Back to Journals » Blood and Lymphatic Cancer: Targets and Therapy » Volume 4
Developments in anaplastic large-cell lymphoma: targeting the anaplastic lymphoma kinase
Authors Farina F, Stasia A, Gambacorti-Passerini C
Received 16 December 2013
Accepted for publication 28 February 2014
Published 21 August 2014 Volume 2014:4 Pages 69—79
DOI https://doi.org/10.2147/BLCTT.S35349
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Francesca Farina, Alessandra Stasia, Carlo Gambacorti-Passerini
Department of Health Sciences, University of Milano Bicocca, Monza, Italy
Abstract: Anaplastic lymphoma kinase (ALK) is a receptor tyrosine kinase protein implicated in a variety of hematological malignancies and solid tumors. ALK contributes to the development of cancers in different cell lineages through a variety of genetic mechanisms: gene fusions, activating point mutations, and possibly gene amplification. Recent developments led to significant therapeutic advances, including efficient diagnostic tests and ALK-targeting agents. This review addresses some therapeutic considerations with regard to the use of ALK inhibitors in ALK-positive lymphomas where, in spite of the advanced stage of the disease, long-lasting responses could be obtained in a substantial portion of heavily pretreated patients. Data and mechanisms for the development of resistance to ALK inhibitors will also be presented and discussed.
Keywords: ALK, lymphoma, tyrosine kinase, targeted therapy, crizotinib
Introduction
Anaplastic lymphoma kinase (ALK) is a tyrosine kinase (TK) receptor that has been initially identified through its involvement in chromosomal translocations associated with anaplastic large-cell lymphoma (ALCL). De novo systemic ALK-positive ALCLs are clinically aggressive lymphomas and frequently occur within the first 3 decades of life with a male predominance. ALCLs usually present as stage III–IV disease, with B symptoms, high levels of lactate dehydrogenases, and extranodal involvement.1–4 ALK-positive ALCLs often have a good clinical response to cytotoxic drugs, but relapses can occur, determining a worse prognosis.2,5–7
In addition, the use of cytotoxic therapy bears the risk for long-term toxicities, such as second cancers, lung fibrosis, or cardiac failure, which represent important issues, given the young age of most patients, who usually belong to the teenager/young adult groups.
ALK was identified for the first time in 1994 as a result of its fusion to nucleophosmin 1 (NPM1), which represents the predominant partner in ALCL.8,9 The NPM1-ALK results from the t(2;5)(p23;q25) chromosomal translocation, present in 50%–70% of ALCLs. NPM1-ALK is deregulated and transforms cells in vitro, activating the signal in several transduction pathways.10–16
It has been demonstrated, by using small interfering RNA (siRNA) to specifically downregulate the expression of the NPM1-ALK in ALCL cell lines, that the downregulation of NPM1-ALK resulted in decreased cell proliferation and increased cell apoptosis. All these data point toward a causal relationship between the development of ALK-containing fusions and malignant transformation, thus rendering ALK a potential therapeutic target for both pharmacologic and immunologic intervention. In fact, ALK expression is physiologically restricted to the immune privileged nervous system.15,17
Industrial interest in ALK’s role in cancer and the therapeutic potential of its specific inhibition has increased since 2007 with the discovery of echinoderm microtubule associated protein 4 (EML4) ALK in non-small-cell lung cancer (NSCLC). Subsequently, other tumors, such as inflammatory myofibroblastic tumors, neuroblastomas, and some carcinomas, both in adults and children, were shown to be driven by an altered ALK signal.18 Recently, several ALK inhibitors have been developed, offering new treatment options in tumors driven by abnormal ALK signaling.19–22
In 2011, crizotinib, a small-molecule inhibitor of the receptor tyrosine kinases of the hepatocyte growth factor (c-Met) and ALK, was approved by the Food and Drug Administration in ALK-positive NSCLC. Dramatic activity of crizotinib in ALK-positive ALCL was also demonstrated.23–25 New and more potent ALK inhibitors are likely to follow shortly. These molecules represent another excellent proof of principle for targeted therapy.26 As has been observed with other tyrosine kinase inhibitors, resistance has also recently emerged in patients treated with ALK inhibitors.27–31
ALK biology
Most of the knowledge of ALK expression in cancer comes from its pathological expression in NSCLCs and neuroblastomas that are more common in adults or children than ALCL or ALK-positive diffuse large-B-cell lymphoma (DLBCL). Nevertheless, what we know now is still useful for ALK-positive lymphomas and will help scientists and clinicians in approaching this disease.
ALK is normally expressed only in the nervous system (thalamus, hypothalamus, midbrain, olfactory bulb, selected cranial, dorsal root, and ganglial cells). It has a role in neural development and differentiation.32–34 ALK is involved in oncogenesis in both nonhematopoietic and hematopoietic malignancies. The full-length form of ALK is expressed in different types of cancers, including glioblastoma,32,35 breast cancer, neuroblastoma,36 Ewing sarcoma,37 retinoblastoma,38 DLBCL,39 and melanoma.40 Except for neuroblastoma, the pathogenic role of ALK is not clear in these tumors.41
A variety of mechanisms leading to aberrant kinase activation and constitutive phosphorylation of downstream pathway components have been identified, including missense mutation, gene amplification, and chromosomal translocation. ALK pathological expression in lymphoma cells is a result of chromosomal translocations that lead to the formation of ALK-containing oncogenic fusion proteins42 also found in several tumors, as mentioned earlier. The fusions interrupt the chromosome at the level of the ALK gene at 2p23. The chromosomal breakpoint involves, in the case of NPM1-ALK fusion, intron 4 of NPM1 and intron 16 of ALK; as result, the N-terminal region of NPM1 is fused to the catalytic domain of ALK, which conserves its protein-kinase domain.9 The fusion partner at the N terminus is usually widely expressed in normal cells and controls chimeric protein expression and localization.43 The ALK partner brings an oligomerization domain that mediates constitutive self-association of the ALK fusion, causing constitutive activation of its kinase domain and controlling its expression levels.41,44 At this time, almost 15 different ALK fusion proteins have been identified (Table 1).45 As a result, ALK fusion proteins are deregulated, ectopically expressed, and constitutively activated in lymphoid and other types of neoplastic cells. In lymphomas, this leads to growth factor-independent proliferation of lymphocytes,46–51 the biological target cells of the lymphoma-associated oncogene. ALK fusion plays a key role in neoplastic transformation by altering the phosphorylation of intracellular substrates, which likely contributes to the molecular pathogenesis of ALK-positive ALCL.15,52 NPM1-ALK is the most common fusion protein in ALK-positive ALCL, whereas clathrin heavy polypeptide (CLTC) ALK is present in ALK-positive DLBCL.53
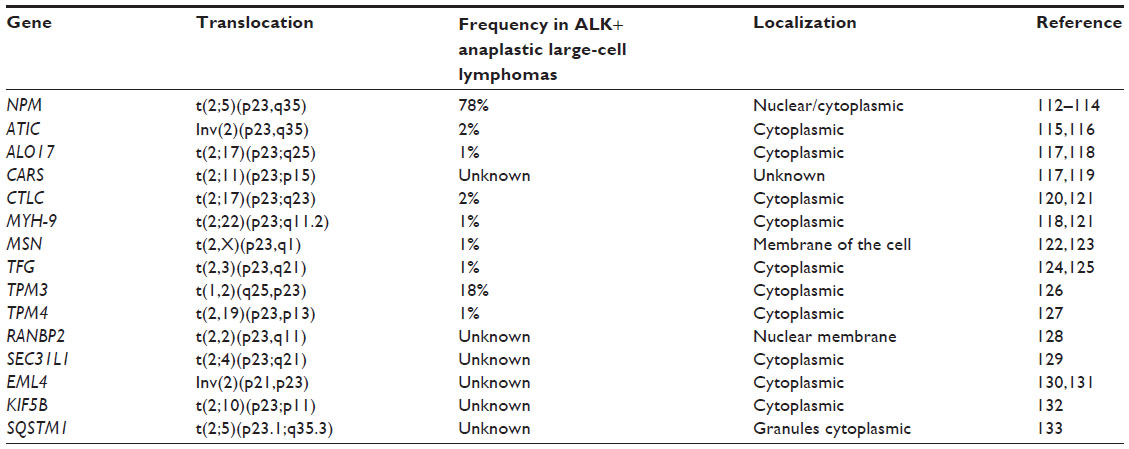 | Table 1 Chromosomal rearrangements involving the ALK gene and producing oncogenic fusion proteins in anaplastic large-cell lymphomas and other cancers |
The NPM1 portion mediates the constitutive dimerization and consequent activation of ALK kinase domain and controls its expression levels.44 NPM1-ALK is able to activate proliferative pathways such as rat sarcoma protein (RAS)/mitogen activated protein kinase (MAPK),46 Phospholipase C gamma (PLCγ).54 Janus kinase (JAK)/ signal transducer and activator of transcription (STAT),51,55,56 and phosphoinositide 3 kinase (PI3K)/ protein kinase B (Akt).10,12,57 Data point to the functional link between hematopoietic oncogenic tyrosine kinases and the G(1) cell cycle regulator Cell division cycle 25 homolog A (CDC25A).58,59 NPM1-ALK can protect cells from apoptosis mediated by this pathway or by inducing the transcription of antiapoptotic genes, including Bcl-2 and Bcl-xL.60–62 Animal models showed that the oncogenic role of NPM1-ALK induced a lymphoproliferative disorder in a short period of time.49,63 The tumorigenicity of NPM1-ALK was first demonstrated in a study by Kuefer et al50 in which retrovirally transfected bone marrow expressing NPM1-ALK was transplanted into lethally irradiated BALB/cByJ mice. These mice developed B-cell lymphomas within 4–6 months, clearly linking aberrant ALK activation with tumorigenesis. Chiarle et al generated transgenic mice in which NPM1-ALK expression was targeted to T cells, using a cluster of differentiation 4 (CD4) transgene cassette.49 These mice developed thymic lymphomas and plasma cell neoplasms from 5 weeks of age.
However, the presence of different ALK fusions confers different sensitivity to ALK inhibitors; for example, in vitro NPM1-ALK-expressing cells exhibit a higher response with lower ALK half maximal inhibitory concentration (IC50) values than SEC31 homolog A (SEC31A)-ALK-expressing cells.64
ALK is also a frequent object of mutation. Gain-of-function point mutations are critical to activate the kinase domain and may be the cause of secondary drug resistance. This was found first in neuroblastoma,65 where mutations affect the ALK intracellular segment that is linked to regulatory and catalytic signals. About 10% of sporadic neuroblastoma have somatic nonsynonymous ALK mutations, including K1062M, F1174L/C/I, F1245C/V/L, and R1275Q amino acid substitutions.66 In this setting, there is a significant correlation between activating mutations in the ALK TK domain and poor clinical outcome.67 ALK mutations are also associated with familial neuroblastoma (eg, T1087I, G1128A, and R1275Q).68–70
It is not clear how multiple copies of the ALK gene can contribute to tumor pathogenesis, even though rare case reports with amplification of aberrant forms of ALK are documented.71,72 Frequently, ALK amplification co-occurs with amplification of v-myc avian myelocytomatosis viral oncogene neuroblastoma derived homolog (MYCN).73 In particular, ALK amplifications without any mutations or gene fusions may not have a strong role in tumor pathogenesis, and thus may not represent a good target for ALK inhibitors.
Different diagnostic methods are available to detect ALK rearrangement. Fluorescence in situ hybridization (FISH), reverse transcriptase-polymerase chain reaction (RT-PCR), chromogen in situ hybridization, and immunohistochemistry (ICH) are currently used.74 FISH is normally used in trials to test ALK fusions, but it does not discriminate between different fusion types. RT-PCR can be used in a rapid and extremely sensitive way, but only when a defined ALK partner is known and novel fusions may be missed. ICH is cheap, but interpretation may be difficult. It can be used as screening in ALK-positive NSCLC, which has a lower incidence than ALK-positive ALCL.75–77 In a large case series of NSCLCs, a comparison of the sensitivity and specificity among ICH, FISH, and RT-PCR was performed. The study revealed that FISH has the best specificity and ICH the best sensitivity.78
ALK testing can be useful not only during diagnosis but also during the monitoring of treatment; in this setting, RT-PCR is the methodology of choice to detect early resistance, which develops with different frequencies in different diseases.
ALK inhibitors and their role in ALCL
ALK-positive ALCLs are currently treated with combined chemotherapy, with an overall response rate (ORR) of 60%–70%.20 Nevertheless, ALK is a good candidate for the development of targeted treatment because of the lack of wide expression in normal adult tissues. For this and the reasons mentioned earlier, blocking ALK function should not give important toxic effects.16,28,79 Potential strategies for targeting ALK include immunotherapy, gene silencing, inhibition of downstream signaling pathways, and direct inhibition of its catalytic activity through small-molecule inhibitors. The aim of this target therapy is to obtain a maximum tumor-specific effect with low toxicity, contrary to the conventional citotoxic chemotherapy.80
After the identification of different constitutively activated forms of ALK, both as fusion proteins and as mutated ALK forms, the attention focused on the development of small molecules targeting the protein kinase activity to abolish the ALK-dependent cancer cell growth.45 Kinase inhibitors are directed against the adenosine triphosphate (ATP)-binding site of the catalytic domain, which is highly conserved in ALK TK, with the goal of obtaining successful ALK inhibition. In preclinical studies, several ALK inhibitors have shown activity against NPM1-ALK and EML4-ALK cell lines.81,82 Initial testing of ALK inhibitors was performed using inhibitors derived from natural products such as staurosporine derivatives, which are not specific inhibitors of ALK.83 Synergies with heat shock protein 90 inhibitors were also observed.84 These and other natural-derived compounds inhibit ALK, increasing the proteosome-mediated degradation of ALK protein binding to heat shock protein 90.85,86 Subsequently, more-potent and more-specific ALK inhibitors have been developed, including at least 18 different classes of small molecule inhibitors of ALK (Table 2). ALK inhibitors have shown activity not only in ALCL but also in ALK-positive DLBCL, inducing tumor growth delay and its regression in murine xenograft.64,87
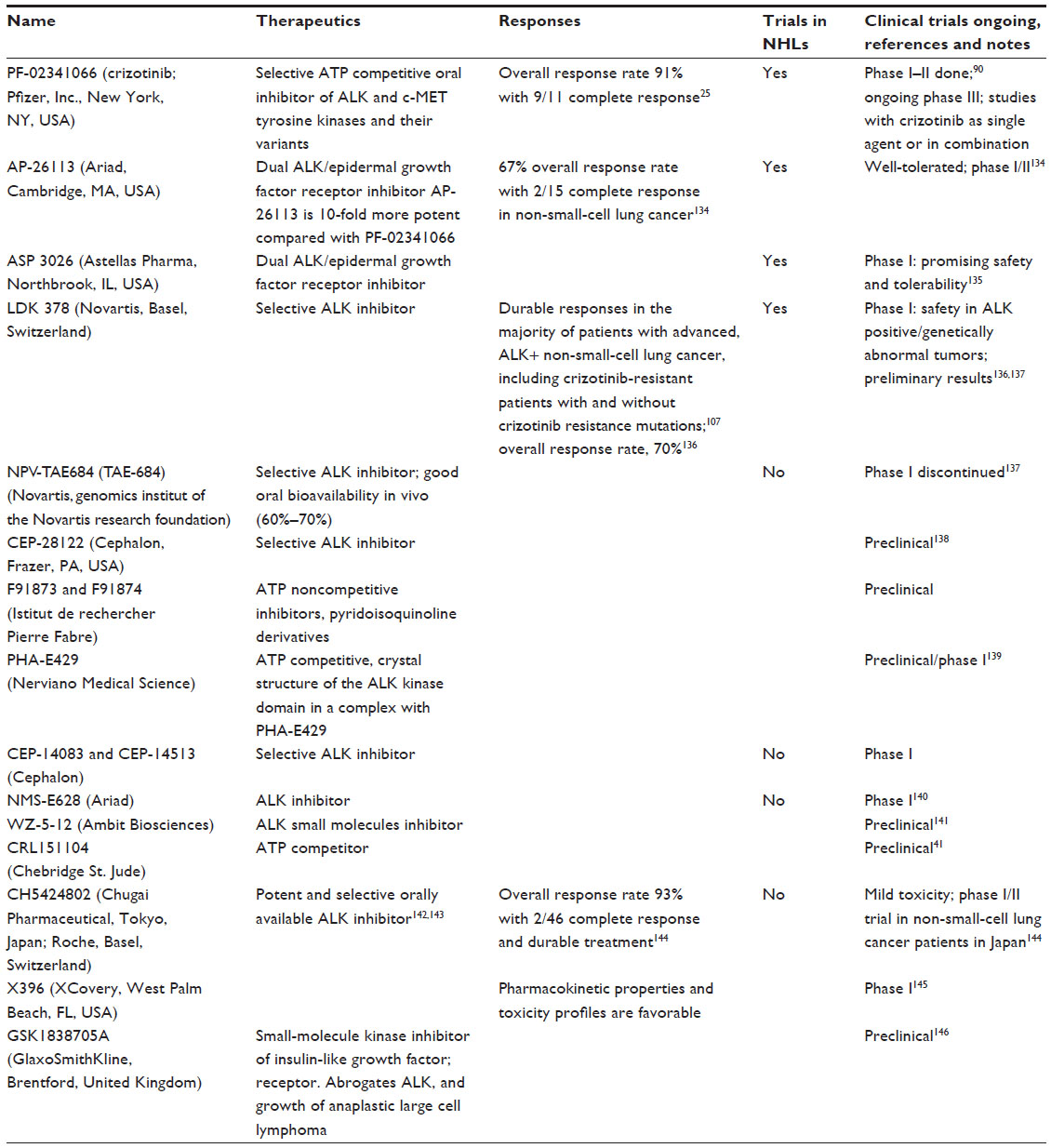 | Table 2 Inhibitors of anaplastic lymphoma kinase in development26,139,147–149 |
The largest volume of available knowledge relates to crizotinib. Crizotinib (PF-02341066) is the first human ALK inhibitor developed. It is a derivative of aminopyridine and was originally developed as a potent, orally bioavailable, ATP-competitive small-molecule inhibitor of mesenchymal epithelial transition growth factor/hepatocyte growth factor receptor/c-MET.88 Crizotinib suppresses the proliferation of ALK-positive ALCLs cell lines.81 This orally available TK inhibitor was being tested in an open-label, multicenter, two-part, dose escalation phase I clinical trial as an MET inhibitor to investigate its safety, tolerability, pharmacokinetics, pharmacodynamics, and activity in 37 patients with advanced cancer (excluding leukemias).89 The first part of this trial established 250 mg twice daily as the maximum tolerated dose. Three dose-limiting toxicities were observed: a grade 3 increase in alanine transaminase (ALT) (one patient at 200 mg once a day) and grade 3 fatigue (two patients at 300 mg twice a day). The most common adverse events were ocular flashes, nausea, emesis, fatigue, and diarrhea, all of which were manageable and reversible.90
The first treatment of an ALK-positive ALCL patient occurred in 2010. A report published in the New England Journal of Medicine described two adult patients with recurrent ALK-positive ALCLs who achieved complete response (CR) shortly after receiving crizotinib as a single agent.23 In June 2012, the authors updated their experience at the European Hematology Association, reporting on additional patients who achieved complete remission or partial response (PR), demonstrating clearly the activity of crizotinib in ALK-positive ALCLs. ORR was 10/11 (91%) and included 9 CR (82%) and 1 PR, with an ORR of 91% (95% confidence interval, 60%–99%); 7 patients had been in CR for more than 22 months, with a 2 year progression-free survival of 64%. All relapses developed within the initial 3 months of treatment.24 No parameter could predict the development of a durable response, with the only indication being that patients who were treated with crizotinib after failing an autologous bone marrow transplantation never achieved a durable response, at difference with patients treated after failing an allogeneic bone marrow transplantation. The final results of this study were recently published.25 These results were recently confirmed in a sponsored trial in which an ORR of 64% among 14 ALK-positive lymphoma patients is reported.91 A recent phase I study also reported this good safety profile with high response rates in children with relapsed ALK-positive ALCL.92
Some experimental evidence suggests that ALK inhibitors could also be efficacious in the treatment of ALK-positive DLBCL.64,87 The compassionate study previously cited24,25 reported 2 patients with ALK-positive DLBCL treated with crizotinib with one rapid but transient response.
As has been seen with other targeted therapies, resistance can emerge in some patients who have demonstrated initial response to ALK inhibition, as happened in the studies mentioned earlier, at quite a rapid pace.
Resistance to ALK inhibitors
Acquired resistance to kinase inhibitors is a serious problem in long-term cancer therapy. Data on ALK inhibitors resistance are, for the majority, reported in the context of NSCLCs. The mechanism can be divided into three groups, very similar to what we know about chronic myeloid leukemia (CML). The first one is an ALK mutation (28%), second is resistance resulting from mechanisms that upregulate ALK (such as gene amplification or copy number gain, which occur in 18% of patients), and last is the activation of other transduction pathways; for example, KRAS or mast/stem cell growth factor receptor (KIT) amplification occurs in 30% of cases.93–95
The first mechanism was initially reported in 2010 in NSCLCs,96 and two point mutations in the kinase domain of EML4-ALK were found.
These mutations, C1156Y and L1196M, conferred clinical resistance to crizotinib in NSCLC patients, and another one, F1174L, causes resistance to the same drug in an inflammatory myofibroblastic tumor patient.96,97 The acquired resistance to ALK-targeted therapy has been distinguished in “ALK-dominant” and “ALK-nondominant” mechanisms. In the first case, the acquired resistance is a result of the development of novel ALK kinase domain mutation alone or in combination with the increase of rearranged ALK gene copies in cancer cells. These are called ALK-dominant mechanisms because ALK signaling remains dominant in the crizotinib-resistant state. Multiple ALK kinase domain mutations that reduce sensitivity to crizotinib were recently identified.98 In vitro, a single amino acid substitution became predominant at higher crizotinib concentrations in two ALCL cell cultures grown at increasing crizotinib concentrations. The authors found that L1196Q substitution conferred resistance to crizotinib, but not to AP26113 and NVP-TAE 694, whereas cells carrying I1171N mutation were resistant to all tested inhibitors.27 In the clinical report described earlier,24 the kinase domain of NPM1-ALK could be amplified from peripheral blood samples obtained at the time of relapse in two ALCL patients. Deep sequencing of these products revealed the presence of different mutations: Q1064R at high prevalence (95%) in one patient and I1171N (33%) plus M1328I (14%) in a second patient. All these mutations were not present in samples obtained before crizotinib treatment, which did not contain either these or any other mutation in the ALK kinase domain. I1171N was already discovered in an in vitro screening17 and commands an intermediate level of resistance to crizotinib (resistance index (RI), 5.8), which, however, is cross-resistant with other anti-ALK TKI such as AP26113 and NVP-TAE684.
In addition, a series of different second oncogenic drivers were also described, coexisting in the same cell with the ALK rearrangement. These cases are ALK-nondominant mechanisms.99–101 These events should, in theory, bypass the dominance of ALK signaling and replace its oncogenic potential.27 To overcome resistance, it will be important to differentiate patients who preserve ALK dominance versus those who have diminished ALK dominance.
Future developments
The complexity of mechanisms of acquired resistance recently described suggests that other therapeutic options, including combination of ALK and other targeted approaches, such as immunotherapy,102,103 will be required in the future.104 ALK can be a target of antitumor vaccination because ALK-positive cells are dependent on ALK activity for survival and proliferation and ALK is spontaneously immunogenic in ALCL with an antibody and a T-cytotoxic response.105 In particular, as we learned in preclinical tests in neuroblastoma cell lines, the combination of ALK inhibitors with ALK antibody may work synergistically.106 Thus, we hypothesize that a combination approach with chemotherapy or vaccination and ALK inhibitors can reduce the burden of the disease obtaining durable responses.102
In addition, substantial effort has been focused on optimizing direct kinase inhibition by developing new ALK inhibitors that are structurally different from crizotinib. LDK378 (Novartis Inc., B Basel, Switzerland), CH5424802 (Chugai Pharmaceutical, Tokyo, Japan), AP26113 (Ariad Pharmaceuticals, Cambridge, MA, USA), and X396 (XCovery, West Palm Beach, FL, USA) are some examples, with many others also in development (Table 2). The experience with these drugs is, for now, limited only to ALK-positive NSCLCs. The first phase I trial in humans of LDK378 (NCT01283516), limited to relapsed/refractory NSCLC patients, demonstrated objective responses in patients with ALK-rearranged NSCLC who were previously treated with crizotinib.107,108 A phase I/II trial of CH5424802 in crizotinib-naïve patients with ALK-positive NSCLC demonstrated objective responses in 43 of 46 patients enrolled at the maximum tolerated dose, with two CRs and 41 PRs.109 AP26113, a dual ALK and epidermal growth factor receptor inhibitor, also showed evidence of clinical activity in crizotinib-naïve and crizotinib-resistant NSCLC, with an ORR of 73%.110
Conclusion
The initial identification of the genetic lesion at the basis of malignant transformation in ALK-positive ALCLs, originally obtained in 1994,9 was successfully exploited and brought to the patient bedside in 2010.23 This compares favorably with the time that elapsed between the discovery of the Philadelphia chromosome in CML and the clinical development of imatinib.111
However, it is sad to recognize that ALK-positive ALCL patients could be treated earlier and that ALK inhibitors became available only because ALK alterations were identified in more common cancers such as NSCLC.
The entry of crizotinib in the treatment of ALK-positive ALCLs marked a new era in the therapy of this rare but highly malignant type of tumor. The demonstration of high response rates and durable responses, even in the setting of advanced and resistant disease, should prompt the development of clinical studies in less advanced conditions and in combination with already-active drugs. A particular emphasis should be placed on the need to decrease as much as possible the use of cytotoxic drugs, given their long-term toxic effects and the young age of most of the patients. Drugs of interest could be steroids, vincristine, and brentuximab, given their lack of cross-resistance with crizotinib.
The possibility of obtaining durable responses in a substantial fraction of advanced, heavily pretreated ALK-positive lymphomas illustrates our present inability to forecast the level of heterogeneity present inside a tumor. The type of presentation and clinical history of these patients would suggest they should be placed in the category of highly heterogeneous diseases, such as a metastatic NSCLC, relapsed blast crisis CML, or relapsed Philadelphia chromosome-positive acute lymphoblastic leukemia, in which monotherapy with TKIs seldom obtains durable responses. Further studies, such as exome sequencing of pre- and posttreatment samples, could hopefully shed light and provide a useful indicator of the level of heterogeneity present inside a tumor at any given time.
The next couple of years will hopefully see the fading of regimens based only on unspecific cytotoxic drugs in favor of more specific and hopefully less toxic approaches.
Disclosure
The authors declare no conflicts of interest in this work.
References
Agostinelli C, Piccaluga PP, Went P, et al. Peripheral T cell lymphoma, not otherwise specified: the stuff of genes, dreams and therapies. J Clin Pathol. 2008;61(11):1160–1167. | |
Stein H, Foss HD, Dürkop H, et al. CD30 anaplastic large cell lymphoma: a review of its histopathologic, genetic, and clinical features. Blood. 2000;96(12):3681–3695. | |
Morris SW, Xue L, Ma Z, Kinney MC. Alk+ CD30+ lymphomas: a distinct molecular genetic subtype of non-Hodgkin’s lymphoma. Br J Haematol. 2001;113(2):275–295. | |
Falini B, Bigerna B, Fizzotti M, et al. ALK expression defines a distinct group of T/null lymphomas (“ALK lymphomas”) with a wide morphological spectrum. Am J Pathol. 1998;153(3):875–886. | |
Ferreri AJ, Govi S, Pileri SA, Savage KJ. Anaplastic large cell lymphoma, ALK-positive. Crit Rev Oncol Hematol. 2012;83(2):293–302. | |
Savage KJ, Harris NL, Vose JM, et al; International Peripheral T-Cell Lymphoma Project. ALK-anaplastic large-cell lymphoma is clinically and immunophenotypically different from both ALK+ ALCL and peripheral T-cell lymphoma, not otherwise specified: report from the International Peripheral T-Cell Lymphoma Project. Blood. 2008;111(12):5496–5504. | |
Mak V, Hamm J, Chhanabhai M, et al. Survival of patients with peripheral T-cell lymphoma after first relapse or progression: spectrum of disease and rare long-term survivors. J Clin Oncol. 2013;31(16):1970–1976. | |
Shiota M, Fujimoto J, Semba T, Satoh H, Yamamoto T, Mori S. Hyperphosphorylation of a novel 80 kDa protein-tyrosine kinase similar to Ltk in a human Ki-1 lymphoma cell line, AMS3. Oncogene. 1994;9(6):1567–1574. | |
Morris SW, Kirstein MN, Valentine MB, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science. 1994;263(5151):1281–1284. | |
Bai RY, Ouyang T, Miething C, Morris SW, Peschel C, Duyster J. Nucleophosmin-anaplastic lymphoma kinase associated with anaplastic large-cell lymphoma activates the phosphatidylinositol 3-kinase/Akt antiapoptotic signaling pathway. Blood. 2000;96(13):4319–4327. | |
Nieborowska-Skorska M, Slupianek A, Xue L, et al. Role of signal transducer and activator of transcription 5 in nucleophosmin/anaplastic lymphoma kinase-mediated malignant transformation of lymphoid cells. Cancer Res. 2001;61(17):6517–6523. | |
Slupianek A, Nieborowska-Skorska M, Hoser G, et al. Role of phosphatidylinositol 3-kinase-Akt pathway in nucleophosmin/anaplastic lymphoma kinase-mediated lymphomagenesis. Cancer Res. 2001;61(5):2194–2199. | |
Zamo A, Chiarle R, Piva R, et al. Anaplastic lymphoma kinase (ALK) activates Stat3 and protects hematopoietic cells from cell death. Oncogene. 2002;21(7):1038–1047. | |
Zhang Q, Raghunath PN, Xue L, et al. Multilevel dysregulation of STAT3 activation in anaplastic lymphoma kinase-positive T/null-cell lymphoma. J Immunol. 2002;168(1):466–474. | |
Passoni L, Gambacorti-Passerini C. ALK a novel lymphoma-associated tumor antigen for vaccination strategies. Leuk Lymphoma. 2003;44(10):1675–1681. | |
Chiarle R, Voena C, Ambrogio C, Piva R, Inghirami G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer. 2008;8(1):11–23. | |
Hsu FY, Zhao Y, Anderson WF, Johnston PB. Downregulation of NPM-ALK by siRNA causes anaplastic large cell lymphoma cell growth inhibition and augments the anti cancer effects of chemotherapy in vitro. Cancer Invest. 2007;25(4):240–248. | |
Lowe EJ, Lim MS. Potential therapies for anaplastic lymphoma kinase-driven tumors in children: progress to date. Paediatr Drugs. 2013;15(3):163–169. | |
Minoo P, Wang HY. ALK-immunoreactive neoplasms. Int J Clin Exp Pathol. 2015(5):397–410. | |
Foyil KV, Bartlett NL. Brentuximab vedotin and crizotinib in anaplastic large-cell lymphoma. Cancer J. 2012;18(5):450–456. | |
Tartari CJ, Scapozza L, Gambacorti-Passerini C. The ALK gene, an attractive target for inhibitor development. Curr Top Med Chem. 20111(11):1406–1419. | |
Lee JA, Bubendorf L, Stahel R, Peters S. Testing for anaplastic lymphoma kinase rearrangement to target crizotinib therapy: oncology, pathology and health economic perspectives. Expert Rev Anticancer Ther. 2013;13(5):625–636. | |
Gambacorti-Passerini C, Messa C, Pogliani EM. Crizotinib in anaplastic large-cell lymphoma. N Engl J Med. 2011;364(8):775–776. | |
Stasia A, Guerra, Bernard L, et al. Crizotinib obtains durable responses in advanced chemoresistant ALK+ lymphoma patients. Oral session presented at: 17th Congress of the European Hematology Association Haematologica. 2012;97(suppl 1):1153. | |
Gambacorti Passerini C, Farina F, Stasia A, et al. Crizotinib in advanced, chemoresistant anaplastic lymphoma kinase-positive lymphoma patients. J Natl Cancer Inst. 2014;106(2):djt378. | |
Mologni L. Inhibitors of the anaplastic lymphoma kinase. Expert Opin Investig Drugs. 2012;21(7):985–994. | |
Ceccon M, Mologni L, Bisson W, Scapozza L, Gambacorti-Passerini C. Crizotinib-resistant NPM-ALK mutants confer differential sensitivity to unrelated Alk inhibitors. Mol Cancer Res. 2013;11(2):122–132. | |
Hallberg B, Palmer RH. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat Rev Cancer. 2013;13(10):685–700. | |
Heuckmann JM, Hölzel M, Sos ML, et al. ALK mutations conferring differential resistance to structurally diverse ALK inhibitors. Clin Cancer Res. 2011;17(23):7394–7401. | |
Huang D, Kim DW, Kotsakis A, et al. Multiplexed deep sequencing analysis of ALK kinase domain identifies resistance mutations in relapsed patients following crizotinib treatment. Genomics. 2013; 102(3):157–162. | |
Voena C, Chiarle R. The battle against ALK resistance: successes and setbacks. Expert Opin Investig Drugs. 2012;21(12):1751–1754. | |
Dirks WG, Fähnrich S, Lis Y, Becker E, MacLeod RA, Drexler HG. Expression and functional analysis of the anaplastic lymphoma kinase (ALK) gene in tumor cell lines. Int J Cancer. 2002;100(1):49–56. | |
Powers C, Aigner A, Stoica GE, McDonnell K, Wellstein A. Pleiotrophin signaling through anaplastic lymphoma kinase is rate-limiting for glioblastoma growth. J Biol Chem. 2002;277(16):14153–14158. | |
Li R, Morris SW. Development of anaplastic lymphoma kinase (ALK) small-molecule inhibitors for cancer therapy. Med Res Rev. 2008;28(3):372–412. | |
Lu KV, Jong KA, Kim GY, et al. Differential induction of glioblastoma migration and growth by two forms of pleiotrophin. J Biol Chem. 2005;280(29):26953–26964. | |
Lamant L, Pulford K, Bischof D, et al. Expression of the ALK tyrosine kinase gene in neuroblastoma. Am J Pathol. 2000;156(5):1711–1721. | |
Zoubek A, Simonitsch I, Panzer-Grümayer ER, et al. Ewing tumor after treatment of Ki-1+ anaplastic large cell lymphoma. Therapy-associated secondary neoplasm or unrelated coincidence? Cancer Genet Cytogenet. 1995;83(1):5–11. | |
Rassidakis GZ, Lai R, Herling M, Cromwell C, Schmitt-Graeff A, Medeiros LJ. Retinoblastoma protein is frequently absent or phosphorylated in anaplastic large-cell lymphoma. Am J Pathol. 2004;164(6):2259–2267. | |
Delsol G, Lamant L, Mariamé B, et al. A new subtype of large B-cell lymphoma expressing the ALK kinase and lacking the 2; 5 translocation. Blood. 1997;89(5):1483–1490. | |
Czubayko F, Schulte AM, Berchem GJ, Wellstein A. Melanoma angiogenesis and metastasis modulated by ribozyme targeting of the secreted growth factor pleiotrophin. Proc Natl Acad Sci U S A. 1996;93(25):14753–14758. | |
Webb TR, Slavish J, George RE, et al. Anaplastic lymphoma kinase: role in cancer pathogenesis and small-molecule inhibitor development for therapy. Expert Rev Anticancer Ther. 2009;9(3):331–356. | |
Ladanyi M. The NPM/ALK gene fusion in the pathogenesis of anaplastic large cell lymphoma. Cancer Surv. 1997;30:59–75. | |
Armstrong F, Duplantier MM, Trempat P, et al. Differential effects of X-ALK fusion proteins on proliferation, transformation, and invasion properties of NIH3T3 cells. Oncogene. 2004;23(36):6071–6082. | |
Pulford K, Lamant L, Espinos E, et al. The emerging normal and disease-related roles of anaplastic lymphoma kinase. Cell Mol Life Sci. 2004;61(23):2939–2953. | |
Ardini E, Magnaghi P, Orsini P, Galvani A, Menichincheri M. Anaplastic Lymphoma Kinase: role in specific tumours, and development of small molecule inhibitors for cancer therapy. Cancer Lett. 2010;299(2):81–94. | |
Fujimoto J, Shiota M, Iwahara T, et al. Characterization of the transforming activity of p80, a hyperphosphorylated protein in a Ki-1 lymphoma cell line with chromosomal translocation t(2;5). Proc Natl Acad Sci U S A. 1996;93(9):4181–4186. | |
Bischof D, Pulford K, Mason DY, Morris SW. Role of the nucleophosmin (NPM) portion of the non-Hodgkin’s lymphoma-associated NPM-anaplastic lymphoma kinase fusion protein in oncogenesis. Mol Cell Biol. 1997;17(4):2312–2325. | |
Mason DY, Pulford KA, Bischof D, et al. Nucleolar localization of the nucleophosmin-anaplastic lymphoma kinase is not required for malignant transformation. Cancer Res. 1998;58(5):1057–1062. | |
Chiarle R, Gong JZ, Guasparri I, et al. NPM-ALK transgenic mice spontaneously develop T-cell lymphomas and plasma cell tumors. Blood. 2003;101(5):1919–1927. | |
Kuefer MU, Look AT, Pulford K, et al. Retrovirus-mediated gene transfer of NPM-ALK causes lymphoid malignancy in mice. Blood. 1997;90(8):2901–2910. | |
Wan W, Albom MS, Lu L, et al. Anaplastic lymphoma kinase activity is essential for the proliferation and survival of anaplastic large-cell lymphoma cells. Blood. 2006;107(4):1617–1623. | |
Amin HM, Lai R. Pathobiology of ALK+ anaplastic large-cell lymphoma. Blood. 2007;110(7):2259–2267. | |
Morgan EA, Nascimento AF. Anaplastic lymphoma kinase-positive large B-cell lymphoma: an underrecognized aggressive lymphoma. Adv Hematol. 2012;2012:529572. | |
Bai RY, Dieter P, Peschel C, Morris SW, Duyster J. Nucleophosmin-anaplastic lymphoma kinase of large-cell anaplastic lymphoma is a constitutively active tyrosine kinase that utilizes phospholipase C-gamma to mediate its mitogenicity. Mol Cell Biol. 1998;18(12):6951–6961. | |
Marzec M, Kasprzycka M, Ptasznik A, et al. Inhibition of ALK enzymatic activity in T-cell lymphoma cells induces apoptosis and suppresses proliferation and STAT3 phosphorylation independently of Jak3. Lab Invest. 2005;85(12):1544–1554. | |
Galkin AV, Melnick JS, Kim S, et al. Identification of NVP-TAE684, a potent, selective, and efficacious inhibitor of NPM-ALK. Proc Natl Acad Sci U S A. 2007;104(1):270–275. | |
Polgar D, Leisser C, Maier S, et al. Truncated ALK derived from chromosomal translocation t(2;5)(p23;q35) binds to the SH3 domain of p85-PI3K. Mutat Res. 2005;570(1):9–15. | |
Thornber K, Colomba A, Ceccato L, Delsol G, Payrastre B, Gaits-Iacovoni F. Reactive oxygen species and lipoxygenases regulate the oncogenicity of NPM-ALK-positive anaplastic large cell lymphomas. Oncogene. 2009;28(29):2690–2696. | |
Fernandez-Vidal A, Mazars A, Gautier EF, Prévost G, Payrastre B, Manenti S. Upregulation of the CDC25A phosphatase down-stream of the NPM/ALK oncogene participates to anaplastic large cell lymphoma enhanced proliferation. Cell Cycle. 2009;8(9):1373–1379. | |
Coluccia AM, Perego S, Cleris L, et al. Bcl-XL down-regulation suppresses the tumorigenic potential of NPM/ALK in vitro and in vivo. Blood. 2004;103(7):2787–2794. | |
Ergin M, Denning MF, Izban KF, et al. Inhibition of tyrosine kinase activity induces caspase-dependent apoptosis in anaplastic large cell lymphoma with NPM-ALK (p80) fusion protein. Exp Hematol. 2001;29(9):1082–1090. | |
Reed JC, Jurgensmeier JM, Matsuyama S. Bcl-2 family proteins and mitochondria. Biochim Biophys Acta. 1998;1366(1–2):127–137. | |
Lange K, Uckert W, Blankenstein T, et al. Overexpression of NPM-ALK induces different types of malignant lymphomas in IL-9 transgenic mice. Oncogene. 2003;22(4):517–527. | |
Van Roosbroeck K, Cools J, Dierickx D, et al. ALK-positive large B-cell lymphomas with cryptic SEC31A-ALK and NPM1-ALK fusions. Haematologica. 2010;95(3):509–513. | |
Murugan AK, Xing M. Anaplastic thyroid cancers harbor novel oncogenic mutations of the ALK gene. Cancer Res. 2011;71(13):4403–4411. | |
Chen Y, Takita J, Choi YL, et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature. 2008;455(7215):971–974. | |
Passoni L, Longo L, Collini P, et al. Mutation-independent anaplastic lymphoma kinase overexpression in poor prognosis neuroblastoma patients. Cancer Res. 2009;69(18):7338–7346. | |
George RE, Sanda T, Hanna M, et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature. 2008;455(7215):975–978. | |
Janoueix-Lerosey I, Lequin D, Brugières L, et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature. 2008;455(7215):967–970. | |
Mossé YP, Laudenslager M, Longo L, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008;455(7215):930–935. | |
Salido M, Pijuan L, Martínez-Avilés L, et al. Increased ALK gene copy number and amplification are frequent in non-small cell lung cancer.J Thorac Oncol. 2011;6(1):21–27. | |
Carén H, Abel F, Kogner P, Martinsson T. High incidence of DNA mutations and gene amplifications of the ALK gene in advanced sporadic neuroblastoma tumours. Biochem J. 2008;416(2):153–159. | |
van Gaal JC, Flucke UE, Roeffen MH, et al. Anaplastic lymphoma kinase aberrations in rhabdomyosarcoma: clinical and prognostic implications. J Clin Oncol. 2012;30(3):308–315. | |
Kim H, Yoo SB, Choe JY, et al. Detection of ALK gene rearrangement in non-small cell lung cancer: a comparison of fluorescence in situ hybridization and chromogenic in situ hybridization with correlation of ALK protein expression. J Thorac Oncol. 2011;6(8):1359–1366. | |
Selinger CI, Rogers TM, Russell PA, et al. Testing for ALK rearrangement in lung adenocarcinoma: a multicenter comparison of immunohistochemistry and fluorescent in situ hybridization. Mod Pathol. 2013;26(12):1545–1553. | |
Wallander ML, Geiersbach KB, Tripp SR, Layfield LJ. Comparison of reverse transcription-polymerase chain reaction, immunohistochemistry, and fluorescence in situ hybridization methodologies for detection of echinoderm microtubule-associated proteinlike 4-anaplastic lymphoma kinase fusion-positive non-small cell lung carcinoma: implications for optimal clinical testing. Arch Pathol Lab Med. 2012;136(7):796–803. | |
Shan L, Lian F, Guo L, Yang X, Ying J, Lin D. Combination of conventional immunohistochemistry and qRT-PCR to detect ALK rearrangement. Diagn Pathol. 2014;9:3. | |
Wu YC, Chang IC, Wang CL, et al. Comparison of IHC, FISH and RT-PCR methods for detection of ALK rearrangements in 312 non-small cell lung cancer patients in Taiwan. PLoS One. 2013;8(8):e70839. | |
Piva R, Chiarle R, Manazza AD, et al. Ablation of oncogenic ALK is a viable therapeutic approach for anaplastic large-cell lymphomas. Blood. 2006;107(2):689–697. | |
Coluccia AM, Gunby RH, Tartari CJ, Scapozza L, Gambacorti-Passerini C, Passoni L. Anaplastic lymphoma kinase and its signalling molecules as novel targets in lymphoma therapy. Expert Opin Ther Targets. 2005;9(3):515–532. | |
Christensen JG, Zou HY, Arango ME, et al. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol Cancer Ther. 2007;6(12 Pt 1):3314–3322. | |
Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561–566. | |
Gunby RH, Tartari CJ, Porchia F, Donella-Deana A, Scapozza L, Gambacorti-Passerini C. An enzyme-linked immunosorbent assay to screen for inhibitors of the oncogenic anaplastic lymphoma kinase. Haematologica. 2005;90(7):988–990. | |
Turturro F, Arnold MD, Frist AY, Pulford K. Model of inhibition of the NPM-ALK kinase activity by herbimycin A. Clin Cancer Res. 2002;8(1):240–245. | |
Georgakis GV, Li Y, Rassidakis GZ, Medeiros LJ, Younes A. The HSP90 inhibitor 17-AAG synergizes with doxorubicin and U0126 in anaplastic large cell lymphoma irrespective of ALK expression. Exp Hematol. 2006;34(12):1670–1679. | |
Sang J, Acquaviva J, Friedland JC, et al. Targeted inhibition of the molecular chaperone Hsp90 overcomes ALK inhibitor resistance in non-small cell lung cancer. Cancer Discov. 2013;3(4):430–443. | |
Cerchietti L, Damm-Welk C, Vater I, et al. Inhibition of anaplastic lymphoma kinase (ALK) activity provides a therapeutic approach for CLTC-ALK-positive human diffuse large B cell lymphomas. PLoS One. 2011;6(4):e18436. | |
Zou HY, Li Q, Lee JH, et al. An orally available small-molecule inhibitor of c-Met, PF-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Cancer Res. 2007;67(9):4408–4417. | |
Kwak EL, Camidge DR, Clark J, et al. Clinical activity observed in a phase I dose escalation trial of an oral c-MET and ALK inhibitor, PF-02341066. Presented at: 2009 ASCO annual meeting. J Clin Oncol. 2009;15(Suppl):3509. | |
Camidge DR, Bang YJ, Kwak EL, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol. 2012;13(10):1011–1019. | |
Gambacorti-Passerini C, Horibe K, Braiteh F, et al. Safety and clinical activity of crizotinib in patients with ALK-rearranged hematologic malignancies. Poster presented at: 55th ASH Annual Meeting and Exposition; December 9, 2013; New Orleans, LA. Blood. 2013,122(21)4342. | |
Mossé YP, Lim MS, Voss SD, et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: a Children’s Oncology Group phase 1 consortium study. Lancet Oncol. 2013;14(6):472–480. | |
Doebele RC, Pilling AB, Aisner DL, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res. 2012;18(5):1472–1482. | |
Katayama R, Shaw AT, Khan TM, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci Transl Med. 2012;4(120):120ra17. | |
Mossé YP, Wood A, Maris JM. Inhibition of ALK signaling for cancer therapy. Clin Cancer Res. 2009;15(18):5609–5614. | |
Choi YL, Soda M, Yamashita Y, et al; ALK Lung Cancer Study Group. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med. 2010;363(18):1734–1739. | |
Hallberg B, Palmer RH. Crizotinib – latest champion in the cancer wars? N Engl J Med. 2010;363(18):1760–1762. | |
Camidge DR, Doebele RC. Treating ALK-positive lung cancer – early successes and future challenges. Nat Rev Clin Oncol. 2012;9(5):268–277. | |
Awad MM, Katayama R, McTigue M, et al. Acquired resistance to crizotinib from a mutation in CD74-ROS1. N Engl J Med. 2013;368(25):2395–2401. | |
Schwab R, Petak I, Kollar M, et al. Major partial response to crizotinib, a dual MET/ALK inhibitor, in a squamous cell lung (SCC) carcinoma patient with de novo c-MET amplification in the absence of ALK rearrangement. Lung Cancer. 2014;83(1):109–111. | |
Yamaguchi N, Lucena-Araujo AR, Nakayama S, et al. Dual ALK and EGFR inhibition targets a mechanism of acquired resistance to the tyrosine kinase inhibitor crizotinib in ALK rearranged lung cancer. Lung Cancer. 2014;83(1):37–43. | |
Chiarle R, Martinengo C, Mastini C, et al. The anaplastic lymphoma kinase is an effective oncoantigen for lymphoma vaccination. Nat Med. 2008;14(6):676–680. | |
Mastini C, Martinengo C, Inghirami G, Chiarle R. Anaplastic lymphoma kinase: an oncogene for tumor vaccination. J Mol Med (Berl). 2009;87(7):669–677. | |
Kruczynski A, Delsol G, Laurent C, Brousset P, Lamant L. Anaplastic lymphoma kinase as a therapeutic target. Expert Opin Ther Targets. 2012;16(11):1127–1138. | |
Pulford K, Falini B, Banham AH, et al. Immune response to the ALK oncogenic tyrosine kinase in patients with anaplastic large-cell lymphoma. Blood. 2000;96(4):1605–1607. | |
Carpenter EL, Mossé YP. Targeting ALK in neuroblastoma – preclinical and clinical advancements. Nat Rev Clin Oncol. 2012;9(7):391–399. | |
Li N, MP-Y, Sungjon K, Culazzo Pferdekamper A, et al. Activity of a potent and selective phase I ALK inhibitor LDK378 in naïve and crizotinib-resistant preclinical tumor models. Presented at: AACR-NCI-EORTC International Conference:San Francisco, CA. Molecular Targets and Cancer Therapeutics. 2001;10(suppl 11):B232. | |
Mehra R. First-in-human phase I study of the ALK inhibitor LDK378 in advanced solid tumors. Presented at: 2012 ASCO Annual Meeting; Chicago, IL. JCO. 2012;30(suppl):3007. | |
Seto T, Kiura K, Nishio M, et al. CH5424802 (RO5424802) for patients with ALK-rearranged advanced non-small-cell lung cancer (AF-001JP study):a single-arm, open-label, phase 1–2 study. Lancet Oncol. 2013;14(7):590–598. | |
Gettinger SWG, Salgia R, et al. A first-in-human dose-finding study of the ALK/EGFR inhibitor AP26113 in patients with advanced malignancies. Poster presented at: 37th Annual ESMO Conference; 2012; Vienna. | |
Gambacorti-Passerini C. Part I: Milestones in personalised medicine – imatinib. Lancet Oncol. 2008;9(6):600. | |
Pulford K, Lamant L, Morris SW, et al. Detection of anaplastic lymphoma kinase (ALK) and nucleolar protein nucleophosmin (NPM)-ALK proteins in normal and neoplastic cells with the monoclonal antibody ALK1. Blood. 1997;89(4):1394–1404. | |
Jäger R, Hahne J, Jacob A, et al. Mice transgenic for NPM-ALK develop non-Hodgkin lymphomas. Anticancer Res. 2005;25(5):3191–3196. | |
Kadin ME, Morris SW. The t(2;5) in human lymphomas. Leuk Lymphoma. 1998;29(3–4):249–256. | |
Ma Z, Cools J, Marynen P, et al. Inv(2)(p23q35) in anaplastic large-cell lymphoma induces constitutive anaplastic lymphoma kinase (ALK) tyrosine kinase activation by fusion to ATIC, an enzyme involved in purine nucleotide biosynthesis. Blood. 2000;95(6):2144–2149. | |
Trinei M, Lanfrancone L, Campo E, et al. A new variant anaplastic lymphoma kinase (ALK)-fusion protein (ATIC-ALK) in a case of ALK-positive anaplastic large cell lymphoma. Cancer Res. 2000;60(4):793–798. | |
Cools J, Wlodarska I, Somers R, et al. Identification of novel fusion partners of ALK, the anaplastic lymphoma kinase, in anaplastic large-cell lymphoma and inflammatory myofibroblastic tumor. Genes Chromosomes Cancer. 2002;34(4):354–362. | |
Lamant L, Gascoyne RD, Duplantier MM, et al. Non-muscle myosin heavy chain (MYH9):a new partner fused to ALK in anaplastic large cell lymphoma. Genes Chromosomes Cancer. 2003;37(4):427–432. | |
Debelenko LV, Arthur DC, Pack SD, Helman LJ, Schrump DS, Tsokos M. Identification of CARS-ALK fusion in primary and metastatic lesions of an inflammatory myofibroblastic tumor. Lab Invest. 2003;83(9):1255–1265. | |
Touriol C, Greenland C, Lamant L, et al. Further demonstration of the diversity of chromosomal changes involving 2p23 in ALK-positive lymphoma: 2 cases expressing ALK kinase fused to CLTCL (clathrin chain polypeptide-like). Blood. 2000;95(10):3204–3207. | |
Gascoyne RD, Lamant L, Martin-Subero JI, et al. ALK-positive diffuse large B-cell lymphoma is associated with Clathrin-ALK rearrangements: report of 6 cases. Blood. 2003;102(7):2568–2573. | |
Tort F, Campo E, Pohlman B, Hsi E. Heterogeneity of genomic breakpoints in MSN-ALK translocations in anaplastic large cell lymphoma. Hum Pathol. 2004;35(8):1038–1041. | |
Tort F, Pinyol M, Pulford K, et al. Molecular characterization of a new ALK translocation involving moesin (MSN-ALK) in anaplastic large cell lymphoma. Lab Invest. 2001;81(3):419–426. | |
Hernández L, Pinyol M, Hernández S, et al. TRK-fused gene (TFG) is a new partner of ALK in anaplastic large cell lymphoma producing two structurally different TFG-ALK translocations. Blood. 1999;94(9):3265–3268. | |
Rosenwald A, Ott G, Pulford K, et al. t(1;2)(q21;p23) and t(2;3)(p23;q21):two novel variant translocations of the t(2;5)(p23;q35) in anaplastic large cell lymphoma. Blood. 1999;94(1):362–364. | |
Lamant L, Dastugue N, Pulford K, Delsol G, Mariamé B. A new fusion gene TPM3-ALK in anaplastic large cell lymphoma created by a (1;2)(q25;p23) translocation. Blood. 1999;93(9):3088–3095. | |
Meech SJ, McGavran L, Odom LF, et al. Unusual childhood extramedullary hematologic malignancy with natural killer cell properties that contains tropomyosin 4 – anaplastic lymphoma kinase gene fusion. Blood. 2001;98(4):1209–1216. | |
Ma Z, Hill DA, Collins MH, et al. Fusion of ALK to the Ran-binding protein 2 (RANBP2) gene in inflammatory myofibroblastic tumor. Genes Chromosomes Cancer. 2003;37(1):98–105. | |
Panagopoulos I, Nilsson T, Domanski HA, et al. Fusion of the SEC31L1 and ALK genes in an inflammatory myofibroblastic tumor. Int J Cancer. 2006;118(5):1181–1186. | |
Soda M, Takada S, Takeuchi K, et al. A mouse model for EML4-ALK-positive lung cancer. Proc Natl Acad Sci U S A. 2008;105(50):19893–19897. | |
Martelli MP, Sozzi G, Hernandez L, et al. EML4-ALK rearrangement in non-small cell lung cancer and non-tumor lung tissues. Am J Pathol. 2009;174(2):661–670. | |
Takeuchi K, Choi YL, Togashi Y, et al. KIF5B-ALK, a novel fusion oncokinase identified by an immunohistochemistry-based diagnostic system for ALK-positive lung cancer. Clin Cancer Res. 2009;15(9):3143–3149. | |
Takeuchi K, Soda M, Togashi Y, et al. Identification of a novel fusion, SQSTM1-ALK, in ALK-positive large B-cell lymphoma. Haematologica. 2011;96(3):464–467. | |
Camidge DR. First-in-human dose-finding study of the ALK/EGFR inhibitor AP26113 in patients with advanced malignancies: Updated results. Presented at: 2013 ASCO Annual Meeting; Chicago, IL. JCO. 2013;31(suppl):2524. | |
Patnaik A. Pharmacokinetics and safety of an oral ALK inhibitor, ASP3026, observed in a phase I dose escalation trial. Presented at: 2013 ASCO Annual Meeting; Chicago, IL. JCO. 2013;31(suppl):2602. | |
Shaw AT. Clinical activity of the ALK inhibitor LDK378 in advanced, ALK-positive NSCLC. Presented at: 2013 ASCO Annual Meeting; Chicago, IL. JCO. 2013;31(suppl):8010. | |
Marsilje TH, Pei W, Chen B, et al. Synthesis, structure-activity relationships, and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (ALK) inhibitor 5-chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulfonyl)phenyl)pyrimidine-2,4-diamine (LDK378) currently in phase 1 and phase 2 clinical trials. J Med Chem. 2013;56(14):5675–5690. | |
Cheng M, Quail MR, Gingrich DE, et al. CEP-28122, a highly potent and selective orally active inhibitor of anaplastic lymphoma kinase with antitumor activity in experimental models of human cancers. Mol Cancer Ther. 2012;11(3):670–679. | |
Bossi RT, Saccardo MB, Ardini E, et al. Crystal structures of anaplastic lymphoma kinase in complex with ATP competitive inhibitors. Biochemistry. 2010;49(32):6813–6825. | |
Grande E, Bolós MV, Arriola E. Targeting oncogenic ALK: a promising strategy for cancer treatment. Mol Cancer Ther. 2011;10(4):569–579. | |
McDermott U, Iafrate AJ, Gray NS, et al. Genomic alterations of anaplastic lymphoma kinase may sensitize tumors to anaplastic lymphoma kinase inhibitors. Cancer Res. 2008;68(9):3389–3395. | |
Sakamoto H, Tsukaguchi T, Hiroshima S, et al. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell. 2011;19(5):679–690. | |
Latif M, Saeed A, Kim SH. Journey of the ALK-inhibitor CH5424802 to phase II clinical trial. Arch Pharm Res. 2013;36(9):1051–1054. | |
Nakagawa K. A phase I/II study with a highly selective ALK inhibitor CH5424802 in ALK-positive non-small cell lung cancer (NSCLC) patients: Updated safety and efficacy results from AF-001JP. Presented at: 2013 ASCO Annual Meeting; Chicago, IL. JCO. 2013;31(suppl):8033. | |
Lovly CM, Heuckmann JM, de Stanchina E, et al. Insights into ALK-driven cancers revealed through development of novel ALK tyrosine kinase inhibitors. Cancer Res. 2011;71(14):4920–4931. | |
Sabbatini P, Korenchuk S, Rowand JL, et al. GSK1838705A inhibits the insulin-like growth factor-1 receptor and anaplastic lymphoma kinase and shows antitumor activity in experimental models of human cancers. Mol Cancer Ther. 2009;8(10):2811–2820. | |
Gingrich DE, Lisko JG, Curry MA, et al. Discovery of an orally efficacious inhibitor of anaplastic lymphoma kinase. J Med Chem. 2012 24;55(10):4580–4593. | |
Lewis RT, Bode CM, Choquette DM, et al. The discovery and optimization of a novel class of potent, selective, and orally bioavailable anaplastic lymphoma kinase (ALK) inhibitors with potential utility for the treatment of cancer. J Med Chem. 2012;55(14):6523–6540. | |
Morales La Madrid A, Campbell N, Smith S, Cohn SL, Salgia R. Targeting ALK: a promising strategy for the treatment of non-small cell lung cancer, non-Hodgkin’s lymphoma, and neuroblastoma. Target Oncol. 2012;7(3):199–210. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.