide probes.10 Denatured DNA products are hybridized to oligonucleotide probes coupled to color-coded beads.10 The beads are separated within a Luminex analyzer and interrogated with two lasers- one for the identification of the bead and the other for quantification of bound reporter fluorophore.10 Similar technology is used in cancer research11 or for the simultaneous detection of several respiratory viruses.12
Materials And Methods
Clinical Samples
We used dried blood spot (DBS) samples (AlphaKits® GE Healthcare Ltd, Cardiff, CF147YT, UK) shipped to our laboratory at the University of Marburg for the routine diagnosis of AATD. The physicians confirmed that patients approved and signed informed consent for the samples shipped to our laboratory for genetic analysis. Because our study reflects a retrospective analysis of those routine data, an ethics approval is not necessary.
In a phase 1 of the algorithm evaluation, we compared the results of 1979 samples with suspected AATD, analyzed by both, the conventional diagnostic algorithm (Figure 1A) and a modified algorithm based on the Luminex xMAP (Luminex Corporation, 12212 Technology Boulevard Austin, Texas) (Figure 1B). Phase 1 took place from July 2016 to May 2017. In a phase 2 (May 2017 until November 2017), the conventional PCR was not conducted any longer, and all 1133 AlphaKits were analyzed only by Luminex xMAP.
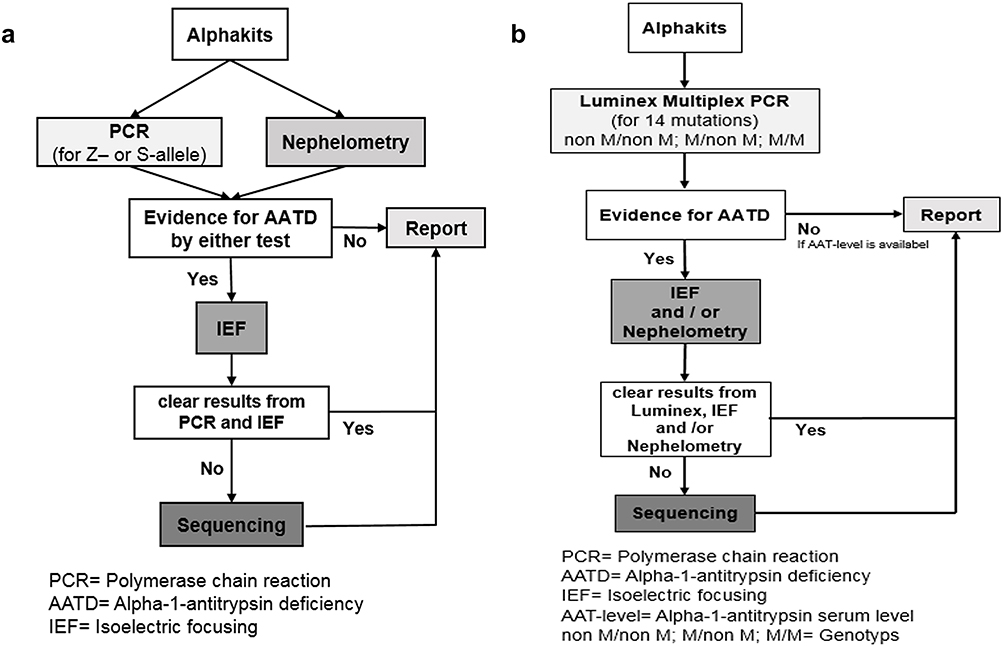 |
Figure 1 (A) The traditional workflow (modified after 9) and (B) the Luminex-based workflow. (A) If there were indications for the presence of AATD in the PCR (presence of Z- or S allele) or in the nephelometry (AAT level below a threshold) an IEF was performed. The analysis of 1979 samples by PCR and nephelometry revealed deviating results in 1220 samples (presence of Z- or S-allele; AAT-level <1.7 mg/dl). Out of 1220, in 93 samples no clear result was obtained from the IEF, and therefore these samples were sequenced. (B) Possible Luminex results (“non M/non M”, “M/non M” and “M/M”) and the following procedures depending on the results. The algorithm is based on two principles: To exclude AATD, Luminex result had to be negative and the AAT level had to be in the normal range. To diagnose heterozygote or homozygote deficiency, the result had to be confirmed on a second biological level (genes and proteins). Abbreviations: PCR, Polymerase chain reaction; AATD, Alpha-1-antitrypsin deficiency; IEF, Isoelectric focusing; AAT-level, Alpha-1-antitrypsin serum level non M/non M; M/non M; MM= Genotype. |
Combining both periods from July 2016 to November 2017, 818 senders mailed 3112 AlphaKits to our laboratory and all were analyzed.
Traditional Workflow
We used nephelometry for the semi quantitative determination of the plasma AAT levels and PCR for the detection of the S- and Z-allele, which was eventually followed by the IEF and gene sequencing. Two independent readers validated the results of PCR and IEF. We have described this workflow in detail elsewhere.9,13
Luminex-Based Workflow
All samples were first processed for the detection of the putative 14 mutations (Table 1). We established a workflow as follows:
 |
Table 1 Allelic Variants And Associated Alleles Which Were Tested With The AAT Genotyping Kit |
Testing Procedure
Genotyping was performed through Luminex. We defined three possible Luminex results (Figure 2A–C):
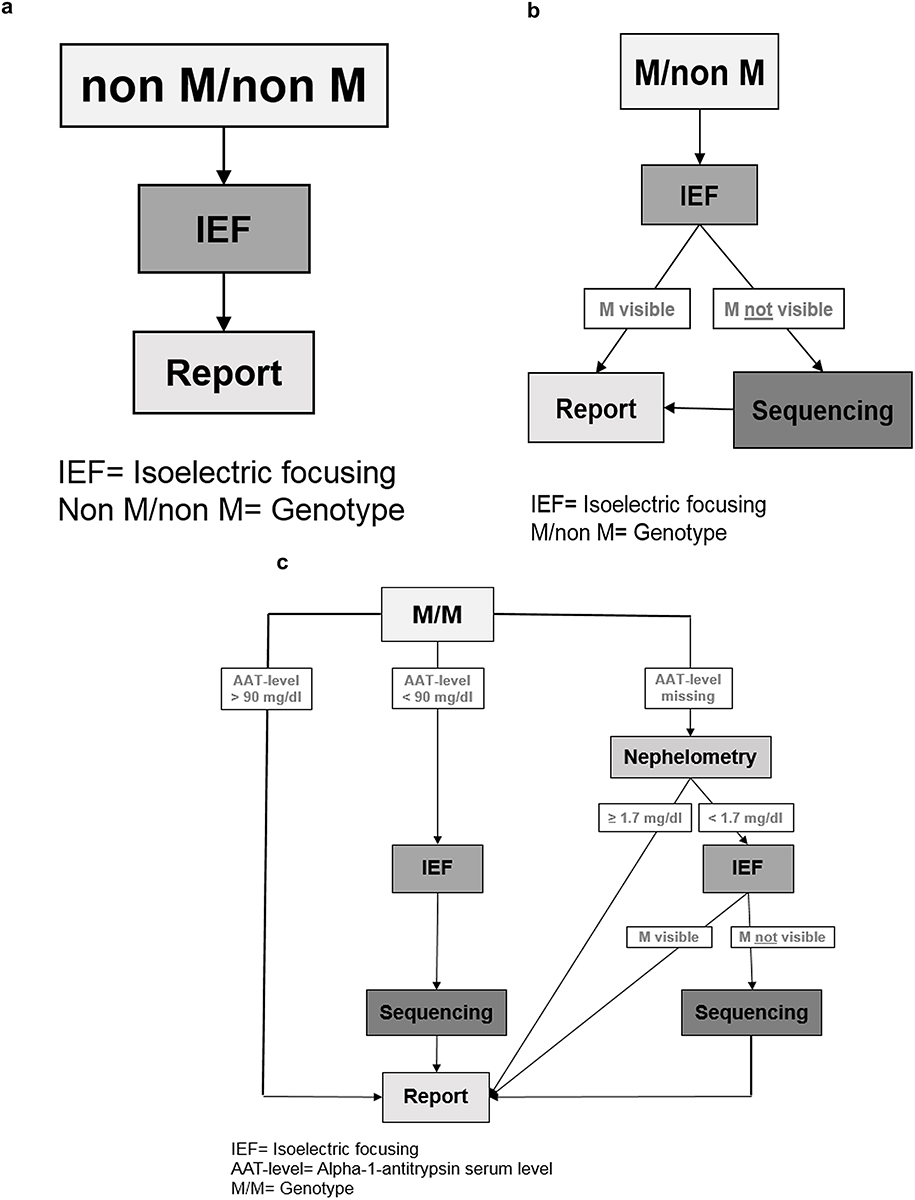 |
Figure 2 (A–C) Schematic of the Luminex-based workflow shown at this figure separately. (A) “Non M/non M” means that out of the 14 mutations which are listed in Table 1, a combination of heterozygote or homozygote form was detected. An additional phenotyping was performed through isoelectric focusing (IEF). (B) “M/non M”: M means that Luminex could not detect one of the 14 mutations (listed in Table1), so it was necessary to verify the results by IEF. “non M” means that one of the 14 mutations was detected. So a combination of M and one of the listed 14 mutations could be verified. (C) (1–3) “M/M” means no mutation could be detected. In this case the AAT-level analysis has a great importance. (C) (1) If the AAT-level was ≥ 90 mg/dl there was no need for further verification. (C) (2) If the AAT-level was < 90 mg/dl, IEF was performed to detect M-allele and samples were sequenced if M-allele was not found. (C) (3) If the external AAT-level was missing, the AAT-level (derived from dried blood spot (DBS)) was measured through nephelometry. If AAT level was below a corresponding serum level of 104 mg/dl, phenotyping was carried out. If there was no M allele visible through IEF, we sequenced the sample to get a final result. If there was an M allele visible through IEF the result was directly sent to the physician. If the result of nephelometry was ≥ 1.7 mg/dl a mutation was excluded, and the result was sent to the physician. Abbreviations: IEF, Isoelectric focusing M/non M; MM= Genotype; IEF, Isoelectric focusing; AAT-level, Alpha-1-antitrypsin serum level MM= Genotype |
The first was “nonM/nonM” (Figure 2A) meaning that any combination (heterozygote or homozygote) out of the 14 mutations listed in Table 1, was detected. To verify the result, the additional phenotyping was performed by IEF. Concordant results were reported to the physician.
The second was “M/non M” (Figure 2B). “M” means that Luminex could not detect any of the 14 mutations listed in Table 1 while “non M” means that one of the 14 mutations was detected. Thus, a combination of M allele with one of the listed 14 mutations was the suspected genotype. Afterwards, we performed phenotyping and if the M allele was visible in IEF (concordant to “M/non M”), the result was sent to the physician. If the M allele was not visible in the IEF, we sequenced the sample to achieve a final diagnosis. This result was then sent to the physician.
The third was “M/M” (Figure 2C[1–3]. With “M/M” we termed samples in which none of the 14 mutations was found. In that case, we followed this algorithm: if the reported (externally) measured serum AAT-level was > 90 mg/dl, a mutation was excluded, and the result was sent to the physician (Figure 2C[1]). However, if the serum level was < 90 mg/dl, we performed additional IEF and sequencing (Figure 2C[2]). If there was no external AAT-level, then the level of AAT (derived from dried blood spot, DBS) was determined semi-quantitatively by nephelometry (Figure 2C[3]). In case the result of the nephelometry was below a predetermined cut-off (corresponding to a serum level of AAT of 104 mg/dl),8 we continued with phenotyping; additional sequencing was performed if the M protein was not visible in IEF. If the result of the nephelometry was above the threshold of AAT levels, a mutation was excluded.
Laboratory Work
Dried Blood Spots (DBS) in Whatmann 903 cards were placed on the automatic puncher (DBS Puncher. Perkin Elmer [1296-071]) and 3.2 mm diameter punch was automatically made in a 96-well reaction plate (Micro Amp Optical 96 well reaction plates, Life Technologies [N801-0560]).
Each DBS punch was first lysed with 20 µl lysis buffer (SIGMA), shortly centrifuged and incubated at 55 °C for 15=min. Afterwards, 180µl of neutralization buffer (Neutralization solution for blood. SIGMA [N9784/SRE0087]) was added and mixed thoroughly. The extracted DNA could be directly used in the next step (DNA amplification).
Extracted DNA was amplified using multiplex PCR with a set of 14 primers recognizing mutations in the SERPINA1 gene (Table 1). The amplification was performed in 384-well reaction plates (Micro Amp Optical 384 reaction well plates, Life Technologies (4317236) in a final volume of 15 µl (10 µl master mix [Progenika AAT genotyping kit], including primers, HotStar Taq DNA Polymerase [QIAGEN 203203], and 5 µL extracted DNA).
Reactions were heated for 15 min at 95 °C, followed by 45 repeating cycles of 95 °C for 30 s, 62.5 °C for 30 s, 72 °C for 1 min, and a final extension step at 72 °C for 7 min. The PCR was performed in the Verti Dx 384-Well Thermal Cycler (4452300) according to the manufacturer’s instructions.
Following the PCR amplification, 4 µl of PCR products and 46 µL of Bead Master Mix (Progenika A1AT Genotyping Test) were transferred to 96-well plates (Micro Amp Optical 96 well reaction plates, Life Technologies [N801-0560]). The hybridization cycling conditions consisted of a denaturation 2 min 95 °C cycle followed by 30 min 52°C hybridization.
To the hybridization mix was added 80 µl of labeling mix (SAPE and SAPE Dilution Buffer [Progenika A1AT Genotyping Test] and mixed gently by pipetting up and down [keeping the plate on the thermal cycler, 52 °C]).
For measurement of the AAT amount on the filter paper, a circular disk was punched out and incubated with 200 mL of phosphate buffer (PBS) overnight at 4 °C. Afterwards, the samples were applied to nephelometry (Nephelometer Analyzer 2, Dade Behring, Frankfurt, Germany). These semi-quantitative data were not included in the report to be sent for the physician but served as an internal control.9
For the qualitative detection and characterization of the different AAT phenotypes, we used the Hydragel 18 AAT Isofocusing® kit (Sebia, Diagnostic Department, Evry, France). The procedure includes IEF on ready-to-use agarose gels in the semi-automatic Hydrasys® System (Sebia, Diagnostic Department, Evry, France).14 The analysis was carried out directly on DBS punch. Each DBS punch was incubated with 20 µl of distil water and centrifuged at 3700 rpm for 10 min and applied automatically on the isoelectric focusing gel (18 samples can be applied simultaneously). After sample migration, the detection of AAT was performed with specific anti-AAT antiserum labelled with peroxidase. The gel was visually evaluated by two observers.
For the sequencing, the DBS samples we shipped to a reference laboratory (Eurofins Genomics Germany GmbH).
Data Analysis
Raw data were processed by the A1AT Genotyping Test ANALYSIS SOFTWARE (Progenika Biopharma S.A.) to obtain a genotype for each mutation detected.
All procedures were quality controlled and documented by the standard operating procedures (SOPs). All laboratory procedures included appropriate negative and positive controls (different samples with known pathological genotypes). The PCR-based genetic analysis and Luminex were controlled by an external inter-laboratory test.
Results
During phase 1, 1979 samples were analyzed by the traditional (conventional PCR) (Figure 1A) and by the Luminex-based workflow (Luminex, Figure 1B). While it was necessary to perform nephelometry assays with all samples following the traditional workflow, in the Luminex-based workflow the nephelometry measurements were reduced to 497 (by 79%). Moreover, the number of samples that underwent sequencing was reduced from 93 to 34 (by 63.4%). The number of samples where IEF was performed remained stable (1220 vs 1225 samples).
In phase 2, only the Luminex based workflow was used. 1133 AlphaKits arrived at the laboratory of Marburg, 231 samples were analyzed by nephelometry, 757 samples- by IEF, and 22 samples had to be sequenced.
During the combined period (phase 1 and 2), a total number of 3112 samples were analyzed by Luminex-based flow-work. Data showed 432 (13.9%) samples as “non M/non M”, 1473 (47.3%) samples “M/non M” and 1207 (38.8%) samples “M/M”. The six most common genotypes, M/M (n=1207; 38.8%), M/Z (n=1233; 39.6%), M/S (n=147; 4.7%), Z/Z (n=246; 7.9%), S/Z (n=114; 3.7%) and S/S (n=130; 4%) were identified. In addition, Luminex analysis revealed rare genotypes among 152 (4.9%) samples (Table 2).
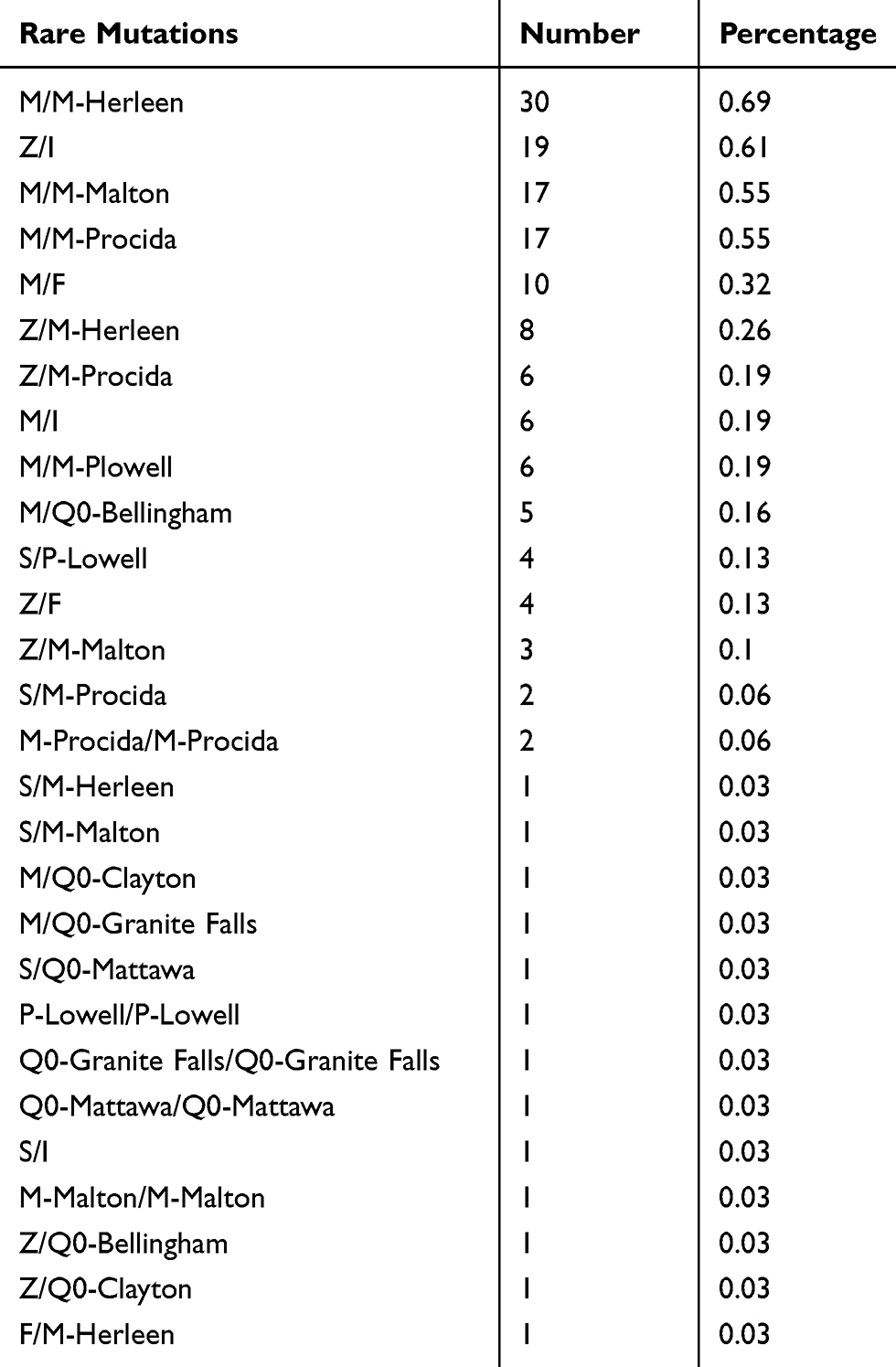 |
Table 2 Luminex Analysis Revealed Rare Genotypes In 152 Samples Directly By Multiplex PCR |
For these, 152 samples traditional analysis including sequencing took a median time of 83 days (35 days to 159 days) while samples analyzed with the Luminex-based workflow required a median of 14 days (7 days to 21 days).
We evaluated the Luminex system alone (without the combination with another test) against the traditional algorithm (gold standard). The sensitivity and specificity of the Luminex results are the following:
Sensitivity= number of true positives/(number of true positives + number of false negatives)= 1910/(1910+34)= 98.2%; specificity= number of true negatives/(number of true negatives + number of false positives)= 1168/(1168 + 0)= 100%.
Positive predictive value= number of true positives/(number of true positives + number of false positives)= 1910/(1910+0)= 100%; negative predictive value= number of true negatives/(number of true negatives + number of false negatives)= 1168/(1168+34)= 97.2%.
Discussion
By introducing a Luminex xMAP based algorithm, we were able simultaneously identify 14 mutations in SERPINA1 gene (instead of two- S and Z-by using our old algorithm). Although the quantity of IEF assays remained unchanged, the nephelometric measurements and sequencing were reduced by 79% and 63.4%, respectively. The Luminex xMAP system is a multiplexed microsphere-based suspension array technology allowing simultaneous detection of multiple nucleic acid sequences in a single reaction. While this method has been used in various fields of genotyping (eg, inflammatory cytokines, bacterial pathogens, and single nucleotide polymorphisms),10–12 it has only recently been applied to AAT genotyping. This method is designed for use of genomic DNA extracted from human whole blood samples collected as DBS or in tubes coated with spray dried K2EDTA (anticoagulant). Extracted DNA is amplified and biotinylated by multiplex PCR. The PCR products are hybridized to oligonucleotide probes coupled to color-coded beads, fluorescent labelled and detected with a Luminex system. To obtain the final data, we used AAT genotyping software algorithm that converts the allelic variant genotypes into associated alleles, based on the current literature. The lower limit of DNA detection by this method is 0.0310 ng/ul.
As a reference laboratory, we based the new algorithm on two principles. Firstly, to exclude AATD, Luminex result had to be negative (M/M) and the AAT-level had to be within the normal range. Most laboratories in Germany define 90–200 mg/dl as the normal range. Concordantly, an analysis > 6000 individuals enrolled in a large population based Swiss cohort study (SAPALDIA), revealed 90 mg/dl as the level discriminating intermediate deficiency.15 Therefore, we used this cutoff. Secondly, to provide a definitive diagnosis of heterozygote or homozygote AAT deficiency, the result has to be confirmed conventional techniques as recommended by the recent guidelines. Samples with abnormal AAT serum levels, should undergo qualitative testing to exclude the possibility that these levels do not correspond to heterozygous phenotypes.16 Samples reported to have a low serum level of AAT were analyzed by genotyping and IEF. Samples that arrived to the laboratory without reported serum levels of AAT were analyzed semi-quantitatively for DBS-derived AAT level. In a previous study, we found that an DBS-derived AAT level of 1.7 mg/dl correlates to an AAT serum level of 104 mg/dl.8
The introduction of a Luminex-based algorithm in our clinical diagnostic laboratory, allowed us to reduce the amount of work required for the final diagnosis of AATD. Instead of two mutations (S and Z) detected by the traditional algorithm, now we were able to detect 14 rare mutations simultaneously. Thus, Luminex based algorithm increased the number of simultaneously identified mutations from 2 to 14, enabling a faster clinical diagnosis of AATD. This reduced the time-expenditure for theses samples from 83 days to 14 days. The new method is convenient, fast and user-friendly, and obtained results show a perfect correlation with data obtained by the traditional workflow. An easier laboratory workflow would be to use Luminex PCR only; however, the decision will depend on costs and practicability. On the other hand, current guidelines recommend the combination of at least two different methods for the final diagnosis of AATD.
The direct comparison of our new algorithm with an externally used “standard” algorithm is not possible because there is no gold standard. Each specialized clinical laboratory has developed its own diagnostic flow for AATD detection, starting its sample collection either from blood, plasma/serum or DBS. Laboratories in Germany, Spain and Italy use DBS, in France blood analysis is preferred, and in Ireland plasma/serum.4,6,17,18
Usually, the initial step is a measurement of the AAT plasma level, followed by phenotyping, genotyping, or whole gene sequencing depending on availability and/or the need for more detailed interpretation of the results.6 These steps are carried out differently from country to country.17,18
Currently, only the analysis of serum/plasma AAT levels is well established. However, little is known about the functional activity of many rare variants of AAT. As a matter of fact, a serum level of 60 mg/dl in a Pi*MZ mutation might have different functional properties than a serum level of 60 mg/dl in a Pi*M/rare mutation. Consequently, methods are needed that allow a functional characterization of the AAT protein.
Serum separator cards may be used, enabling the laboratory to conduct a multiple step analysis (AAT level, CRP level, PCR, IEF, gene sequencing) with only one patient visit.19 Furthermore, “Next Generation Sequencing” is becoming widely available and may become affordable. A potential future algorithm would consist of a screening test with a very high sensitivity, and test-positive individuals could then directly undergo sequencing of the SERPINA1 gene. A major strength of next-generation sequencing is that it can detect abnormalities across the entire genome using less DNA than required for other DNA sequencing approaches. However, this method requires sophisticated bioinformatics systems to analyze and clinically interpret the data.
Conclusion
In conclusion, we employed a new technology to detect 14 different AATD mutations simultaneously. Our results indicate that this Luminex-based multiplexed assay has the potential for a rapid and cost-effective diagnostic tool for AATD. In the future, the assay could be expanded to include more specific probes to identify other mutations
Acknowledgments
We thank all employees of the alpha-1 antitrypsin laboratory at the University of Marburg for their continuous effort they put into this project. The alpha-1 antitrypsin laboratory at the University of Marburg has been supported by Grifols and Progenika Biopharma (A Grifols Company).
Disclosure
Martina Veith reports grants from Grifols, during the conduct of the study; grants from Grifols, outside the submitted work. Iker Anton, Rachid El Hamss, and Noelia Rapun are employees of Progenika Biopharma-Grifols. Viktor Kotke reports grants from Grifols, outside the submitted work. Dr. Robert Bals reports grants, personal fees from CSL Behring and Grifols, grants from Boehinger Ingelheim and Novartis, during the conduct of the study. Claus Franz Vogelmeier reports grants, personal fees from AstraZeneca, GlaxoSmithKline, Grifols, Novartis, and Boehringer Ingelheim, personal fees from CSL Behring, Menarini, Mundipharma, Teva, Cipla, Nuvaira, and Chiesi, grants from Bayer Schering Pharma AG, MSD, and Pfizer, outside the submitted work. Timm Greulich reports grants from Grifols, during the conduct of the study; personal fees from AstraZeneca, Berlin-Chemie, Boehringer-Ingelheim, Chiesi, CSL-Behring, Grifols, GSK, and Novartis, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Stoller JK, Aboussouan LS, . Alpha1-antitrypsin deficiency. Lancet. 2005;365(9478):2225–36.
2. Kohnlein T, Janciauskiene S, Welte T. Diagnostic delay and clinical modifiers in alpha-1 antitrypsin deficiency. Ther Adv Respir Dis. 2010;4(5):279–87.
3. Campos MA, Wanner A, Zhang G, Sandhaus RA. Trends in the diagnosis of symptomatic patients with alpha1-antitrypsin deficiency between 1968 and 2003. Chest. 2005;128(3):1179–86. doi:10.1183/09031936.06.00062305
4. Stoller JK, Sandhaus RA, Turino G, Dickson R, Rodgers K, Strange C. Delay in diagnosis of alpha1-antitrypsin deficiency: a continuing problem. Chest. 2005;128(4):1898–94. doi:10.1183/13993003.00610-2017
5. WHO. Alpha 1-antitrypsin deficiency: memorandum from a WHO meeting. Bull World Health Organ. 1997;75(5):397–415. doi:10.1016/j.rmed.2011.02.002
6. Miravitlles M, Dirksen A, Ferrarotti I, et al. European Respiratory Society statement: diagnosis and treatment of pulmonary disease in alpha1-antitrypsin deficiency. Eur Respir J. 2017;50(5).
7. Barrecheguren M, Monteagudo M, Simonet P, et al. Diagnosis of alpha-1 antitrypsin deficiency: a population-based study. Int J Chron Obstruct Pulmon Dis. 2016;11:999–1004. doi:10.2147/COPD.S108505
8. Greulich T, Ottaviani S, Bals R, et al. Alphai-antitrypsin deficiency - diagnostic testing and disease awareness in Germany and Italy. Respir Med. 2013;107(9):1400–1408. doi:10.1016/j.rmed.2013.04.023
9. Bals R, Koczulla R, Kotke V, Andress J, Blackert K, Vogelmeier C. Identification of individuals with alpha-1-antitrypsin deficiency by a targeted screening program. Respir Med. 2007;101(8):1708–1714. doi:10.1016/j.rmed.2007.02.024
10. Dunbar SA. Applications of Luminex (R) xMAP (TM) technology for rapid, high-throughput multiplexed nucleic acid detection. Clinica Chimica Acta. 2006;363(1–2):71–82. doi:10.1016/j.cccn.2005.06.023
11. Ozaki S, Kato K, Abe Y, et al. Analytical performance of newly developed multiplex human papillomavirus genotyping assay using Luminex xMAP (TM) technology (Mebgen((TM)) HPV kit). J Virol Methods. 2014;204:73–80. doi:10.1016/j.jviromet.2014.04.010
12. Yan Y, Luo JY, Chen Y, et al. A multiplex liquid-chip assay based on Luminex xMAP technology for simultaneous detection of six common respiratory viruses. Oncotarget. 2017;8(57):96913–96923. doi:10.18632/oncotarget.18533
13. Greulich T, Nell C, Herr C, et al. Results from a large targeted screening program for alpha-1-antitrypsin deficiency: 2003–2015. Orphanet J Rare Dis. 2016;11.
14. Zerimech F, Hennache G, Bellon F, et al. Evaluation of a new Sebia isoelectrofocusing kit for alpha(1)-antitrypsin phenotyping with the Hydrasys (R) system. Clin Chem Lab Med. 2008;46(2):260–263. doi:10.1515/CCLM.2008.036
15. Ferrarotti ITG, Zorzetto M, Ottaviani S, et al. Serum levels and genotype distribution of α1-antitrypsin in the general population. Br Thorac Soc. 2012;67(8):669–674.
16. Amer Thoracic Soc European R. American Thoracic Society/European Respiratory Society Statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med. 2003;168(7):818–855. doi:10.1164/rccm.168.7.818
17. Miravitlles M, Herr C, Ferrarotti I, et al. Laboratory testing of individuals with severe alpha(1)-antitrypsin deficiency in three European centres. Eur Respir J. 2010;35(5):960–968. doi:10.1183/09031936.00069709
18. McElvaney NG. Diagnosing alpha 1-antitrypsin deficiency: how to improve the current algorithm. Eur Respir Rev. 2015;24(135):52–57. doi:10.1183/09059180.10010814
19. Sanders C, Kim J. Frequencies of alpha-1 antitrypsin deficiency phenotypes detected using a clinical testing strategy that reflexes to next-generation sequencing. Am J Respir Crit Care Med. 2015;191.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.