Back to Journals » Neuropsychiatric Disease and Treatment » Volume 14
Dexmedetomidine attenuates the propofol-induced long-term neurotoxicity in the developing brain of rats by enhancing the PI3K/Akt signaling pathway
Authors Xiao Y, Zhou L, Tu Y, Li Y, Liang Y, Zhang X, Lv J, Zhong Y, Xie Y ![]()
Received 23 March 2018
Accepted for publication 24 May 2018
Published 28 August 2018 Volume 2018:14 Pages 2191—2206
DOI https://doi.org/10.2147/NDT.S169099
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Wai Kwong Tang
Yong Xiao,1,* Lifang Zhou,2,* Youbing Tu,1 Yuantao Li,3 Yubing Liang,4 Xu Zhang,1 Jing Lv,1 Yu Zhong,1 Yubo Xie1
1Department of Anesthesiology, The First Affiliated Hospital of Guangxi Medical University, Nanning, People’s Republic of China; 2Department of Anesthesiology, Institute of Cardiovascular Diseases, The First Affiliated Hospital of Guangxi Medical University, Nanning, People’s Republic of China; 3Department of Anesthesiology, Shenzhen Maternity & Child Healthcare Hospital, Southern Medical University, Shenzhen, People’s Republic of China; 4Department of Anesthesiology, Affiliated Tumor Hospital of Guangxi Medical University, Nanning, People’s Republic of China
*These authors contributed equally to this work
Background: Propofol induces short- and long-term neurotoxicity. Our previous study showed that dexmedetomidine (Dex) can attenuate the propofol-induced acute neurotoxicity in rodents by enhancing the PI3K/Akt signaling. However, whether treatment of young rats with Dex could protect them from long-term neurotoxicity induced by propofol is unclear.
Materials and methods: Seven-day-old male Sprague Dawley rats were randomized and injected intraperitoneally with saline (100 µL, NS), propofol (100 mg/kg), Dex (75 µg/kg), propofol (100 mg/kg) plus Dex (25, 50 or 75 µg/kg), 10% dimethyl sulfoxide (DMSO, 100 µL) or TDZD-8 (a GSK3β inhibitor, 1 mg/kg), or intracerebroventricularly with DMSO (5 µL) or LY294002 (a PI3K inhibitor, 25 µg/5 µL DMSO). Other rats in the experimental group were injected with the same doses of propofol, Dex and LY294002 or TDZD-8. All the rats were monitored until they were 9 weeks old. Their spatial learning and memory were tested by Morris water maze. The neuronal apoptosis, expression of PSD95, expression and phosphorylation of Akt and GSK3β and synaptic ultrastructures were determined by terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling, immunohistochemistry, Western blot and transmission electron microscopy assays, respectively.
Results: Compared with the NS control group, young rats injected with intralipid, Dex, TDZD-8, LY294002 or DMSO alone did not show any significant change as they aged. Propofol significantly increased the escape latency time, hippocampal neuroapoptosis and synaptic ultrastructural changes but decreased the relative levels of PSD95 expression, and Akt and GSK3β phosphorylation in the developing hippocampus of the rats. The neuronal toxic effects of propofol were significantly mitigated by the pretreatment with a higher dose of Dex. The neuroprotective effect of Dex was enhanced by the treatment with TDZD-8, but was completely abrogated by the treatment with LY294002.
Conclusion: Our results indicated that the pretreatment of young rats with Dex attenuated the propofol-induced long-term neurotoxicity in their developing hippocampus by enhancing the PI3K/Akt signaling.
Keywords: propofol, dexmedetomidine, long-term neurotoxicity, PI3K, hippocampus
Introduction
Propofol (2,6-diisopropylphenol) has been commonly used in pediatric and obstetric patients as an intravenous anesthetic and sedative drug because of its rapid onset, continuous infusion without accumulation and quick recovery.1,2 However, fetal and neonatal exposure to antagonists for N-methyl-D-aspartate (NMDA) receptors or agonists for γ-aminobutyric acid (GABA) receptors may lead to accelerated neurodegeneration.3 Propofol acts as a positive allosteric modulator of GABAA receptors and produces adverse effects on immature neurons in different species.4–6 At a clinical-relevant dose, propofol can enhance neuronal differentiation, but cause long-term changes in the dendritic development of immature GABAergic neurons in vitro.7,8 Furthermore, propofol induces dose-dependent changes leading to the damage of neonatal neurons and embryonic neural stem cells, and permanent neurological impairment in rodents.4,9–11 The potential side effects of propofol affecting the development of infant brain have also raised concerns about its safety.12,13 Hence, discovery of new therapeutic reagents to prevent the neuronal side effects of propofol will be of significance.
Dexmedetomidine (Dex) is a highly specific agonist of the α2-adrenoceptor and has been used clinically as a potent anesthetic adjuvant for sedation, analgesia and anxiolytic therapy without respiratory suppression.14,15 Recent studies have reported that the engagement of α2-adrenoceptors by Dex reduces the ischemia- and hypoxia-induced neuronal damages by increasing the expression of antiapoptotic protein Bcl-2 and decreasing the expression of pro-apoptotic proteins Bax and p53 in the brain.16–18 A study showed that Dex induced FAK activation and supported the survival of neurons by inhibiting the caspase-3 activation in rat hippocampal sections cultured in an oxygen- and glucose-deprived condition.19 Dex has been shown to attenuate the isoflurane-induced neuronal injury in the hippocampus of neonatal rats in a dose-dependent manner and prevent the isoflurane-induced long-term memory impairment.20,21 Dex also exerts a protective effect on lipopolysaccharide-induced acute lung injury by mediating the PI3K/Akt/mTOR pathway.22 However, it is unclear whether treatment with Dex can mitigate the propofol-induced long-term neurotoxicity.
The PI3K/Akt signaling is crucial for the proliferation, survival and differentiation of cells.23 The activation of the PI3K/Akt signaling can induce antiapoptotic Bcl-2 expression and inhibit pro-apoptotic Bax expression and caspase activation.24 The PI3K/Akt signaling is negatively regulated by GSK3β, while activated Akt can induce GSK3β phosphorylation, leading to its degradation.25 Previous studies have shown that propofol can inhibit the Akt activation in the neurons and induce neuronal apoptosis via PI3K/Akt signaling pathway.26,27 Our previous study indicates that treatment with Dex can attenuate the Akt activation inhibition of propofol in neonatal rats.28 However, whether treatment of neonatal rats with Dex can prevent the Akt activation inhibition by propofol when they reach adulthood is unknown.
PSD95 is one of the most abundant components in synapses. It can regulate the synaptic plasticity and affect the synaptic structures and functions in the developing hippocampus.29 In Alzheimer’s disease, the impairment of synaptic function is related to the decrease of PSD95 levels.30 It is possible that Dex treatment may preserve the levels of PSD95 in rats after exposure to propofol.
More recently, studies confirmed that repeated exposure to propofol induced long-term neurotoxicity in neonatal rats.9,31 In our previous study, Dex preconditioning attenuated the propofol-induced formation of acute lesions in hippocampal neurons in postnatal 7-day-old rats by upregulating the phosphorylation of Akt and GSK3β.28 Hence, we assumed that pretreatment with Dex might attenuate the propofol-induced long-term neurotoxicity in the developing brain of neonatal rats by enhancing the PI3K/Akt signaling pathway. To verify this hypothesis, postnatal 7-day-old rats were treated with Dex, propofol and the antagonists of PI3K/Akt signaling pathway. When the rats reached the age of 9 weeks, changes in the spatial learning ability, histomorphology and expression level of PSD95 in their hippocampus were investigated to explore the mechanisms and possible effects of Dex on the propofol-induced long-term neurotoxicity.
Materials and methods
Animals and study design
The experimental protocols were approved by the Laboratory Animal Use and Care Ethical Committee of Guangxi Medical University (Approval No 201702003). The experiments were performed according to the animal care and use guidance (the guidelines outlined by the National Institutes of Health, Bethesda, MD, USA).32 Male Sprague Dawley rats at 7 days of age and weighing 10–15 g and their mothers were obtained from the Laboratory Animal Center of Guangxi Medical University and housed in a specific pathogen-free facility at 23°C–25°C and 50%±10% humidity with a 12-h light/dark cycle and free access to food and water ad libitum.4,33
All rats were randomly divided into 13 groups (30 animals per group) using a computer-generated random number table: some groups of rats were injected intraperitoneally (i.p.) with vehicle saline (NS group), 100 mg/kg intralipid (F group, medium- and long-chain fat emulsion; Yao Pharma, Chongqing, People’s Republic of China), 75 μg/kg Dex (Dex group; Hengrui Medicine, Jiangsu, People’s Republic of China) or 50 mg/kg propofol twice at an interval of 40–60 min when the righting reflex of rats was recovered (P group; Corden Pharma SPA, Liestal, Switzerland); some groups of rats were pretreated with 25, 50 or 75 μg/kg Dex and then injected with 50 mg/kg propofol twice (PD25, PD50 and PD75 groups, respectively); some groups of rats were injected intracerebroventricularly (i.c.) with 5 μL of 10% DMSO vehicle (DS1 group), 25 μg/5 μL LY294002 (a PI3K inhibitor) alone (L group; Sigma-Aldrich, St Louis, MO, USA) or the same dose of LY294002 and 30 min later with Dex and propofol (LPD group); some groups of rats were injected with 1 mg/kg TDZD-8 (a GSK3β inhibitor) (T group; Sigma-Aldrich) or the same dose of TDZD-8 and 30 min later with Dex and propofol (TPD group); and one group of rats were injected i.p. with 100 μL of 10% DMSO (DS2 group). One hour post-recovery, blood gas analysis was performed in five pups from each group, and the other rats were returned to their mothers until weaning at 4 weeks of age. At 9 weeks of age, the experimental and control groups of rats were tested simultaneously in a blinded manner. All adult rats were anesthetized by intraperitoneal injection with 10% chloral hydrate (3.5 mL/kg) before sacrifice by cervical dislocation. The experimental design and all the steps are shown in Figure 1.
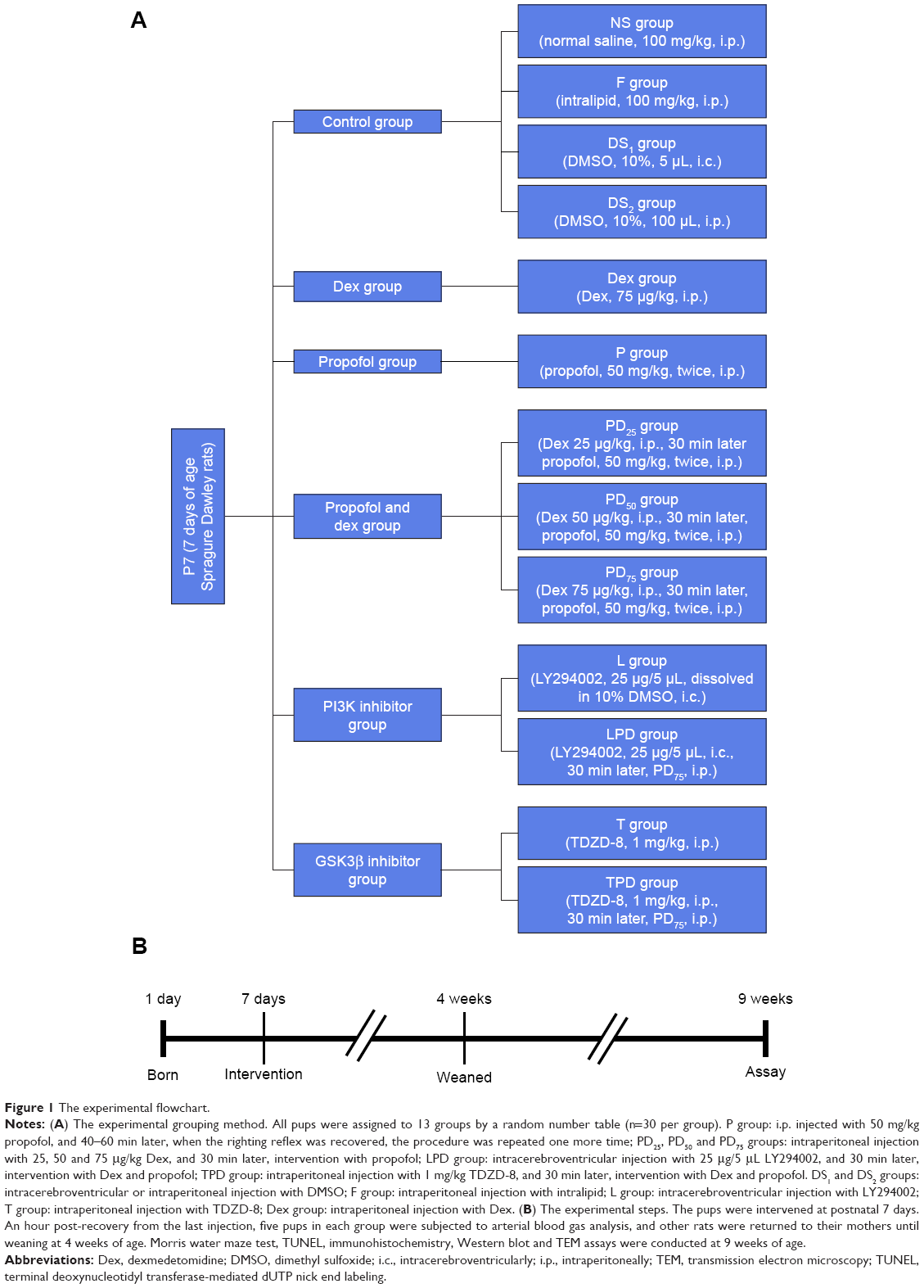 | Figure 1 The experimental flowchart. |
Intracerebroventricular injection
Individual rats were injected i.c. with drug, as described previously.34 Briefly, the rats were kept on the stereotaxic instrument on a warm soft pad, and with an ultrabright flashlight to illuminate the anatomical structures of the skull, the injection site of each rat was marked at three-fifths of the distance from each eye to the lambda suture with a nontoxic marker. After their skins were disinfected with 70% ethanol, the rats were carefully injected with 5 μL of vehicle or drug using a Hamilton syringe and a 27-gauge needle into the ventricle about 2 mm from the skin surface at a speed of 1 μL/s. After injection for 20 s, the needle was withdrawn slowly to prevent backflow. The contralateral ventricle was injected using the same procedure.
Arterial blood gas analysis and body weight monitoring
To determine whether the hypoxia and hypercapnia induced during propofol anesthesia would affect the development of the brain, an hour after recovery from the last injection, five pups from each group were anesthetized i.p. with 10% chloral hydrate (3.5 mL/kg), and the arterial blood was drawn out from the left ventricle to analyze the pH, PaO2, PaCO2 and HCO3- (i-STAT 300; Abbott, Chicago, IL, USA). The other rats were weighted at every week until they were 9 weeks old.
Morris water maze (MWM) test
To evaluate the functions of spatial learning and memory, the rats (n=8 per group) were assessed by MWM test, as described previously.35 Briefly, the test was performed in a round, flat-bottomed black plastic pool (Institute of Materia Medica, Chinese Academy of Medical Sciences, Peking, People’s Republic of China) with a diameter of 150 cm and a height of 50 cm. The pool was divided into four quadrants and filled with water at 23°C–25°C and had no visible internal marker. A round escape platform (diameter 10 cm, height 35 cm) was set hidden 2 cm underwater in one of the quadrants (target quadrant) of the pool. Each rat was trained to find the hidden platform in four trials daily for four consecutive days. Each rat was placed in the pool in the four quadrants, and the escape latency time (time to find the platform) was recorded using a camera (3 m above the pool) connected to a computerized recording system equipped with an automated analysis system (Smart V3.0; Panlab Harvard, Barcelona, Spain). The rats were allowed to rest on the escape platform for 15 s. If a rat failed to find the platform within 90 s, it was placed on the platform. On the fifth day, 24 h after the last training, all the rats were placed in the quadrant facing the target quadrant and allowed to swim freely for 90 s when the escape platform was removed. The number of times each rat attempted to reach the previous platform location and the time spent on the previous platform quadrant were recorded.
Western blot
The hippocampus of individual rats (n=5 per group) was dissected, frozen in liquid nitrogen and lysed in a lysis buffer (Beyotime Biotech, Shanghai, People’s Republic of China), followed by centrifugation. After quantification of their protein concentrations using bicinchoninic acid protein assay (Sangon Biotech, Shanghai, People’s Republic of China), the lysates (50 μg/lane) were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis on 10% gels and transferred onto a polyvinylidene fluoride membrane (0.22 μm pore size; EMD Millipore, Billerica, MA, USA). The membranes were blocked with 5% skim dry milk in Tris-buffered saline with Tween 20 at 37°C for 2 h and incubated with primary antibodies against Akt (Akt pan C67E7, rabbit monoclonal antibody [mAb]; Cell Signaling, Danvers, MA, USA), pAkt (Ser473, D9E XP, rabbit mAb; Cell Signaling), GSK3β (D5C5Z XP, rabbit mAb; Cell Signaling) and pGSK3β (Ser9 5B3, rabbit mAb, 1:1,000; Cell Signaling), anti-PSD95 antibody (ab18258, rabbit polyclonal antibody [pAb], 1:500; Abcam, Cambridge, UK) and GAPDH antibody (rabbit pAb, 1:10,000; Proteintech, Rosemont, IL, USA) overnight at 4°C. After being washed, the bound antibodies were detected with secondary antibody (IRDye® 680RD, goat anti-rabbit IgG, 1:5,000; LI-COR, Lincoln, NE, USA) and visualized using the enhanced chemiluminescent reagents using the Odyssey infrared imaging system (LI-COR) and Image Lab software (Bio-Rad Laboratories Inc., Hercules, CA, USA). The relative levels of target protein to the control GAPDH were determined by densitometric analysis using the Quantity One V5.0 software (Bioz, Los Altos, CA, USA).28
Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay
The hippocampus of individual rats (n=5 per group) was fixed in 4% paraformaldehyde and paraffin-embedded. The brain coronal sections (5 μm) were selected randomly from each rat, and the percentages of apoptotic neurons in the hippocampus were determined using the TUNEL Apoptosis Assay Kit (TUN11684817; Roche Applied Science, Penzberg, Germany), according to the manufacturer’s instruction. Briefly, the hippocampal tissue sections were deparaffinized, rehydrated and treated with proteinase K (Roche Applied Science), followed by quenching with 3.0% hydrogen peroxide. After being washed, the sections were incubated in a terminal deoxynucleotidyl transferase (TdT) reaction mix for 1 h at 37°C, incubated for 30 min in a solution of fluorescein isothiocyanate–anti-digoxigenin conjugate and counterstained with 4′,6-diamidino-2-phenylindole. The control section had no TdT reaction. The sections were imaged at ×200 magnification under an Olympus BX51 fluorescent microscope using an OLYMPUS DP71 digital camera and DP controller software.28 The TUNEL-positive cells were counted in five sampling fields selected randomly in a blinded manner.
Immunohistochemistry
The rats (n=5 per group) were anesthetized by chloral hydrate, and were perfused transaorticly with normal saline and 4% ice-cold paraformaldehyde. The brain was dissected and paraffin-embedded. The consecutive brain coronal sections (4 μm) were dewaxed, rehydrated and subjected to antigen retrieval in citric acid (Solarbio Life Sciences, Beijing, People’s Republic of China). The sections were blocked with goat serum (Beyotime Biotech) and incubated with anti-PSD95 antibody (Abcam) at 4°C overnight. After being washed, the bound antibodies were detected with horseradish peroxidase-conjugated secondary antibody (LI-COR) and visualized with 3,3′-diaminobenzidine tetrahydrochloride (ZSGB Bio, Beijing, People’s Republic of China), followed by counterstaining with hematoxylin. The cornu ammonis areas CA1, CA2 and CA3, and dentate gyrus regions of the hippocampus were photoimaged under a light microscope (magnification ×200). The integrated optical density in the positively stained areas on the section was statistically analyzed using Image-Pro Plus 6.0 software (Media Cybernetics, Rockville, MD, USA).36
Transmission electron microscopy (TEM)
The synaptic ultrastructures of hippocampal neurons in adult rats (n=2 per group) were examined by TEM. After perfusion, the hippocampus of each rat was dissected and fixed in 1% osmium tetroxide, dehydrated and embedded in epon-araldite. The middle third of the CA1 stratum radiatum of the hippocampus was cut into serial sections (50 nm) using an ultramicrotome. The sections were imaged using a TEM (Hitachi-7650) operating at 80 keV.28
Statistical analysis
Data are expressed as mean±standard error of the mean. Multiple comparisons were performed by one-way analysis of variance (ANOVA) and post hoc Dunnett’s test or repeated ANOVA where applicable using SPSS version 20.0 (IBM Corporation, Armonk, NY, USA). A p-value of <0.05 was considered statistically significant.
Results
Treatment with Dex mitigates the propofol-induced neurological function impairment in adult rats
Previous studies have shown that propofol can induce neonatal neuronal damages and Dex has a neuroprotective effect.4,6,7,12,37 To investigate the long-term neurotoxicity of propofol, young rats were injected with propofol and/or Dex or other specific inhibitors or vehicle controls. These rats were monitored up to 9 weeks of age. There was no significant difference in the arterial blood gas (Table 1) and body weight among these groups of rats (Figure 2). In addition, no skin injury was observed in the rats. These suggest that treatment with propofol, Dex or other compounds did not affect the development and maturation of these rats. To test the potential protective effect of Dex on propofol-induced neuronal impairment, the spatial learning and memory functions of the adult rats were tested by MWM assay. As shown in Figure 3, during the training trials, the escape latency time of the different groups of rats gradually decreased (Figure 3A). Although there was no significant difference in the swimming speeds of the different groups of rats, the escape latency times of these groups of rats were significantly different (Figure 3B). In comparison with the NS control group, rats injected with propofol, but not intralipid, Dex, TDZD-8 (a GSK3β inhibitor), LY294002 (a PI3K inhibitor) or DMSO at young age showed significantly increased escape latency period in their adult age (Figure 3D). Pretreatment with Dex significantly shortened the propofol-prolonged escape latency period, relative to the propofol-alone treatment, and this effect was enhanced by treatment with TDZD-8, but was completed abrogated by intracerebroventricular injection with LY294002. These results indicated that treatment with Dex mitigated the propofol-induced long-term neuronal impairment in adult rats, which appeared to depend on the PI3K/Akt signaling pathway.
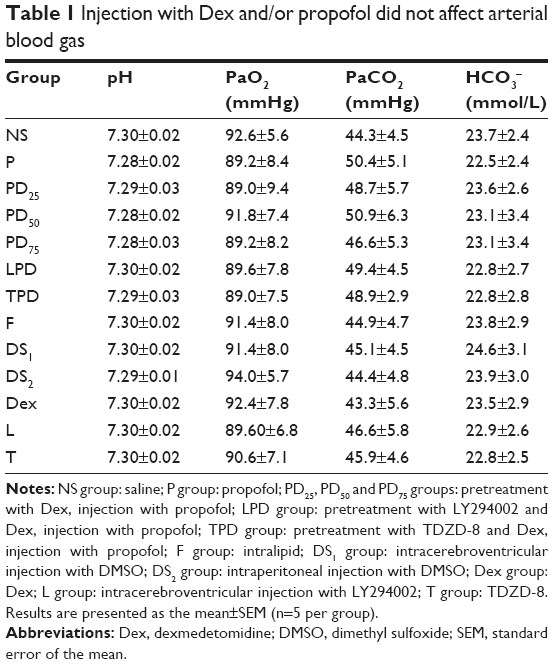 | Table 1 Injection with Dex and/or propofol did not affect arterial blood gas |
 | Figure 2 Injection of young rats with Dex and/or propofol did not affect their body weights in adult age. |
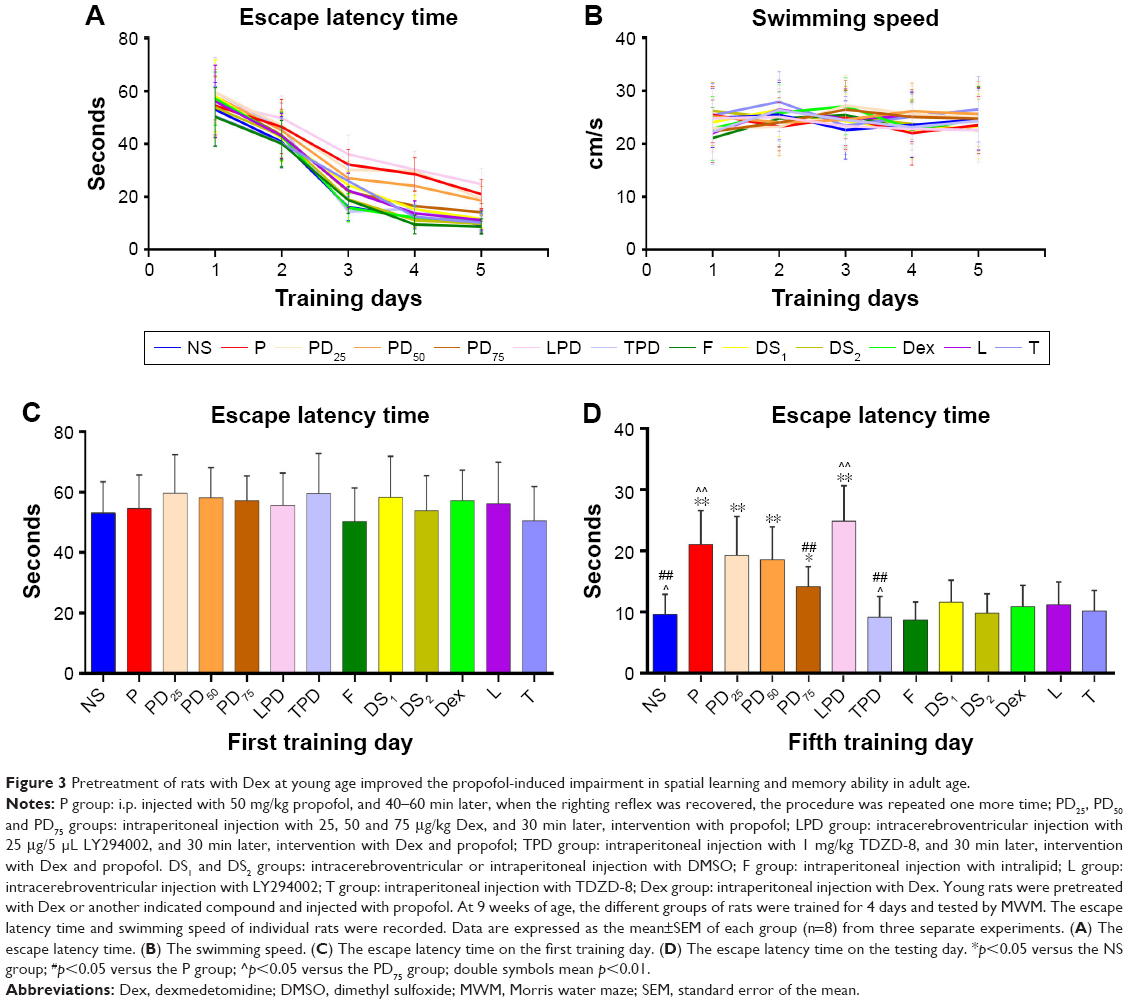 | Figure 3 Pretreatment of rats with Dex at young age improved the propofol-induced impairment in spatial learning and memory ability in adult age. |
Treatment with Dex reduces the propofol-induced neuronal apoptosis in the hippocampus of adult rats
To understand the structural changes associated with the brain function impairment, we evaluated the neuronal apoptosis of the hippocampus in the different groups of rats by TUNEL assays. As shown in Figure 4, in comparison with the NS control rats, young rats injected with intralipid, Dex, TDZD-8, LY294002 or DMSO did not show any increase in the frequency of neuronal apoptosis in the hippocampus as they aged. In contrast, injection with propofol increased the frequency of apoptosis in the hippocampus of rats (p<0.01), which was significantly mitigated by pretreatment with Dex at a dose of 75 μg/kg, but not at a lower dose. In addition, treatment with TDZD-8, together with Dex and propofol, further reduced the frequency of neuronal apoptosis (p<0.01), while treatment with LY294002, together with Dex and propofol, abrogated the protective effect of Dex and increased the neuronal apoptosis in the hippocampus of adult rats. Hence, treatment with Dex significantly mitigated the propofol-mediated neuronal apoptosis in adult rats, which was positively regulated by the PI3K/Akt signaling pathway.
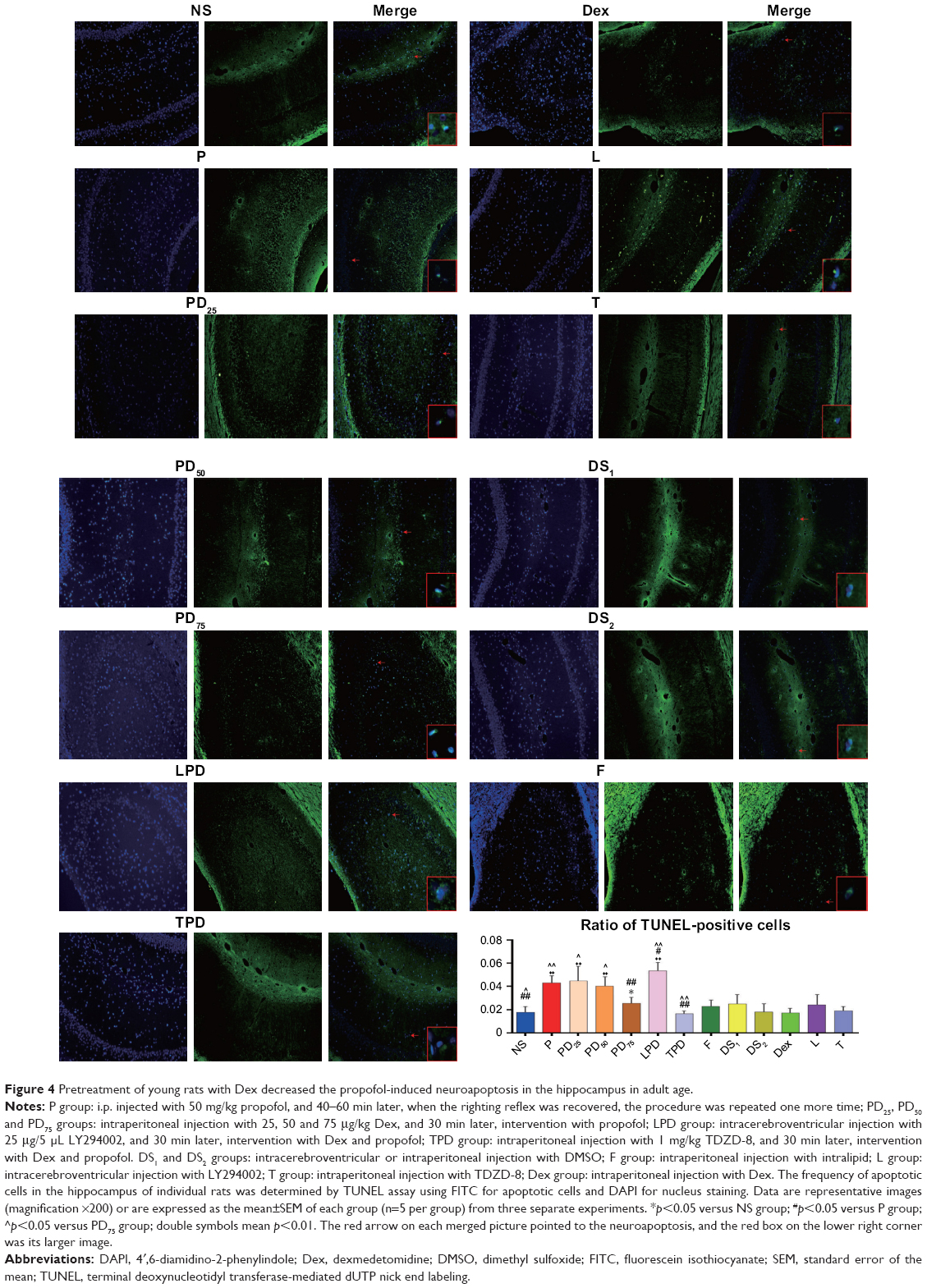 | Figure 4 Pretreatment of young rats with Dex decreased the propofol-induced neuroapoptosis in the hippocampus in adult age. |
Treatment with Dex mitigates the propofol-induced neuronal damages in the hippocampus of adult rats
PSD95 is a scaffold protein which is crucial for memory, synaptic plasticity and other physiological functions in the developing hippocampus.29,38 We tested the protective effect of Dex on the propofol-induced hippocampal tissue damages by detecting the levels of PSD95 expression using immunohistochemistry. As shown in Figure 5, in comparison with the NS control group, rats treated with intralipid, Dex, TDZD-8, LY294002 or DMSO did not show any significant change in the levels of PSD95 expression in the hippocampus. In contrast, the levels of PSD95 expression in the hippocampus of rats treated with propofol at young age were significantly reduced, while treatment with Dex and propofol prevented the propofol-decreased PSD95 expression in the hippocampus of rats (Figure 6A and B). While treatment with TDZD-8 enhanced the protective effect of Dex, treatment with LY294002 completely eliminated the protective effect of Dex and further decreased the levels of PSD95 expression in the hippocampus of rats. A similar pattern of PSD95 expression was detected in the hippocampus of the different groups of rats by Western blot assays (Figure 6).
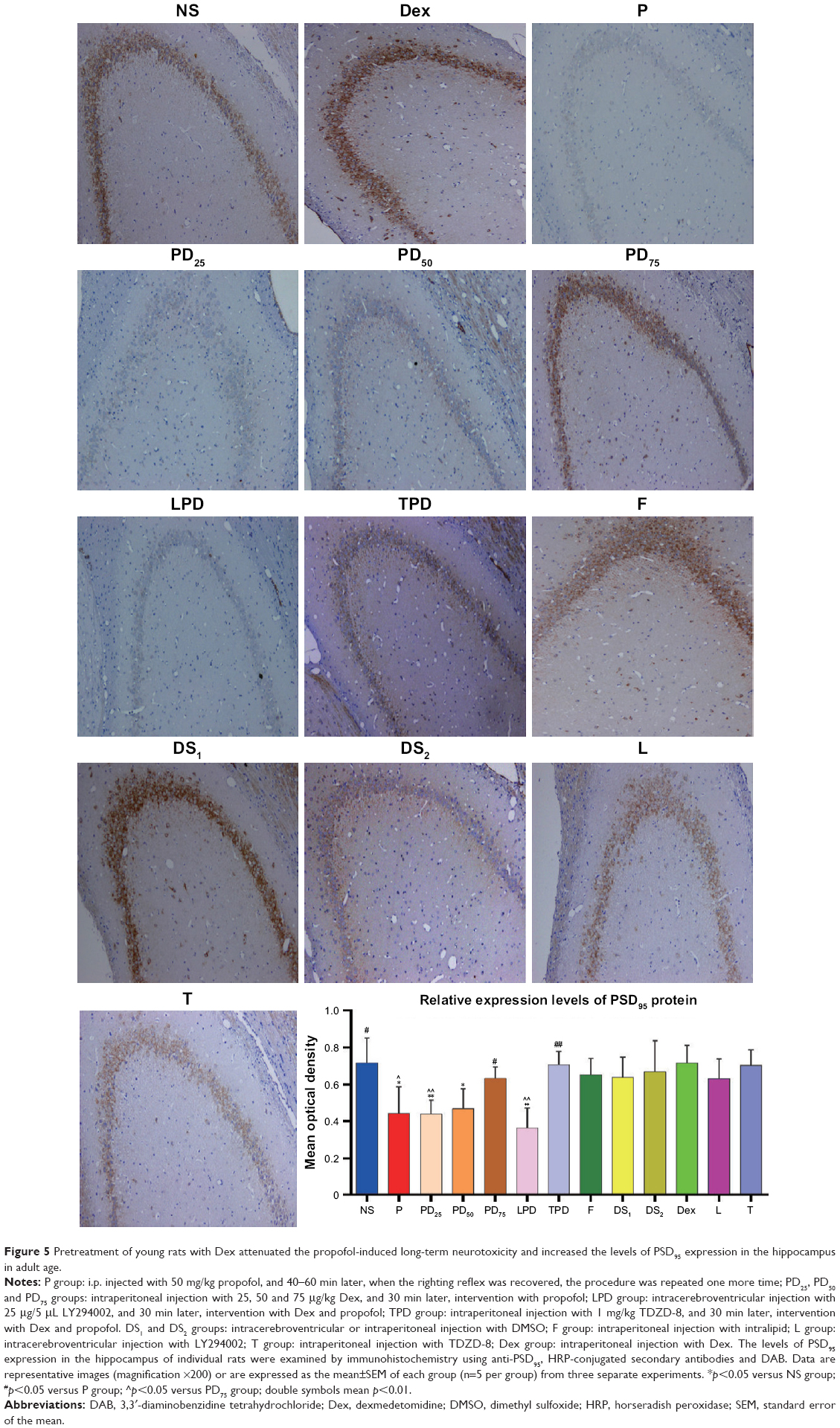 | Figure 5 Pretreatment of young rats with Dex attenuated the propofol-induced long-term neurotoxicity and increased the levels of PSD95 expression in the hippocampus in adult age. |
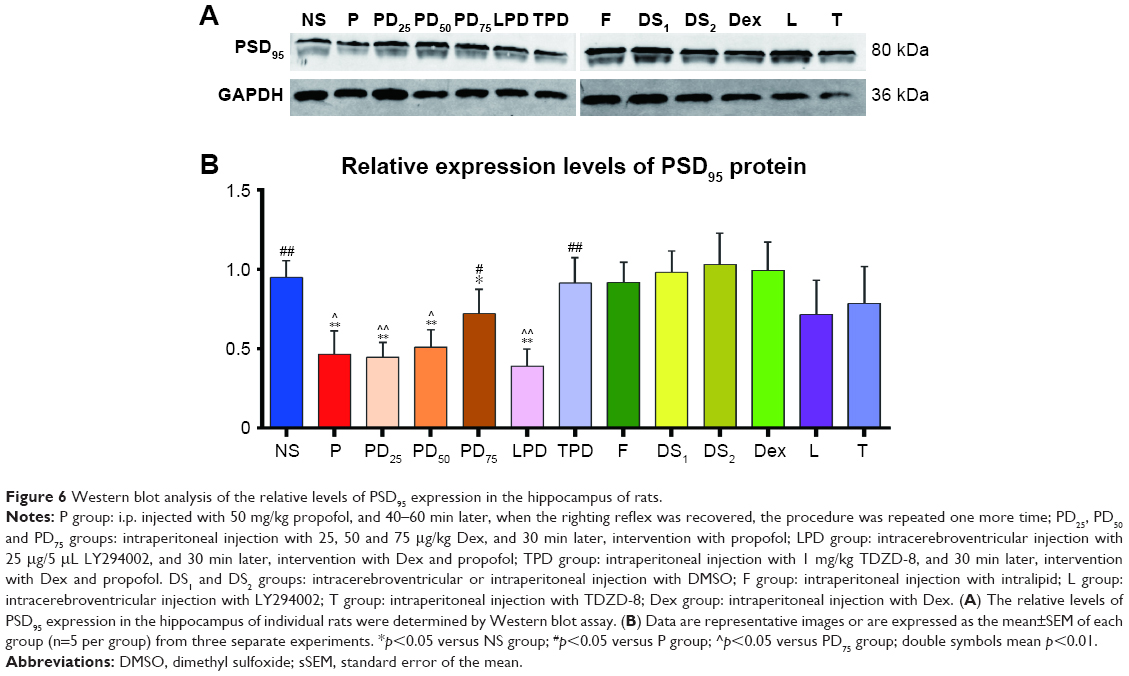 | Figure 6 Western blot analysis of the relative levels of PSD95 expression in the hippocampus of rats. |
Further TEM observation revealed that there were obvious changes in the hippocampal synaptic ultrastructure, such as a decrease in the thickness of hippocampal PSD, the synaptic vesicles, vague presynaptic and postsynaptic membranes and obvious synaptic clefts in the hippocampus of rats injected with propofol alone (Figure 7). In contrast, injection with intralipid, Dex, TDZD-8, LY294002 or DMSO did not cause obvious change in rats. Furthermore, injection with Dex attenuated the propofol-induced synaptic changes in the hippocampus, and injection with TDZD-8 enhanced the protective effect of Dex. However, injection with LY294002 abrogated the effect of Dex and further increased the synaptic changes in the hippocampus of rats. These results indicated that injection of young rats with propofol induced neuronal damages that remained at adult age and treatment with Dex prevented the propofol-induced neuronal damages in rats, dependent on the PI3K/Akt signaling pathway.
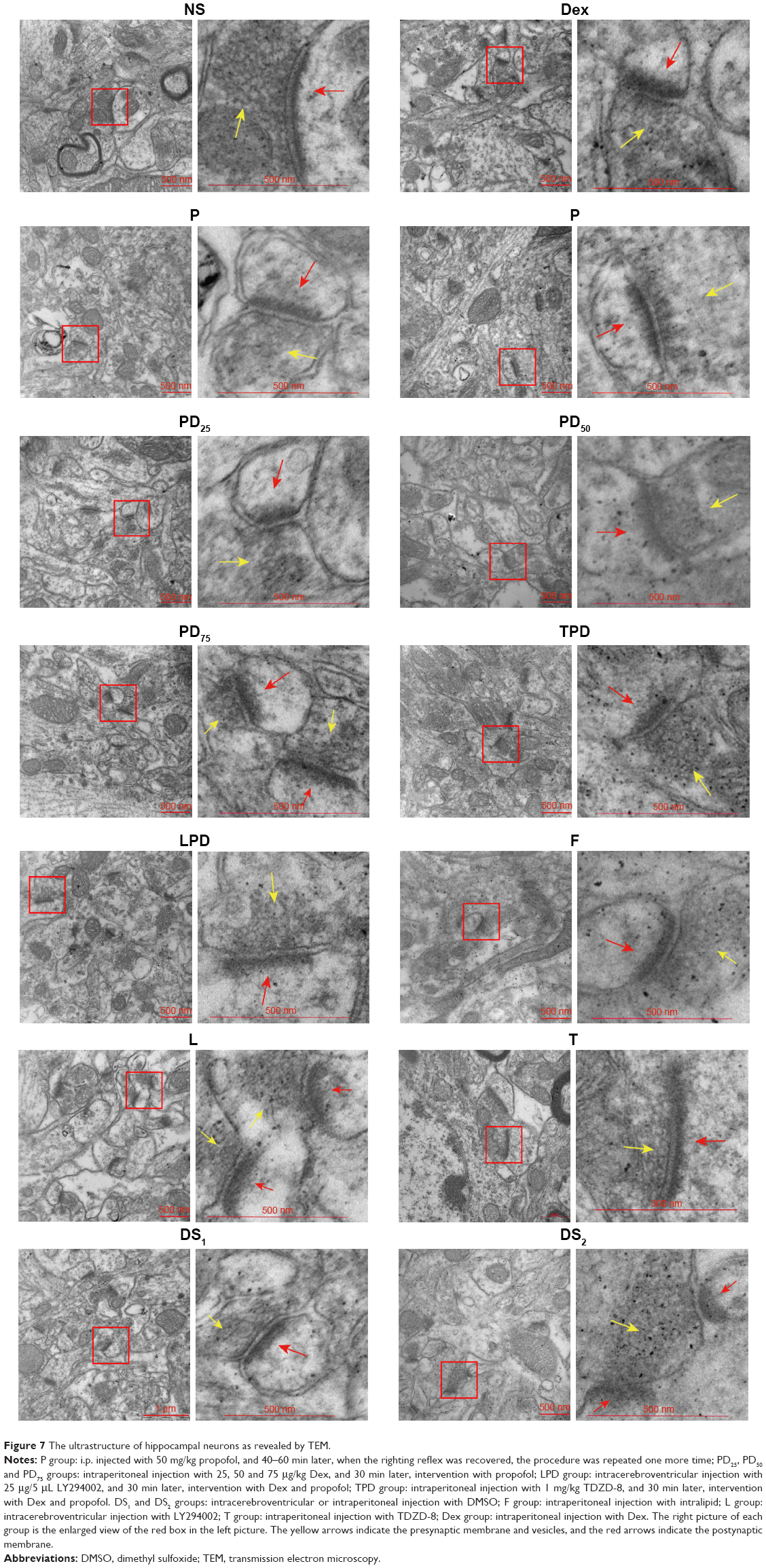 | Figure 7 The ultrastructure of hippocampal neurons as revealed by TEM. |
Dex enhances the PI3K/Akt signaling in the hippocampus of adult rats
Activation of the PI3K/Akt signaling can support the survival of neurons, which is positively regulated by GSK3β phosphorylation. Given that our results indicated the protective effect of Dex on the propofol-induced long-term neurotoxicity was dependent on the PI3K/Akt activation, we finally examined the relative levels of Akt and GSK3β expression and phosphorylation in the hippocampus of the different groups of adult rats by Western blot assays. As shown in Figure 8, there was no significant difference in the relative levels of Akt and GSK3β expression in the hippocampus of the different groups of rats. In comparison with the NS control rats, rats injected with intralipid, Dex, TDZD-8, LY294002 or DMSO did not show any change in the relative levels of Akt and GSK3β phosphorylation in the hippocampus. In contrast, the relative levels of Akt and GSK3β phosphorylation in the hippocampus were significantly reduced in the rats treated with propofol, while treatment with Dex at a higher dose significantly mitigated the propofol-reduced Akt and GSK3β phosphorylation in the hippocampus of rats. Furthermore, while treatment with TDZD-8 further increased the Akt and GSK3β phosphorylation to a level similar to that of the NS group, injection with LY294002 together with Dex and propofol further decreased the relative levels of Akt and GSK3β phosphorylation in the hippocampus of adult rats. Collectively, these results clearly demonstrated that treatment with Dex mitigated the propofol-induced long-term neurotoxicity in the hippocampus of adult rats by enhancing the PI3K/Akt signaling activation.
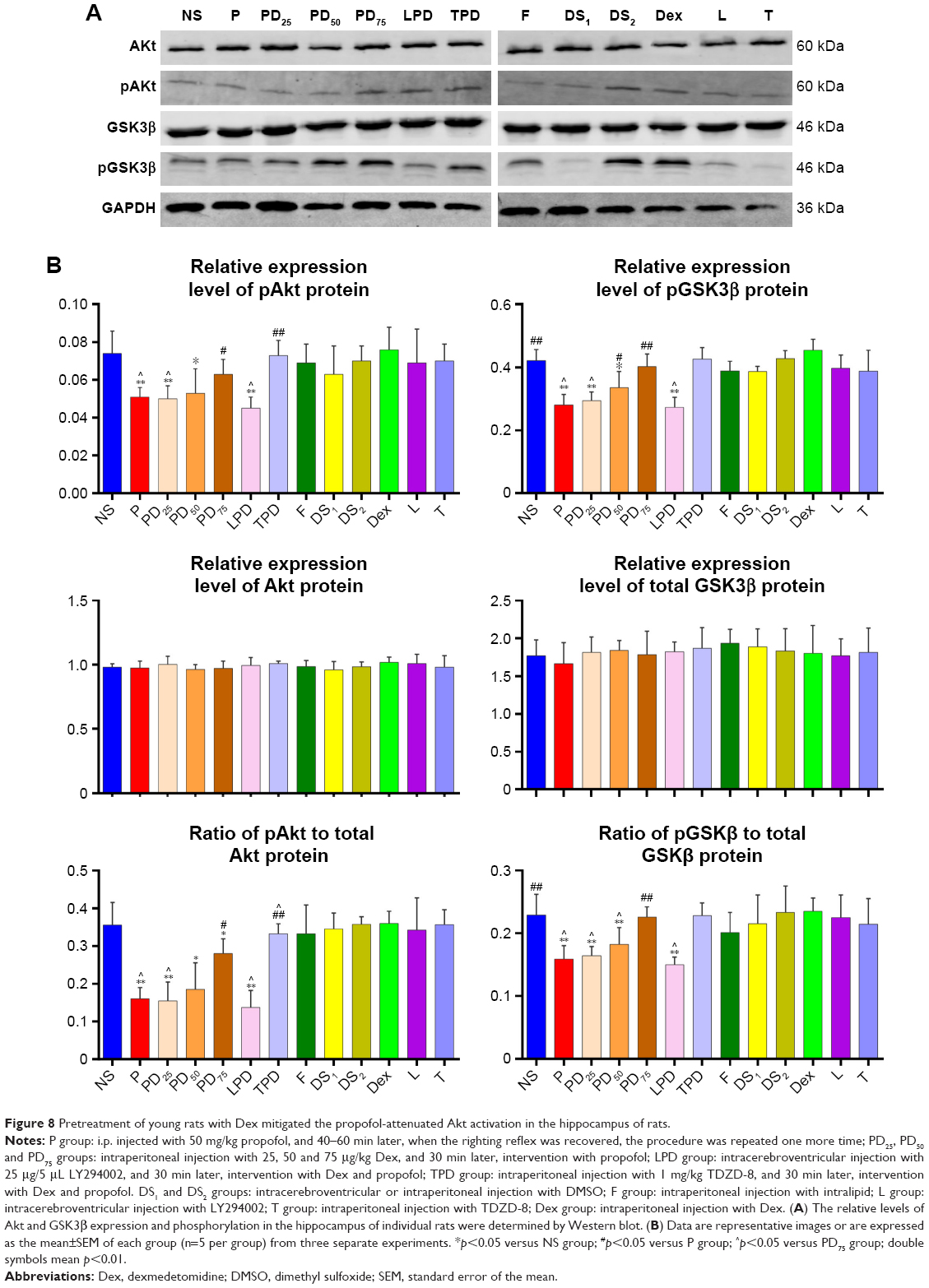 | Figure 8 Pretreatment of young rats with Dex mitigated the propofol-attenuated Akt activation in the hippocampus of rats. |
Discussion
Our previous studies have demonstrated that propofol induced acute neurotoxicity in young rats and Dex attenuated the propofol-induced neuroapoptosis by enhancing the Akt activation in a dose-dependent manner.28,39 In our study, excluding the effect of hypoxia and hypercapnia on the development of brain, the results indicated that propofol injected into young rats induced neurotoxicity, leading to impairment of spatial learning and memory in their adult age. These suggest that the neurotoxicity induced by propofol at young age in rats may remain up to their adult age. Pretreatment with Dex mitigated and prevented the propofol-induced long-term neurotoxicity and improved the spatial learning and memory by increasing the Akt activation in the hippocampus of rats. Our finding might provide new insights into the underlying toxic pathogenesis of propofol and aid in the design of new therapies for prevention of propofol-related neurotoxicity.
Previous studies have shown that propofol has an adverse effect on immature neurons in different species and treatment with even a sub-anesthetic dose of propofol induces neuronal apoptosis in the cerebral cortex, caudate and putamen of neonatal rats.4–6,40 In our study, we observed that the number of TUNEL-positive neurons in the hippocampus increased in the adult rats that had been treated with propofol at young age. These, together with ultrastructural changes, including decreased levels of PSD95 expression, widening synaptic cleft and hazing postsynaptic membrane as well as impaired spatial learning and memory, indicated that administration of high doses of propofol for a short time period induced long-term neurotoxicity, structural damages and functional impairments, similar to a previous observation with similar doses of propofol administered for a longer period.31 Indeed, injection with propofol affects the expression and proteolytic processing of crucial presynaptic and postsynaptic proteins, such as GAP-43 and MAP-2, in the cortex and thalamus of the developing brain in rats, which leads to permanent impairment of neuroplasticity and emotional behaviors.41 These also support the notion that healthy neurons and their intact synaptic structure are the basis of neurological function.
The mechanisms underlying the propofol-induced neurotoxicity in the developing brain are still unclear.42,43 Previous studies have suggested that propofol-induced neuronal toxicity is associated with increased levels of TNF-α in the developing brain and increases intracellular free calcium by activating the GABAA receptor and subsequent opening of the L-type voltage-gated calcium channels.4,44 Our previous studies have shown that induction of hippocampal neuron apoptosis is associated with decrease of the NF-κB and Bcl-2 expression in vitro and inhibition of the PI3K/Akt signaling pathway in the hippocampus of rats.28,39 Activation of the PI3K/Akt signaling can promote neuronal survival and synaptic plasticity.45 However, we observed that the long-term neurotoxicity of propofol was related to decrease of the levels of Akt phosphorylation and worsened by injection with a PI3K/Akt inhibitor, similar to a previous observation.46 It is known that all animals undergo physiological neuronal remodeling during which many neurons undergo spontaneous apoptosis. It is possible that in such a condition the neurons are especially sensitive to propofol. We will further investigate how propofol induces the long-term neurotoxicity and functional impairment after drug clearance in future.
PSD95 is a cytoskeletal protein and regulates the learning, memory and synaptic plasticity.29 In our study, we observed that exposure of young rats to propofol significantly decreased the levels of PSD95 and induced the synaptic ultrastructural damages in their hippocampus as they aged. It is important that PSD95 binds to the NMDA receptors to control bidirectional synaptic plasticity and learning.47 The decreased levels of PSD95 and the structural damages may be associated with the impairment of spatial learning and memory functions. Although the mechanisms underlying the action of propofol in downregulating PSD95 expression are unclear, they may be associated with that propofol induced neuronal apoptosis and inhibited the synapse formation.31 It is known that activation of NMDA receptor can promote calcium influx and activate the CaMK-II and F-actin to induce the phosphorylation and structural reorganization of PSD.45,48,49 Hence, PSD95 is an excellent biomarker for measuring the integrity of postsynaptic membrane in animals.
A previous study has shown that Dex supports neurogenesis and plasticity.50 Our results showed that Dex modulated the neuronal remodeling by increasing the thickness of hippocampal PSD and decreasing the synaptic cleft. Moreover, pretreatment of young rats with a higher dose of Dex significantly attenuated the propofol-induced neuronal apoptosis and decreased PSD95 expression and structural damages as well as learning and memory impairment in their adult age, which was enhanced by co-injection with TDZD-8, but abrogated by injection with LY294002. More importantly, treatment with Dex before the intervention of propofol significantly increased the relative levels of Akt and GSK3β phosphorylation in the hippocampus of adult rats. These results demonstrated that treatment with Dex mitigated the propofol-induced long-term neurotoxicity in adult rats, dependent on the activation of PI3K/Akt signaling pathway in the hippocampus. Our findings support the notion that activation of the PI3K/Akt signaling pathway can promote neuronal survival and synaptic plasticity.45 The protective effect of Dex may be attributed to its promotion of neurogenesis and plasticity as well as neuronal survival by preserving the neurons from propofol-induced apoptosis.50 Alternatively, Dex may promote the restoration from the neuronal injury following exposure to propofol and/or modulate the propofol-mediated neuronal remodeling in young rats. Therefore, our findings may aid in the design of new therapies for intervention and prevention of propofol-induced neuronal toxicity.
There are some limitations in the current study that need to be clarified. We did not separate the pups in the treatment groups from the pups in the control groups, for we worried that mother rats would abandon and hurt the adopted pups. The treatment drug might be released into the urine or feces from the treated rats which might affect the control rats in the same cage. Though the metabolites of propofol and Dex are thought to be inactive,51 the impact on the result was unclear. In this study, chloral hydrate was used in the rats’ sacrifice, for it did not have such effects that the volatile anesthetics and ketamine might affect apoptosis and the expression levels of proteins of the hippocampus of rats.52–55 In addition, the rats were sacrificed immediately after anesthesia without any recovery procedures. It was consistent with the animal ethical guidelines of the UK.56 In this study, though we had examined the exterior of the rats, the vision of rats was hard to measure, and it might have interfered with the results of the MWM test. The design of this study did not take account of the time gradient which needs further research. Despite these above limitations, the results of our study are still reliable and also support our conclusions.
Conclusion
In summary, our results indicated that injection of young rats with propofol induced neurotoxicity of the hippocampal neurons when the rats reached adulthood, demonstrating the long-term neurotoxicity to the developing brains. Pretreatment with Dex significantly mitigated the long-term neuronal damages by enhancing the PI3K/Akt activation in the hippocampus of adult rats. Our findings may provide new insights into the toxicity of propofol and aid in the design of new therapies for intervention and prevention of propofol-related neuronal damages and may be of value in clinical anesthesia management. However, our results still need further confirmation.
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (No 81373498 and No 81560500) to Yubo Xie and the Natural Science Foundation for Youth of Guangxi (No 2017GXNSFBA198108) to Yubing Liang and the Program of Shenzhen Science and Technology Innovation Committee, China (knowledge innovation program, CYJ2015040209413018) to Li Yuantao. All authors thank Mr Xiaole Liang, from the Basic Medical Experimental Centre of Guangxi Medical University, for his technical assistance in experiments. In addition, we would like to acknowledge the helpful comments on this article received from our reviewers.
Disclosure
The authors report no conflicts of interest in this work.
References
Bolkenius D, Dumps C, Halbeck E. Medikamente zur intravenösen Narkoseinduktion: Barbiturate. Anaesthesist. 2018;67(2):147–162. | ||
Biricik E, Karacaer F, Gulec E, Surmelioglu O, Ilginel M, Ozcengiz D. Comparison of TIVA with different combinations of ketamine-propofol mixtures in pediatric patients. J Anesth. 2018;32(1):104–111. | ||
McCann ME, Soriano SG. General anesthetics in pediatric anesthesia: influences on the developing brain. Curr Drug Targets. 2012;13(7):944–951. | ||
Kahraman S, Zup SL, McCarthy MM, Fiskum G. GABAergic mechanism of propofol toxicity in immature neurons. J Neurosurg Anesthesiol. 2008;20(4):233–240. | ||
Guo P, Huang Z, Tao T, et al. Zebrafish as a model for studying the developmental neurotoxicity of propofol. J Appl Toxicol. 2015;35(12):1511–1519. | ||
Huang J, Jing S, Chen X, et al. Propofol administration during early postnatal life suppresses hippocampal neurogenesis. Mol Neurobiol. 2016;53(2):1031–1044. | ||
Vutskits L, Gascon E, Tassonyi E, Kiss JZ. Clinically relevant concentrations of propofol but not midazolam alter in vitro dendritic development of isolated gamma-aminobutyric acid-positive interneurons. Anesthesiology. 2005;102(5):970–976. | ||
Sall JW, Stratmann G, Leong J, Woodward E, Bickler PE. Propofol at clinically relevant concentrations increases neuronal differentiation but is not toxic to hippocampal neural precursor cells in vitro. Anesthesiology. 2012;117(5):1080–1090. | ||
Yu D, Jiang Y, Gao J, Liu B, Chen P. Repeated exposure to propofol potentiates neuroapoptosis and long-term behavioral deficits in neonatal rats. Neurosci Lett. 2013;534:41–46. | ||
Spahr-Schopfer I, Vutskits L, Toni N, Buchs PA, Parisi L, Muller D. Differential neurotoxic effects of propofol on dissociated cortical cells and organotypic hippocampal cultures. Anesthesiology. 2000;92(5):1408–1417. | ||
Zou WW, Xiao HP, Gu MN, Liu KX, Liu ZQ. Propofol induces rat embryonic neural stem cell apoptosis by activating both extrinsic and intrinsic pathways. Mol Med Rep. 2013;7(4):1123–1128. | ||
Sun L. Early childhood general anaesthesia exposure and neurocognitive development. Br J Anaesth. 2010;105 (Suppl 1):i61–i68. | ||
US Food and Drug Administration. Safety Announcement: FDA Review Results in New Warnings about Using General Anesthetics and Sedation Drugs in Young Children and Pregnant Women; 2016. Available from: http://www.fda.gov/downloads/Drugs/DrugSafety/UCM533197.pdf. Accessed August 21, 2017. | ||
Djaiani G, Silverton N, Fedorko L, et al. Dexmedetomidine versus propofol sedation reduces delirium after cardiac surgery: a randomized controlled trial. Anesthesiology. 2016;124(2):362–368. | ||
Andersen JH, Grevstad U, Siegel H, Dahl JB, Mathiesen O, Jaeger P. Does dexmedetomidine have a perineural mechanism of action when used as an adjuvant to ropivacaine? A paired, blinded, randomized trial in healthy volunteers. Anesthesiology. 2017;126(1):66–73. | ||
Ma D, Hossain M, Rajakumaraswamy N, et al. Dexmedetomidine produces its neuroprotective effect via the alpha 2A-adrenoceptor subtype. Eur J Pharmacol. 2004;502(1–2):87–97. | ||
Engelhard K, Werner C, Eberspacher E, et al. The effect of the alpha 2-agonist dexmedetomidine and the N-methyl-D-aspartate antagonist S(+)-ketamine on the expression of apoptosis-regulating proteins after incomplete cerebral ischemia and reperfusion in rats. Anesth Analg. 2003;96(2):524–531, table of contents. | ||
Dahmani S, Paris A, Jannier V, et al. Dexmedetomidine increases hippocampal phosphorylated extracellular signal-regulated protein kinase 1 and 2 content by an alpha 2-adrenoceptor-independent mechanism: evidence for the involvement of imidazoline I1 receptors. Anesthesiology. 2008;108(3):457–466. | ||
Dahmani S, Rouelle D, Gressens P, Mantz J. Effects of dexmedetomidine on hippocampal focal adhesion kinase tyrosine phosphorylation in physiologic and ischemic conditions. Anesthesiology. 2005;103(5):969–977. | ||
Sanders RD, Xu J, Shu Y, et al. Dexmedetomidine attenuates isoflurane-induced neurocognitive impairment in neonatal rats. Anesthesiology. 2009;110(5):1077–1085. | ||
Sanders RD, Sun P, Patel S, Li M, Maze M, Ma D. Dexmedetomidine provides cortical neuroprotection: impact on anaesthetic-induced neuroapoptosis in the rat developing brain. Acta Anaesthesiol Scand. 2010;54(6):710–716. | ||
Meng L, Li L, Lu S, et al. The protective effect of dexmedetomidine on LPS-induced acute lung injury through the HMGB1-mediated TLR4/NF-kappaB and PI3K/Akt/mTOR pathways. Mol Immunol. 2018;94:7–17. | ||
Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129(7):1261–1274. | ||
Song G, Ouyang G, Bao S. The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med. 2005;9(1):59–71. | ||
Juhaszova M, Zorov DB, Kim SH, et al. Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest. 2004;113(11):1535–1549. | ||
Twaroski DM, Yan Y, Olson JM, Bosnjak ZJ, Bai X. Down-regulation of microRNA-21 is involved in the propofol-induced neurotoxicity observed in human stem cell-derived neurons. Anesthesiology. 2014;121(4):786–800. | ||
Deng X, Chen B, Wang B, Zhang J, Liu H. TNF-alpha mediates the intrinsic and extrinsic pathway in propofol-induced neuronal apoptosis via PI3K/Akt signaling pathway in rat prefrontal cortical neurons. Neurotox Res. 2017;32(3):409–419. | ||
Lv J, Wei Y, Chen Y, et al. Dexmedetomidine attenuates propofol-induce neuroapoptosis partly via the activation of the PI3k/Akt/GSK3beta pathway in the hippocampus of neonatal rats. Environ Toxicol Pharmacol. 2017;52:121–128. | ||
Kim E, Sheng M. PDZ domain proteins of synapses. Nat Rev Neurosci. 2004;5(10):771–781. | ||
Gardoni F, Marcello E, Di Luca M. Postsynaptic density-membrane associated guanylate kinase proteins (PSD-MAGUKs) and their role in CNS disorders. Neuroscience. 2009;158(1):324–333. | ||
Chen B, Deng X, Wang B, Liu H. Persistent neuronal apoptosis and synaptic loss induced by multiple but not single exposure of propofol contribute to long-term cognitive dysfunction in neonatal rats. J Toxicol Sci. 2016;41(5):627–636. | ||
Demers G, Griffin G, De Vroey G, Haywood JR, Zurlo J, Bedard M. Animal research. Harmonization of animal care and use guidance. Science. 2006;312(5774):700–701. | ||
Durand GM, Kovalchuk Y, Konnerth A. Long-term potentiation and functional synapse induction in developing hippocampus. Nature. 1996;381(6577):71–75. | ||
Kim JY, Grunke SD, Jankowsky JL. Widespread neuronal transduction of the rodent CNS via neonatal viral injection. Methods Mol Biol. 2016;1382:239–250. | ||
Zhang D, Zhang Z, Liu Y, et al. The short- and long-term effects of orally administered high-dose reduced graphene oxide nanosheets on mouse behaviors. Biomaterials. 2015;68:100–113. | ||
Fu DA, Campbell-Thompson M. Immunohistochemistry staining for human alpha-1 antitrypsin. Methods Mol Biol. 2017;1639:139–143. | ||
Wu GJ, Chen JT, Tsai HC, Chen TL, Liu SH, Chen RM. Protection of dexmedetomidine against ischemia/reperfusion-induced apoptotic insults to neuronal cells occurs via an intrinsic mitochondria-dependent pathway. J Cell Biochem. 2017;118(9):2635–2644. | ||
Bayes A, van de Lagemaat LN, Collins MO, et al. Characterization of the proteome, diseases and evolution of the human postsynaptic density. Nat Neurosci. 2011;14(1):19–21. | ||
Zhong Y, Liang Y, Chen J, et al. Propofol inhibits proliferation and induces neuroapoptosis of hippocampal neurons in vitro via downregulation of NF-kappaB p65 and Bcl-2 and upregulation of caspase-3. Cell Biochem Funct. 2014;32(8):720–729. | ||
Cattano D, Young C, Straiko MM, Olney JW. Subanesthetic doses of propofol induce neuroapoptosis in the infant mouse brain. Anesth Analg. 2008;106(6):1712–1714. | ||
Milanovic D, Pesic V, Loncarevic-Vasiljkovic N, et al. Neonatal propofol anesthesia changes expression of synaptic plasticity proteins and increases stereotypic and anxyolitic behavior in adult rats. Neurotox Res. 2017;32(2):247–263. | ||
Pesic V, Milanovic D, Tanic N, et al. Potential mechanism of cell death in the developing rat brain induced by propofol anesthesia. Int J Dev Neurosci. 2009;27(3):279–287. | ||
Yon JH, Daniel-Johnson J, Carter LB, Jevtovic-Todorovic V. Anesthesia induces neuronal cell death in the developing rat brain via the intrinsic and extrinsic apoptotic pathways. Neuroscience. 2005;135(3):815–827. | ||
Chen B, Deng X, Wang B, Liu H. Etanercept, an inhibitor of TNF-a, prevents propofol-induced neurotoxicity in the developing brain. Int J Dev Neurosci. 2016;55:91–100. | ||
Chandler LJ. Ethanol and brain plasticity: receptors and molecular networks of the postsynaptic density as targets of ethanol. Pharmacol Ther. 2003;99(3):311–326. | ||
Wang Y, Wu C, Han B, et al. Dexmedetomidine attenuates repeated propofol exposure-induced hippocampal apoptosis, PI3K/Akt/Gsk-3beta signaling disruption, and juvenile cognitive deficits in neonatal rats. Mol Med Rep. 2016;14(1):769–775. | ||
Migaud M, Charlesworth P, Dempster M, et al. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature. 1998;396(6710):433–439. | ||
Kuriu T, Inoue A, Bito H, Sobue K, Okabe S. Differential control of postsynaptic density scaffolds via actin-dependent and -independent mechanisms. J Neurosci. 2006;26(29):7693–7706. | ||
Dosemeci A, Jaffe H. Regulation of phosphorylation at the postsynaptic density during different activity states of Ca2+/calmodulin-dependent protein kinase II. Biochem Biophys Res Commun. 2010;391(1):78–84. | ||
Endesfelder S, Makki H, von Haefen C, Spies CD, Buhrer C, Sifringer M. Neuroprotective effects of dexmedetomidine against hyperoxia-induced injury in the developing rat brain. PLoS One. 2017;12(2):e0171498. | ||
Miller RD. Miller’s Anesthesia. 8th ed. Philadelphia, PA: Elsevier Saunders; 2015. | ||
Zhou X, Li W, Chen X, et al. Dose-dependent effects of sevoflurane exposure during early lifetime on apoptosis in hippocampus and neurocognitive outcomes in Sprague-Dawley rats. Int J Physiol Pathophysiol Pharmacol. 2016;8(3):111–119. | ||
Erasso DM, Camporesi EM, Mangar D, Saporta S. Effects of isoflurane or propofol on postnatal hippocampal neurogenesis in young and aged rats. Brain Res. 2013;1530:1–12. | ||
Kalenka A, Gross B, Maurer MH, Thierse HJ, Feldmann RE Jr. Isoflurane anesthesia elicits protein pattern changes in rat hippocampus. J Neurosurg Anesthesiol. 2010;22(2):144–154. | ||
Hu K, Xie YY, Zhang C, et al. microRNA expression profile of the hippocampus in a rat model of temporal lobe epilepsy and miR-34a-targeted neuroprotection against hippocampal neurone cell apoptosis post-status epilepticus. BMC Neurosci. 2012;13:115. | ||
Grundy D. Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology. J Physiol. 2015;593(12):2547–2549. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.