Back to Journals » Neuropsychiatric Disease and Treatment » Volume 14
Developmental lead (Pb)-induced deficits in hippocampal protein translation at the synapses are ameliorated by ascorbate supplementation
Authors Ahmad F, Salahuddin M ![]() , Alsamman K, AlMulla AA
, Alsamman K, AlMulla AA ![]() , Salama KF
, Salama KF ![]()
Received 14 May 2018
Accepted for publication 12 October 2018
Published 29 November 2018 Volume 2018:14 Pages 3289—3298
DOI https://doi.org/10.2147/NDT.S174083
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Roger Pinder
Faraz Ahmad,1 Mohammad Salahuddin,2 Khaldoon Alsamman,3 Abdulaziz A AlMulla,4 Khaled F Salama4
1School of Life Science, BS Abdur Rahman Crescent Institute of Science & Technology, Vandulur, Chennai 600048, India; 2Animal House Department, Institute for Research and Medical Consultations, Imam Abdurrahman Bin Faisal University, Dammam 31441, Saudi Arabia; 3Department of Clinical Laboratory Sciences, College of Applied Medical Sciences, Imam Abdurrahman Bin Faisal University, Dammam 31441, Saudi Arabia; 4Department of Environmental Health, College of Public Health, Imam Abdurrahman Bin Faisal University, Dammam 31441, Saudi Arabia
Background: Lead (Pb) is a persistent environmental neurotoxin and its exposure even in minute quantities has been known to induce neuronal defects. The immature brain is singularly sensitive to Pb neurotoxicity, and its exposure during development has permanent detrimental effects on the brain developmental trajectory and neuronal signaling and plasticity, culminating into compromises in the cognitive and behavioral attributes which persists even later in adulthood. Several molecular pathways have been implicated in the Pb-mediated disruption of neuronal signaling, including elevated oxidative stress, alterations in neurotransmitter biology, and mitochondrial dysfunction. Nevertheless, the neuronal targets and biochemical pathways underlying these Pb-mediated alterations in synaptic development and function have not been completely deduced. In this respect, recent studies have shown that synaptic signaling and its maintenance and plasticity are critically dependent on localized de novo protein translation at the synaptic terminals.
Materials and methods: The present study hence aimed to assess the alterations in the synapse-specific translation induced by developmental Pb exposure. To this end, in vitro protein translation rate was analyzed in the hippocampal synaptoneurosomal fractions of rat pups pre- and postnatally exposed to Pb using a puromycin incorporation assay. Moreover, we evaluated the therapeutic effects of ascorbic acid supplementation against Pb-induced deficits in synapse-localized protein translation.
Results: We observed a significant loss in the rates of de novo protein translation in synaptoneurosomes of Pb-exposed pups compared to age-matched control pups. Interestingly, ascorbate supplementation lead to an appreciable recovery in Pb-induced translational deficits. Moreover, the deficit in activity-dependent synaptic protein translation was found to correlate significantly with the increase in the blood Pb levels.
Conclusion: Dysregulation of synapse-localized de novo protein translation is a potentially critical determinant of Pb-induced synaptic dysfunction and the consequent deficits in behavioral, social, and psychological attributes of the organisms. In addition, our study establishes ascorbate supplementation as a key ameliorative agent against Pb-induced neurotoxicity.
Keywords: synaptoneurosomes, heavy metal neurotoxicity, neuropsychiatric, blood lead level, puromycin
Introduction
Widespread industrial use of lead (Pb), a naturally occurring toxic heavy metal, has resulted in an elevated risk of its exposure in both animals and humans, making it a prominent environmental and occupational health hazard.1,2 In spite of measures to limit its use and contain its exposure, Pb remains a high-risk environmental toxin and a major threat to public health, particularly in developing countries.1 Several factors are responsible for this. First, Pb exposure can occur through contaminated air, water as well as food. Second, exposure of almost all forms of Pb (metallic, organic, and inorganic) is toxic.2
The nervous system constitutes as a prominent target of Pb toxicity which is evident by behavioral abnormalities, neuromuscular disabilities, and cognitive deficits in events of Pb exposure.2–4 Of note, exposure to even low levels of Pb, previously thought to be permissible, can be neurotoxic and have deleterious and irreversible cognitive and psychological outcomes.5–8 Brain functions including higher order sensory motor, cognitive and behavioral functions rely principally on the inter-neuronal communication at the synapses.9,10 Deleterious effects of Pb exposure on synapse function and their maintenance and plasticity have been confirmed by many studies7,11,12 and seem to involve alterations in several intricate interacting physiological processes including redox homeostasis, mitochondrial functions and dynamics, and signaling of calcium and other secondary messenger molecules, transcription and gene expression, membrane biophysics, neurotrophic signaling, neurotransmitter synthesis, and release and biology of their receptors.7,13
The developing nervous system is particularly vulnerable to early life lead exposure.14 Pb-induced changes in the synaptic functions during brain development have dire consequences on the function and plasticity of the brain,7,14,15 culminating into permanent alterations in higher order brain functions, including sensory motor, cognitive and social attributes as well as response to psychological stressors.5,16,17 Hence, a thorough assessment of the molecular and cellular players involved in synaptic dysfunction induced by developmental Pb exposure in the developing nervous system is warranted. Among several mechanisms implicated in maintenance and plasticity of proper synaptic signaling, the role of localized de novo protein translation is only beginning to be appreciated.18–21 In addition to the tightly controlled target mRNA transport to the synapses, presence of a functional translational machinery at the synaptic terminals empowers individual synapses to regulate the strengths of their signaling independently. Of note, changes in the synaptic proteome can be robustly induced by neuronal activity in part by alterations in the protein translation profiles.20,22 Consequently, localized protein translation at the synapses plays an important role in the proper re-configuration of neuronal circuitry in events of neuronal depolarization.18,20–23 In view of the critical roles of localized protein translation in the regulation of their function, maintenance and plasticity, alterations in synaptic protein translation are increasingly being perceived as major contributors to neuropathological outcomes in a wide variety of neurological disorders.20,24–28
In this study, we analyze the effects of early life Pb exposure on synapse-localized de novo protein translation in the rat hippocampus. The hippocampus is a limbic structure that is closely involved in learning and memory function. As such, neuronal circuitry of the hippocampi is particularly susceptible to activity-dependent alterations in synaptic strength. In fact, the best known mechanisms of activity-dependent neuronal plasticity, long-term potentiation, and long-term depression have been extensively characterized in the hippocampus and are implicated as potential mechanisms underlying the higher order brain functions, such as learning and memory, the formation and modification of cognitive maps, and the ability of organisms to react to stressful experiences.29 Of note, several recent studies have implicated dysfunction at the hippocampal circuitry as among the most critical mechanisms that govern brain deficits induced by Pb neurotoxicity.7,13,30–34
The present study also assesses the therapeutic effects of supplementation of ascorbic acid or vitamin C in preventing the Pb-induced alterations in the hippocampal synaptic protein translation. Ascorbic acid is a water soluble vitamin and has been shown to have tremendous neuromodulatory and neuroprotective properties in several neuropathologies, including ethanol-induced neuroinflammation, cerebral ischemia, oxidative damage, excitotoxicity, kainate-induced seizures as well as neuropsychiatric diseases.35–39 Neuroprotective effects of ascorbate supplementation in heavy metal toxicity are also well documented and stem from a multifaceted array of therapeutic mechanisms including metal chelation, antioxidant and anti-apoptotic actions, and modulation of neurotransmitter signaling.37,40,41 In particular, therapeutic effects of ascorbate in the events of Pb exposure and toxicity have been well-documented by a number of groups.42–47 Of note, our recent study has shown appreciable recovery of early life Pb-induced synapse-specific mitochondrial bioenergetic defects by ascorbate supplementation.48
Materials and methods
Chemicals, reagents, and antibodies
Puromycin dihydrochloride (CAS no. 58-58-2) and chloramphenicol (CAS no. 56-75-7) were procured from Millipore Merck (Billerica, MA, USA). Protease and phosphatase inhibitor cocktails were from Thermo Fisher Scientific (product no. 1861748; Waltham, MA, USA) and Sigma-Aldrich Co. (catalog no. P5726; St Louis, MO, USA). 100 μm and 10 μm nylon membrane filters were obtained from Merck Millipore (catalog nos. NY1H02500 and NY1002500, respectively). Antibodies against PSD-95 and α-tubulin were from Thermo Fisher Scientific (catalog nos. MA1046 and 322500, respectively). The antibody against puromycin was procured from Merck Millipore (catalog no. MABE341). The horse-radish peroxidase-linked secondary antibodies against rabbit (catalog no. 31460) and mouse immunoglobulin G (catalog no. 31430) were from Thermo Fisher Scientific. Clarity™ Western ECL substrate was obtained from Bio-Rad Laboratories Inc. (catalog no. 170-5061; Hercules, CA, USA). All other chemicals purchased were of analytical grade and from either Merck Millipore or Sigma-Aldrich Co.
Animals and experimental paradigms
All experiments involving animals were carried out in accordance with the institutional guidelines for animal care and use for scientific research and after approval by the institutional review board, Imam Abdulrahman Bin Faisal University, Dammam. The experimental paradigm of pre- and postnatal lead exposure and ascorbate supplementation employed has been previously reported by us.48 In brief, female Wistar rats were housed in cages with sexually mature males (2:1; male to female ratio) under a light/day 12/12 hours regime in rooms with a controlled temperature of 25°C. Food chow and drinking water were provided ad libitum. At gestation day 15 (GD15), the pregnant females were randomly divided into four groups: Ctrl (control), Pb (lead), Pb+Asc (lead and ascorbic acid) and Asc (ascorbic acid). Dams of the Ctrl and Asc groups were provided with normal drinking water and Pb and Pb+Asc groups received 0.2% lead acetate (2,000 ppm) in drinking water from GD15 until the day of weaning of pups at postnatal day 21 (P21). Mothers of Pb+Asc and Asc groups were provided with 500 mg/kg body weight of ascorbic acid using an oral gavage from GD15 until weaning of pups (P21). Treatment with ascorbic acid and/or lead acetate was stopped immediately after weaning of the pups. To abrogate the effects of gender-specific responses and outcomes to early life Pb exposure particularly in the hippocampus,49–52 only male pups were randomly selected from each group for the study and sacrificed at P30. This model of oral Pb administration in drinking water has been widely employed as it is thought to mimic the environmental exposure to this heavy metal.34,53–55
Measurement of blood Pb levels
Digestion of blood obtained immediately before sacrificing the rat pups was performed as described by Chaurasia et al with slight modifications.56 Briefly, blood (0.5 mL each sample) was digested in 5 mL of a nitric acid-perchloric acid solution (ratio of concentrated HNO3:HClO4 was 1:6) at 110°C for 1 hour. After filtering through a Whatman filter paper no. 41, the digested blood was analyzed for Pb levels by using an iCAP 6300 Duo inductively coupled plasma optical emission spectrometer (ICP-OES; Thermo Fisher Scientific).
Isolation of synaptoneurosomes
A protocol involving sequential filtration steps was employed for biochemical isolation of synaptoneurosomes. This method was chosen because it has been shown to be suitable for studying in vitro protein translation by us and others.24,27,57–59 Briefly, the hippocampi isolated from the pups were homogenized using a Potter-Elvehjem tissue grinder (Kimble, Rockwood, TN, USA) in 10× volume of ice-cold translation buffer (118 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 2.5 mM CaCl2, 1.53 mM KH2PO4, 212.7 mM glucose, 1 mM 1,4-dithiothreitol (DTT), pH 7.4) supplemented with protease inhibitor cocktail, phosphatase inhibitor cocktail, 30 U/mL RNAse inhibitor, and 200 μg·mL−1 chloramphenicol. The homogenate (denoted as Hgt) obtained was then sequentially passed through 100 μm (twice) and 10 μm nylon membrane filters. A small part of the filtrate obtained after passing through two 100 μm filters (designated filtrate 1 or F1) was saved to assess enrichment of post-synaptic density 95 (PSD-95), a synaptic marker protein58,59 for quality-control analysis of synaptoneurosomes. The final filtrate obtained after passing through the 10 μm filter was centrifuged at 1,500× g at 4°C for 10 minutes to obtain the supernatant (denoted Sup) and the synaptoneurosomal pellet (denoted as SN). The SN pellet was resuspended in translation buffer and processed immediately for in vitro protein translation assay. A part of the resuspended SN pellet was solubilized in SDS-PAGE sample buffer (62.5 mM Tris-HCl, pH 6.8, 2% SDS, 5% 2-mercaptoethanol, 20% glycerol, and 0.0006% bromophenol blue) and stored at −20°C for immunoblotting to assess PSD-95 levels.
Stimulation of synaptoneurosomes and puromycin incorporation assay
Subcellular preparations of synaptoneurosomes obtained using the sequential filtration protocol have been shown to be a robust in vitro system for assaying local synthesis of synaptic proteins.27,57–59 Because this protocol allows preservation of both the soluble factors and the energy sources (synaptic mitochondria) in their native form in the synaptoneurosomal fractions, their addition prior to in vitro protein translation assays is not required.60 We employed a puromycin-based nonradioactive surface sensing of translation, the SUnSET method,61 for the assessment of de novo protein synthesis in the synaptoneurosomal samples. The SUnSET protocol has been previously employed by us and others24,62,63 and was chosen because of its obvious advantage over the conventional radioactivity-based 35S-methionine incorporation assay. In brief, synaptoneurosomes were diluted in translation buffer to a concentration of 1 mg·mL−1 protein and preincubated at 37°C for 5 minutes. Stimulation of synaptoneurosomes was performed in the presence of 50 mM KCl and 10 μg·mL−1 puromycin at 37°C for 15 minutes.24,27,57,64 Unstimulated samples were incubated with 10 μg·mL−1 puromycin alone. Chloramphenicol in the translation buffer ensured assay of protein translation in a synapse-specific manner by inhibiting any protein translation in the synaptic mitochondria present in the synaptoneurosomal fraction.24,57,60 Following puromycin incorporation into nascent peptide chains for 15 minutes, synaptoneurosomal samples were pelleted at high speed (14,300 rpm), resuspended in SDS-PAGE sample buffer, and stored at −20°C for immunoblotting.
Immunobloting
Synaptoneurosomal and other neuronal subcellular fractions were separated using 4%–15% gradient SDS-PAGE, electroblotted onto a polyvinylidene difluoride membrane, and immunostained using appropriate primary and secondary antibodies. Immunoreactive chemiluminescent signals were detected on a ChemiDoc™ MP Imaging System (Bio-Rad Laboratories Inc.) and quantified using Image Lab software (version 5.2; Bio-Rad Laboratories Inc.).
Statistical analysis
Results are represented as mean ± standard error of the mean (SEM), unless stated otherwise, and expressed as a multiple of the control unstimulated (Ctrl US) sample for each immunoblot. The multiple groups were compared using one-way ANOVA followed by post hoc tests with Newman–Keuls correction. The correlation between the ratio of stimulated to basal protein translation and blood Pb levels was calculated using Pearson’s correlation analysis. All analyses were performed using GraphPad Prism 5 software (GraphPad Software, Inc., La Jolla, CA, USA). Data were considered significant if P<0.05.
Results
Blood Pb levels are elevated upon pre- and postnatal delivery of lead acetate to rat pups
Although there were no significant differences in the volume of water consumed between the groups, the blood Pb levels in pups of the Pb group as assessed by ICP-OES were found to be around 50-folds in excess of those in the Ctrl pups. In addition, ascorbate supplementation significantly reduced the levels of blood Pb (Figure 1), as has been previously observed in both rodent and human subjects.47,65–71 However, it still remained higher than Ctrl values.
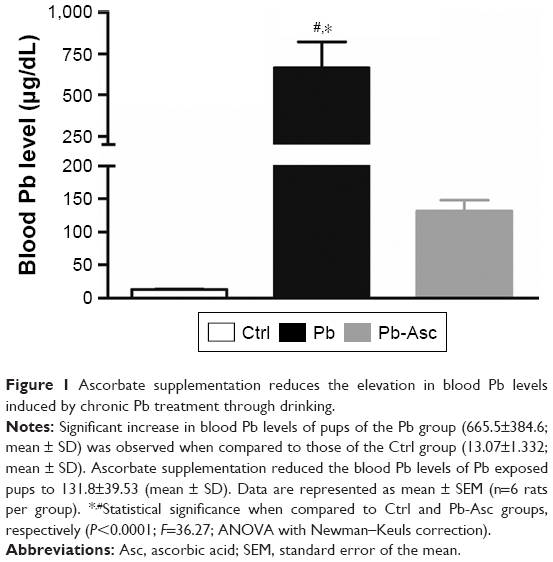 | Figure 1 Ascorbate supplementation reduces the elevation in blood Pb levels induced by chronic Pb treatment through drinking. |
Hippocampal synaptoneurosomes are enriched in PSD-95
The protein levels of PSD-95 were measured using immunoblotting for various cellular fractions to assess the level of enrichment of the synaptoneurosomal preparations obtained by the filtration method. Protein expression of PSD-95 in the synaptoneurosomes was found to be more than fourfolds greater than the starting homogenate material, indicating a robust enrichment of synaptic components (Figure 2).
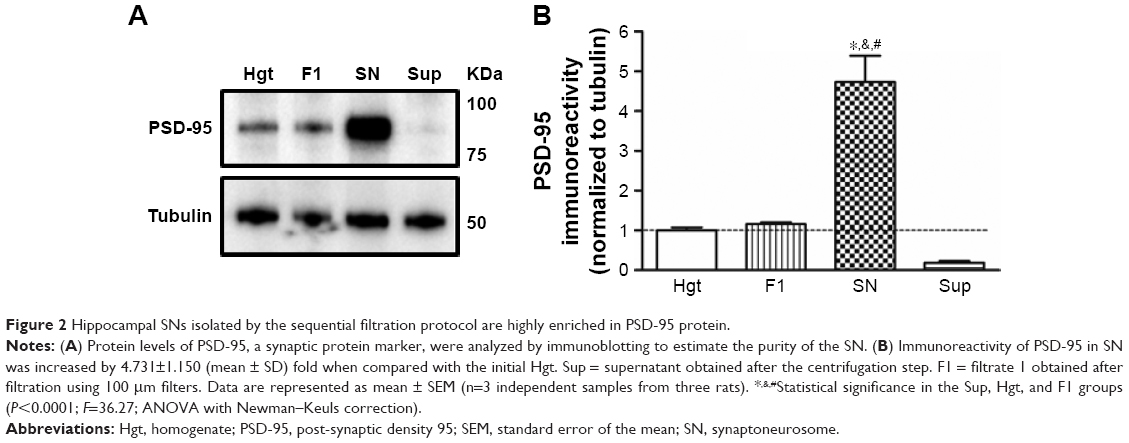 | Figure 2 Hippocampal SNs isolated by the sequential filtration protocol are highly enriched in PSD-95 protein. |
Ascorbic acid supplementation rescues the Pb-induced alterations in localized protein translation in the synaptoneurosomes of rat pups
Appreciable increase in the rate of de novo protein synthesis, as assessed by puromycin incorporation, was observed when synaptoneurosomes isolated from Ctrl animals were stimulated with KCl (Figure 3), as previously reported.27,57,64 However, high K+-mediated depolarization did not stimulate a similar increase in protein translation in synaptoneurosomes of Pb-exposed pups. On the other hand, Pb induced a small decrease in the basal protein translation rate; the decrease however did not reach statistical significance when compared to Ctrl basal levels. Pb-induced decrease in both basal and K+-stimulated protein translation was recovered in Pb-Asc animals (Figure 3), indicating the ameliorative effects of ascorbate supplementation in Pb-induced protein translational deficits. However, ascorbate supplementation alone did not result in any changes in basal or activity driven synaptic translation when compared to control animals (Figure S1). Interestingly, the detrimental effects of Pb toxicity on protein translation were specific to synaptosomal preparations as no alterations were observed in global protein translation rates in the homogenate samples (Figure S2).
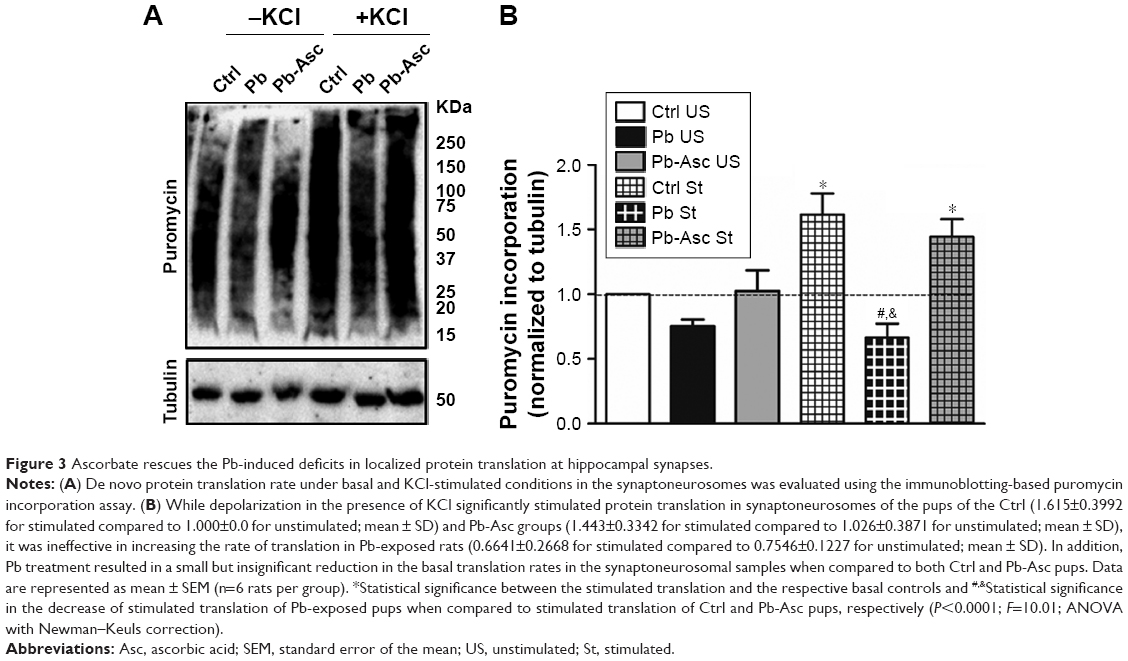 | Figure 3 Ascorbate rescues the Pb-induced deficits in localized protein translation at hippocampal synapses. |
Deficits in activity-dependent protein translation correlate with the blood Pb levels of the rat pups
Reduction in localized activity-dependent protein translation in synaptoneurosomes upon exposure of Pb was confirmed when analyzing the ratio of KCl-induced puromycin and the respective unstimulated basal puromycin incorporation rates for each of animals of the three groups (Figure 4A). Lastly, the correlation between activity-driven protein translation and blood Pb levels was also analyzed. Hippocampal Pb levels were not used as a correlative measure for the Pb-mediated neurotoxic effects because of the obvious advantage of evaluating the blood Pb levels as a diagnostic measure of Pb-mediated neurotoxicity. A strong and significant correlation was observed between the loss in K+-stimulated synaptoneurosomal translation (expressed as the ratio of stimulated to unstimulated translation) and increase in blood Pb levels (Figure 4B).
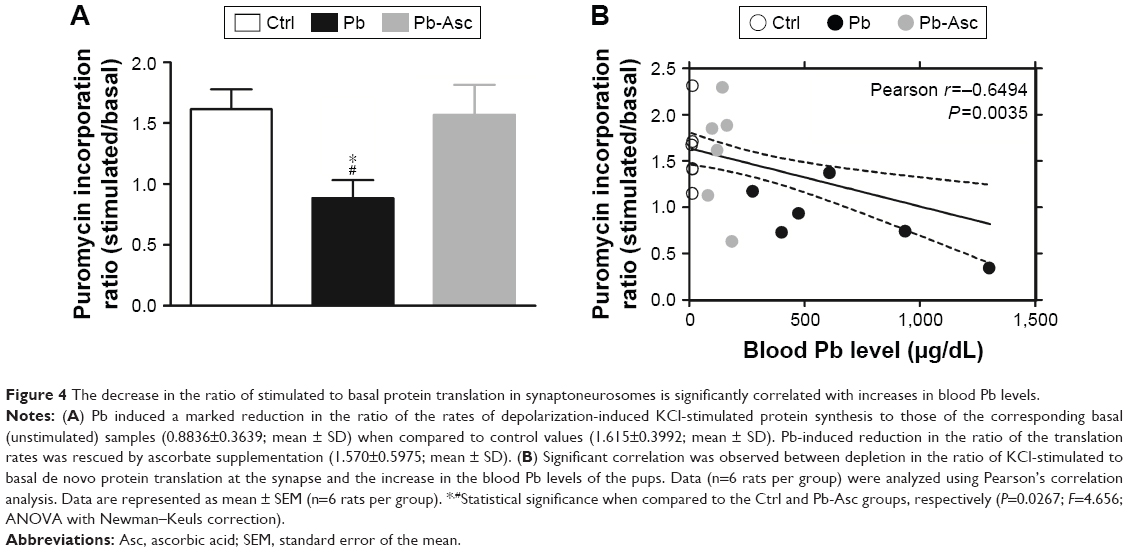 | Figure 4 The decrease in the ratio of stimulated to basal protein translation in synaptoneurosomes is significantly correlated with increases in blood Pb levels. |
Discussion
Because of the presence of a still-developing blood–brain barrier, the immature nervous system is particularly vulnerable to Pb exposure.14 Importantly, early life chronic Pb exposure has been shown to induce irreversible deleterious effects on the developmental trajectory and maintenance and function of the synapses, which in turn accounts for long-term deficits in sensory motor and cognitive skills as well vulnerability to neuropsychiatric stress.5,6,29
Pb neurotoxicity is most eminent in prefrontal cortex, hippocampus, and cerebellum3 where it results in morphological, structural, and pathological alterations in neuronal cells and their synaptic connections.29,75 Because of its interference with cellular processes which require divalent cations Ca2+ and Zn2+, mechanisms of lead neurotoxicity are both multifaceted and complex.76 Nevertheless, detrimental alterations in synaptic signaling and its maintenance and plasticity are among the prime factors underlying Pb neurotoxicity.7,11,12 Although changes in neurotransmitter (glutamate) release and N-methyl-D-aspartate receptor physiology are the best known pathways implicated in Pb-induced synaptic dysfunction, a complex interplay of several factors is probably at work in synchrony. Indeed, recent studies have shed light on some of the deleterious effects of Pb on redox and calcium homeostasis, cell signaling and death pathways, and membrane receptor trafficking and gene expression as candidate mechanisms of Pb-induced synaptic dysfunction.13,14
Recent studies have suggested that localized protein translation, and particularly activity-dependent protein translation, at the synapses play critical roles in the precise control of their function and plasticity.18,19,21–23 Indeed, synaptic plasticity and its long-term consolidation are critically dependent on both explicit and dynamic changes in the spatiotemporal regulation of localized protein translation, trafficking, and organization within the micro-domain of the synaptic contacts. Localized protein translation has significant effects on the expression and function of a wide variety of critical synaptic proteins, including neurotransmitter receptors, signaling proteins, and cytoskeletal and scaffold elements.23 Furthermore, any dysfunction of the synaptic machinery for protein translation could potentially lead to behavioral and cognitive deficits.23,77,78 Despite this, the role of the dysregulation of synapse-specific protein translation in neuropathological states is only being started to be acknowledged.24,27,57,59,79–81 In view of this, the present study aimed 1) to study the alterations of de novo synaptic protein translation induced by developmental Pb exposure, if any and 2) to evaluate the capacity of ascorbate supplementation as a beneficial therapeutic strategy in mitigation of this dysregulation. Our results suggest a significant breakdown of localized protein translation machinery at the hippocampal synapses in juvenile rats pre- and postnatally exposed to Pb. Local protein translation at the synapse is regulated by several signaling cascades, Akt/mTOR and Erk/MAP kinase pathways being the two most prominent among them.23,78,82 Interestingly, dysregulation of the signaling pathways of both the kinases has been observed in the brain, possibly by the induction of oxidative stress.73,83
Interestingly, the deficits in the KCl-stimulated protein translation observed in our study correlated significantly with blood Pb levels, indicating that synaptic protein translation is a critical molecular pathway linking developmental adversities, such as Pb exposure to their detrimental effects on neuronal communication at the pre- and post-synapse. It should be noted that early life Pb exposure paradigms through drinking water similar to that used in our study are also known to cause significant accumulation of Pb in hippocampal tissues34,72,73 and show a strong and significant correlation with blood Pb levels.74 Moreover, in support of previous studies in human subjects,1,6,8,17,32,84,85 our results suggest that blood Pb levels serve as a valuable diagnostic measure of Pb-mediated dysregulation of neurological functions. In addition, while our study implicates alterations in synapse-specific protein translation as a potential mechanism underlying Pb-induced alterations in synapse maintenance, plasticity; and consequently on memory and cognition as well as vulnerability to psychological stress, it also establishes ascorbic acid as an important therapeutic agent against Pb neurotoxicity,44,45,47,48 The mechanism of ascorbate-mediated neuroprotection in Pb toxicity seems to be multifactorial, stemming from both its antioxidant properties and its Pb chelating function. Being a lactone containing an enediol group, ascorbate can form a soluble complex with Pb, enhancing its urinary excretion while also preventing its gastrointestinal absorption, and eventually lowering its blood levels and reducing its bioavailability.70,86,87
Conclusion
Our results suggest that alteration in synaptic protein translation, particularly that induced by neuronal depolarization is observed upon exposure to Pb and potentially contributes to the neurotoxic effects of this environmental toxin in the developing brain with consequent long term effects on higher order brain functions of behavior and cognition. Moreover, the study proposes the utilization of ascorbate based therapeutic measures in mitigation of Pb-induced brain toxicity.
Acknowledgments
The study was partly funded by Deanship of Scientific Research, Imam Abdulrahman Bin Faisal University, Saudi Arabia (project no. 2016-087-IRMC).
Disclosure
The authors report no conflicts of interest in this work.
References
Meyer PA, Brown MJ, Falk H. Global approach to reducing lead exposure and poisoning. Mutat Res. 2008;659(1–2):166–175. | ||
Assi MA, Hezmee MN, Haron AW, Sabri MY, Rajion MA. The detrimental effects of lead on human and animal health. Vet World. 2016;9(6):660–671. | ||
Sharma P, Chambial S, Shukla KK. Lead and neurotoxicity. Indian J Clin Biochem. 2015;30(1):1–2. | ||
Flora G, Gupta D, Tiwari A. Toxicity of lead: A review with recent updates. Interdiscip Toxicol. 2012;5(2):47–58. | ||
Allen KA. Is prenatal lead exposure a concern in infancy? What is the evidence? Adv Neonatal Care. 2015;15(6):416–420. | ||
Chiodo LM, Jacobson SW, Jacobson JL. Neurodevelopmental effects of postnatal lead exposure at very low levels. Neurotoxicol Teratol. 2004;26(3):359–371. | ||
Neal AP, Guilarte TR. Molecular neurobiology of lead (Pb(2+)): effects on synaptic function. Mol Neurobiol. 2010;42(3):151–160. | ||
Bellinger DC, Bellinger AM. Childhood lead poisoning: The torturous path from science to policy. J Clin Invest. 2006;116(4):853–857. | ||
Sweatt JD. Neural plasticity and behavior – sixty years of conceptual advances. J Neurochem. 2016;139(Suppl 2):179–199. | ||
Lepeta K, Lourenco MV, Schweitzer BC, et al. Synaptopathies: synaptic dysfunction in neurological disorders – A review from students to students. J Neurochem. 2016;138(6):785–805. | ||
Toscano CD, Guilarte TR. Lead neurotoxicity: from exposure to molecular effects. Brain Res Brain Res Rev. 2005;49(3):529–554. | ||
Sadiq S, Ghazala Z, Chowdhury A, Büsselberg D. Metal toxicity at the synapse: presynaptic, postsynaptic, and long-term effects. J Toxicol. 2012;2012:1–42. | ||
Verstraeten SV, Aimo L, Oteiza PI. Aluminium and lead: molecular mechanisms of brain toxicity. Arch Toxicol. 2008;82(11):789–802. | ||
Neal AP, Guilarte TR. Mechanisms of lead and manganese neurotoxicity. Toxicol Res. 2013;2(2):99–114. | ||
Hagberg H, Mallard C, Rousset CI, Thornton C. Mitochondria: Hub of injury responses in the developing brain. Lancet Neurol. 2014;13(2):217–232. | ||
Hsiang J, Díaz E. Lead and developmental neurotoxicity of the central nervous system. Curr Neurobiol. 2011;2(1):35–42. | ||
Grandjean P, Herz KT. Trace elements as paradigms of developmental neurotoxicants: Lead, methylmercury and arsenic. J Trace Elem Med Biol. 2015;31:130–134. | ||
Di Liegro CM, Schiera G, Di Liegro I. Regulation of mRNA transport, localization and translation in the nervous system of mammals (Review). Int J Mol Med. 2014;33(4):747–762. | ||
Klann E, Antion MD, Banko JL, Hou L. Synaptic plasticity and translation initiation. Learn Mem. 2004;11(4):365–372. | ||
Kelleher RJ, Bear MF. The autistic neuron: troubled translation? Cell. 2008;135(3):401–406. | ||
Richter JD, Klann E. Making synaptic plasticity and memory last: mechanisms of translational regulation. Genes Dev. 2009;23(1):1–11. | ||
Steward O, Schuman EM. Protein synthesis at synaptic sites on dendrites. Annu Rev Neurosci. 2001;24(1):299–325. | ||
Wang DO, Martin KC, Zukin RS. Spatially restricting gene expression by local translation at synapses. Trends Neurosci. 2010;33(4):173–182. | ||
Ahmad F, Salahuddin M, Alsamman K, Herzallah HK, Al-Otaibi ST. Neonatal maternal deprivation impairs lkocalized de novo activity-induced protein translation at the synapse in the rat hippocampus. Biosci Rep. 2018;138(3). pii: BSR20180118. | ||
Li N, Lee B, Liu RJ, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329(5994):959–964. | ||
Stoppel LJ, Auerbach BD, Senter RK, Preza AR, Lefkowitz RJ, Bear MF. β-arrestin2 couples metabotropic glutamate receptor 5 to neuronal protein synthesis and is a potential target to treat fragile X. Cell Rep. 2017;18(12):2807–2814. | ||
Ahmad F, Singh K, Das D, et al. Reactive oxygen species-mediated loss of synaptic akt1 signaling leads to deficient activity-dependent protein translation early in alzheimer’s disease. Antioxid Redox Signal. 2017;27(16):1269–1280. | ||
Bettegazzi B, Bellani S, Roncon P, et al. EIF4B phosphorylation at Ser504 links synaptic activity with protein translation in physiology and pathology. Sci Rep. 2017;7(1):10563. | ||
White LD, Cory-Slechta DA, Gilbert ME, et al. New and evolving concepts in the neurotoxicology of lead. Toxicol Appl Pharmacol. 2007;225(1):1–27. | ||
Baranowska-Bosiacka I, Listos J, Gutowska I, et al. Effects of perinatal exposure to lead (Pb) on purine receptor expression in the brain and gliosis in rats tolerant to morphine analgesia. Toxicology. 2016;339:19–33. | ||
An J, Cai T, Che H, et al. The changes of miRNA expression in rat hippocampus following chronic lead exposure. Toxicol Lett. 2014;229(1):158–166. | ||
Sanders T, Liu Y, Buchner V, Tchounwou PB. Neurotoxic effects and biomarkers of lead exposure: a review. Rev Environ Health. 2009;24(1):15–45. | ||
Soleimani E, Goudarzi I, Abrari K, Lashkarbolouki T. Maternal administration of melatonin prevents spatial learning and memory deficits induced by developmental ethanol and lead co-exposure. Physiol Behav. 2017;173:200–208. | ||
Baranowska-Bosiacka I, Strużyńska L, Gutowska I, et al. Perinatal exposure to lead induces morphological, ultrastructural and molecular alterations in the hippocampus. Toxicology. 2013;303:187–200. | ||
Ahmad A, Shah SA, Badshah H, et al. Neuroprotection by vitamin C against ethanol-induced neuroinflammation associated neurodegeneration in the developing rat brain. CNS Neurol Disord Drug Targets. 2016;15(3):360–370. | ||
de Freitas P, Zanoni JN, Alves AM, de Miranda Neto MH. Neuroprotection and neurodegeneration in submucosal VIP-IR neurons in the jejunum of ascorbic acid supplemented aging Wistar rats. Nutr Neurosci. 2012;15(6):283–288. | ||
Moretti M, Fraga DB, Rodrigues ALS. Ascorbic acid to manage psychiatric disorders. CNS Drugs. 2017;31(7):571–583. | ||
Rice ME. Ascorbate regulation and its neuroprotective role in the brain. Trends Neurosci. 2000;23(5):209–216. | ||
Kim HJ, Song W, Jin EH, et al. Combined low-intensity exercise and ascorbic acid attenuates kainic acid-induced seizure and oxidative stress in mice. Neurochem Res. 2016;41(5):1035–1041. | ||
Hsu PC, Guo YL. Antioxidant nutrients and lead toxicity. Toxicology. 2002;180(1):33–44. | ||
Patrick L. Toxic metals and antioxidants: Part II. The role of antioxidants in arsenic and cadmium toxicity. Altern Med Rev. 2003;8(2):106–128. | ||
Karamian R, Komaki A, Salehi I, et al. Vitamin C reverses lead-induced deficits in hippocampal synaptic plasticity in rats. Brain Res Bull. 2015;116:7–15. | ||
Acharya UR, Rathore RM, Mishra M. Role of vitamin C on lead acetate induced spermatogenesis in swiss mice. Environ Toxicol Pharmacol. 2003;13(1):9–14. | ||
Chang BJ, Jang BJ, Son TG, et al. Ascorbic acid ameliorates oxidative damage induced by maternal low-level lead exposure in the hippocampus of rat pups during gestation and lactation. Food Chem Toxicol. 2012;50(2):104–108. | ||
Han JM, Chang BJ, Li TZ, et al. Protective effects of ascorbic acid against lead-induced apoptotic neurodegeneration in the developing rat hippocampus in vivo. Brain Res. 2007;1185:68–74. | ||
Patra RC, Swarup D, Dwivedi SK. Antioxidant effects of alpha tocopherol, ascorbic acid and L-methionine on lead induced oxidative stress to the liver, kidney and brain in rats. Toxicology. 2001;162(2):81–88. | ||
Sadeghi A, Ebrahimzadeh Bideskan A, Alipour F, Fazel A, Haghir H. The effect of ascorbic acid and garlic administration on lead-induced neural damage in rat offspring’s hippocampus. Iran J Basic Med Sci. 2013;16(2):157–164. | ||
Ahmad F, Salahuddin M, Alamoudi W, Acharya S. Dysfunction of cortical synapse-specific mitochondria in developing rats exposed to lead and its amelioration by ascorbate supplementation. Neuropsychiatr Dis Treat. 2018;14:813–824. | ||
Singh G, Singh V, Wang ZX, et al. Effects of developmental lead exposure on the hippocampal methylome: Influences of sex and timing and level of exposure. Toxicol Lett. 2018;290:63–72. | ||
Kasten-Jolly J, Lawrence DA. Sex-specific effects of developmental lead exposure on the immune-neuroendocrine network. Toxicol Appl Pharmacol. 2017;334:142–157. | ||
Schneider JS, Anderson DW, Talsania K, Mettil W, Vadigepalli R. Effects of developmental lead exposure on the hippocampal transcriptome: influences of sex, developmental period, and lead exposure level. Toxicol Sci. 2012;129(1):108–125. | ||
Varma G, Sobolewski M, Cory-Slechta DA, Schneider JS. Sex- and brain region-specific effects of prenatal stress and lead exposure on permissive and repressive post-translational histone modifications from embryonic development through adulthood. Neurotoxicology. 2017;62:207–217. | ||
Chang W, Chen J, Wei QY, Chen XM. Effects of Brn-3a protein and RNA expression in rat brain following low-level lead exposure during development on spatial learning and memory. Toxicol Lett. 2006;164(1):63–70. | ||
Gottipolu RR, Davuljigari CB. Perinatal exposure to lead: reduction in alterations of brain mitochondrial antioxidant system with calcium supplement. Biol Trace Elem Res. 2014;162(1–3):270–277. | ||
Harry GJ, Schmitt TJ, Gong Z, Brown H, Zawia N, Evans HL. Lead-induced alterations of glial fibrillary acidic protein (GFAP) in the developing rat brain. Toxicol Appl Pharmacol. 1996;139(1):84–93. | ||
Chaurasia N, Pandey SK, Mohan D. Determination of arsenic content in the water and blood samples of ballia region using hydride generation atomic absorption spectrophotometer. Res J Forensic Sci Res J Forensic Sci. 2013;1(4):2321–1792. | ||
Muddashetty RS, Kelić S, Gross C, Xu M, Bassell GJ. Dysregulated metabotropic glutamate receptor-dependent translation of AMPA receptor and postsynaptic density-95 mRNAs at synapses in a mouse model of fragile X syndrome. J Neurosci. 2007;27(20):5338–5348. | ||
Iliff AJ, Renoux AJ, Krans A, Usdin K, Sutton MA, Todd PK. Impaired activity-dependent FMRP translation and enhanced mGluR-dependent LTD in Fragile X premutation mice. Hum Mol Genet. 2013;22(6):1180–1192. | ||
Williams C, Mehrian Shai R, Wu Y, et al. Transcriptome analysis of synaptoneurosomes identifies neuroplasticity genes overexpressed in incipient Alzheimer’s disease. PLoS One. 2009;4(3):e4936. | ||
Eyman M, Cefaliello C, Bruno AP, Bruno A, Crispino M, Giuditta A. Synaptosomal protein synthesis in P2 and Ficoll purified fractions. J Neurosci Methods. 2012;203(2):335–337. | ||
Schmidt EK, Clavarino G, Ceppi M, Pierre P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods. 2009;6(4):275–277. | ||
Dalet A, Argüello RJ, Combes A, et al. Protein synthesis inhibition and GADD34 control IFN-β heterogeneous expression in response to dsRNA. EMBO J. 2017;36(6):761–782. | ||
Henrich CJ. A microplate-based nonradioactive protein synthesis assay: application to TRAIL sensitization by protein synthesis inhibitors. PLoS One. 2016;11(10):e0165192. | ||
Scheetz AJ, Nairn AC, Constantine-Paton M. NMDA receptor-mediated control of protein synthesis at developing synapses. Nat Neurosci. 2000;3(3):211–216. | ||
Simon JA, Hudes ES. Relationship of ascorbic acid to blood lead levels. JAMA. 1999;281(24):2289. | ||
Tandon SK, Chatterjee M, Bhargava A, Shukla V, Bihari V. Lead poisoning in Indian silver refiners. Sci Total Environ. 2001;281(1–3):177–182. | ||
Dalley JW, Gupta PK, Lam FC, Hung CT. Interaction of L-ascorbic acid on the disposition of lead in rats. Pharmacol Toxicol. 1989;64(4):360–364. | ||
Cheng Y, Willett WC, Schwartz J, Sparrow D, Weiss S, Hu H. Relation of nutrition to bone lead and blood lead levels in middle-aged to elderly men. The Normative Aging Study. Am J Epidemiol. 1998;147(12):1162–1174. | ||
Vij AG, Satija NK, Flora SJ. Lead induced disorders in hematopoietic and drug metabolizing enzyme system and their protection by ascorbic acid supplementation. Biomed Environ Sci. 1998;11(1):7–14. | ||
Dawson EB, Evans DR, Harris WA, Teter MC, McGanity WJ. The effect of ascorbic acid supplementation on the blood lead levels of smokers. J Am Coll Nutr. 1999;18(2):166–170. | ||
Shalan MG, Mostafa MS, Hassouna MM, El-Nabi SE, El-Refaie A. Amelioration of lead toxicity on rat liver with Vitamin C and silymarin supplements. Toxicology. 2005;206(1):1–15. | ||
Liu F, Xue Z, Li N, et al. Effects of lead exposure on the expression of amyloid β and phosphorylated tau proteins in the C57BL/6 mouse hippocampus at different life stages. J Trace Elem Med Biol. 2014;28(2):227–232. | ||
Seddik L, Bah TM, Aoues A, Slimani M, Benderdour M. Elucidation of mechanisms underlying the protective effects of olive leaf extract against lead-induced neurotoxicity in Wistar rats. J Toxicol Sci. 2011;36(6):797–809. | ||
Baranowska-Bosiacka I, Falkowska A, Gutowska I, et al. Glycogen metabolism in brain and neurons – astrocytes metabolic cooperation can be altered by pre- and neonatal lead (Pb) exposure. Toxicology. 2017;390:146–158. | ||
Gąssowska M, Baranowska-Bosiacka I, Moczydłowska J, et al. Perinatal exposure to lead (Pb) induces ultrastructural and molecular alterations in synapses of rat offspring. Toxicology. 2016;373:13–29. | ||
Garza A, Vega R, Soto E. Cellular mechanisms of lead neurotoxicity. Med Sci Monit. 2006;12(3):RA57–RA65. | ||
Buffington SA, Huang W, Costa-Mattioli M. Translational control in synaptic plasticity and cognitive dysfunction. Annu Rev Neurosci. 2014;37(1):17–38. | ||
Kindler S, Kreienkamp HJ. Dendritic mRNA targeting and translation. Adv Exp Med Biol. 2012;970:285–305. | ||
Darnell JC. Defects in translational regulation contributing to human cognitive and behavioral disease. Curr Opin Genet Dev. 2011;21(4):465–473. | ||
Pavitt GD, Proud CG. Protein synthesis and its control in neuronal cells with a focus on vanishing white matter disease. Biochem Soc Trans. 2009;37(Pt 6):1298–1310. | ||
Darnell JC, Klann E. The translation of translational control by FMRP: therapeutic targets for FXS. Nat Neurosci. 2013;16(11):1530–1536. | ||
Gong R, Park CS, Abbassi NR, Tang SJ. Roles of glutamate receptors and the mammalian target of rapamycin (mTOR) signaling pathway in activity-dependent dendritic protein synthesis in hippocampal neurons. J Biol Chem. 2006;281(27):18802–18815. | ||
Liu CM, Zheng GH, Cheng C, Sun JM. Quercetin protects mouse brain against lead-induced neurotoxicity. J Agric Food Chem. 2013;61(31):7630–7635. | ||
Vorvolakos T, Arseniou S, Samakouri M. There is no safe threshold for lead exposure: Α literature review. Psychiatriki. 2016;27(3):204–214. | ||
Shefa ST, Héroux P. Both physiology and epidemiology support zero tolerable blood lead levels. Toxicol Lett. 2017;280:232–237. | ||
Goyer RA, Cherian MG. Ascorbic acid and EDTA treatment of lead toxicity in rats. Life Sci. 1979;24(5):433–438. | ||
Dhawan M, Kachru DN, Tandon SK. Influence of thiamine and ascorbic acid supplementation on the antidotal efficacy of thiol chelators in experimental lead intoxication. Arch Toxicol. 1988;62(4):301–304. |
Supplementary materials
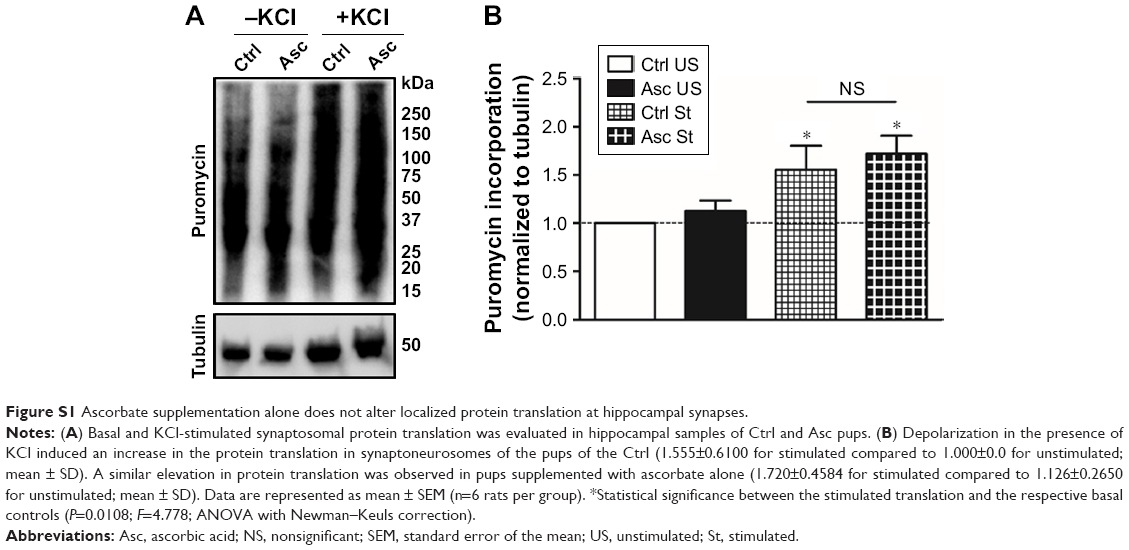 | Figure S1 Ascorbate supplementation alone does not alter localized protein translation at hippocampal synapses. |
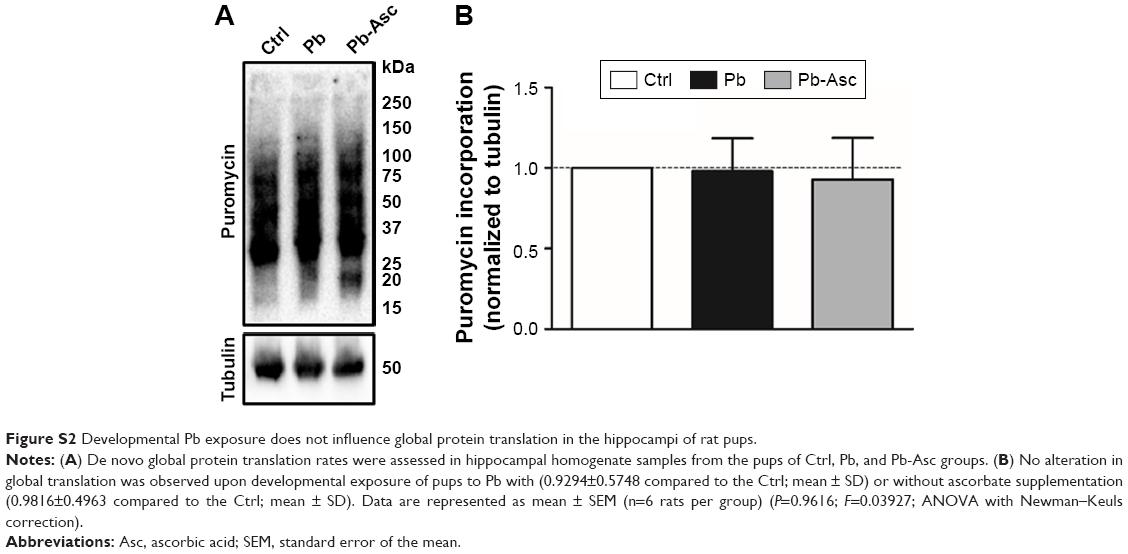 | Figure S2 Developmental Pb exposure does not influence global protein translation in the hippocampi of rat pups. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.