Back to Journals » Drug Design, Development and Therapy » Volume 10
Dexamethasone-(C21-phosphoramide)-[anti-EGFR]: molecular design, synthetic organic chemistry reactions, and antineoplastic cytotoxic potency against pulmonary adenocarcinoma (A549)
Authors Coyne CP, Narayanan L
Received 8 December 2015
Accepted for publication 9 March 2016
Published 12 August 2016 Volume 2016:10 Pages 2575—2597
DOI https://doi.org/10.2147/DDDT.S102075
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Cody P Coyne,1 Lakshmi Narayanan2
1Department of Basic Sciences, 2Department of Clinical Sciences, College of Veterinary Medicine, Mississippi State University, Starkville, MS, USA
Purpose: Corticosteroids are effective in the management of a variety of disease states, such as several forms of neoplasia (leukemia and lymphoma), autoimmune conditions, and severe inflammatory responses. Molecular strategies that selectively “target” delivery of corticosteroids minimize or prevents large amounts of the pharmaceutical moiety from passively diffusing into normal healthy cell populations residing within tissues and organ systems.
Materials and methods: The covalent immunopharmaceutical, dexamethasone-(C21-phosphoramide)-[anti-EGFR] was synthesized by reacting dexamethasone-21-monophosphate with a carbodiimide reagent to form a dexamethasone phosphate carbodiimide ester that was subsequently reacted with imidazole to create an amine-reactive dexamethasone-(C21-phosphorylimidazolide) intermediate. Monoclonal anti-EGFR immunoglobulin was combined with the amine-reactive dexamethasone-(C21-phosphorylimidazolide) intermediate, resulting in the synthesis of the covalent immunopharmaceutical, dexamethasone-(C21-phosphoramide)-[anti-EGFR]. Following spectrophotometric analysis and validation of retained epidermal growth factor receptor type 1 (EGFR)-binding avidity by cell-ELISA, the selective anti-neoplasic cytotoxic potency of dexamethasone-(C21-phosphoramide)-[anti-EGFR] was established by MTT-based vitality stain methodology using adherent monolayer populations of human pulmonary adenocarcinoma (A549) known to overexpress the tropic membrane receptors EGFR and insulin-like growth factor receptor type 1.
Results: The dexamethasone:IgG molar-incorporation-index for dexamethasone-(C21-phosphoramide)-[anti-EGFR] was 6.95:1 following exhaustive serial microfiltration. Cytotoxicity analysis: covalent bonding of dexamethasone to monoclonal anti-EGFR immunoglobulin did not significantly modify the ex vivo antineoplastic cytotoxicity of dexamethasone against pulmonary adenocarcinoma at and between the standardized dexamethasone equivalent concentrations of 10-9 M and 10-5 M. Rapid increases in antineoplastic cytotoxicity were observed at and between the dexamethasone equivalent concentrations of 10-9 M and 10-7 M where cancer cell death increased from 7.7% to a maximum of 64.9% (92.3%–35.1% residual survival), respectively, which closely paralleled values for “free” noncovalently bound dexamethasone.
Discussion: Organic chemistry reaction regimens were optimized to develop a multiphase synthesis regimen for dexamethasone-(C21-phosphoramide)-[anti-EGFR]. Attributes of dexamethasone-(C21-phosphoramide)-[anti-EGFR] include a high dexamethasone molar incorporation-index, lack of extraneous chemical group introduction, retained EGFR-binding avidity (“targeted” delivery properties), and potential to enhance long-term pharmaceutical moiety effectiveness.
Keywords: dexamethasone, anti-EGFR, organic chemistry reactions, synthesis, selective “targeted” delivery, covalent immunopharmaceuticals, EGFR
Introduction
Dexamethasone (8S,9R,10S,11S,13S,14S,16R,17R)-9-fluoro-11,17-dihydroxy-17-(2-hydroxyacetyl)-10,13,16-trimethyl-6,7,8,9,10,11,12,13,14,15,16,17-dodecahydro-3H-cyclopenta[α]phenanthren-3-one is a corticosteroid with profound anti-inflammatory properties that are attributed to several different molecular mechanisms of action that involves inhibition of several synthesis pathways that includes 1) phospholipase-A2 biochemical activity inhibition resulting in a diminished arachidonic acid substrate availability for prostaglandin and leukotriene synthesis; 2) NF-κB resulting in a reduced production of tumor necrosis factor-α and Th1 interleukins (eg, IL-1, IL-6, and IL-2); 3) reduced IL-5 production (IL-5 >> IL-2 inhibition); 4) suppression of IFN-γ-induced major histocompatibility antigen Type II expression accompanied by 5) an induced synthesis of endogenous IL-10 that potently exerts profound anti-inflammatory properties. Influences of dexamethasone on immune cellular function are in part related to their 6) prevention or reduction of leukocyte degranulation; 7) inhibition of macrophage phagocytosis; and 8) promotion of overt lymphocyte cytolysis. Each of these properties to a varying degree represents a justification for corticosteroid administration in the therapeutic management of the hematopoietic neoplastic conditions of leukemia and lymphoma in addition to a spectrum of autoimmune disorders. In clinical regimens for the treatment of B-cell chronic lymphocytic leukemia (B-CLL), corticosteroids are coadministered in the cyclophosphamide–doxorubicin–vincristine–prednisone (CHOP) treatment regimen of cyclophosphamide, doxorubicin, vincristine, and prednisolone.1
Common complications associated with corticosteroid administration include relatively rapid development of resistance in addition to substantial immunosuppression (susceptibility to septic complications) where each of these confounding disadvantages can restrict the duration of administration and limit the successful resolution of aggressive or advanced conditions of neoplastic disease. Alternatively, if a corticosteroid-like dexamethasone is covalently bound to a molecular platform-like immunoglobulin G (IgG) that possesses properties of selective binding avidity, it then becomes possible to 1) “selectively” “target” their delivery at a single specific cell type, 2) attain or promote highly elevated cytosol dexamethasone or chemotherapeutic concentrations, and 3) activate multiple host immune responses that can evoke selectively “targeted” cytotoxicity.
Significant advances have been made in identifying trophic membrane receptors uniquely overexpressed in many adenocarcinoma and carcinoma neoplastic cell types affecting the breast, prostate, intestine, ovary, and kidney. Unique overexpression of trophic membrane receptors directly influences cancer cell biology and affects cancer cell biology2 as it pertains to viability,3,4 proliferation rate,4,5 local invasiveness,6 metastatic potential,7,8 and chemotherapeutic resistance (eg, P-glycoprotein coexpression).6,9,10 Trophic membrane receptor, epidermal growth factor receptor type 1 (EGFR), is overexpressed in non-small-cell lung cancer at a case frequency of 40%–80% and is particularly common in the cell types of squamous cell carcinoma and bronchoalveolar carcinoma.11 Analogous to many adenocarcinoma and carcinoma cell types that overexpress EGFR and HER2/neu, conditions of leukemia and lymphoma frequently overexpress several cell differentiation antigens and receptors on their exterior surface membrane. In an effect similar to herceptin (anti-HER2/neu) and cetuximab (anti-EGFR) on adenocarcinoma and carcinoma cell types, the monoclonal IgG fractions, anti-CD20 (rituxumab and ofatumumab), and anti-CD52 (alemtuzumab) suppress growth and vitality of leukemia and lymphoma, while some subtypes (chronic lymphocytic leukemia [CLL]) also can express insulin-like growth factor receptor type 1 (IGF-1R) membrane receptors.12 Therapeutically, anti-CD20 (rituxumab and ofatumumab) and anti-CD52 (alemtuzumab) have efficacy against B-CLL. Anti-CD20 (rituximab) in simultaneous combination with CHOP increases survival over CHOP alone in conditions of high-grade lymphomas.1 In contrast to anti-HER2/neu, anti-EGFR, and anti-IGF-1R, however, the predominate mechanism by which vitality and growth of leukemia or lymphoma populations is compromised by anti-CD20 and anti-CD52 occurs through the formation of surface membrane Ag:IgG complexes that subsequently activate the endogenous-based immune mechanisms of 1) antibody-dependent cell cytotoxicity (ADCC), 2) complement-mediated cytolysis (CMC), and 3) opsonization/phagocytosis.13–18 Resistance to anti-CD20 and anti-CD52 may develop through multiple mechanisms, such as rapid receptor-mediated endocytosis prior to ADCC/CMC/opsonization,13,19 monocyte/macrophage CD20/CD52 “shaving” (trogocytosis),20 and immune evasion imposed through immunosuppressive mediators emanating from cancer cell populations.21,22 Interestingly, ofatumumab (anti-CD20) has been approved for B-CLL resistant to alemtuzumab and fludarabine.
Minimizing the systemic corticosteroid immunosuppression, the rapid development of corticosteroid resistance, and the propensity for IgG-based monotherapies to primarily suppress cancer cell growth but not to exert profound potent antineoplastic cytotoxicity can be attained by molecular strategies that entail covalent bonding pharmaceuticals with a steroid motif to a biologically relevant molecular platform possessing properties of selective targeted delivery. Corticosteroids, such as dexamethasone, have previously been bound covalently to serum albumin23–25 and various carbohydrate or glycosaminoglycan analogs,26–29 but the molecular design and organic chemistry reaction regimens for the synthesis of covalent dexamethasone immunopharmaceuticals have rarely if ever been described extensively in scientific literature where their efficacy has most commonly been directed toward reducing inflammatory responses.30,31 Distinct attributes of a covalent dexamethasone immunopharmaceuticals include their potential to promote and facilitate 1) continual, progressive, and selective deposition of therapeutic steroid moieties on the exterior surface membrane of targeted cell populations; 2) decreased innocent exposure and reduced distribution of steroid analogs into normal healthy cells residing within tissues and organ systems; 3) prolongation of steroid moiety plasma pharmacokinetic profiles; and 4) progressive steroid moiety accumulation within the cytosol of targeted cell populations facilitated by the active transport mechanism of ligand-initiated receptor-mediated endocytosis. The latter biological phenomenon can potentially facilitate one of the therapeutic advantages of a covalent dexamethasone immunopharmaceutical because it modulates continual selective deposition and intracellular steroid moiety accumulation that can result in achieving cytosol concentrations 8.5-fold32 to >100-fold33,34 higher than those safely attainable by simple passive diffusion of a “free” noncovalently bound steroid analog from the extravascular fluid compartment postintravenous injection at clinically relevant dosages. In corticosteroid-sensitive leukemia and lymphoma cell populations, membrane-associated antigens and receptor complexes that are uniquely or highly overexpressed and are also known to be internalized by the active transport mechanism of receptor-mediated endocytosis include CCR7,35 CXCR5,36 TNFR1 (CD120a),37 CD19,38,39 CD20,13,19 and CD52.40
The molecular design and a corresponding organic chemistry reaction scheme have been delineated to enable a multiphase regimen for synthesizing a covalent dexamethasone-(C21-phosphoramide)-[anti-EGFR] immunocorticosteroid. Analogous organic chemistry reaction schemes have been used to synthesize the covalent immunochemotherapeutics, fludarabine-(C2-methylhydroxyphosphoramide)-[anti-IGF-1R],41 and gemcitabine-(C2-phosphoramide)-[anti-IGF-1R]. Removal of residual unreacted dexamethasone or reagents by serial microfiltration of a highly concentrated reaction mixture formulation of dexamethasone-(C21-phosphoramide)-[anti-EGFR] was determined by standardized high-performance thin layer chromatography (HP-TLC) analysis. Lack of anti-EGFR fragmentation or IgG–IgG polymerization was established by mass separation analysis using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) in combination with affinity blotting and chemiluminescent autoradiography. Retained biological activity of dexamethasone-(C21-phosphoramide)-[anti-EGFR] as a function of EGFR-binding avidity was determined by cell-ELISA utilizing monolayer populations of pulmonary adenocarcinoma (A549). Selectively targeted anti-neoplastic cytotoxicity of dexamethasone-(C21-phosphoramide)-[anti-EGFR] was then determined by the assessment of residual cell vitality/survival utilizing pulmonary adenocarcinoma (A549) as an ex vivo neoplastic disease model.
Materials and methods
Covalent dexamethasone immunopharmaceutical synthesis
Phase I synthesis format for amine-reactive chemotherapeutic intermediates
Dexamethasone-(C21-monophosphate) was formulated at a concentration of 3.85×10−2 M in a modified phosphate-buffered saline (PBS) buffer (phosphate 5.0 mM, NaCl 75 mM, ethylene diamine tetra-acetic acid [EDTA] 5.0 mM, pH 7.4) and reacted with 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide at a 5:1 molar ratio. The Phase I and Phase II reaction mixture was then allowed to gently stir at 25°C for 10–15 minutes.
Phase II and Phase III synthesis format for covalent dexamethasone immunochemotherapeutics utilizing an amine-reactive chemotherapeutic intermediate
Monoclonal IgG fractions of anti-EGFR (3.0 mg, 2.0×10−5 mmol) devoid of molecular stabilizing agents were formulated in imidazole buffer (100 mM, pH 6.0) and combined at a 1:50 molar ratio with the amine-reactive dexamethasone-(C21)-phosphorylimidazolide intermediate generated as the end product from the Phase I synthesis reaction scheme. The Phase II reaction mixture was then gently stirred continuously for 2 hours at 25°C to maximize the synthesis yield of the Phase III covalent dexamethasone-(C21-phosphoramide)-[anti-EGFR] immunopharmaceutical end product. Residual unreacted dexamethasone was removed from the covalent dexamethasone-(C21-phosphoramide)-[anti-EGFR] immunochemotherapeutic by exhaustive serial microfiltration (molecular weight cut-off [MWCO] =10 kDa) and buffer exchange utilizing conventional PBS buffer (phosphate 100 mM, NaCl 150 mM, pH 7.4).
Molecular analysis and characterization of properties
Covalently bound dexamethasone content
Detection and monitoring the relative amount of residual unreacted (noncovalently bound) dexamethasone contained in the Phase III reaction end product in the form of covalent dexamethasone-(C21-phosphoramide)-[anti-EGFR] immunopharmaceutical was determined by analytical-scale HP-TLC (silica gel, 250 μm thickness, UV 254 nm indicator). Sensitivity of detecting residual unreacted dexamethasone by analytical-scale HP-TLC was enhanced by analyzing highly concentrated formulations of the Phase III dexamethasone-(C21-phosphoramide)-[anti-EGFR] end product and the application of standardized dexamethasone reference controls formulated at matched reference control concentrations. Individual analytical-scale HP-TLC silica gel plates were subsequently developed utilizing a mobile phase solvent system composed of propanol/ethanol/ddH2O/glacial acetic acid (17:5:5:1, v/v). Detection of residual unreacted dexamethasone in dexamethasone-(C21-phosphoramide)-[anti-EGFR] and standardized dexamethasone reference controls following analytical-scale HP-TLC development was subsequently determined by direct UV illumination. Total dexamethasone concentration within the dexamethasone-(C21-phosphoramide)-[anti-EGFR] following exhaustive serial microfiltration was ≥10−4 M which is well within the range of detection for corticosteroids42 and chemotherapeutic agents43,44 by analytical-scale HP-TLC analysis. Complementary methods involve combining the covalent immunopharmaceutical 1:5 (v/v) with cold methanol or cold chloroform:isopropanol (2:1, v/v) and measurement of free noncovalently bound chemotherapeutic in the resulting supernatant.
Measurement of covalently bound dexamethasone
Total individual absorbance values at 265 nm were measured for dexamethasone-C21-monophosphate, immunoglobulin, and immunoglobulin/dexamethasone-C21-monophosphate-standardized reference controls in addition to the covalent dexamethasone-(C21-phosphoramide)-[anti-EGFR] immunopharmaceutial. Concentrations of the IgG component contained within the Phase III dexamethasone-(C21-phosphoramide)-[anti-EGFR] end product and IgG standardized reference controls were measured at 660 nm utilizing a metal-dye complex reagent (Pierce 660 nm Protein Assay; Thermo Fisher Scientific, Waltham, MA, USA). Concentration of the IgG component within the Phase III end product established by measurements at 660 nm was then utilized to calculate the corresponding 265 nm absorbance measurement. Differences between the total absorbance for dexamethasone-(C21-phosphoramide)-[anti-EGFR] measured at 265 nm and the calculated 265 nm absorbance for the immunoglobulin content of dexamethasone-(C21-phosphoramide)-[anti-EGFR] were then applied to calculate the total dexamethasone equivalent concentration within the Phase III covalent dexamethasone-(C21-phosphoramide)-[anti-EGFR] immunopharmaceutical end product.
Mass separation analysis for the detection of polymerization and fragmentation
Covalent dexamethasone-(C21-phosphoramide)-[anti-EGFR] immunochemotherapeutic in addition to reference control anti-EGFR immunoglobulin fractions formulated at a standardized protein concentration of 60 μg/mL were combined 50/50 (v/v) with conventional SDS-PAGE sample preparation buffer (Tris/glycerol/bromphenyl blue/SDS) without 2-mercaptoethanol or boiling. Covalent dexamethasone-(C21-phosphoramide)-[anti-EGFR] immunochemotherapeutic, reference control IgG (0.9 μg/well), and a mixture of prestained molecular weight marker reference controls were then developed individually by nonreducing SDS-PAGE (11% acrylamide) performed at 100 V constant voltage at 3°C for 2.5 hours.
Detection analysis for polymerization or fragmentation
Covalent dexamethasone-(C21-phosphoramide)-[anti-EGFR] immunopharmaceutical following mass/size-dependent separation by nonreducing SDS-PAGE was equilibrated in tank buffer devoid of methanol. Mass/size-separated dexamethasone-(C21-phosphoramide)-[anti-EGFR] immunopharmaceutical contained within acrylamide SDS-PAGE gels was then transferred laterally onto sheets of nitrocellulose membrane at 20 V (constant voltage) for 16 hours at 2°C–3°C with the transfer manifold packed in crushed ice.
Covalent dexamethasone-(C21-phosphoramide)-[anti-EGFR] immunopharmaceutical laterally transferred onto nitrocellulose membrane was then equilibrated in Tris-buffered saline (TBS; Tris HCl 0.1 M, NaCl 150 mM, pH 7.5, 40 mL) at 4°C for 15 minutes followed by an incubation period at 2°C–3°C for 16 hours in TBS blocking buffer (Tris 0.1 M, pH 7.4, 40 mL) containing bovine serum albumin (BSA: 5%) applied in combination with gentle horizontal agitation. Prior to further processing, nitrocellulose membranes were vigorously rinsed with TBS (Tris 0.1 M, pH 7.4, 40 mL, n=3).
Rinsed BSA-blocked nitrocellulose membranes developed for Western blot (immunodetection) analyses were incubated with horseradish peroxidase biological reagent protein G conjugate (0.25 μg/mL) at 4°C for 18 hours on a horizontal orbital shaker. Nitrocellulose membranes following vigorous rinsing in TBS (pH 7.4, 4°C, 50 mL, n=3) were incubated in blocking buffer (Tris 0.1 M, pH 7.4, with BSA 5%, 40 mL). Blocking buffer was decanted from nitrocellulose membrane blots that were again vigorously rinsed with TBS (pH 7.4, 4°C, 50 mL, n=3) before incubation with horseradish peroxidase biological reagent chemiluminescent substrate (25°C, 5–10 minutes). Under dark conditions, chemiluminescent autoradiography images were acquired by exposing radiographic film (BioMax XAR, Eastman Kodak, Rochester, NY, USA) to nitrocellulose membranes sealed within transparent ultraclear resealable plastic envelops.
Pulmonary adenocarcinoma (A549) cell tissue culture
Pulmonary adenocarcinoma ex vivo cell culture
Pulmonary adenocarcinoma (A549: ATCC American Tissue Cell Culture) populations were propagated until monolayers were ≥85% confluent in 150 cc2 tissue culture flasks containing F-12K growth media supplemented with fetal bovine serum (10%, v/v) and penicillin–streptomycin at a temperature of 37°C under a gas atmosphere of carbon dioxide (CO2 5%) and air (95%). Trypsin or any other biochemically active enzyme fractions were not used to facilitate the harvest of pulmonary adenocarcinoma (A549) cell suspensions for the seeding of tissue culture flasks or multiwell tissue culture plates. Growth media were not supplemented with growth factors, growth hormones, or any other type of growth stimulant. Pulmonary adenocarcinoma (A549) monolayer populations utilized for cell-ELISA analyses were uniformly propagated to a ≥85% level of confluency.
The human pulmonary adenocarcinoma/alveolar basal epithelial cell line A549 which was derived in 1972 from a 58-year-old Caucasian male, was utilized as an ex vivo model for neoplastic disease. Characteristic features and biological properties of the pulmonary adenocarcinoma (A549) cell line include 1) multidrug/chemotherapeutic resistance, 2) corticosteroid sensitivity, and 3) overexpression of membrane endogenous trophic receptors or antigenic sites. Most prominent in this regard include 1) EGFR (ErbB-1 and HER1; 170–180 kDa); 2) HER2/neu (EGFR2, ERBB2, CD340, HER2, MLN19, Neu, NGL, and TKR1); 3) IGF-1R (CD221, IGFIR, IGFR, and JTK13; 320 kDa); 4) IL-7 receptor; 5) β1-integrin (CD29, ITGB1, FNRB, GPIIA, MDF2, MSK12, VLA-BETA, and VLAB; 110–130 kDa); and 6) folate receptors (100 kDa). The EGFR trophic membrane receptor is also overexpressed in non-small-cell lung cancer at a frequency of 40%–80% and most commonly in squamous cell and bronchoalveolar carcinoma subtypes.11 Other neoplastic cells that overexpress EGFR include Chinese hamster ovary cell (Chinese hamster ovary =1.01×105 EGFR/cell), gliomas (2.7–6.8×105 EGFR/cell), epidermoid carcinoma (A431 =2.7×106/cell), and malignant glioma (U87MG =5.0×105/cell).
Cell-ELISA detection of total external membrane-bound IgG
Pulmonary adenocarcinoma (A549) cell suspensions were seeded into 96-well microtiter plates in aliquots of 2×105 cells/well and allowed to form a confluent adherent monolayer over a period of 24–48 hours. The growth media content in each individual well was removed manually by pipette, and the cellular monolayers were then serially rinsed (n=3) with PBS followed by their stabilization onto the plastic surface of 96-well microtiter plates with paraformaldehyde (0.4% in PBS, 15 minutes). Stabilized cellular monolayers were then incubated in triplicate with gradient concentrations of covalent dexamethasone-(C21-phosphoramide)-[anti-EGFR] immunopharmaceutical formulated at IgG equivalent concentrations of 0.01 μg/mL, 0.10 μg/mL, 1.00 μg/mL, and 10.00 μg/mL in tissue culture growth media (200 μL/well). Direct contact incubation between pulmonary adenocarcinoma (A549) monolayers and dexamethasone-(C21-phosphoramide)-[anti-EGFR] was performed at 37°C over a 3-hour incubation period under a gas atmosphere of carbon dioxide (5% CO2) and air (95%). Following serial rinsing with PBS (n=3), the development of stabilized pulmonary adenocarcinoma (A549) monolayers entailed incubation with β-galactosidase-conjugated goat antimouse IgG (1:500 dilution) for 2 hours at 25°C with residual unbound IgG removed by serial rinsing with PBS (n=3). Final development of the cell-ELISA required serial rinsing (n=3) of stabilized pulmonary adenocarcinoma (A549) monolayers with PBS followed by incubation with ortho- nitrophenyl-β-D-galactopyranoside substrate (ONPG 100 μL/well formulated fresh at 0.9 mg/mL in PBS, pH 7.2, containing 10 mM MgCl2 and 0.1 M 2-mercaptoethanol). Absorbance within each individual well was measured at 410 nm (630 nm reference wavelength) after incubation at 37°C for a period of 15 minutes.
Antineoplastic cytotoxic potency evaluation in an ex vivo cancer disease model
Cell proliferation–vitality assay for measuring cytotoxic antineoplastic potency
Individual preparations of dexamethasone-(C21-phosphoramide)-[anti-EGFR] were formulated in growth media at final standardized dexamethasone equivalent concentrations of 10−10 M, 10−9 M, 10−8 M, 10−7 M, and 10−6 M. Each standardized dexamethasone equivalent concentration of the covalent immunopharmaceutical was then transferred in triplicate into 96-well microtiter plates containing adherent pulmonary adenocarcinoma (A549: 2,000 cells/well) monolayers and growth media (200 μL/well). Covalent dexamethasone-(C21-phosphoramide)-[anti-EGFR] immunopharmaceutical was then incubated in direct contact with pulmonary adenocarcinoma (A549) monolayer populations for a period of 192 hours at 37°C under a gas atmosphere of carbon dioxide (CO2 5%) and air (95%). Following the initial 96-hour incubation period and then again 144 hours after initial challenge, pulmonary adenocarcinoma (A549) populations were replenished with fresh tissue culture media with or without covalent dexamethasone-(C21-phosphoramide)-[anti-EGFR] immunopharmaceutical.
Antineoplastic cytotoxic potency of dexamethasone-(C21-phosphoramide)-[anti-EGFR] was measured by removing all contents within the 96-well microtiter plates manually by pipette followed by serial rinsing of stabilized monolayers (n=3) with PBS followed by incubation with 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide vitality stain reagent formulated in RPMI-1640 growth media devoid of pH indicator or bovine fetal calf serum (MTT: 5 mg/mL). During an incubation period of 3–4 hours at 37°C under a gas atmosphere of carbon dioxide (CO2 5%) and air (95%) the enzyme mitochondrial succinate dehydrogenase and/or NADH/NADPH-dependent cellular oxidoreductase convert MTT vitality stain reagent to navy blue formazone crystals within the cytosol of pulmonary adenocarcinoma (A549) cell populations.45,46 Contents were then removed from each of the 96 wells in the microtiter plate, followed by serial rinsing with PBS (n=3). The resulting blue intracellular formazone crystals were dissolved with dimethyl sulfoxide (DMSO) (300 μL/well) and then spectrophotometric absorbance of the resulting blue-colored supernatant measured at 570 nm using a computer-integrated microtiter plate reader.
Results
Covalently bound dexamethasone content
The predominant Phase I end product in PBS at pH 7.4 is a reactive dexamethasone carbodiimide phosphate ester intermediate complex (Figure 1). Addition of the reactive dexamethasone phosphate carbodiimide ester intermediate to IgG formulated in imidazole buffer at pH 6.0 preferentially produces a transient Phase II amine-reactive dexamethasone-phosphorylimidazolide intermediate (Figure 1). The aliphatic ε-monoamine of lysine residue side chains within the amino acid sequence of anti-IGF-1R monoclonal IgG then preferentially reacts with the Phase II dexamethasone-phosphorylimidazolide intermediate (Figure 1). Preferential reaction with the ε-monoamine of lysine amino acid residues is attributed to their significantly greater base characteristics compared to aromatic amines due to the electron sink effect imposed by organic ring structures. The covalent phosphoramide bond structure is highly stable at 4°C or in whole plasma or tissue culture media-like environments containing 5% plasma or 5% serum albumin47 in contrast to strictly aqueous buffer solutions devoid of biological proteins where at 37°C ~12% of total liberation rate occurs over a 100-hour period.48
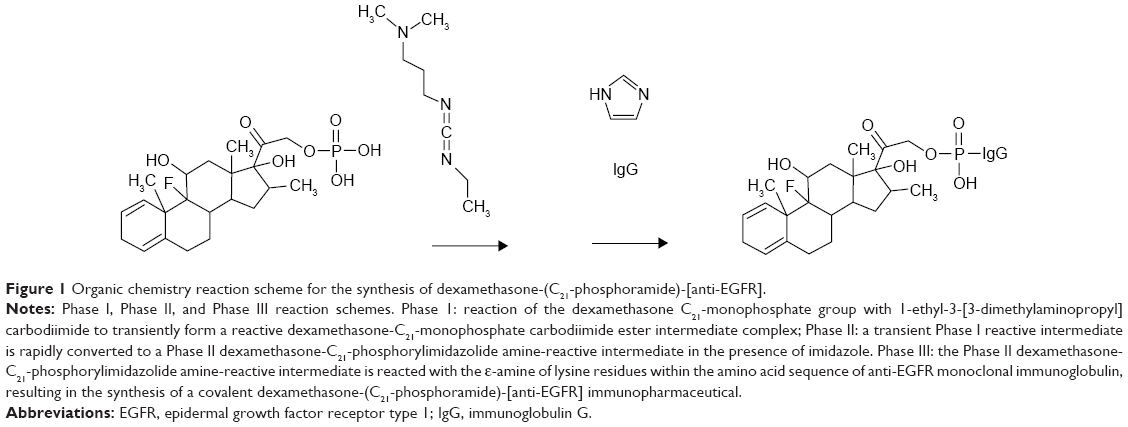 | Figure 1 Organic chemistry reaction scheme for the synthesis of dexamethasone-(C21-phosphoramide)-[anti-EGFR]. |
Serial microfiltrations (MWCO =10 kDa) of dexamethasone-(C21-phosphoramide)-[anti-EGFR] consistently yielded a Phase III covalent immunopharmaceutical end product that was devoid of any residual free noncovalently bound dexamethasone detectable by standardized analytical-HP-TLC (UV 254 nm) analysis of highly concentrated formulations (Figure 2).49–53 Results from these analyses were highly analogous to findings attained in previous investigations for covalent epirubicin49,51,52 and gemcitabine50,53 immunopharmaceutical that contained only ≤3%–4% of the total chemotherapeutic content as noncovalently bound chemotherapeutic, which cannot be removed by further serial applications of either microscale size-exclusion column chromatography or microfiltration methodologies.54
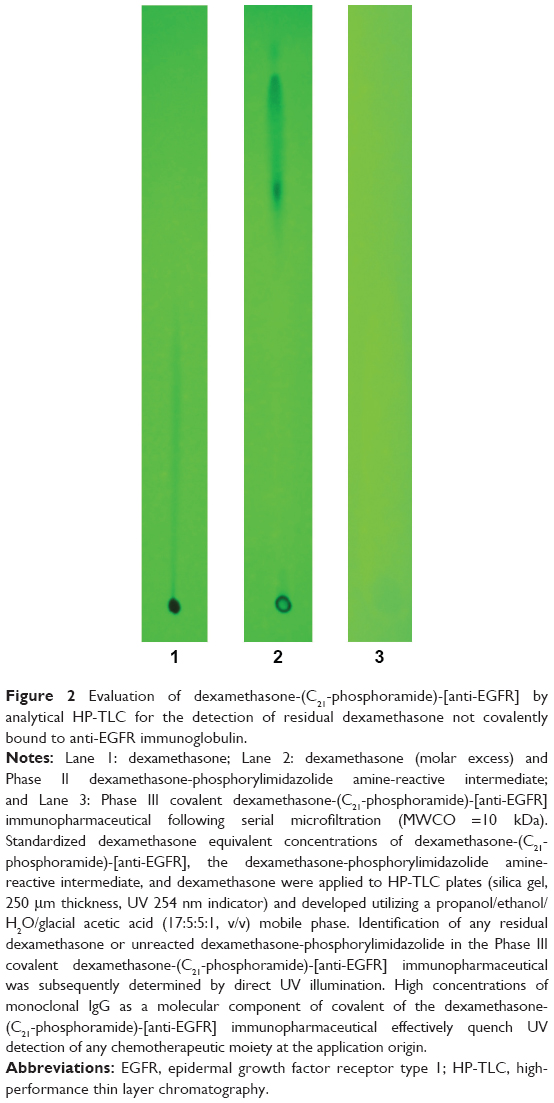 | Figure 2 Evaluation of dexamethasone-(C21-phosphoramide)-[anti-EGFR] by analytical HP-TLC for the detection of residual dexamethasone not covalently bound to anti-EGFR immunoglobulin. |
Molar incorporation index
The calculated dexamethasone:IgG molar-incorporation-index for covalent dexamethasone-(C21-phosphoramide)-[anti-EGFR] was 6.95:1 utilizing the organic chemistry reaction scheme to form a covalent phosphoramide bond at the C21-monophosphate group of dexamethasone. Microfiltration (MWCO =10 kDa) provided substantially greater yield levels for dexamethasone-(C21-phosphoramide)-[anti-EGFR] than did the removal of residual unreacted chemotherapeutic/corticosteroid and reactive intermediates by microscale size-exclusion column chromatography.
Mass separation analysis for the detection of polymerization and fragmentation
Molecular weight profile analysis of covalent dexamethasone-(C21-phosphoramide)-[anti-EGFR] immunopharmaceutical mass-separated by SDS-PAGE in combination with immunodetection analysis (Western blot) and chemiluminescent autoradiography recognized a single primary condensed band of 150 kDa between a molecular weight range of 5.0 kDa and 450 kDa (Figure 3). Profiles consistent with low molecular weight fragmentation (proteolytic/hydrolytic degradation) or high molecular weight IgG–IgG polymerization were not detected (Figure 3). The observed molecular weight of 150 kDa for dexamethasone-(C21-phosphoramide)-[anti-EGFR] directly corresponds with the known molecular weight/mass of reference control anti-EGFR/anti-IGF-1R monoclonal IgG fractions (Figure 3). Analogous results have been reported for similar covalent immunochemotherapeutics.49–53,55,56
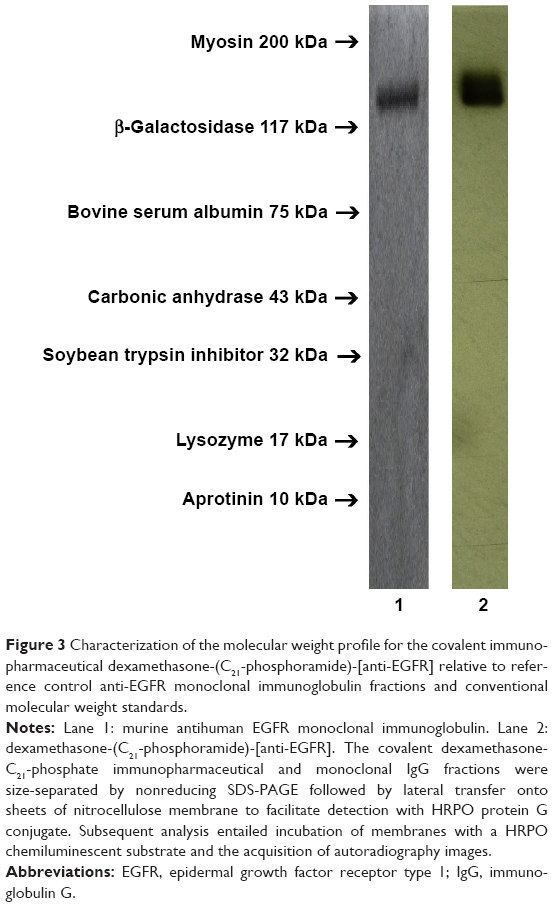 | Figure 3 Characterization of the molecular weight profile for the covalent immunopharmaceutical dexamethasone-(C21-phosphoramide)-[anti-EGFR] relative to reference control anti-EGFR monoclonal immunoglobulin fractions and conventional molecular weight standards. |
Cell-ELISA total membrane IgG-binding analysis
Total IgG in the form of dexamethasone-(C21-phosphoramide)-[anti-EGFR] bound on the external surface membrane of adherent pulmonary adenocarcinoma (A549) monolayer populations was detected and measured by cell-ELISA (Figure 4). Increases in dexamethasone-(C21-phosphoramide)-[anti-EGFR] formulated at the standardized IgG equivalent concentrations of 0.010 μg/mL, 0.10 μg/mL, 1.00 μg/mL, and 10.00 μg/mL corresponded with progressive elevations in the total amount of membrane-bound IgG (Figure 4). Collectively, results from cell-ELISA analyses validated the retained selective binding avidity of dexamethasone-(C21-phosphoramide)-[anti-EGFR] for external membrane EGFR sites highly overexpressed on the exterior surface membrane of pulmonary adenocarcinoma (A549) monolayer populations (Figure 4). Detection of essentially no significant antineoplastic cytotoxic effect by anti-EGFR monoclonal immunoglobulin in an ex vivo cell culture environment over a relatively brief period of time very closely correlated with previously reported results.
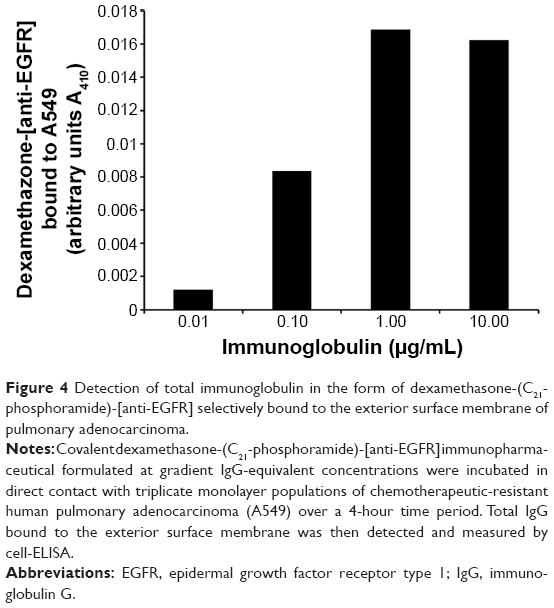 | Figure 4 Detection of total immunoglobulin in the form of dexamethasone-(C21-phosphoramide)-[anti-EGFR] selectively bound to the exterior surface membrane of pulmonary adenocarcinoma. |
Antineoplastic cytotoxic potency
Nearly identical levels of antineoplastic cytotoxic potency were detected individually for dexamethasone-(C21-phosphoramide)-[anti-EGFR] and dexamethasone against pulmonary adenocarcinoma (A549) when challenged with dexamethasone equivalent concentrations at and between 10−9 M and 10−6 M over a 192-hour incubation period (Figure 5). Antineoplastic cytotoxicity of dexamethasone-(C21-phosphoramide)-[anti-EGFR] increased rather dramatically at and between the standardized dexamethasone equivalent concentrations of 10−9 M, 10−8 M, and 10−7 M, which corresponded with lethal cancer cell death values of 7.7%, 26.9%, and 64.9% (92.3%, 73.1%, and 35.1% residual survival), respectively (Figure 5). A much more gradual increase in antineoplastic cytotoxicity was detected at and between the standardized dexamethasone equivalent concentrations of 10−7 M, 10−6 M, and 10−5 M for dexamethasone-(C21-phosphoramide)-[anti-EGFR], which were associated with the lethal cancer cell death values of 64.9%, 69.9%, and a maximum of 73.0% (35.1%, 30.1%, and 27.0% residual survival), respectively (Figure 5).
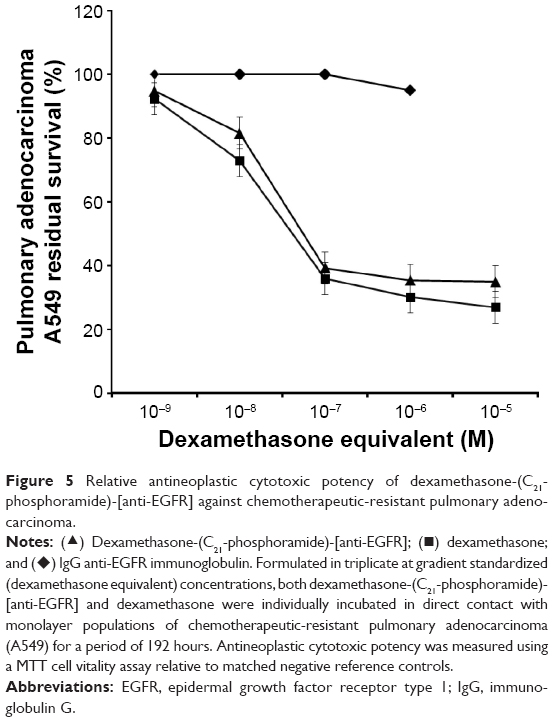 | Figure 5 Relative antineoplastic cytotoxic potency of dexamethasone-(C21-phosphoramide)-[anti-EGFR] against chemotherapeutic-resistant pulmonary adenocarcinoma. |
Discussion
Covalent biopharmaceuticals designed and synthesized to evoke antineoplastic cytotoxicity has primarily involved the implementation of anthracyclines,49,51,52,55,57–79 gemcitabine,50,53 methotrexate,80,81 mitomycin,80 vinca alkaloid analogs,82–84 bleomycine,85,86 chlorambucil,87,88 cyclophosphamide,89,90 paclitaxel (non-IgG),91–93 ozogamicin,94,95 calicheamicins,94 and monomethyl auristatin E (MMAE).96–99 A relatively modest array of organic chemistry reaction schemes have been described for covalently bonding dexamethasone and other steroid core pharmaceuticals to biologically relevant molecular platforms. Besides covalent compounding with other low molecular weight pharmaceuticals,100 corticosteroids have most commonly been covalently bound to a spectrum of high molecular weight platforms of natural origin, such as albumin,23–25,101–106 cardiolipin,107,108 chondroitin sulfate (sulfated glycosaminoglycan),29 glucose-6-phosphate dehydrogenase,109 horseradish peroxidase,110,111 spermine,112 and cloned fusion proteins.113 Pharmaceuticals with a steroid motif have additionally been covalently bound to artificial or semiartificial molecular platforms, such as amino-PEG (eg, α-methoxy-ω-amino-PEG),27 chitosan,26 dextran,114 N-(2-hydroxypropyl)methacrylamide,115 amine-modified polysaccharides,116 poly-L-glutamic acid (polypeptide configuration),117 1-dodecylthio-2-decyloxypropyl-3-phophatidic acid,118,119 lipid nucleosides,120 N-(2-hydroxypropyl)methacrylamide polymer,75 benzodiazepine receptor ligands,121,122 4-(N)-valeroyl, 4-(N)-lauroyl, and 4-(N)-stearoyl,123 4-fluoro[18F]-benzaldehyde derivatives (diagnostic positron-emitting radionucleotide),124 polyamidoamine dendrimer,28 and peptide hormone antagonists.125 The intent and purpose of covalently bonding corticosteroids to biologically relevant molecular platforms has most frequently been for diagnostic-related applications24,80,101–104,110,111,113,116 and much less often for the development of advanced therapeutics.28,112 In rare instances where covalent corticosteroid biopharmaceutical therapeutics have been designed, synthesized, and evaluated for efficacy28,112 it has most frequently been to determine their capacity to suppress cellular inflammatory responses utilizing molecular delivery platforms that include or are analogous to anti-E selectin and anti-CD183.30,31 Covalent bonding of dexamethasone or other steroid motif pharmaceuticals to IgG, IgG fragments (eg, F(ab′)2 and Fab′), receptor ligands (eg, epidermal growth factor [EGF] → EGFR), or other high molecular weight biological proteins has to date not been extensively described or has the design, the molecular structure, or organic chemistry reaction regimes been reported that describe the synthesis of covalent corticosteroid biopharmaceuticals that exert properties of selectively targeted antineoplastic cytotoxic potency relevant to conditions of leukemia or lymphoma.
Precarboxylated corticosteroid/steroid intermediate analogs
Precarboxylation of corticosteroids and steroid core pharmaceuticals is an initial requirement for some organic chemistry reactions which is implemented prior to synthetically bonding them covalently to biologically relevant molecular platforms.101 Corticosteroid/steroid precarboxylation can be achieved utilizing several different organic chemistry reaction schemes. Hydrazine acetic acid ethyl ester and heating to 120°C produce a hydrazino acetic acid analog of a corticosteroid/steroid and introduces a functionally available carboxyl group and an acid-cleavable hydrazino at the C21 position.27 Precarboxylation of corticosteroids/steroids through the introduction of –(O-carboxymethyl)oxime is achieved by first converting a hydroxyl (–CH2OH) group into an aldehyde (–CHO) through the application of chromium (VI) oxide, anhydrous magnesium sulfate, pyridine, dichloromethane, and ether.126 The corticosteroid/steroid aldehyde is then combined with O-(carboxymethyl)hydroxylamine hemihydrochloride and is further processed utilizing toluene, ether, ethyl acetate, methanol, NaHCO3, and H2SO4 to yield an –(O-carboxymethyl)oxime analog.126 Corticosteroids/steroids in combination with a dicarboxylic organic acid-like glutarate can be converted to a carboxyl ester analog at an available hydroxyl group (eg, C21-OH) in the presence of a carbodiimide following the initial formation of a reactive glutarate O-acylisourea ester intermediate.28 Some corticosteroid/steroid hemisuccinate analogs (eg, C21 position) are commercially available or they can be synthesized utilizing succinic anhydride reagent in combination with sodium sulfate, pyridine, chloroform, acetone, and benzene/hexane.24,105,106,109,125 Similarly, covalent corticosteroid/steroid –(O-carboxymethyl)oxime analogs can be synthesized “in-house” utilizing a wide spectrum of reagents including –(O-carboxymethyl)hydroxyamine in combination with diazomethane, N-nitroso-N-methylurea, aluminum isopropoxide, ethyl acetate, ether, acetone, benzene, KOH, and HCl reagents.105,106 Carboxylation of corticosteroids/steroids through the introduction of glutarate,28 hemisuccinate,24,109,125 or –(O-carboxymethyl)oxime24,109 can potentially occur at hydroxyl groups located at the C3, C6, C11, C17, or C21 positions within the chemical structure of steroid core pharmaceuticals. Alternatively, some but not all corticosteroids/steroids can be obtained commercially as hemisuccinate24,26,109 or –(O-carboxymethyl)oxime analogs.
Preaminated corticosteroid/steroid intermediate analogs
Corticosteroid/steroid monoamine analogs can be produced utilizing N-trityl-glycine and a carbodiimide resulting in the production of a trityl-glycine-steroid intermediate that is then converted by AcOH to a glycyl steroid (eg, glycyl-prednisolone).29 In a second reaction, the monoamine group of the glycyl steroid is transformed into a covalent amide bond structure at a carboxyl group associated with a biologically relevant molecular platform (eg, chondroitin sulfate) in the presence of a carbodiimide and N-hydroxysuccinimide (NHS).29 Given this general synthesis strategy, corticosteroids/steroids initially can be converted to either a monoamine or a monocarboxyl analog that is then covalently bound to a biologically relevant molecular platform at an available primary carboxyl group or primary amine group, respectively.
Covalent bonding of carboxylated corticosteroid/steroid analogs independent of carbodiimide
A limited number of carbodiimide-independent organic chemistry reaction schemes have been developed for covalent bonding of carboxylated corticosteroids/steroids to biologically relevant molecular platforms. Carboxylated corticosteroids/steroids in the form of either hemisuccinate analogs or –(O-carboxymethyl)oxime analogs can be covalently bound to proteins through a mixed anhydride reaction when performed in combination with acetic anhydride, 2-methyl-6-nitrobenzoic anhydride, tri-n-butylamine, isobutylchlorocarbonate, gamma amino-n-butyric acid, dioxane, acetone, NaOH, and HCL reagents.23–25,31,105,106,116,127,128 During mixed anhydride reactions of the Dalziel Hammick type (RCO2H + H2C=C=O → RCO2C(O)CH3), corticosteroids become covalently bound to a second biologically relevant molecular platform at an available primary carboxyl group. A disadvantage of some mixed anhydride reactions is the associated increased risk of generating high molecular weight end products when the organic chemistry scheme is allowed to progress, resulting in the occurrence of polymer-forming side reactions.25
Carbodiimide-dependent covalent bonding of carboxylated corticosteroid/steroid analogs
Many, if not majority, of the methods for covalent bonding of corticosteroids and other pharmaceuticals with a steroid core to a biologically relevant molecular platform use a carbodiimide reagent that forms a covalent amide bond structure between primary carboxyl and primary amine groups. A characteristic of carbodiimide reagents is their formation of a transient O-acylisourea intermediate at primary carboxyl groups that can be either immediately reacted with a primary amine group or reacted with NHS to create a more stable and amine-reactive corticosteroid/steroid NHS ester intermediate.24 Carboxylated corticosteroids/steroids, such as hemisuccinate,24,109,125 glutarate,28 and –(O-carboxymethyl)oxime24,109 analogs, in anhydrous DMSO or dimethylformamide (DMF) are converted by carbodiimides to a transient O-acylisourea intermediate.24,26,109 Some reaction schemes simultaneously utilize 1-hydroxybenzotriazole, 1-hydroxy-7-azabenzotriazole, or similar reagents to suppress racemization.125 Electrophilic properties of the monocarboxyl group of modified corticosteroid/steroid analogs increase following their reaction with carbodiimide reagents. The negatively charged carboxyl oxygen has nucleophile characteristics and forms a covalent bond with the central carbon atom of the functional group (R–N=C=N–R) of carbodiimides. Carboxylated corticosteroid/steroid O-acylisourea ester intermediates ultimately can be reacted with several different chemical groups associated with a biologically relevant molecular platform: 1) primary amine groups resulting in the formation of a covalent amide bond structure;125 2) hydroxyl groups resulting in the formation of a covalent ester bond structure;28 and alternatively with 3) NHS resulting in the production of a relatively stable amine-reactive corticosteroid/steroid NHS ester intermediate in the presence of dioxane and DMF.24,26,109 When O-acylisourea ester125 and NHS ester24,27 intermediates of corticosteroids/steroids are reacted with the ε-monoamine of lysine amino acid residues within the sequence of biologically relevant peptides, polypeptides, and proteins, they form a stable covalent amide bond structures. Such synthesis regimens may require performing this phase of the reaction scheme in anhydrous dioxane, DMSO, or DMF in the presence of 1-hydroxybenzotriazole (suppresses racemization).24,125
Preliminary synthesis of the amine-reactive dexamethasone-(C21-phophorylimidazole) intermediate
The molecular design of dexamethasone-(C21-phosphoramide)-[anti-IGF-1R] and organic chemistry reactions embedded within a multiphase synthesis regime represents a notable departure from previously described methodologies for covalent immunochemotherapeutics.24,26,28,109,125 The organic chemistry reactions used in the multiphase synthesis scheme for dexamethasone-(C21-phosphoramide)-[anti-EGFR] involved the initial generation of a transient Phase I dexamethasone-C21-phosphate ester-carbodiimide reactive intermediate that rapidly transforms in the presence of imidazole into a more stable Phase II dexamethasone amine-reactive phophorylimidazole intermediate (Figure 1). Dexamethasone-C21-phosphate was formulated at a relatively large molar excess to the carbodiimide reagent in order to 1) maximize the yield of the Phase II amine-reactive dexamethasone-C21-phosphate intermediate, 2) accelerate the rate of the organic chemistry reaction, 3) promote the maximal depletion of the carbodiimide reagent (also unstable for prolonged incubation periods in aqueous-based buffer systems), and 4) minimize the risk of high molecular weight IgG–IgG polymerization.
Covalent bonding of an amine-reactive dexamethasone-(C21-phophorylimidazole) intermediate to immunoglobulin
In Phase III of the organic chemistry synthesis scheme, the Phase II dexamethasone-(C21-phophorylimidazole) was ultimately reacted with the ε-monoamine group of lysine residues within the amino acid sequence of anti-EGFR monoclonal IgG, resulting in the formation of a covalent phosphoramide bond structure at the dexamethasone C21 position (Figure 1). A relatively limited number of organic chemistry reactions can be utilized to covalently bond chemotherapeutic moieties to biologically relevant molecular platforms utilizing conditions and reagents that do not detrimentally modify their function and integrity. A covalent amide bond structure is most commonly formed at the ε-monoamine group of lysine residues within the amino acid sequence of most common biologically relevant molecular platforms that are peptide subunits or proteins. Alternatively, either IgG and other glycoproteins can also be prethiolated (introduction of reduced sulfhydryl R-SH groups) at ε-monoamine of lysine amino acid residues or covalent bond structures can be formed at aldehyde groups within the carbohydrate component created by limited oxidation.
Multiphase organic chemistry synthesis scheme qualities and advantages
In addition to the classical variables of temperature, concentrations, and reaction time duration that can be modified to enhance efficiency and yield of organic chemistry reactions, there are several other parameters that likely contributed to achieving a dexamethasone:IgG molar-incorporation-index of 6.95:1 for dexamethasone-(C21-phosphoramide)-[anti-EGFR], including 1) lack of a prethiolation requirement for anti-IGF-1R immunoglobulin; 2) potentially greater relative chemical reactivity of carbodiimide analogs compared to other previously applied covalent bond forming reagents; 3) enhanced preferential phosphate reactivity of the carbodiimide reagent in the presence of imidazole; 4) formation of a covalent bond at the single available monophosphate group located at the C21 position in contrast to a phosphate (Ar-PO4−), carboxyl (Ar-CO2−), amine (Ar-NH2+), or sulfhydryl (Ar-SH) chemical group located directly on any one of the four aromatic ring structures; 5) restricting the initial chemical reaction of the carbodiimide reagent with only the dexamethasone-C21-monophosphate in a manner that enhanced yield and minimized generation of side reaction end products (IgG-based determination); 6) formulation of dexamethasone-C21-monophosphate group in molar excess to the carbodiimide in order to promote maximal depletion of the covalent bond-forming reagent; 7) selective amine reactivity of the Phase II dexamethasone-(C21-phophorylimidazole) intermediate; and 8) presence of only a single phosphate group within the chemical composition of dexamethasone-C21-monophosphate.
Presence of the single phosphate group at the C21 instead of the C11 or C17 position of dexamethasone decreases both the influence of steric-hindrance phenomenon at the C21-monophosphate of dexamethasone-C21-monophosphate during initial chemical reactions with a carbodiimide and the unique influences from aromatic electron orbital properties associated with any one of the four planar ring structures which often modifies the chemical characteristics of phosphate (Ar-PO4−2), carboxyl (Ar-CO2−), and amine (Ar-NH3+) groups. Complementing the effectiveness of the organic chemistry reactions, the final yield of dexamethasone-(C21-phosphoramide)-[anti-EGFR] was substantially improved by implementing both a multiphase organic chemistry reaction scheme (in preference to a single-phase “co-mingled” reagent regimen) in concert with the removal of residual dexamethasone-C21-monophosphate and unreacted reagents from dexamethasone-(C21-phosphoramide)-[anti-EGFR] by serial microfiltration (MWCO =10 kDa) in preference to the application of microscale column chromatography for the separation and purification of the final Phase III covalent immunopharmaceutical end product. Logistical attributes of the multiphase organic chemistry reaction scheme utilized for the synthesis of dexamethasone-(C21-phosphoramide)-[anti-EGFR] consisted of 1) comparatively brief incubation time periods for organic chemistry reactions utilized in the multiphase synthesis regimen; 2) relatively efficient execution of organic chemistry reaction regimens; 3) option of producing an amine-reactive Phase II dexamethasone intermediate that is stable enough for short- to long-term preservation/storage; 4) flexibility of substituting other pharmaceutical agents in the organic chemistry reaction scheme in place of dexamethasone; 5) flexibility of substituting other biologically relevant molecular platforms for anti-EGFR monoclonal IgG; 6) option of modifying organic chemistry reaction scheme within the multiphase synthesis regimen in a manner that affords higher or lower molar incorporation ratios; 7) moderate-to-high levels of laboratory technical convenience; and 8) a low degree of dependence on the utilization of advanced forms of laboratory instrumentation.
Dexamethasone-(C21-phosphoramide)-[anti-EGFR] selectively targeted antineoplastic cytotoxic potency
The selective antineoplastic cytotoxic potency of dexamethasone-(C21-phosphoramide)-[anti-EGFR] against pulmonary adenocarcinoma (A549) was nearly identical to dexamethasone when formulated at and between the standardized dexamethasone equivalent concentrations of 10−9 M and 10−6 M (Figure 4). Acknowledgment of both the molecular weight of dexamethasone-(C21-phosphoramide)-[anti-EGFR] and the known mechanism of action for dexamethasone collectively serve to validate the concept that covalent the dexamethasone immunopharmaceutical was effectively internalized by the active transport mechanism of IgG-induced receptor-mediated endocytosis following selective targeted binding at EGFR uniquely overexpressed on the external surface membrane of pulmonary adenocarcinoma (A549). Ultimately, the total combined selective targeted antineoplastic cytotoxicity afforded by dexamethasone-(C21-phosphoramide)-[anti-EGFR] is logically presumed to be substantially enhanced in vivo by IgG–antigen complex stimulation of endogenous host immune responses that are difficult to simultaneously access ex vivo utilizing a tissue culture-based model for neoplastic disease.
Attributes of covalent dexamethasone-(C21-phosphoramide)-[anti-EGFR] immunopharmaceutical
The molecular design of dexamethasone-(C21-phosphoramide)-[anti-EGFR] provides several distinct qualities pertaining to chemical composition and molecular structure. Importantly, the multiphase organic chemistry reaction scheme utilized to synthesize dexamethasone-(C21-phosphoramide)-[anti-EGFR] generated a Phase III end product with a 6.95:1 dexamethasone:IgG molar-incorporation-index. A dexamethasone:IgG molar-incorporation-index of 6.95:1 for dexamethasone-(C21-phosphoramide)-[anti-EGFR] was modestly greater than values obtained in previous investigations using other covalent bond-forming agents for the synthesis of 1) gemcitabine-(C5-carbamate)-[anti-HER2/neu] (Gem:IgG =1.1:1),50 2) gemcitabine-(C4-amide)-[anti-HER2/neu] (Gem:IgG =2.78:1),53 3) epirubicin-(C3-amide)-[anti-HER2/neu] (Epi:IgG =0.275:1),49 4) epirubicin-(C3-amide)-[anti-EGFR] (Epi:IgG =0.407:1),49 and 5) epirubicin-(C13-imino)-[anti-HER2/neu] (Epi:IgG =0.400:1).51 In the chemical configuration of dexamethasone-(C21-phosphoramide)-[anti-EGFR], the corticosteroid moiety is covalently bound to anti-EGFR through a C21-phosphoramide bond structure that at least in theory provides potentially higher levels of bioavailability for the dexamethasone moiety after it enters the acidic microenvironment of the phagolysosome following selective targeted delivery and internalization by active transport mechanisms of IgG-induced receptor-mediated endocytosis. A complementary quality of dexamethasone-(C21-phosphoramide)-[anti-EGFR] is the insertion or addition of no “foreign” chemical groups into dexamethasone-(C21-phosphoramide)-[anti-EGFR] during synthetic formation of the C21-phosphoramide bond structure which decreases the risk of inducing host humoral immune responses. Other innate attributes include retained molecular weight and more importantly retained biological activity of the anti-EGFR component within dexamethasone-(C21-phosphoramide)-[anti-EGFR] in the form of binding avidity for EGFR overexpressed on the external surface membrane of pulmonary adenocarcinoma (A549).
The molecular design and organic chemistry reactions implemented for the synthesis of dexamethasone-(C21-phosphoramide)-[anti-EGFR] used anti-EGFR monoclonal IgG as a molecular delivery platform because of its selective binding avidity for epidermal growth factor membrane receptors (EGFR, ErbB-1, and HER1). Motivation for utilizing EGFR as a site to facilitate the selective targeted delivery of a covalent dexamethasone-(C21-phosphoramide)-[IgG] immunopharmaceutical was to a large part dependent on the utilization of pulmonary adenocarcinoma (A549) as an ex vivo neoplastic disease model, which is known to uniquely overexpress both EGFR and IGF-1R trophic membrane receptors. A wide spectrum of adenocarcinomas and carcinomas uniquely overexpress each of these two trophic receptors in addition to HER2/neu on their exterior surface membrane. EGFR (ErbB-1 and HER1) is a 170 kDa glycoprotein within the ErbB epidermal growth factor family of receptors. The nonprotein component of EGFR is located on the external surface of cell membranes and consists of an N-linked glycan with a GlcNAc terminus. The ligands, such as EGF and transforming growth factor-α, activate EGFR, and upon stimulation, it is transformed from an EGFR monomer complex to an activated homodimer. The transformation results in marked increases in intrinsic intracellular protein tyrosine kinase activity and autophosphorylation of tyrosine residues. Such changes initiate downstream activation and signaling of several proteins that in turn induce the mitogen-activated protein kinases, Akt, and JNK signal transduction cascades that ultimately lead to DNA synthesis and increased cellular proliferation. Mutations characterized by EGFR overexpression promote persistent stimulation and patterns of uncontrolled cellular division. Immunotherapeutics in the form of monoclonal antibody inhibitors with binding avidity for the EGFR oncogene receptor block the extracellular ligand-binding domain, thereby blocking signal transduction. Other monoclonal antibody inhibitors have been developed that inhibit activity of the cytoplasmic tyrosine kinase segment of EGFR resulting in the receptor not being able to selfactivate.
Uniquely or highly overexpressed trophic membrane receptors (eg, EGFR, IGF-1R, HER2/neu, and vascular endothelial growth factor receptor) associated with many forms of adenocarcinoma and carcinoma, in addition to certain cell differentiating antigens (eg, CD19, CD20, CD22, and CD30) found on leukemia and lymphoma cell types, are each capable of facilitating both the selective targeted delivery and receptor-mediated endocytosis32–34,129 of a wide spectrum of covalent immunopharmaceuticals analogous in the form and function to dexamethasone-(C21-phosphoramide)-[anti-EGFR]. Monoclonal IgG with binding avidity for endogenous trophic membrane receptors have the potential to exert other additional biological properties that are independent of the activity associated with pharmaceutical moieties when incorporated as a component of covalent immunopharmaceuticals, such as dexamethasone-(C21-phosphoramide)-[anti-EGFR]. Monoclonal IgG with binding avidity for EGFR, HER2/neu, IGF-1R, VEGFR, or other endogenous trophic membrane receptors can therefore 1) competitively inhibit binding and stimulation by endogenous trophic ligands at shared epitopes (eg, EGF  IgG-EGFR), 2) transiently reduce surface membrane expression densities as a desirable consequence of induced receptor-mediated endocytosis, and 3) reduce the biochemical function of certain receptor subtypes (eg, anti-HER2/neu → HER2/neu tyrosine kinase activity). Monoclonal IgG with binding avidity for trophic receptors, such as EGFR, IGF-1R, and HER2/neu that are uniquely or highly overexpressed on the external surface membrane of neoplastic cell types, can therefore suppress the proliferation rate and viability of various neoplastic cell types, affecting the breast, prostate, lung, and some sarcomas. Competitive inhibition of overexpressed endogenous trophic receptors, such as EGFR, in neoplastic cell types can also reduce metastatic transformation, mobility, and metastatic potential. Inhibition of overexpressed endogenous trophic membrane receptor, therefore, affords an approach to suppressing neoplastic conditions refractory (resistant) to conventional low molecular weight chemotherapeutics while at the same time avoiding the risk of many serious sequellae.
IgG-EGFR), 2) transiently reduce surface membrane expression densities as a desirable consequence of induced receptor-mediated endocytosis, and 3) reduce the biochemical function of certain receptor subtypes (eg, anti-HER2/neu → HER2/neu tyrosine kinase activity). Monoclonal IgG with binding avidity for trophic receptors, such as EGFR, IGF-1R, and HER2/neu that are uniquely or highly overexpressed on the external surface membrane of neoplastic cell types, can therefore suppress the proliferation rate and viability of various neoplastic cell types, affecting the breast, prostate, lung, and some sarcomas. Competitive inhibition of overexpressed endogenous trophic receptors, such as EGFR, in neoplastic cell types can also reduce metastatic transformation, mobility, and metastatic potential. Inhibition of overexpressed endogenous trophic membrane receptor, therefore, affords an approach to suppressing neoplastic conditions refractory (resistant) to conventional low molecular weight chemotherapeutics while at the same time avoiding the risk of many serious sequellae.
In addition to facilitating selective pharmaceutical targeted delivery and blocking endogenous ligand binding at trophic receptor sites, the covalent bonding of dexamethasone, classical low molecular weight chemotherapeutics, or other types of anticancer agents specifically to monoclonal IgG with binding avidity for uniquely or highly overexpressed endogenous trophic receptors or cell differentiation proteins can serve an effective means for recruiting and selectively “targeting” multiple host immune responses. Formation of membrane IgG:Ag complexes on the external surface of neoplastic cell types can evoke the selectively “targeted” endogenous host immune responses that collectively involve the activation of 1) ADCC, 2) CMC, and 3) opsonization/phagocytosis. Secondary antineoplastic properties from the activation of antibody-dependent cell-mediated cytotoxicity in general are more efficient and have greater effectiveness when the monoclonal immunoglobulin utilized is either the IgG1 isotype or the IgG2 isotype. Monoclonal IgG fractions with binding avidity for trophic membrane receptors that have most extensively been utilized in clinical oncology for the therapeutic management of adenocarcinomas and carcinomas affecting the breast, prostate, intestine, and lung include anti-HER2/neu (trastuzumab and pertuzumab),130–134 anti-EGFR (cetuximab),135–138 combined anti-HER2/neu and anti-EGFR (panitumumab),137–140 and anti-IGF-1R (figitumumab and dalotuzumab).141–144 In contrast to many nonhematopoietic neoplastic conditions, leukemia and lymphoma either uniquely or highly overexpress sites on their external surface membrane that do not function as classical endogenous trophic receptor complexes and include the cell differentiation antigens, such as CD20, CD22, CD30 (TNFRSF8), CD33 (SIGLEC: sialic acid-binding lectin), and CD54. Each of these cell differentiation antigens has served as the basis for the development of therapeutic monoclonal immunoglobulins, such as anti-CD20 (ibritumomab, ofatumumab, rituximab, trubion, and veltuzumab) for B-cell non-Hodgkin’s lymphoma, resistant CLL, other lymphomas, leukemia, transplant rejection, and autoimmune disease; anti-CD22 (inotuzumab) for non-Hodgkin’s lymphoma; anti-CD33 (gemtuzumab) for acute myeloid leukemia (AML); and anti-CD52 (alemtuzumab) for CLL, cutaneous T-cell lymphoma, and T-cell lymphoma. Nonhematopoietic neoplastic cells can similarly uniquely overexpress cell differentiation antigens including CD44 (bivatuzumab) relevant to breast cancer and CD66e (carcinoembryonic antigen-related cell adhesion molecule: labetuzumab) associated with intestinal carcinoma. Most of the cell differentiation antigens overexpressed by leukemia and lymphoma cell types do not function as endogenous receptors and are not known to bind endogenous hormone-like ligands so monoclonal immunoglobulins with binding avidity at these sites do not exert extensive degrees of neoplastic cell inhibition through the same processes as those documented for anti-EGFR, anti-IGF-1R, anti-HER2/neu, or anti-VEGFR in adenocarcinomas, carcinomas, or other nonhematopoietic neoplastic cell types. Alternatively, the antineoplastic properties of anti-CD20, anti-CD22, anti-CD33, and anti-CD54 attained in vivo against populations of leukemia and lymphoma cell types are instead highly dependent upon if not largely restricted to the activation of endogenous immune responses.13–18,20,38,145,146 Majority, if not all, of the in vivo antineoplastic cytotoxic properties of anti-CD20, anti-CD33, and anti-CD54 is therefore predominately attained through their ability to induce multiple endogenous host immune responses.147–152
Despite the inhibitory characteristics of anti-HER2/neu, anti-EGFR, anti-IGF-1R, and similar monoclonal IgG-based modalities on the function of membrane trophic receptors, they frequently only suppress the in vivo proliferative growth and vitality of cancer cells but they are almost invariably incapable independently of evoking a degree of cytotoxic activity sufficient enough to successfully resolve most aggressive or advanced forms of neoplastic disease.130,131,153–167 Inability of most immunoglobulins with binding avidity for trophic membrane receptors to exert significant cytotoxic efficacy in vivo coincides with the detection of increases in cell-cycle G1 arrest, cancer cell transformation into states of apoptosis resistance,154 and preferential selection for resistant subpopulations.130,131 In addition, this scenario can be further complicated by frequent reversal of tumor growth inhibition130 and relapse of trophic receptor overexpression153 upon cessation and withdrawal. Greater levels of antineoplastic cytotoxicity are attainable when antitrophic receptor IgG is utilized in dual combination with conventional chemotherapeutics or other cancer treatment modalities.168–170 Development of resistance has also been detected for monoclonal IgG with binding avidity for cell differentiation proteins, such as anti-CD20 (veltuzumab and ofatumumab) and anti-CD52 (alemtuzumab). Mechanisms of resistance associated with monoclonal IgG fractions with binding avidity for these and other membrane-associated cell differentiation antigens are attributed to 1) accelerated rates of receptor-mediated endocytosis prior to ADCC/CMC/opsonization,13,19 2) monocyte/macrophage CD20/CD52 shaving or trogocytosis,20 and 3) immune evasion as a consequence of immunosuppressive mediators liberated from cancer cell populations.21,22 Interestingly, ofatumumab has been approved for B-CLL resistant to alemtuzumab and fludarabine.
In an ex vivo tissue culture environment, most of the therapeutic monoclonal IgGs with binding avidity for overexpressed trophic membrane receptors evoke very limited or a total lack of any measurable selectively targeted antineoplastic cytotoxicity or detectable inhibition of vitality and viability.49,56,80,171–173 Multiple variables contribute to this observation, but some of the most important and relevant in this regard include the 1) comparatively low concentration of endogenous trophic ligands present in conventional tissue culture media (eg, 5%–10% bovine serum), 2) relatively brief incubation periods used to access efficacy and potency (eg, 3–8 days), and 3) absence of significant activation of or influence from any of the three endogenous host immune responses. Monoclonal IgG, including anti-HER2/neu, anti-EGFR, and anti-IGF-1R, with binding avidity for trophic membrane receptors in vivo produces detectable decline in neoplastic cell proliferation and vitality. However, monoclonal IgG bound to antigenic sites on the external surface membrane of neoplastic cells can also induce selectively targeted host immune responses that can produce a significant cytotoxic effect. Most notable in this regard are ADCC, CMC, and opsonization/phagocytosis that serve as the immunology mechanisms primarily responsible for anti-CD20 and anti-CD52 efficacy and potency against leukemia neoplastic disease states.
Despite limited anticancer cytotoxicity and a general inability in vivo to independently resolve many neoplastic disease states, monoclonal IgG with binding avidity for unique or highly overexpressed “sites” on the external surface membrane of many neoplastic cell types makes them nearly ideally suited for selectively targeting delivery of therapeutic or diagnostic moieties. The mechanism of action for diagnostic radioimmunopharmaceutical agents, or anticancer therapeutic agents that exert biological activity through physical and functional disruptions of cancer cell membrane integrity, does not require direct entry into cytosol or nuclear environments (eg, [213Bi or 211At or 224Ra]-anti-TAG-72 for colon carcinoma). In contrast, passive or active transmembrane transport is particularly relevant and essentially a requirement when pharmaceutical moieties are utilized in the synthesis of covalent immunopharmaceuticals that have a mechanism of action that is entirely dependent on their entry into cytosol or nuclear environments. Because of this consideration, a second critically important biological function of surface membrane sites that can make them invaluable as “targets” for the facilitation of selective delivery of pharmaceutical moieties is an ability to be internalized intracellularly by mechanisms of receptor-mediated endocytosis32–34,129 following and in response to selective binding of immunoglobulin or endogenous ligands. Intracellular internalization of dexamethasone-(C21-phosphoramide)-[anti-EGFR] by the transmembrane active transport mechanism of receptor-mediated endocytosis32–34,129 provides the advantage of minimizing or avoiding the simple “coating” of the external surface membranes of neoplastic cell populations. Highly desirable sites on the external surface membrane of neoplastic cell types for facilitating selectively targeted pharmaceutical delivery include the endogenous trophic membrane receptors, such as EGFR, IGF-1R, HER2/neu, and VEGFR, in addition cell differentiating antigens, such as CD19, CD20, CD22, and CD30, because they are both uniquely and highly overexpressed and are internalized by processes of induced receptor-mediated endocytosis. The functional implications of both unique and high overexpression in addition to an ability to undergo internalization induced by mechanisms of receptor-mediated endocytosis collectively allows such membrane sites to directly influence the efficacy and potency of covalent immunopharmaceuticals and analogous biopharmaceutical agents.
The selective targeted binding of dexamethasone-(C21-phosphoramide)-[anti-EGFR] to uniquely or highly overexpressed sites on the external surface membrane of neoplastic cell types that have the capacity to facilitate the active transmembrane transport of the covalent immunopharmaceutical by induced receptor-mediated endocytosis contributes to attaining several critically important attributes that directly influence efficacy. The IgG component of covalent immunopharmaceuticals that possesses selective binding avidity for uniquely or highly overexpressed endogenous receptors therefore can promote 1) selective targeted pharmaceutical moiety delivery, 2) continual deposition of a pharmaceutical moiety on external surface membranes of cancer cells as a function of trophic receptor expression/reexpression, 3) persistent high levels of transmembrane active pharmaceutical transport by mechanisms of receptor-mediated endocytosis, and 4) progressive cytosol pharmaceutical accumulation resulting in concentrations that can exceed those possible by simple passive diffusion (eg, postintravenous injection of classical low molecular weight chemotherapeutics at therapeutically relevant dosages). Monoclonal antibody with binding avidity for CD74 (myeloma), CEA (colon carcinoma), and CD33 (promyelocytic leukemia and myeloid leukemia) is known to be internalized by mechanism analogous to receptor-mediated endocytosis detected for EGFR- and HER2/neu-positive adenocarcinoma and carcinoma cell populations. Although specific data for IGF-1R-mediated endocytosis are somewhat limited for pulmonary adenocarcinoma (A549), other neoplastic cell types, such as Lewis Lung carcinoma (H-59: highly metastatic subculture with hepatic propensity) and mammary adenocarcinoma (MCF-7), are known to actively internalize membrane IGF-1Rs by mechanisms of receptor-mediated endocytosis at a rate of ≅2.1×104/cell (54%) and ≅4.5×104/cell (45%) within an 1-hour IGF incubation period.174 Related investigations have demonstrated that metastatic multiple myeloma internalizes and metabolizes ~8×106 molecules of anti-CD74 monoclonal antibody per day.175 Given this perspective, the three most critical numerical variables related to cancer cell biology that determines the antineoplastic cytotoxic potency of covalent immunopharmaceuticals, such as dexamethasone-(C21-phosphoramide)-[anti-EGFR], gemcitabine-(C5-carbamate)-[anti-HER2/neu],50 gemcitabine-(C4-amide)-[anti-HER2/neu],53 epirubicin-(C3-amide)-[anti-HER2/neu],49 epirubicin-(C3-amide)-[anti-EGFR],49 and epirubicin-(C13-imino)-[anti-HER2/neu]51 are the 1) external surface membrane expression density of endogenous trophic receptor targets relative to normal healthy cells residing in tissues and organ systems, 2) rate of internalization by mechanisms of receptor-mediated endocytosis, and 3) rate that receptors on the external surface membrane are subsequently replenished following internalization by receptor-mediated endocytosis.
The capacity of dexamethasone-(C21-phosphoramide)-[anti-EGFR] to selectively target dexamethasone delivery at EGFR membrane receptor sites and subsequently promote IgG-induced receptor-mediated endocytosis functionally represents an active transport mechanism activated in neoplastic cell populations that can promote and facilitate substantial intracellular accumulation of pharmaceutical moieties. A direct outcome of receptor-mediated endocytosis of membrane-bound covalent immunocorticosteroid can be an increase in the intracellular cytosol concentrations of pharmaceutical moieties that are 8.5-fold32 to >100-fold33,34 greater than those attainable by simple passive diffusion of conventional low molecular weight pharmaceuticals from the extracellular fluid compartment following intravenous injection at clinically relevant dosages. Active transport of dexamethasone or other pharmaceutical agents across intact cancer cell membranes by mechanisms of IgG- or ligand-induced receptor-mediated endocytosis following selective targeted delivery at endogenous membrane receptors can therefore serve as a strategy capable of 1) maximizing therapeutic efficacy and potency, 2) accelerating cytotoxic resolution of neoplastic cell populations, 3) reducing the influence of chemotherapeutic resistance,176–181 and 4) decreasing the time frame during which acquired chemotherapeutic resistance can potentially develop. The latter consideration is particularly relevant to many conditions of leukemia and lymphoma that are capable of developing resistance to corticosteroid therapy over a relatively brief time period.
Therapeutic properties potentially afforded by monoclonal immunoglobulin with binding avidity for sites on the external surface membrane of neoplastic cell types are frequently complemented by the efficacy of chemotherapeutic agents. Antineoplastic properties of anti-HER2/neu are additively or synergistically increased by cyclophosphamide,169,182 docetaxel,182 doxorubicin,169,182 etoposide,182 methotrexate,182 paclitaxel,169,182 or vinblastine.182 Similar to anti-HER2/neu,169,182–186 the antineoplastic properties of other monoclonal immunoglobulin inhibitors of trophic membrane receptors, including anti-EGFR,187–189 anti-IGF-1R,190,191 and anti-VEGFR,168,192,193 are also additively and synergistically complemented by conventional chemotherapeutic agents. In conditions of leukemia and lymphoma, monoclonal anti-CD22 immunoglobulin has been applied in the development of both inotuzumab ozogamicin (calicheamicin: DNA strand scission) and moxetumomab pasudotox (CAT-8015: active Pseudomonas-origin exotoxin). Each of these anti-CD22-based biotherapeutics has demonstrated efficacy against non-Hodgkin’s lymphoma or hairy cell leukemia, respectively. Monoclonal anti-CD30 covalently bound to highly toxic MMAE (brentuximab vedotin) similarly possesses antineoplastic efficacy that is associated with the MMAE moiety, which has a mechanism of action involving inhibition of tubulin polymerization and has been found to be effective for therapeutic management of Hodgkin’s lymphoma and systemic anaplastic large cell lymphoma. AML has been therapeutically managed with variable degrees of effectiveness utilizing the monoclonal anti-CD33-based immunochemotherapeutics, gemtuzumab ozogamicin (2000–2010 United States Federal Drug Administration withdrawal), and SGN-CD33, which contain pyrrolobenzodiazepine (promotes intrastrand DNA cross-linking) as a chemotherapeutic moiety. In anti-CD33 therapeutic monoclonal IgGs or anti-CD33-based covalent immunochemotherapeutics, some of the anticancer properties attained are associated with the tyrosine-based inhibitory motif located intracellularly and its relationship or influence on inhibiting the cellular activity. Monoclonal anti-CD56 immunoglobulin has been covalently bound to a mertansine (maytansinoid analog) chemotherapeutic moiety to form lorvotuzumab mertansine, which has demonstrated efficacy against CD56-positive multiple myeloma.
Many attributes of antitrophic receptor IgGs (eg, anti-EGFR) in dual combination with the biological activity of pharmaceutical moieties (eg, dexamethasone) provide a strategy for establishing the molecular design and organic chemistry synthesis of covalent immunopharmaceuticals, such as dexamethasone-(C21-phosphoramide)-[anti-EGFR], that exerts synergistic or additive levels of therapeutic potency, which is attained collectively through multiple mechanisms of action. Additionally, molecular platforms, such as IgG and endogenous ligands, impart other properties and beneficial attributes that substantially complement their potential to facilitate selective targeted pharmaceutical delivery by processes that are distinctly different from their innate properties of selective binding avidity or an ability to induce receptor-mediated endocytosis. The molecular weight and overall size of IgG and many endogenous trophic receptor ligands (eg, IgG molecular weight [MW] =150 kDa and EGF MW =6.05 kDa) are much greater than the size of the vast majority of conventional low molecular weight pharmaceuticals and chemotherapeutic agents (dexamethasone MW =392.461, fludarabine MW =365.212, and gemcitabine MW =263.198). Due to this innate biological characteristic, dexamethasone-(C21-phosphoramide)-[anti-EGFR], fludarabine-(C2-methylhydroxylphosphoramide)-[anti-IGF-1R], gemcitabine-(C2-methylhydroxylphosphoramide)-[anti-IGF-1R], or other covalent biopharmaceuticals neither bind selectively to nor do extensively diffuse passively across intact external surface membrane structures of normal cell populations residing within healthy tissues and organ systems. Such characteristics frequently are unrecognized and underappreciated as one of the most important molecular mechanisms contributing to the greater margin of safety potentially afforded by dexamethasone-(C21-phosphoramide)-[anti-EGFR] and analogous covalent biopharmaceutical, including immunopharmaceuticals and immunochemotherapeutics.49–53 In the context of dexamethasone and other corticosteroid agents, minimizing their passive diffusion into normal cell populations within healthy tissues and organ systems substantially reduces this risk and severity of immunosuppression, compromised memory function, and neuropsychological side effects.194–196 Immunoglobulin (IgG MW =150 kDa) or other biologically relevant molecular platforms that possess a relatively high molecular weight are of sufficient physical size to also effectively delay and reduce acute elimination burdens of pharmaceutical moieties on the process of renal glomerular filtration (MWCO =60 kDa) or metabolism by biochemical pathways within hepatocytes. Reducing the rate and extent of renal excretion and hepatic metabolism of the dexamethasone moiety also in effect prolongs its intravascular pharmacokinetic profile in concert with substantially lowering the pharmaceutical moiety total volume of distribution. In this manner, total dosage requirements can potentially be reduced while simultaneously providing another variable that can improve in vivo margin of safety.
Corticosteroids are frequently a component of therapeutic protocols for hematopoietic neoplastic conditions in addition to autoimmune disease and severe inflammatory responses. In clinical oncology, corticosteroids have additional utility for decreasing general inflammation and tissue edema associated with burdens from expanding neoplastic lesions (eg, central nervous system) and for reducing the severity of general morbidity and inappetence. Given this perspective, dexamethasone-(C21-phosphoramide)-[IgG] represents a prototypical covalent immunopharmaceutical that potentially has a wide range of clinically relevant applications. The molecular mechanisms of action for corticosteroid class of pharmaceuticals directly correlate with their effectiveness as a significant “cornerstone” class of therapeutic agents for leukemia and lymphoma,197–199 especially for conditions of multiple myeloma. Although corticosteroids are commonly administrated for the therapeutic management of lymphoma,200 CLL,201,202 and acute lymphoblastic leukemia,203–205 the molecular design, organic chemistry synthesis reaction schemes, and the analysis of the antineoplastic cytotoxic potencies of covalent biocorticosteroids with selective antineoplastic cytotoxic efficacy against leukemia and lymphoma have not previously been described extensively. Covalent milatuzumab–dexamethasone has, however, been incorporated into liposomes to facilitate selective targeted delivery for therapeutic purposes against multiple myeloma and non-Hodgkin’s lymphoma when classified as CD74+ B-cell malignancies.201 The multiphase organic chemistry reaction scheme used in the synthesis of dexamethasone-(C21-phosphoramide)-[anti-EGFR] can potentially serve as a template guide for producing a modest array of covalent biotherapeutics that contain different corticosteroid/steroid-based moieties or other high molecular weight and biologically relevant molecular platforms besides anti-EGFR monoclonal immunoglobulin. Prednisone-(C21-phosphoramide)-[anti-CD19], prednisolone-(C21-phosphoramide)-[anti-CD20], and betamethazone-(C21-phosphoramide)-[anti-CD52] represent prototypical covalent immunopharmaceuticals that can be synthesized for such purposes utilizing dexamethasone-(C21-phosphoramide)-[anti-EGFR] as a reference template for designing their molecular structure and organic chemistry reactions for multiphase synthesis schemes. Conceptually, the molecular design and the multiphase organic chemistry reaction scheme described could therefore similarly be implemented for the synthesis of steroid hormone-based immunopharmaceuticals that contain agonist or antagonist analogs of estrogen/estradiol, androgen/testosterone, or progesterone functioning as pharmaceutical moieties with applicability relevant to steroid-dependent neoplasia or steroid-responsive disease states. Molecular platforms that can potentially facilitate selective targeted delivery of dexamethasone or other corticosteroids relevant to CLL include monoclonal immunoglobulin, or ligands with selective binding avidity for CCR7, CXCR5, CD120a (TNFR1), CD19, CD20, and CD52 found on membrane exterior surfaces. Similarly, membrane sites potentially effective for selective targeted pharmaceutical moiety delivery for AML include CD7, CD33, CD44, CD47, CD123 (IL-31), CLL-1, and survivin.
Disclosure
The investigators and authors have no material, financial, or personal relationships external to College of Veterinary Medicine or Mississippi State University that would represent a conflict of interest in a manner that might bias the factual and ethical content of the published article. The authors report no conflicts of interest in this work.
References
Habermann TM, Weller EA, Morrison VA, et al. Rituximab-CHOP versus CHOP alone or with maintenance rituximab in older patients with diffuse large B-cell lymphoma. J Clin Oncol. 2006;24(19):3121–3127. | ||
Xu JW, Li QQ, Tao LL, et al. Involvement of EGFR in the promotion of malignant properties in multidrug resistant breast cancer cells. Int J Oncol. 2011;39(6):1501–1509. | ||
Hurrell T, Outhoff K. The in-vitro influences of epidermal growth factor and heregulin-â1 on the efficacy of trastuzumab used in Her-2 positive breast adenocarcinoma. Cancer Cell Int. 2013;13(1):97. | ||
Liu K, Chen H, You Q, Shi H, Wang Z. The siRNA cocktail targeting VEGF and HER2 inhibition on the proliferation and induced apoptosis of gastric cancer cell. Mol Cell Biochem. 2014;386(1–2):117–124. | ||
Moerkens M, Zhang Y, Wester L, et al. Epidermal growth factor receptor signalling in human breast cancer cells operates parallel to estrogen receptor α signalling and results in tamoxifen insensitive proliferation. BMC Cancer. 2014;14:283. | ||
Schneider J, Rubio MP, Barbazán MJ, Rodriguez-Escudero FJ, Seizinger BR, Castresana JS. P-glycoprotein, HER-2/neu, and mutant p53 expression in human gynecologic tumors. J Natl Cancer Inst. 1994;86(11):850–856. | ||
Lu Y, Jingyan G, Baorong S, Peng J, Xu Y, Cai S. Expression of EGFR, Her2 predict lymph node metastasis (LNM)-associated metastasis in colorectal cancer. Cancer Biomark. 2012;11(5):219–226. | ||
Carlsson J, Shen L, Xiang J, Xu J, Wei Q. Tendencies for higher co-expression of EGFR and HER2 and downregulation of HER3 in prostate cancer lymph node metastases compared with corresponding primary tumors. Oncol Lett. 2013;5(1):208–214. | ||
Lehne G, Grasmo-Wendler UH, Berner JM, et al. Upregulation of stem cell genes in multidrug resistant K562 leukemia cells. Leuk Res. 2009;33(10):1379–1385. | ||
Munoz JL, Rodriguez-Cruz V, Greco SJ, Nagula V, Scotto KW, Rameshwar P. Temozolomide induces the production of epidermal growth factor to regulate MDR1 expression in glioblastoma cells. Mol Cancer Ther. 2014;13(10):2399–2411. | ||
Salomon DS, Brandt R, Ciardiello F. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol. 1995;19(3):183–232. | ||
Yaktapour N, Übelhart R, Schüler J, et al. Insulin-like growth factor-1 receptor (IGF1R) as a novel target in chronic lymphocytic leukemia. Blood. 2013;122(9):1621–1633. | ||
Li ZH, Zhang Q, Zhang Q, et al. Preclinical studies of targeted therapies for CD20-positive B lymphoid malignancies by ofatumumab conjugated with auristatin. Invest New Drugs. 2014;32(1):75–86. | ||
Lapalombella R, Yu B, Triantafillou G, et al. Lenalidomide down-regulates the CD20 antigen and antagonizes direct and antibody-dependent cellular cytotoxicity of rituximab on primary chronic lymphocytic leukemia cells. Blood. 2008;112(13):5180–5189. | ||
Stanglmaier M, Reis S, Hallek M. Rituximab and alemtuzumab induce a nonclassic, caspase-independent apoptotic pathway in B-lymphoid cell lines and in chronic lymphocytic leukemia cells. Ann Hematol. 2004;83(10):634–645. | ||
Weitzman J, Betancur M, Boissel L, Rabinowitz AP, Klein A, Klingemann H. Variable contribution of monoclonal antibodies to ADCC in patients with chronic lymphocytic leukemia. Leuk Lymphoma. 2009;50(8):1361–1368. | ||
Lv M, Lin Z, Qiao C, et al. Novel anti-CD20 antibody TGLA with enhanced antibody-dependent cell-mediated cytotoxicity mediates potent anti-lymphoma activity. Cancer Lett. 2010;294(1):66–73. | ||
Asgeirsdóttir SA, Kok RJ, Everts M, Meijer DK, Molema G. Delivery of pharmacologically active dexamethasone into activated endothelial cells by dexamethasone-anti-E-selectin immunoconjugate. Biochem Pharmacol. 2003;65(10):1729–1739. | ||
Lim SH, Vaughan AT, Ashton-Key M, et al. Fc gamma receptor IIb on target B cells promotes rituximab internalization and reduces clinical efficacy. Blood. 2011;118(9):2530–2540. | ||
Beum PV, Peek EM, Lindorfer MA, et al. Loss of CD20 and bound CD20 antibody from opsonized B cells occurs more rapidly because of trogocytosis mediated by Fc receptor-expressing effector cells than direct internalization by the B cells. J Immunol. 2011;187(6):3438–3447. | ||
Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–296. | ||
Mao Y, Poschke I, Kiessling R. Tumor-induced immune suppression: role of inflammatory mediators released by myelomonocytic cells. J Intern Med. 2014;276(2):154–170. | ||
Melgert BN, Olinga P, Jack VK, Molema G, Meijer DK, Poelstra K. Dexamethasone coupled to albumin is selectively taken up by rat nonparenchymal liver cells and attenuates LPS-induced activation of hepatic cells. J Hepatol. 2000;32(4):603–611. | ||
Basu A, Shrivastav TG, Maitra SK. A direct antigen heterologous enzyme immunoassay for measuring progesterone in serum without using displacer. Steroids. 2006;71(3):222–230. | ||
De Goeij AF, van Zeeland JK, Beek CJ, Bosman FT. Steroid-bovine serum albumin conjugates: molecular characterization and their interaction with androgen and estrogen receptors. J Steroid Biochem. 1986;24(5):1017–1031. | ||
He XK, Yuan ZX, Wu XJ, Xu CQ, Li WY. Low molecular weight hydroxyethyl chitosan-prednisolone conjugate for renal targeting therapy: synthesis, characterization and in vivo studies. Theranostics. 2012;2(11):1054–1063. | ||
Funk D, Schrenk HH, Frei E. Development of a novel polyethylene glycol-corticosteroid-conjugate with an acid-cleavable linker. J Drug Target. 2011;19(6):434–445. | ||
Inapagolla R, Guru BR, Kurtoglu YE, et al. In-vivo efficacy of dendrimer-methylprednisolone conjugate formulation for the treatment of lung inflammation. Int J Pharm. 2010;399(1–2):140–147. | ||
Onishi H, Matsuyama M. Conjugate between chondroitin sulfate and prednisolone with a glycine linker: preparation and in vitro conversion analysis. Chem Pharm Bull. 2013;61(9):902–912. | ||
Granfeldt A, Hvas CL, Graversen JH, et al. Targeting dexamethasone to macrophages in a porcine endotoxemic model. Crit Care Med. 2013;41(11):e309–e318. | ||
Everts M, Kok RJ, Asgeirsdóttir SA, et al. Selective intracellular delivery of dexamethasone into activated endothelial cells using an E-selectin-directed immunoconjugate. J Immunol. 2002;168(2):883–889. | ||
Stan AC, Radu DL, Casares S, Bona CA, Brumeanu TD. Antineoplastic efficacy of doxorubicin enzymatically assembled on galactose residues of a monoclonal antibody specific for the carcinoembryonic antigen. Cancer Res. 1999;59(1):115–121. | ||
Pimm MV, Paul MA, Ogumuyiwa T, et al. Biodistribution and tumour localization of a daunomycin-monoclonal antibody conjugate in nude mice and human tumour xenografts. Cancer Immunol Immunother. 1988;27(3):267–271. | ||
Weaver DJ, Voss EW. Analysis of rates of receptor-mediated endocytosis and exocytosis of a fluorescent hapten-protein conjugate in murine macrophage: implications for antigen processing. Biol Cell. 1998;90(2):169–181. | ||
Alfonso-Pérez M, López-Giral S, Quintana NE, Loscertales J, Martín-Jiménez P, Muñoz C. Anti-CCR7 monoclonal antibodies as a novel tool for the treatment of chronic lymphocyte leukemia. J Leukoc Biol. 2006;79(6):1157–1165. | ||
Bürkle A, Niedermeier M, Schmitt-Gräff A, Wierda WG, Keating MJ, Burger JA. Overexpression of the CXCR5 chemokine receptor, and its ligand, CXCL13 in B-cell chronic lymphocytic leukemia. Blood. 2007;110(9):3316–3325. | ||
Söderberg A, Hossain A, Rosén A. A protein disulfide isomerase/thioredoxin-1 complex is physically attached to exofacial membrane tumor necrosis factor receptors: overexpression in chronic lymphocytic leukemia cells. Antioxid Redox Signal. 2013;18(4):363–375. | ||
Awan FT, Lapalombella R, Trotta R, et al. CD19 targeting of chronic lymphocytic leukemia with a novel Fc-domain-engineered monoclonal antibody. Blood. 2010;115(6):1204–1213. | ||
Stephens DM, Byrd JC. Improving the treatment outcome of patients with chronic lymphocytic leukemia through targeted antibody therapy. Hematol Oncol Clin North Am. 2013;27(2):303–327. | ||
Klabusay M, Sukova V, Coupek P, Brychtova Y, Mayer J. Different levels of CD52 antigen expression evaluated by quantitative fluorescence cytometry are detected on B-lymphocytes, CD 34+ cells and tumor cells of patients with chronic B-cell lymphoproliferative diseases. Cytometry B Clin Cytom. 2007;72(5):363–370. | ||
Coyne CP, Narayanan L. Fludarabine-(C2-methylhydroxyphosphoramide)-[anti-IGF-1R]: synthesis and selectively “targeted” anti-neoplastic cytotoxicity against pulmonary adenocarcinoma (A549). J Pharm Drug Deliv Res. 2015;4(1):129. | ||
Golf SW, Graef V, Schiller JT, Hischer H, Funk W. Thin-layer chromatography – the forgotten alternative for the quantitative determination of steroids. Biomed Chromatogr. 1987;2(5):189–192. | ||
Paw B, Misztal G, Dzwonnik K. Thin-layer chromatographic analysis of fludarabine and formycin A in human plasma. Acta Pol Pharm. 2000;57(5):341–343. | ||
Patel H. A validated stability-indicating HPTLC method for the estimation of gemcitabine HCl in its dosage form. J Planar Chromatogr. 2012;25:77–80. | ||
Berridge MV, Herst PM, Tan AS. Tetrazolium dyes as tools in cell biology: new insights into their cellular reduction. Biotechnol Annu Rev. 2005;11:127–152. | ||
Berridge MV, Tan AS. Characterization of the cellular reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT): subcellular localization, substrate dependence, and involvement of mitochondrial electron transport in MTT reduction. Arch Biochem Biophys. 1993;303(2):474–482. | ||
Watson E, Dea P, Chan KK. Kinetics of phosphoramide mustard hydrolysis in aqueous solution. J Pharm Sci. 1985;74(12):1283–1292. | ||
Surabhi P. Synthesis and Evaluation of Phosphoramide Mustard Prodrugs for Site-Specific Activation [Master Dissertation]. Rutgers University, Medicinal Chemistry Graduate Program, New Brunswick, NJ; 2007. | ||
Coyne CP, Ross M, Bailey J, et al. Dual potency anti-HER2/neu and anti-EGFR anthracycline-immunoconjugates in chemotherapeutic-resistant mammary carcinoma combined with cyclosporin-A and verapamil P-glycoprotein inhibition. J Drug Target. 2009;17(6):474–489. | ||
Coyne CP, Jones T, Pharr T. Synthesis of a covalent gemcitabine-(carbamate)-[anti-HER2/neu] immunochemotherapeutic and cytotoxic anti-neoplastic activity against chemotherapeutic-resistant SKBr-3 mammary carcinoma. Bioorg Med Chem. 2011;19(1):67–76. | ||
Coyne CP, Jones T, Sygula A, Bailey J, Pinchuk L. Epirubicin-[anti-HER2/neu] synthesized with an epirubicin-(C13-imino)-EMCS analog: anti-neoplastic activity against chemotherapeutic-resistant SKBr-3 mammary carcinoma in combination with organic selenium. J Cancer Ther. 2011;2(1):22–39. | ||
Coyne CP, Jones T, Bear R. Synthesis of epirubicin-(C3-amide)-[anti-HER2/neu] utilizing a UV-photoactivated epirubicin. Cancer Biother Radiopharm. 2012;27(1):41–55. | ||
Coyne CP, Jones T, Bear R. Synthesis of gemcitabine-(C4-amide)-[anti-HER2/neu] utilizing a UV-photoactivated gemcitabine intermediate: cytotoxic anti-Neoplastic activity against chemotherapeutic-resistant mammary adenocarcinoma SKBr-3. J Cancer Ther. 2012;3(5A):689–711. | ||
Beyer U, Rothen-Rutishauser B, Unger C, et al. Difference in the intracellular distribution of acid-sensitive doxorubicin-protein conjugates in comparison to free and liposomal-formulated doxorubicin as shown by confocal microscopy. Pharmacol Res. 2001;18(1):29–38. | ||
Di Stefano G, Lanza M, Kratz F, Merina L, Fiume L. A novel method for coupling doxorubicin to lactosaminated human albumin by an acid sensitive hydrazone bond: synthesis, characterization. and preliminary biological properties of the conjugate. Eur J Pharm Sci. 2004;23(4–5):393–397. | ||
Sinkule JA, Rosen ST, Radosevich JA. Monoclonal antibody 44-3A6 doxorubicin immunoconjugates: comparative in-vitro anti-tumor efficacy of different conjugation methods. Tumour Biol. 1991;12(4):198–206. | ||
Kaneko T, Willner D, Knipe JO, et al. New hydrazone derivatives of adriamycin and their immunoconjugates: a correlation between acid-stability and cytotoxicity. Bioconjug Chem. 1991;2(3):133–141. | ||
Kratz F, Warnecke A, Scheuermann K, et al. Probing the cysteine-34 position of endogenous serum albumin with thiol-binding doxorubicin derivatives. Improved efficacy of an acid-sensitive doxorubicin derivative with specific albumin-binding properties compared to that of the parent compound. J Med Chem. 2002;45(25):5523–5533. | ||
Unger C, Häring B, Medinger M, et al. Phase I and pharmacokinetic study of the (6-maleimidocaproyl)hydrazone derivative of doxorubicin. Clin Cancer Res. 2007;13(16):4858–4866. | ||
Mazuel C, Grove J, Gerin G, Keenan KP. HPLC-MS/MS determination of a peptide conjugate prodrug of doxorubicin, and its active metabolites, leucine-doxorubicin and doxorubicin, in dog and rat plasma. J Pharm Biomed Anal. 2003;33(5):1093–1102. | ||
Greenfield RS, Kaneko T, Daues A, et al. Evaluation in-vitro of adriamycin immunoconjugates synthesized using an acid-sensitive hydrazone linker. Cancer Res. 1990;50(20):6600–6607. | ||
Lau A, Berube G, Ford CHJ, et al. Novel doxorubicin-monoclonal anti-carcinoembryonic antigen antibody immunoconjugate activity in-vivo. Bioorg Med Chem. 1995;3(10):1305–1312. | ||
Kruger M, Beyer U, Schumacher P, et al. Synthesis and stability of four maleimide derivatives of the anti-cancer drug doxorubicin for the preparation of chemoimmunoconjugates. Chem Pharm Bull. 1997;45(2):399–401. | ||
Furgeson DY, Dreher MR, Chilkoti A. Structural optimization of a “smart” doxorubicin-polypeptide conjugate for thermally targeted delivery to solid tumors. J Control Release. 2006;110(2):362–369. | ||
Liang JF, Yang VC. Synthesis of doxorubicin-peptide conjugate with multidrug resistant tumor cell killing activity. Bioorg Med Chem Lett. 2005;15(22):5071–5075. | ||
Sirova M, Strohalm J, Subr V, et al. Treatment with HPMA copolymer-based doxorubicin conjugate containing human immunoglobulin induces long-lasting systemic anti-tumor immunity in mice. Cancer Immunol Immunother. 2007;56(1):35–47. | ||
Wong BK, Defeo-Jones D, Jones RE, et al. PSA-specific and non-PSA-specific conversion of a PSA-targeted peptide conjugate of doxorubicin to its active metabolite. Drug Metab Dispos. 2001;29(3):313–318. | ||
Bidwell GL 3rd, Davis AN, Fokt I, Priebe W, Raucher D. A thermally targeted elastin-like polypeptide-doxorubicin conjugate overcomes drug resistance. Invest New Drugs. 2007;25(4):313–326. | ||
Ajaj KA, Graeser R, Fichtner I, et al. In-vitro and in-vivo study of an albumin-binding prodrug of doxorubicin that is cleaved by cathepsin B. Cancer Chemother Pharmacol. 2009;64(2):413–418. | ||
Ryppa C, Mann-Steinberg H, Fichtner I, et al. In-vitro and in-vivo evaluation of doxorubicin conjugates with the divalent peptide E-[c(RGDfK)2] that targets integrin aVb3. Bioconjug Chem. 2008;19(7):1414–1422. | ||
Huang YF, Shangguan D, Liu H, et al. Molecular assembly of an aptamer-drug conjugate for targeted drug delivery to tumor cells. Chem Bio Chem. 2009;10(5):862–868. | ||
Ren Y, Wei D, Zhan X. Inhibition of P-glycoprotein and increasing of drug-sensitivity of a human carcinoma cell line (KB-A-1) by an anti-sense oligodeoxynucleotide-doxorubicin conjugate in vitro. Biotechnol Appl Biochem. 2005;41(pt 2):137–143. | ||
Ren Y, Zhan X, Wei D, Liu J. In-vitro reversal MDR of human carcinoma cell line by an antisense oligodeoxynucleotide-doxorubicin conjugate. Biomed Pharmacother. 2004;58(9):520–526. | ||
Kovar L, Etrych T, Kabesova M, et al. Doxorubicin attached to HPMA copolymer via amide bond modifies the glycosylation pattern of EL4 cells. Tumour Biol. 2010;31(4):233–242. | ||
Lammers T, Subr V, Ulbrich K, et al. Simultaneous delivery of doxorubicin and gemcitabine to tumors in vivo using prototypic polymeric drug carriers. Biomaterials. 2009;30(20):3466–3475. | ||
Krakovicova H, Ethch T, Ulbrich K. HPMA-based polymerconjugates with drug combinations. Eur J Pharmacol. 2009;37(3–4):4050–4412. | ||
Cao N, Feng SS. Doxorubicin conjugated to D-alpha-tocopheryl polyethylene glycol 1000 succinate (TPGS): conjugation chemistry, characterization, in-vitro and in-vivo evaluation. Biomaterials. 2008;29(28):3856–3865. | ||
Rodrigues PC, Beyer U, Schumacher P, et al. Acid-sensitive polyethylene glycol conjugates of doxorubicin: preparation, in vitro efficacy and intracellular distribution. Bioorg Med Chem. 1999;7(11):2517–2524. | ||
Kratz F. Albumin as a drug carrier: design of prodrugs, drug conjugates and nanoparticles. J Control Release. 2008;132(3):171–183. | ||
Dillman RO, Johnson DE, Ogden J, Beidler D. Significance of antigen, drug, and tumor cell targets in the preclinical evaluation of doxorubicin, daunorubicin, methotrexate, and mitomycin-C monoclonal antibody immunoconjugates. Mol Biotherapy. 1989;1(5):250–255. | ||
Elias DJ, Kline LE, Robbins BA, et al. Monoclonal antibody KS1/4-methotrexate immunoconjugate studies in non-small cell lung carcinoma. Am J Respir Crit Care Med. 1994;150(4):1114–1122. | ||
Lelievre E, Guillaudeux J, Cardona H, et al. Human pharmacokinetics of a new Vinca alkaloid S 12363 with use of a monoclonal antibody-based radio- or enzyme immunoassay. Cancer Res. 1993;53(15):3536–3540. | ||
Starling JJ, Maciak RS, Law KL, et al. In-vivo antitumor activity of a monoclonal antibody-Vinca alkaloid immunoconjugate directed against a solid tumor membrane antigen characterized by heterogeneous expression and noninternalization of antibody-antigen complexes. Cancer Res. 1991;51(11):2965–2972. | ||
Johnson DA, Zimmermann JL, Laguzza BC, et al. In-vivo antitumor activity demonstrated with squamous carcinoma reactive monoclonal antibody-Vinca immunoconjugates. Cancer Immunol Immunother. 1988;27(3):241–245. | ||
Voznesenskii AI, Galanova YuV, Archakov AI. Covalent binding of bleomycin to concanavalin A and immunoglobulin G enhances the ability of the bleomycin-Fe(II) complex to destroy the erythrocyte membrane. Biomed Sci. 1991;2(2):147–150. | ||
Manabe Y, Tsubota T, Haruta Y, et al. Production of a monoclonal antibody-bleomycin conjugate utilizing dextran T-40 and the antigen-targeting cytotoxicity of the conjugate. Biochem Biophys Res Commun. 1983;115(3):1009–1014. | ||
Beyer U, Roth T, Schumacher P, et al. Synthesis and in-vitro efficacy of transferring conjugates of the anticancer drug chlorambucil. J Med Chem. 1998;41(15):2701–2708. | ||
Kratz F, Beyer U, Roth T, et al. Albumin conjugates of the anticancer drug chlorambucil: synthesis, characterization, and in-vitro efficacy. Arch Pharm (Weinheim). 1998;331(2):47–53. | ||
Yang DJ, Liu CW, Yu DF, et al. Bisaminoethanethiol-targeting ligand conjugates and compositions. U S Patent US8236279 B2. Aug 7, 2012. | ||
Bonny C, Coquoz D, Chen J. Conjugates with enhanced cell uptake activity. U S Patent WO2006050930 A2. May 18, 2006. | ||
Jaime J, Pagé M. Paclitaxel immunoconjugate for the specific treatment of ovarian cancer in-vitro. Anticancer Res. 2001;21(2A):1119–1128. | ||
Safavy A, Raisch KP. Synthesis and biological evaluation of a paclitaxel immunoconjugate. Methods Mol Med. 2005;109:375–388. | ||
Jaime J, Pagé M. Paclitaxel antibody conjugates and trehalose for preserving the immunological activity after freeze-drying. Curr Med Chem. 2004;11(4):439–446. | ||
DiJoseph JF, Dougher MM, Evans DY, Zhou BB, Damle NK. Preclinical anti-tumor activity of antibody-targeted chemotherapy with CMC-544 (inotuzumab ozogamicin), a CD22-specific immunoconjugate of calicheamicin, compared with non-targeted combination chemotherapy with CVP or CHOP. Cancer Chemother Pharmacol. 2011;67(4):741–749. | ||
Tanaka M, Kano Y, Akutsu M, et al. The cytotoxic effects of gemtuzumab ozogamicin (mylotarg) in combination with conventional antileukemic agents by isobologram analysis in-vitro. Anticancer Res. 2009;29(11):4589–4596. | ||
Dornan D, Bennett F, Chen Y, et al. Therapeutic potential of an anti-CD79b antibody-drug conjugate, anti-CD79b-vc-MMAE, for the treatment of non-Hodgkin lymphoma. Blood. 2009;114(13):2721–2729. | ||
Pollack VA, Alvarez E, Tse KF, et al. Treatment parameters modulating regression of human melanoma xenografts by an antibody-drug conjugate (CR011-vcMMAE) targeting GPNMB. Cancer Chemother Pharmacol. 2007;60(3):423–435. | ||
Okeley NM, Miyamoto JB, Zhang X, et al. Intracellular activation of SGN-35, a potent anti-CD30 antibody-drug conjugate. Clin Cancer Res. 2010;16(3):888–897. | ||
Cheng VC, Ho PL, Yuen KY. Two probable cases of serious drug interaction between clarithromycin and colchicine. South Med J. 2005;98(8):811–813. | ||
Berger AS, Cheng CK, Pearson PA, et al. Intravitreal sustained release corticosteroid-5-fluoruracil conjugate in the treatment of experimental proliferative vitreoretinopathy. Invest Ophthalmol Vis Sci. 1996;37(11):2318–2325. | ||
Shrivastav TG, Chaube SK, Kariya KP, Prasad PK, Kumar D. Influence of different length spacers containing enzyme conjugate on functional parameters of progesterone ELISA. J Immunoassay Immunochem. 2013;34(1):94–108. | ||
Ogihara T, Miyal K, Nishi K, Ishibashi K, Kumahara Y. Enzyme-labelled immunoassay for plasma cortisol. J Clin Endocrinol Metab. 1977;44(1):91–95. | ||
Nakao T, Tamamura F, Tsunoda N, Kawata K. Double antibody enzyme immunoassay of cortisol in bovine plasma. Steroids. 1981;38(1):111–120. | ||
Nakao T. Practical procedure for enzyme immunoassay of progesterone in bovine serum. Acta Endocrinol. 1980;93(2):223–227. | ||
Erlanger BE, Borek F, Beiser SM, et al. Steroid-protein conjugates. II. Preparation and characterization of conjugates of bovine serum albumin with progesterone, deoxycorticosterone, and estrone. J Biol Chem. 1959;234(5):1090–1094. | ||
Erlanger BE, Borek F, Beiser SM, Lieberman S. Steroid-protein conjugates. I. Preparation and characterization of conjugates of bovine serum albumin with testosterone and with cortisone. J Biol Chem. 1957;228(2):713–727. | ||
Ali SM, Khan AR, Ahmad MU, Chen P, Sheikh S, Ahmad I. Synthesis and biological evaluation of gemcitabine-lipid conjugate (NEO6002). Bioorg Med Chem Lett. 2005;15(10):2571–2574. | ||
Chen P, Chien PY, Khan AR, et al. In-vitro and in-vivo anti-cancer activity of a novel gemcitabine-cardiolipin conjugate. Anticancer Drugs. 2006;17(1):53–61. | ||
Chiu ML, Tseng TTC, Monbouquette HG. A convenient homogeneous enzyme immunoassay for estradiol detection. Biotechnol Appl Biochem. 2011;58(1):75–82. | ||
Hatzidakis G, Stefanakis A, Krambovitis E. Comparison of different antibody-conjugate derivatives for the development of a sensitive and specific progesterone assay. J Reprod Fertil. 1993;97(2):557–561. | ||
Khatun S, Nara S, Tripathi V, et al. Development of ELISA for measurement of progesterone employing 17-alpha-OH-P-HRP as enzyme label. J Immunoassay Immunochem. 2009;30(2):186–196. | ||
Bucki R, Leszczynska K, Byfield FJ, et al. Combined antibacterial and anti-inflammatory activity of a cationic disubstituted dexamethasone-spermine conjugate. Antimicrob Agents Chemother. 2010;54(6):2525–2533. | ||
Kobayashi N, Kato Y, Oyama H, et al. Anti-estradiol-17beta single-chain Fv fragments: generation, characterization, gene randomization, and optimized phage display. Steroids. 2008;73(14):1485–1499. | ||
Pang YN, Zhang Y, Zhang ZR. Synthesis of an enzyme-dependent prodrug and evaluation of its potential for colon targeting. World J Gastroenterol. 2002;8(5):913–917. | ||
Quan LD, Purdue PE, Liu XM, et al. Development of a macromolecular prodrug for the treatment of inflammatory arthritis: mechanisms involved in arthrotropism and sustained therapeutic efficacy. Arthritis Res Ther. 2010;12(5):R170. | ||
Yamashita K, Takahashi M, Tsukamoto S, Numazawa M, Okuyama M, Honma S. Use of novel picolinoyl derivatization for simultaneous quantification of six corticosteroids by liquid chromatography-electrospray ionization tandem mass spectrometry. J Chromatogr A. 2007;1173:120–128. | ||
Kiew LV, Cheong SK, Sidik K, Chung LY. Improved plasma stability and sustained release profile of gemcitabine via polypeptide conjugation. Int J Pharm. 2010;391(1–2):212–220. | ||
Alexander RL, Greene BT, Torti SV, Kucera GL. A novel phospholipid gemcitabine conjugate is able to bypass three drug-resistance mechanisms. Cancer Chemother Pharmacol. 2005;56(1):15–21. | ||
Alexander RL, Morris-Natschke SL, Ishaq KS, et al. Synthesis of cytotoxic activity of two novel 1-dodecylthio-2-decyloxypropyl-3-phophatidic acid conjugates with gemcitabine and cytosine arabinoside. J Med Chem. 2003;46(19):4205–4208. | ||
Alexander RL, Kucera GL. Lipid nucleoside conjugates for the treatment of cancer. Curr Pharm Des. 2005;11(9):1079–1089. | ||
Guo P, Ma J, Li S, Guo Z, Adams AL, Gallo JM. Targeted delivery of a peripheral benzodiazepine receptor ligand-gemcitabine conjugate to brain tumors in a xenograft model. Cancer Chemother Pharmacol. 2001;48(2):169–176. | ||
Guo Z, Gallo JM. Selective protection of 2′,2′-difluorodexoycytidine (gemcitabine). J Org Chem. 1999;64(22):8319–8322. | ||
Castelli F, Sarpietro MG, Ceruti M, Rocco F, Cattel L. Characterization of lipophilic gemcitabine prodrug-liposomal membrane interaction by differential scanning calorimetry. Mol Pharm. 2006;3(6):737–744. | ||
Lagisetty P, Vilekar P, Awasthi V. Synthesis of radiolabeled cytarabine conjugates. Bioorganic Med Chem Lett. 2009;19(16):4764–4767. | ||
Ratcliffe KE, Fraser HM, Sellar R, Rivier J, Millar RP. Bifunctional gonadotropin-releasing hormone antagonist-progesterone analogs with increased efficacy and duration of action. Endocrinology. 2006;147(1):571–579. | ||
Cerný I, Pouzar V, Hill M, Havlíková H, Hampl R. Syntheses of 19-[O-(carboxymethyl)oxime] haptens of epipregnanolone and pregnanolone. Steroids. 2006;71(2):120–128. | ||
Jones CD, Mason NR. The use of 6alpha-and 6beta-carboxymethyl testosterone-bovine serum albumin conjugates in radioimmunoassay for testosterone. Steroids. 1975;25(1):23–32. | ||
Nambara T, Ohkubo T, Shimada K. Antigenic properties of estriol 3-glucuronide-[C-6]-bovine serum albumin conjugates having oxime bridges. J Pharmacobiodyn. 1983;6(9):692–697. | ||
Shih LB, Goldenberg DM, Xuan H, Lu HW, Mattes MJ, Hall TC. Internalization of an intact doxorubicin immunoconjugate. Cancer Immunol Immunother. 1994;38(2):92–98. | ||
Sliwkowski MX, Lofgren JA, Lewis GD, Hotaling TE, Fendly BM, Fox JA. Nonclinical studies addressing the mechanism of action of trastuzumab (Herceptin). Semin Oncol. 1999;26(4 suppl 12):60–70. | ||
Lewis Phillips GD, Li G, Dugger DL, et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008;68(22):9280–9290. | ||
Gong C, Yao Y, Wang Y, et al. Up-regulation of miR-21 mediates resistance to trastuzumab therapy for breast cancer. J Biol Chem. 2011;286(21):19127–19137. | ||
Scaltriti M, Eichhorn PJ, Cortés J, et al. Cyclin E amplification/overexpression is a mechanism of trastuzumab resistance in HER2+ breast cancer patients. Proc Natl Acad Sci U S A. 2011;108(9):3761–3766. | ||
Pandya K, Meeke K, Clementz A, et al. Targeting both Notch and ErbB-2 signalling pathways is required for prevention of ErbB-2-positive breast tumour recurrence. Br J Cancer. 2011;105(6):796–806. | ||
Morgillo F, Kim W, Kim E, et al. Implications of the insulin-like growth factor-IR pathway in the resistance of non-small cell lung cancer cells to treatment with gefitinib. Clin Cancer Res. 2007;13(9):2795–2803. | ||
Morgillo F, Woo JK, Kim ES, et al. Heterodimerization of insulin-like growth factor receptor/epidermal growth factor receptor and induction of surviving expression counteract the anti-tumor action of Erlotinib. Cancer Res. 2006;66(20):10100–10111. | ||
Sartore-Bianchi A, Di Nicolantonio F, Nichelatti M, et al. Multi-determinants analysis of molecular alterations for predicting clinical benefit to EGFR-targeted monoclonal antibodies in colorectal cancer. PLoS One. 2009;4(10):e7287. | ||
Weickhardt A, Tebbutt N, Mariadason J. Strategies for overcoming inherent and acquired resistance to EGFR inhibitors by targeting downstream effectors in the RAS/PI3K pathway. Curr Cancer Drug Targets. 2010;10(8):824–833. | ||
Modjtahedi H, Essapen S. Epidermal growth factor receptor inhibitors in cancer treatment: advances, challenges and opportunities. Anticancer Drugs. 2009;20(10):851–855. | ||
Dempke W, Heinemann V. Ras mutational status is a biomarker for resistance to EGFR inhibitors in colorectal carcinoma. Anticancer Res. 2010;30(11):4673–4677. | ||
Schmitz S, Kaminsky-Forrett MC, Henry S, et al. Phase II study of figitumumab in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck: clinical activity and molecular response (GORTEC 2008-02). Ann Oncol. 2012;23(8):2153–2161. | ||
Chi KN, Gleave ME, Fazli L, et al. A phase II pharmacodynamic study of preoperative figitumumab in patients with localized prostate cancer. Clin Cancer Res. 2012;18(12):3407–3413. | ||
Atzori F, Tabernero J, Cervantes A, et al. A phase I pharmacokinetic and pharmacodynamic study of dalotuzumab (MK-0646), an anti-insulin-like growth factor-1 receptor monoclonal antibody, in patients with advanced solid tumors. Clin Cancer Res. 2011;17(19):6304–6312. | ||
Bitelman C, Sarfstein R, Sarig M, et al. IGF1R-directed targeted therapy enhances the cytotoxic effect of chemotherapy in endometrial cancer. Cancer Lett. 2013;335(1):153–159. | ||
Klitgaard JL, Koefoed K, Geisler C, et al. Combination of two anti-CD5 monoclonal antibodies synergistically induces complement-dependent cytotoxicity of chronic lymphocytic leukaemia cells. Br J Haematol. 2013;163(2):182–193. | ||
Lefebvre ML, Krause SW, Salcedo M, et al. Ex-vivo-activated human macrophages kill chronic lymphocytic leukemia cells in the presence of rituximab: mechanism of antibody-dependent cellular cytotoxicity and impact of human serum. J Immunother. 2006;29(4):388–397. | ||
Manches O, Lui G, Chaperot L, et al. In-vitro mechanisms of action of rituximab on primary non-Hodgkin lymphomas. Blood. 2003;101(3):949–954. | ||
Stevenson FK, Bell AJ, Cusack R, et al. Preliminary studies for an immunotherapeutic approach to the treatment of human myeloma using chimeric anti-CD38 antibody. Blood. 1991;77(5): 1071–1079. | ||
Li M, Xiao X, Zhang W, Liu L, Xi N, Wang Y. AFM analysis of the multiple types of molecular interactions involved in rituximab lymphoma therapy on patient tumor cells and NK cells. Cell Immunol. 2014;290(2):233–244. | ||
Zhang L, Qian Z, Cai Z, et al. Synergistic antitumor effects of lenalidomide and rituximab on mantle cell lymphoma in vitro and in vivo. Am J Hematol. 2009;84(9):553–559. | ||
Fei F, Lim M, George AA, et al. Cytotoxicity of CD56-positive lymphocytes against autologous B-cell precursor acute lymphoblastic leukemia cells. Leukemia. 2015;29(4):788–797. | ||
Caron PC, Lai LT, Scheinberg DA. Interleukin-2 enhancement of cytotoxicity by humanized monoclonal antibody M195 (anti-CD33) in myelogenous leukemia. Clin Cancer Res. 1995;1(1):63–70. | ||
Pietras RJ, Pegram MD, Finn RS, Maneval DA, Slamon DJ. Remission of human breast cancer xenografts on therapy with humanized monoclonal antibody to HER-2 receptor and DNA-reactive drugs. Oncogene. 1998;17(17):2235–2249. | ||
Marches R, Uhr JW. Enhancement of the p27Kip1-mediated antiproliferative effect of trastuzumab (Herceptin) on HER2-overexpressing tumor cells. Int J Cancer. 2004;112(3):492–501. | ||
Lin NU, Carey LA, Liu MC, et al. Phase II trial of lapatinib for brain metastases in patients with human epidermal growth factor receptor 2-positive breast cancer. J Clin Oncol. 2008;26(12):1993–1999. | ||
Cobleigh MA, Vogel CL, Tripathy D, et al. Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J Clin Oncol. 1999;17(9):2639–2648. | ||
Vogel CL, Cobleigh MA, Tripathy D, et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J Clin Oncol. 2002;20(3):719–726. | ||
Kute TE, Savage L, Stehle JR, et al. Breast tumor cells isolated from in vitro resistance to trastuzumab remain sensitive to trastuzumab anti-tumor effects in vivo and to ADCC killing. Cancer Immunol Immunother. 2009;58(11):1887–1896. | ||
Narayan M, Wilken JA, Harris LN, Baron AT, Kimbler KD, Maihle NJ. Trastuzumab-induced HER reprogramming in “resistant” breast carcinoma cells. Cancer Res. 2009;69(6):2191–2194. | ||
Chen FL, Xia W, Spector NL. Acquired resistance to small molecule ErbB2 tyrosine kinase inhibitors. Clin Cancer Res. 2008;14(21):6730–6734. | ||
Ritter CA, Perez-Torres M, Rinehart C, et al. Human breast cancer cells selected for resistance to trastuzumab in-vivo overexpress epidermal growth factor receptor and ErbB ligands and remain dependent on the ErbB receptor network. Clin Cancer Res. 2007;13(16):4909–4919. | ||
Nanda R. Targeting the human epidermal growth factor receptor 2 (HER2) in the treatment of breast cancer: recent advances and future directions. Rev Recent Clin Trials. 2007;2(2):111–116. | ||
Mitra D, Brumlik MJ, Okamgba SU, et al. An oncogenic isoform of HER2 associated with locally disseminated breast cancer and trastuzumab resistance. Mol Cancer Ther. 2009;8(8):2152–2162. | ||
Köninki K, Barok M, Tanner M, et al. Multiple molecular mechanisms underlying trastuzumab and lapatinib resistance in JIMT-1 breast cancer cells. Cancer Lett. 2010;294(2):211–219. | ||
Oliveras-Ferraros C, Vazquez-Martin A, Cufí S, et al. Inhibitor of Apoptosis (IAP) survivin is indispensable for survival of HER2 gene-amplified breast cancer cells with primary resistance to HER1/2-targeted therapies. Biochem Biophys Res Commun. 2011;407(2):412–419. | ||
Barok M, Tanner M, Köninki K, et al. Trastuzumab-DM1 causes tumour growth inhibition by mitotic catastrophe in trastuzumab-resistant breast cancer cells in-vivo. Breast Cancer Res. 2011;13(2):R46. | ||
Oliveras-Ferraros C, Vazquez-Martin A, Martin-Castilló B, et al. Pathway-focused proteomic signatures in HER2-overexpressing breast cancer with a basal-like phenotype: new insights into de novo resistance to trastuzumab (Herceptin). Int J Oncol. 2010;37(3):669–678. | ||
García-Sáenz JA, Martín M, Calles A, et al. Bevacizumab in combination with metronomic chemotherapy in patients with anthracycline- and taxane-refractory breast cancer. J Chemother. 2008;20(5):632–639. | ||
Slamon DJ, Leyland-Jone B, Shak S, et al. Use of chemotherapy plus monoclonal antibody against HER2 for metastatic breast cancer that overexpress HER2. N Engl J Med. 2001;344(11):786–792. | ||
Harris CA, Ward RL, Dobbins TA, Drew AK, Pearson S. The efficacy of HER2-targeted agents in metastatic breast cancer: a meta-analysis. Ann Oncol. 2011;22(6):1308–1317. | ||
Sivam GP, Martin PJ, Reisfeld RA, Mueller BM. Therapeutic efficacy of a doxorubicin immunoconjugate in a preclinical model of spontaneous metastatic human melanoma. Cancer Res. 1995;55(11):2352–2356. | ||
Yang HM, Reisfeld RA. Doxorubicin conjugated with monoclonal antibody directed to a human melanoma-associated proteoglycan suppresses growth of established tumor xenografts in nude mice. Proc Natl Acad Sci U S A. 1988;85(4):1189–1193. | ||
Sapra P, Stein R, Pickett J, et al. Anti-CD74 antibody-doxorubicin conjugate, IMMU-110, in a human multiple myeloma xenograph and in monkeys. Clin Cancer Res. 2005;11(14):5257–5264. | ||
Navab R, Chevet E, Authier F, Di Guglielmo GM, Bergeron JJ, Brodt P. Inhibition of endosomal insulin-like growth factor-I processing by cysteine proteinase inhibitors blocks receptor-mediated functions. J Biol Chem. 2001;276(17):13644–13649. | ||
Hansen HJ, Ong GL, Diril H. Internalization and catabolism of radiolabeled antibodies to the MHC class-II invariant chain by B-cell lymphomas. Biochem J. 1996;320(pt 1):293–300. | ||
Wang F, Jiang X, Yang DC, Elliott RL, Head JF. Doxorubicin-gallium-transferrin conjugate overcomes multidrug resistance: evidence for drug accumulation in the nucleus of drug resistant MCF-7/ADR cells. Anticancer Res. 2000;20(2A):799–808. | ||
Régina A, Demeule M, Ché C, et al. Antitumour activity of ANG1005, a conjugate between paclitaxel and the new brain delivery vector Angiopep-2. Br J Pharmacol. 2008;155(2):185–197. | ||
Asakura T, Takahashi N, Takada K, Inoue T, Ohkawa K. Drug conjugate of doxorubicin with glutathione is a potent reverser of multidrug resistance in rat hepatoma cells. Anticancer Drugs. 1997;8(2):199–203. | ||
Mazel M, Clair P, Rousselle C, et al. Doxorubicin-peptide conjugates overcome multidrug resistance. Anticancer Drugs. 2001;12(2):107–116. | ||
Lam W, Leung CH, Chan HL, Fong WF. Toxicity and DNA binding of dextran-doxorubicin conjugates in multidrug-resistant KB-V1 cells: optimization of dextran size. Anticancer Drugs. 2000;11(5):377–384. | ||
Dubikovskaya EA, Thorne SH, Pillow TH, Contag CH, Wender PA. Overcoming multidrug resistance of small-molecule therapeutics through conjugation with releasable octaarginine transporters. Proc Natl Acad Sci U S A. 2008;105(34):12128–12133. | ||
Pegram MD, Lopez A, Konecny G, Slamon DJ. Trastuzumab and chemotherapeutics: drug interactions and synergies. Semin Oncol. 2000;27(6 suppl 11):21–25. [discussion 92–100]. | ||
Slamon D, Pegram M. Rationale for trastuzumab (Herceptin) in adjuvant breast cancer trials. Semin Oncol. 2001;28(1 suppl 3):13–19. | ||
Boone JJ, Bhosle J, Tilby MJ, Hartley JA, Hochhauser D. Involvement of the HER2 pathway in repair of DNA damage produced by chemotherapeutic agents. Mol Cancer Ther. 2009;8(11):3015–3023. | ||
Meden H, Beneke A, Hesse T, Novophashenny I, Wischnewsky M. Weekly intravenous recombinant humanized anti-P185HER2 monoclonal antibody (herceptin) plus docetaxel in patients with metastatic breast cancer: a pilot study. Anticancer Res. 2001;21(2B):1301–1305. | ||
Winer EP, Burstein HJ. New combinations with Herceptin in metastatic breast cancer. Oncology. 2001;61(suppl 2):50–57. | ||
Kim S, Prichard CN, Younes MN, et al. Cetuximab and irinotecan interact synergistically to inhibit the growth of orthotopic anaplastic thyroid carcinoma xenografts in nude mice. Clin Cancer Res. 2006;12(2):600–607. | ||
Landriscina M, Maddalena F, Fabiano A, Piscazzi A, La Macchia O, Cignarelli M. Erlotinib enhances the proapoptotic activity of cytotoxic agents and synergizes with paclitaxel in poorly-differentiated thyroid carcinoma cells. Anticancer Res. 2010;30(2):473–480. | ||
Ciardiello F, Bianco R, Damiano V, et al. Antitumor activity of sequential treatment with topotecan and anti-epidermal growth factor receptor monoclonal antibody C225. Clin Cancer Res. 1999;5(4):909–916. | ||
Quek R, Wang Q, Morgan JA, et al. Combination mTOR and IGF-1R inhibition: phase I trial of everolimus and figitumumab in patients with advanced sarcomas and other solid tumors. Clin Cancer Res. 2011;17(4):871–879. | ||
Stacchiotti S, Negri T, Palassini E, et al. Sunitinib malate and figitumumab in solitary fibrous tumor: patterns and molecular bases of tumor response. Mol Cancer Ther. 2010;9(5):1286–1297. | ||
Lynn KD, Udugamasooriya DG, Roland CL, Castrillon DH, Kodadek TJ, Brekken RA. GU81, a VEGFR2 antagonist peptoid, enhances the anti-tumor activity of doxorubicin in the murine MMTV-PyMT transgenic model of breast cancer. BMC Cancer. 2010;10:397. | ||
Zhang L, Yu D, Hicklin DJ, et al. Combined anti-fetal liver kinase 1 monoclonal antibody (anti-VEGFR) and continuous low-dose doxorubicin inhibits angiogenesis and growth of human soft tissue sarcoma xenografts by induction of endothelial cell apoptosis. Cancer Res. 2002;62(7):2034–2042. | ||
Edelmann MN, Ogg RJ, Scoggins MA, et al. Dexamethasone exposure and memory function in adult survivors of childhood acute lymphoblastic leukemia: a report from the SJLIFE cohort. Pediatr Blood Cancer. 2013;60(11):1778–1784. | ||
Warris LT, van den Heuvel-Eibrink MM, den Hoed MA, Aarsen FK, Pieters R, van den Akker EL. Does dexamethasone induce more neuropsychological side effects than prednisone in pediatric acute lymphoblastic leukemia? A systematic review. Pediatr Blood Cancer. 2014;61(7):1313–1318. | ||
Waber DP, McCabe M, Sebree M, et al. Neuropsychological outcomes of a randomized trial of prednisone versus dexamethasone in acute lymphoblastic leukemia: findings from Dana-Farber Cancer Institute All Consortium Protocol 00-01. Pediatr Blood Cancer. 2013;60(11):1785–1791. | ||
Kanat O, Ozet A, Ataergin S, et al. Modified outpatient dexamethazone, cytarabine and cisplatin regimen may lead to high response rates and low toxicity in lymphoma. Med Princ Pract. 2010;19(5):344–347. | ||
Lerza R, Botta M, Barsotti B, et al. Dexamethazone-induced acute tumor lysis syndrome in a T-cell malignant lymphoma. Leuk Lymphoma. 2002;43(5):1129–1132. | ||
Watanabe N, Takahashi T, Sugimoto N, et al. Excellent response of chemotherapy-resistant B-cell-type chronic lymphocytic leukemia with meningeal involvement to rituximab. Int J Clin Oncol. 2005;10(5):357–361. | ||
Wielckens K, Delfs T, Muth A, Freese V, Kleeberg HJ. Glucocorticoid-induced lymphoma cell death: the good and the evil. J Steroid Biochem. 1987;27(1–3):413–419. | ||
Mao Y, Triantafillou G, Hertlein E, et al. Milatuzumab-conjugated liposomes as targeted dexamethasone carriers for therapeutic delivery in CD74+ B-cell malignancies. Clin Cancer Res. 2013;19(2):347–356. | ||
Pileckyte R, Jurgutis M, Valceckiene V, et al. Dose-dense high-dose methylprednisolone and rituximab in the treatment of relapsed or refractory high-risk chronic lymphocytic leukemia. Leuk Lymphoma. 2011;52(6):1055–1065. | ||
Domenech C, Suciu S, De Moerloose B, et al. Dexamethasone (6 mg/m2/day) and prednisolone (60 mg/m2/day) were equally effective as induction therapy for childhood acute lymphoblastic leukemia in the EORTC CLG 58951 randomized trial. Haematologica. 2014;99(7):1220–1227. | ||
Belz K, Schoeneberger H, Wehner S, et al. Smac mimetic and glucocorticoids synergize to induce apoptosis in childhood ALL by promoting ripoptosome assembly. Blood. 2014;124(2):240–250. | ||
Fleury I, Primeau M, Doreau A, et al. Polymorphisms in genes involved in the corticosteroid response and the outcome of childhood acute lymphoblastic leukemia. Am J Pharmacogenomics. 2004;4(5):331–341. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.