Back to Journals » Infection and Drug Resistance » Volume 12
Development, optimization, and validation of an in-house Dot-ELISA rapid test based on SAG1 and GRA7 proteins for serological detection of Toxoplasma gondii infections
Authors Teimouri A ![]() , Modarressi MH, Shojaee S, Mohebali M, Rezaian M, Keshavarz H
, Modarressi MH, Shojaee S, Mohebali M, Rezaian M, Keshavarz H
Received 12 June 2019
Accepted for publication 27 July 2019
Published 27 August 2019 Volume 2019:12 Pages 2657—2669
DOI https://doi.org/10.2147/IDR.S219281
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sahil Khanna
Aref Teimouri,1,2 Mohammad Hossein Modarressi,3 Saeedeh Shojaee,1 Mehdi Mohebali,1,4 Mostafa Rezaian,1 Hossein Keshavarz1,4
1Department of Medical Parasitology and Mycology, Tehran University of Medical Sciences, Tehran, Iran; 2Students Scientific Research Center, Tehran University of Medical Sciences, Tehran, Iran; 3Department of Medical Genetics, School of Medicine, Tehran University of Medical Sciences, Tehran, Iran; 4Center for Research of Endemic Parasites of Iran, Tehran University of Medical Sciences, Tehran, Iran
Correspondence: Hossein Keshavarz
Department of Medical Parasitology and Mycology, School of Public Health, Tehran University of Medical Sciences, PO Box 1417613191, Pour Sina Street, Ghods Avenue, Enghelab Avenue, Tehran, Iran
Tel +98 218 895 1392
Fax +98 218 895 1392
Email [email protected]
Background: The aim of the present study was to develop a simple, portable, and rapid assay for serodiagnosis of toxoplasmosis based on recombinant Toxoplasma gondii (T. gondii) SAG1 (rSAG1) and GRA7 (rGRA7) proteins.
Methods: The rSAG1 and rGRA7 proteins were expressed in Escherichia coli (E. coli) and purified in a single step by immobilized metal ion affinity chromatography. The immunoreactivity of the recombinant antigens was tested in an in-house IgG and IgM Dot enzyme-linked immunosorbent assay (Dot-ELISA) for potential use in serodiagnosis of T. gondii infection.
Results: Results from the comparison of in-house rSAG1-Dot-ELISA with ELISA for the detection of anti-Toxoplasma IgG and IgM include sensitivity of 83.7% and 81.2%, specificity of 90.2% and 89.3%, positive predictive values of 85.9% and 68.4%, and negative predictive values of 88.6% and 94.3%, respectively. Sensitivity of 66.2%, specificity of 81.2%, positive predictive values of 71.6%, and negative predictive values of 77.1% were concluded from in-house IgG rGRA7-Dot-ELISA. The sensitivity and specificity of IgM rGRA7-Dot-ELISA included 87.5% and 83.9%, respectively. Sensitivity and specificity of in-house Dot-ELISA for a combination of rSAG1 and rGRA7 included 87.5% and 91.1% for IgG and IgM, respectively. Sensitivity and specificity of a combination of rSAG1 and rGRA7 for the detection of IgM in suspected sera to acute toxoplasmosis were higher than those for the detection of IgG in sera with chronic infections (90.6% and 92% instead of 86.2% and 91.6%, respectively).
Conclusion: The highlighted parameters of combined recombinant proteins were more significant than those of single recombinant proteins in in-house Dot-ELISA. These data suggest that the in-house Dot-ELISA based on rSAG1 and rGRA7 combination is a promising diagnostic tool with a similar sensitivity to the native antigens of T. gondii, which can be used for the serodiagnosis of toxoplasmosis in fields as well as less equipped laboratories.
Keywords: Toxoplasma gondii RH strain, in-house Dot-ELISA, rSAG1, rGRA7, soluble tachyzoite antigen (STAg), recombinant proteins
Introduction
Toxoplasma gondii (T. gondii; agent of toxoplasmosis) is a ubiquitous intracellular parasite capable of infecting a broad range of warm-blooded animals and humans.1 Humans can be infected through ingestion of contaminated food and water with oocysts and consumption of undercooked/raw meats containing tissue T. gondii cysts. Vertical transmission of rapidly dividing T. gondii tachyzoites from pregnant mothers to developing fetuses is another route of human infection.2 Generally, T. gondii infection is asymptomatic in immune-competent individuals. However, the infection can result in serious diseases in fetuses and immunocompromised patients, including those with HIV/AIDS, cancer, or organ transplantation.3 To date, various methods have been used in the diagnosis of toxoplasmosis, including microbial isolation, protein analysis, immunological (serological) and molecular techniques. Of these methods, serological methods are most commonly used for the detection of specific antibody classes or antigens against toxoplasmosis.4 Most of the available commercially serological kits for the diagnosis of toxoplasmosis use total parasite antigens prepared from tachyzoites in mice and/or tissue cultures in vitro and possibly contain varying quantities of extra parasitic material.5,6 However, inter-assay variability is sometimes seen due to the lack of standard antigens or proper protocols for the preparation of these antigens. Usually, preparation of these antigens is expensive. Furthermore, preparations commonly include host cell-derived components. In addition, use of live parasites in antigen preparation can result in serious health problems. To solve these problems, antigens are now produced using recombinant DNA technology.7 Relatively, target genes from T. gondii have recently been cloned and expressed using various systems. Of these recombinant proteins, surface antigens (SAGs), matrix antigens (MAGs), microneme proteins (MICs), rhoptry proteins (ROPs), and dense granule antigens (GRAs) are the most commonly used proteins in literature.8 Studies have described the successful use of recombinant antigenic proteins to detect T. gondii-specific antibodies. These studies analyzed recombinant antigens separately and in combination with each other to increase diagnostic sensitivity.9,10 In the current study, SAG1 was chosen because it is one of the most immunogenic and stage-specific T. gondii antigens, present in tachyzoites but not in bradyzoite.11 Another chosen proteins included GRA7 because it causes a powerful antibody response in acute phases of the infection.12
Nowadays, enzyme-linked immunosorbent assays (ELISAs) are widely used in routine diagnoses and seroepidemiological investigations using purified recombinant proteins. However, the assay is expensive, laborious and time-consuming and requires expert operators, special materials and equipment.13,14 These requirements are sometimes unavailable in resource-limited countries. Hence, an easily operated, cost-effective, and rapid assay such as Dot-ELISA is necessary for the early diagnosis of toxoplasmosis in clinics and fields. Dot-ELISA, as a modified ELISA technique in which the antigen–antibody interaction is performed on nitrocellulose (NC) membranes, has been developed to detect antigens or antibodies.15 In 1983, Pappas et al have first introduced Dot-ELISA for the serodiagnosis of human visceral leishmaniasis (VL) and later standardized the assay.16 Dot-ELISA has been used for the diagnosis of helminths such as Fasciola gigantica in experimentally infected sheep, Trichinella spiralis in swine, and Haemonchus contortus in sheep sera. Moreover, this technique has been widely used for the detection of infections such as those by Toxocara canis, Dirofilaria immitis, Leishmania infantum, Babesia bovis, B. bigemina, Anaplasma marginale, and T. gondii.17
To date, a very few studies have been carried out to detect T. gondii antigens or antibodies using Dot-ELISA.15,18,19 To the best of the authors’ knowledge, no studies have been performed to assess Dot-ELISA based on recombinant T. gondii SAG1 (rSAG1) and GRA7 (rGRA7) antigens in human sera. Therefore, the aim of the present study was to assess an in-house Dot-ELISA based on various T. gondii antigens such as rSAG1, rGRA7, combination of rSAG1 and rGRA7, and soluble tachyzoite antigens (STAg) for the serodiagnosis of human toxoplasmosis. Results were compared to those from standard ELISA.
Methods
Bacterial strains, plasmids, enzymes, and reagents
The E. coli BL21 (DE3) pLysS (Promega, USA) was used to express recombinant antigens. The pET28a plasmid (Novagen, USA) was used to construct an expression system. The E. coli cells with plasmids were cultured aerobically at 30°C in Luria-Bertani (LB) media supplemented with 50 μg/ml of kanamycin and 50 μg/mL of chloramphenicol. Restriction enzymes were purchased from New England Biolabs (USA) and reagents of polymerase chain reaction (PCR) from CinnaGen (CinnaGen, Iran). Purification system using nickel nitrilotriacetic acid (Ni-NTA) resin (Qiagen, Germany) was provided by Invitrogen (USA). Isopropyl-D-thiogalactopyranoside (IPTG), agarose, and reagents of protein purification were purchased from Sigma-Aldrich (USA). The NC membrane was purchased from Bio-Rad (USA). Goat anti-human IgG and IgM horseradish peroxidase (HRP) labeled conjugates, diaminobenzidine substrate (DAB), and prestained protein markers were purchased from Sigma-Aldrich (USA).
Ethical consideration
The study was carried out according to the ethical standards by institutional and/or national research committees and the Helsinki Declaration, 1964. Animal procedures were carried out according to Guidelines for the Care and Use of Laboratory Animals published by the United States National Institutes of Health and approved by the Ethical Committee of Tehran University of Medical Sciences, Tehran, Iran. The current study was approved by the Ethics Committee of Tehran University of Medical Sciences on April 11, 2016 (Approval No. 29,999). Furthermore, written informed consents were signed by the participants before starting the study. In case the person is illiterate, informed consent can be given by thumbprint and a signature of an impartial witness. Parental consent was obtained from the parent or guardian for participants less than 16 years old included in this study.
Study design and clinical sample collections
Human serum samples were provided by various clinical laboratories in Tehran and Shahriar, Iran, from May 2016 to November 2017. In the current retrospective study, human serum samples were tested using commercial Toxoplasma IgG and IgM ELISA kits (Trinity Biotech, USA) as reference method for T. gondii-specific IgG (Toxo IgG) and IgM (Toxo IgM) detections according to the manufacturer’s instructions. ELISA was chosen as reference, as it is the classical and most widely used test for detection of T. gondii-specific antibodies in patient sera and the diagnostic standard test in our laboratory.19 A total of 224 sera were divided into 3 major groups according to clinical and serological criteria,20 as follows: 1) Group I, chronic infection sera (CIS) including 80 samples positive for anti-T. gondii IgG and negative for anti-T. gondii IgM with a follow-up sample indicating no increase in IgG or presence of IgM; 2) Group II, acute infection sera (AIS) including 32 samples positive for anti-T. gondii IgM and sera were also had very low avidity in avidity assay (VIDAS Toxo IgG Avidity, bioMerieux, France) and all these patients had the sign of lymphadenopathy; and 3) Group III, negative infection sera (NIS) including 112 samples negative for anti-T. gondii IgG and IgM antibodies. Four samples were excluded from further analysis, due to positive for anti-T. gondii IgM and had high or equivocal avidity in avidity assay without any sign of lymphadenopathy, suggesting that IgM alone is not an accurate acute-phase marker. To ensure a blinded analysis, each sample was given a unique identification code. Samples from each setting (CIS, AIS, NIS) were then used for the performance assessment of in-house Dot-ELISA strips with various antigens for the detection of human Toxoplasma specific antibodies according to the identification number. For cross-reactivity studies, in-house Dot-ELISA strips were tested against human sera corresponding to parasitic infections other than toxoplasmosis, including F. hepatica (n=3), L. infantum (n=3), Echinococcus granulosus (n=4), and malaria (n=5).
Enzyme-linked immunosorbent assay (ELISA)
The ELISA commercial kits (Trinity Biotech Captia, USA) were used to detect Toxo IgG and IgM according to the manufacturer’s instructions. First, serum specific antibodies against T. gondii were bound with T. gondii coated antigen on the surface of reagent wells to form antigen–antibody complexes. Goat anti-human IgG or IgM labeled with HRP was used as the secondary antibody to the antigen–antibody complex. After incubation and wash, a chromogen substrate of 3,3´,5,5´-tetramethylbenzidine (TMB) was added to each well to detect HRP activity. The process was stopped by addition of stop solution (1-N H2SO4) and optical density (OD) was recorded using an automated ELISA reader (Biotek, USA) at 450 nm. Calibrator and control sera were used in each test set. Immune status ratio (ISR) of the samples was calculated by dividing the sample OD by the cutoff value (cutoff=mean OD of the calibrators×correction factor). Results were interpreted as follows according to the manufacturer’s recommendations: sera <0.9 ISR were reported as nonreactive, 0.91–1.09 as equivocal, and >1.1 as reactive.19
Expression of recombinant proteins
Recombinant expression plasmids were successfully constructed and purified in the laboratory as described previously.21,22 Briefly, PCR amplicons were amplified using DNA extracted from RH strains of T. gondii. Then, PCR products were purified and cloned into pET28a vectors at specified restriction sites. Resulting recombinant plasmids containing SAG1 and GRA7 genes were tagged pET28a/SAG1 and pET28a/GRA7, respectively. The E. coli strain BL21 (DE3) pLysS transformed with the pET28a/SAG1 or pET28a/GRA7 was overnight grown in LB broth supplemented with 50 μg/mL of kanamycin and 50 μg/mL of chloramphenicol at 37°C with a 200 rpm shake. Then, 100 mL of LB media, supplemented with the same antibiotics, were inoculated with 2 mL of the overnight culture. Temperature decreased 30°C with vigorous shaking until an OD of 0.5–0.6 was achieved at 600 nm. Protein production was then induced using IPTG to a final concentration of 1 mM. Culture was grown at 30°C for 16 hrs with a 200 rpm shake. Cells were harvested using centrifugation at 5000× g for 5 mins and subjected to protein purification.
Purification of recombinant proteins
The histidine-tagged recombinant protein was purified through purification system using Ni-NTA resin according to the manufacturer’s instructions under denaturing and native conditions for SAG1 and GRA7 proteins, respectively. Briefly, cell pellets were resuspended in lysis buffer A1 for His6-SAG1 protein purification (8 M of urea, 20 mM of NaH2PO4, 500 mM of NaCl [pH 8], 1 mg/mL of lysozyme and protease inhibitor cocktail) or lysis buffer A2 for His6-GRA7 (50 mM of NaH2PO4, 500 mM of NaCl [pH 8], 1 mg/mL of lysozyme and protease inhibitor cocktail) (Roche Applied Science, Switzerland). Suspensions were sonicated for 30 pulses for at least 15 times at 1 min intervals in an ice–water mixture bath using microtip (Branson Ultrasonic Corporation, Danbury, USA). Soluble fractions were collected using centrifugation at 8000× g for 30 mins at 4°C. Washed pellets containing insoluble inclusion body proteins were extracted using 8 M of urea in extraction buffer (20 mM of NaH2PO4 and 500 mM of NaCl, pH 6.3) supplemented with a protease inhibitor cocktail. Solubilized proteins were incubated with Ni-NTA resin for 30–60 mins using gentle agitation to keep the resin suspended in lysate solution at room temperature. Recombinant antigens were washed twice with 8 mL of washing buffer B20 (buffer A1 or A2 containing 20 mM of imidazole) and twice with washing buffer B50 (buffer A1 or A2, containing 50 mM of imidazole). Recombinant proteins were eluted with 8–12 mL elution buffer C (buffer A1 or A2, containing 250 mM of imidazole). Eluted fractions were dialyzed against a phosphate-buffered saline (PBS) buffer (1% of NaCl w/v, 0.075% of KCl w/v, 0.14% of Na2HPO4 w/v, and 0.0125% of KH2PO4 w/v). The sizes of the expressed target proteins were determined using 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and staining with Coomassie blue. The concentration of the purified recombinant proteins was assessed using Bradford assay (Bio-Rad, USA) and bovine serum albumin (BSA) as standard. Protein solutions were adjusted to 2 mg/mL, divided into small aliquots and stored at −80°C until use. Purified denatured SAG1 was refolded through urea gradient dialysis before assessment of the protein content using Bradford assay. Proteins were loaded into a dialysis bag with a membrane molecular mass cutoff of 13,000 Da and dialyzed against 100 volumes of buffer (20 mM of NaH2PO4 and 500 mM of NaCl, pH 8) at 4°C for nearly 20 hrs. Denaturants were slowly removed using serial dialysis with descending concentration of urea from 6 to 3 and to 1.5 and then to 0 M in 10 mmol/L of PBS, pH 7.23,24
Western blot analysis of recombinant proteins
The immunoreactivities of recombinant proteins were determined using Western blot analysis. After electrophoresis, proteins were transferred to NC membrane. Membranes were blocked with PBS containing 5% of skimme milk (PBS-M 5%) for 1 hr at room temperature and then washed 3 times with PBS containing 0.1% of Tween 20 (PBS-T 0.1%) for 5 mins. Then, membranes were incubated with positive T. gondii human sera diluted 1:100 in blocking solution for 2 hrs at room temperature. After three washes, membranes were incubated with anti-human IgM peroxidase-labeled conjugate diluted 1:2000 in skimmed milk for 1 hr with shaking. Then, membranes were washed and proteins were detected using DAB as chromogenic substrate. Prestained protein markers were used for SDS-PAGE and Western blotting.
Preparation of soluble antigens of T. gondii
The STAg was prepared from tachyzoites of T. gondii as previously described.25 Briefly, RH tachyzoites of T. gondii were intraperitoneally inoculated into BALB/c mice. At day 5 after infection, tachyzoites were collected from the abdominal cavity of the mice using peritoneum wash with sterile PBS (pH 7.4). Tachyzoites were washed 3 times with PBS and then sonicated and centrifuged at 14,000× g for 1 hr at 4°C. Supernatants were filtered through Whatman® No. 1 paper filters and then divided into small aliquots and stored at −20°C until use. Protein quantity of the samples was subsequently assessed using Bradford assay.
Preparation of the strips and optimization of in-house Dot-ELISA
Dot-ELISA was carried out based on the standardized protocols described previously with optimization of the recombinant antigen concentrations, dilution of serum samples and secondary antibody conjugates.26 Generally, NCs with 0.22 μm pores were cut into strips of 0.8 mm wide and 5 cm long. Then, various quantities of STAg, purified rSAG1 and rGRA7 from T. gondii and combinations of rSAG1 and rGRA7 (1:1, 2:1 and 1:2) were dotted on NCs followed by incubation for 45 mins at room temperature. After drying, non-specific protein binding sites were blocked by addition of PBS-M 5% (pH 7.4) before the incubation with serum samples. Then, serum samples were diluted with PBS-M 3% and added to strips. After incubation at 37°C for 45 mins, strips were washed 3 times in PBS-T 0.1%. Bound human IgG was detected by adding goat anti-human IgG HRP-labeled conjugates to the strips. Human IgM was detected using goat anti-human IgM conjugated with HRP. Following incubation at 37°C for 45 mins, strips were washed 3 further times and soaked with a fresh solution of DAB and incubated for 10 mins, rinsed with distilled water (DW) and blotted dry. Each in-house Dot-ELISA strip was tested using serum from each category. Samples with brown spots (compared to negative and positive control sera, antigens, and secondary antibody controls) were reported as positive. The optimum concentration of each antigen used in the test was assessed using four concentrations of antigens (1.25, 2.5, 3.75, and 5 μg/mL) and tested using positive and negative control sera.
Evaluation of the in-house Dot-ELISA strip
Sensitivity, specificity, positive predictive value, negative predictive value, validity, and relative agreement of in-house Dot-ELISA strips were calculated for each antigen as follows: sensitivity=TP/TP+FN×100, specificity=TN/TN+FP×100, positive predictive values=TP/TP+FP×100, negative predictive values=TN/TN+FN×100, validity=sensitivity+specificity/2 and relative agreement=TP+TN/TP+TN+FP+FN×100; when TP was true positive (No. of samples positive with both tests), FP was false positive (No. of samples positive with in-house Dot-ELISA strips and negative with ELISA), TN was true negative (No. of samples negative with both tests) and FN was false negative (No. of samples negative with in-house Dot-ELISA strips and positive with ELISA).
Stability of the in-house Dot-ELISA strips
Stability of the strips was assessed using standard positive and negative sera. Ten strips were, respectively, stored for 2, 4, and 6 months at room temperature. Stored strips were re-tested for specificity and sensitivity with known T. gondii positive or negative human sera.
Statistical analysis
Statistical analysis was carried out using SPSS software v.20 (IBM Analytics, USA). Data were reported using calculation of frequencies (%) and 95% confidence intervals. Furthermore, Kappa test (κ) was used to calculate the degree of agreement between the in-house Dot-ELISA and Trinity ELISA according to a method originally described by Landis and Koch.27 The strength of agreement was assessed by κ, with values interpreted as poor (κ≤0), slight (0<κ≤0.20), fair (0.21<κ≤0.40), moderate (0.41<κ≤0.60), substantial (0.61<κ≤0.80), and near-perfect agreement (0.81<κ≤1.0). Accuracy of in-house Dot-ELISA strips for the detection of exposures to T. gondii was assessed using sensitivity and specificity. Positive and negative predictive values were reported according to a method by Jacobson.28 P-values less than 0.05 were considered statistically significant.
Results
Cloning of the SAG1 and GRA7 genes into pET28a vectors
The SAG1 (1011 bp) and GRA7 (711 bp) genes were amplified from tachyzoite genomic DNA and successfully cloned into pET28a vectors. Positive colonies were verified using PCR with gene-specific primers, restriction enzymes, and sequencing. Digestion of pET28a/SAG1 with EcoRI and XhoI endonucleases produced two bands of 5369 and 1011 bp for pET28a and SAG1, respectively (Figure 1A). After digestion of pET28a/GRA7 using BamHI and NotI nucleases, two bands of 5369 bp for pET28a and 711 bp for GRA7 were produced (Figure 1B). Sequencing results revealed 100% similarity with the previously recorded SAG1 and GRA7 gene sequences in GenBank database (Accession Nos. MK250980 and MK250981). Verified recombinant plasmids were transformed into BL21 (DE3) pLysS expression bacterial system.
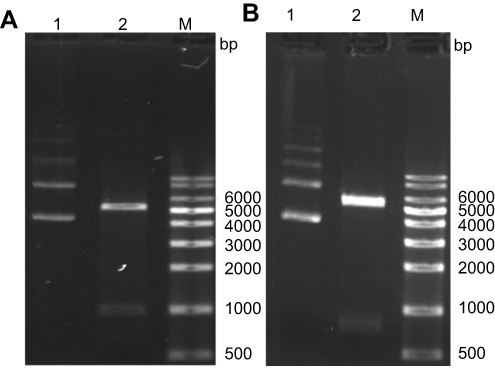 |
Figure 1 The 1.5% agarose gel electrophoreses of digested (A) recombinant pET28a/SAG1 and (B) recombinant pET28a/GRA7. (A) Lane 1, undigested pET28a/SAG1; Lane 2: EcoRI and XhoI digested pET28a/SAG1; Lane M, 1-kb DNA ladder. (B) Lane 1, undigested pET28a/GRA7; Lane 2, BamHI and NotI digested pET28a/GRA7; Lane M, 1-kb DNA ladder. |
Expression and purification of the recombinant antigens
Expression of rSAG1 and rGRA7 were induced using 1 mM of IPTG when cells grew to an OD of 0.6 at 600 nm for 16 hrs. Recombinant T. gondii SAG1 and GRA7 antigens containing 6 histidyl residues at either N- and C-terminals were expressed in E. coli with calculated molecular masses of 37 and 26 kDa, respectively. These recombinant proteins were successfully purified using one-step chromatography and Ni-NTA resin. Furthermore, purified recombinant proteins were verified using Western blot with human sera from Toxoplasma infected patients. In each case, 37 and 26 kDa bands were seen for SAG1 and GRA7 proteins, respectively. No bands were detected in vector control lanes (Figure 2).
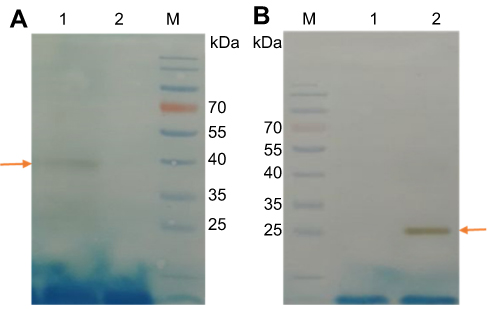 |
Figure 2 Western blot probed with toxoplasmosis patient sera. (A) Lane 1, purified rSAG1 protein; Lane 2, BL21 lysates containing pET28a vector only; Lane M, 10–180 kDa protein marker. (B) Lane 1, BL21 lysates containing pET28a vector only; Lane 2, purified rGRA7 protein; Lane M: 10–180 kDa protein marker. Arrows indicate purified rSAG1 and rGRA7 proteins at approximately 37 and 26 kDa, respectively. |
Optimization of in-house Dot-ELISA strips for detection of anti-T. gondii antibodies
In the present study, optimum concentrations of STAg and recombinant antigens included 2.5 µg for the detection of Toxo IgG and 3.75 µg for the detection of Toxo IgM used in tests appropriate for coating NC strips. The optimum dilution of conjugates included 1:2000 using checker-board titration. During optimization of antigens and conjugates, brown dots on NC strips at antigen-coated sites were clearly visible up to 1:50 dilution values of the positive control serum samples and hence a 1:50 dilution of sera was used for further in-house Dot-ELISA tests.
Reactivity of human serum samples against various antigens of in-house Dot-ELISA strips
Positive reactions were visualized as brown spots. Results were reported as unreactive when no colors were seen (Figure 3). Positive and negative samples of in-house Dot-ELISA strips with various antigens for the detection of Toxo IgG and IgM alone and combined Toxo IgG and IgM antibodies are shown in Tables 1, 2, and 3, respectively. Moreover, intrinsic parameters including sensitivity and specificity, positive and negative predictive values, validity, relative agreement, and kappa (κ) statistics were calculated for each antigen of in-house Dot-ELISA strips, compared to those of commercial ELISA for the detection of anti-Toxoplasma antibodies. Table 4 shows results of in-house Toxo IgG Dot-ELISA strips with various antigens, compared to those of ELISA. Results from the current study on group I sera (CIS) showed that the highest sensitivities were achieved from STAg and combined rSAG1 and rGRA7 antigens of strips for the Toxo IgG with 88.7% and 86.2%, respectively. Furthermore, the highest specificity, validity, relative agreement, and kappa value were achieved from combined rSAG1 and rGRA7 antigens of strips with 91.1%, 88.6%, 89%, and 0.77, respectively (Table 4). Sensitivity achieved with GRA7 antigen was the lowest (66.2%) for Toxo IgG in CIS. Results from in-house IgM Dot-ELISA strips with various antigens are summarized in Table 5. Results of the study on AIS group showed that for detection of Toxo IgM, STAg alone and combined rSAG1 and rGRA7 resulted in a 90.6% sensitivity while GRA7 and SAG1 antigens alone showed 87.5% and 81.2% sensitivity, respectively (Table 5). The relative sensitivity, specificity, validity, and agreement for combined rSAG1 and rGRA7 using in-house Dot-ELISA strip to detect Toxo IgM were 90.6%, 92%, 91.3%, and 91.7%, respectively (Table 5). Results from the comparison of various antigens of in-house Dot-ELISA with ELISA for Toxo IgG and IgM are presented in Table 6. For the detection of Toxo IgG and IgM, STAg alone and a combination of the two antigens (rSAG1+rGRA7) were further sensitive (89.3% and 87.5% respectively). Totally, the following intrinsic parameters were calculated for Toxo IgG and IgM rSAG1+rGRA7 of in-house Dot-ELISA strips, compared to those of ELISA: sensitivity of 87.5%, specificity of 91.1%, validity of 89.3% and further PPV and NPV of 90.7% and 87.9%, respectively (Table 6). No non-Toxo parasitic infection sera reacted with various antigens of in-house Dot-ELISA strips.
 |
Table 1 Summary of results from in-house Dot-ELISA strips with various antigens for the detection of anti-T. gondii IgG in 192 sera, compared to results from ELISA |
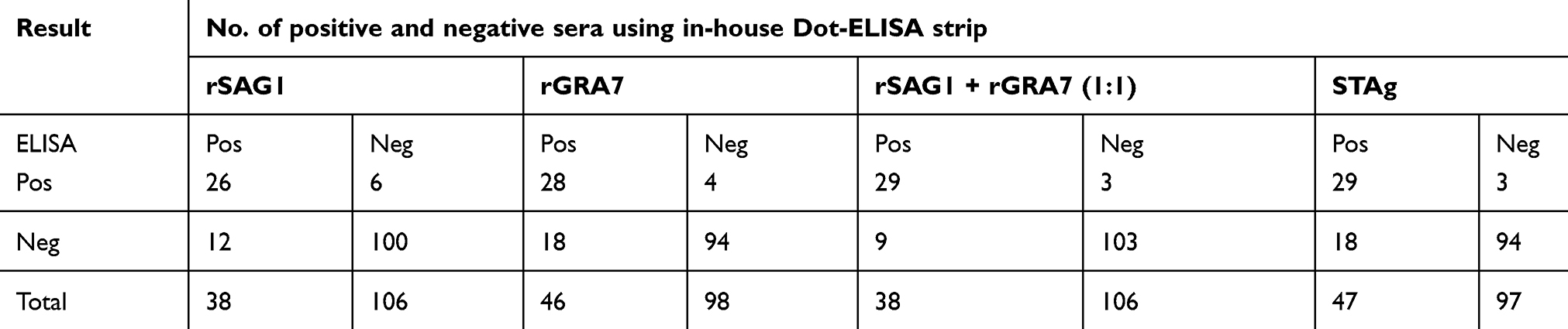 |
Table 2 Summary of results from in-house Dot-ELISA strips with various antigens for the detection of anti-T. gondii IgM in 144 sera, compared to results from ELISA |
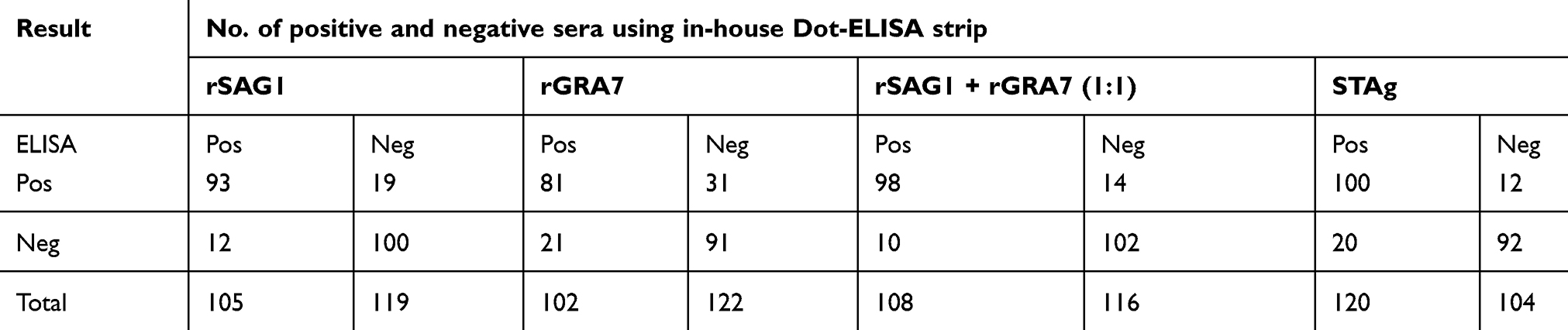 |
Table 3 Summary of results from in-house Dot-ELISA strips with various antigens for detection of anti-T. gondii IgG and IgM in 224 sera, compared to results from ELISA |
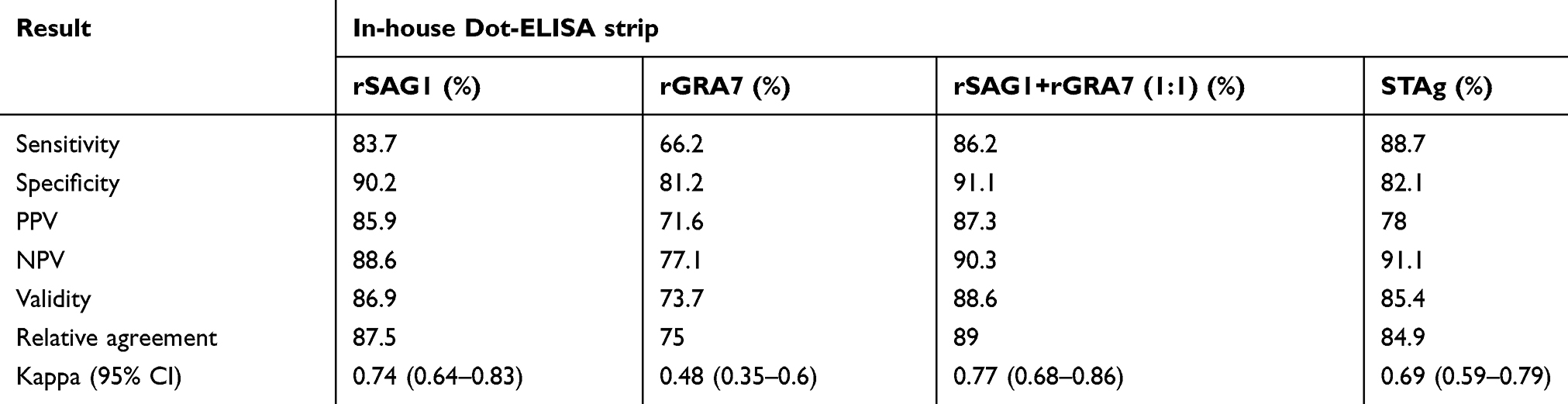 |
Table 4 Assessment of in-house Dot-ELISA strips with various antigens for the detection of anti-T. gondii IgG in 192 sera, compared to that of ELISA |
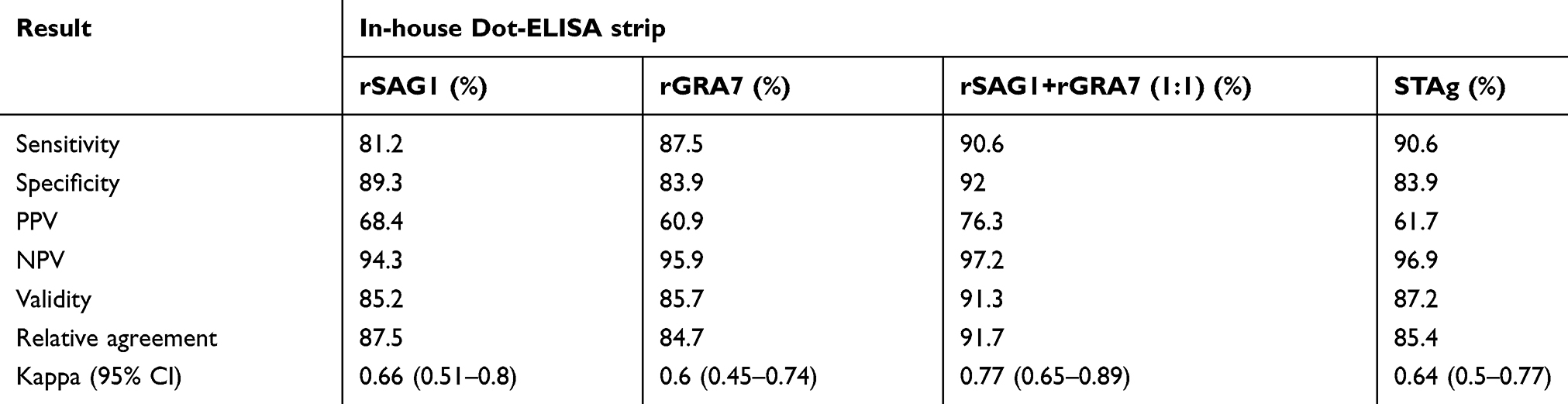 |
Table 5 Assessment of in-house Dot-ELISA strips with various antigens for the detection of anti-T. gondii IgM in 144 sera, compared to that of ELISA |
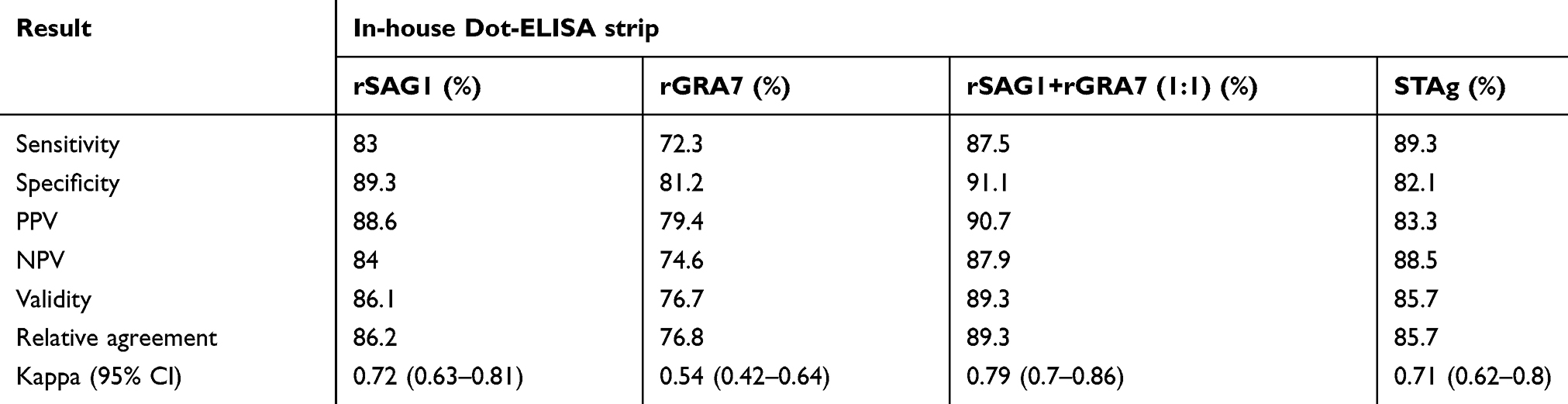 |
Table 6 Assessment of in-house Dot-ELISA strips with various antigens for the detection of anti-T. gondii IgG and IgM in 224 sera, compared to that of ELISA |
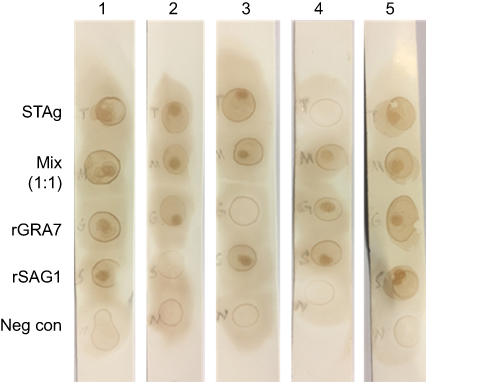 |
Figure 3 Reactivity patterns of in-house Dot-ELISA strip with various antigens for the detection of anti-T. gondii, antibodies. Five strips are shown in the figure as examples. All directions are from top to bottom. Strip 1, positive reaction of STAg, combination of rSAG1 and rGRA7 (1:1), rGRA7 and rSAG1 and negative reaction of negative control with patient sera. Strip 2, positive reaction of STAg, combination of rSAG1 and rGRA7 (1:1), rGRA7 and negative reaction of rSAG1 and negative control with patient sera. Strip 3, positive reaction of STAg, combination of rSAG1 and rGRA7 (1:1), rSAG1 and negative reaction of rGRA7and negative control with patient sera. Strip 4, positive reaction of combination of rSAG1 and rGRA7 (1:1), rSAG1, rGRA7 and negative reaction of STAg and negative control with patient sera. Strip 5, positive reaction of STAg, combination of the rSAG1 and rGRA7 (1:1), rGRA7 and rSAG1 and negative reaction of negative control with patient sera. |
Stability of in-house Dot-ELISA strips
Test results were similar from months 2 to 6 with all known T. gondii positive sera being positive and all known T. gondii negative sera being negative. False positives or negatives were not detected. These results suggest that in-house Dot-ELISA strips can be stored at room temperature for at least 6 months without losing their sensitivity or specificity.
Discussion
Diagnosis of toxoplasmosis is most commonly based on the detection of anti-Toxoplasma specific antibodies in infected sera. Although Dye test (DT) is the gold standard and ELISA is one of the most reliable methods for the detection of specific antibodies against Toxoplasma in sera, use of DT is complicated due to the need of live parasites and ELISA is time-consuming, laborious, expensive, and impractical in fields.29 Available immunoassays are mainly based on tachyzoite lysate (TLA) antigens as coating antigens, which are highly sensitive and specific diagnostic tools.30 However, it is well known that whole somatic antigen-based assays may vary significantly between the laboratories or batches and hence be difficult to standardize. An alternative approach is the use of recombinant antigenic proteins with advantages of precise antigens and easy standardization.10 However, no effective and convenient methods are available for the detection of T. gondii in fields. Therefore, the development of diagnostic kits with affordable prices that do not require special equipment is favorable. In recent years, interests have been increased in immunodiagnosis of microbial and parasitic diseases using fast and inexpensive methodologies with high sensitivity and specificity. Relatively, Dot-ELISA assay has been introduced as an appropriate method for the assessment of antibodies or antigens for human and animal infectious diseases and for the development of seroepidemiological surveys in rural areas and less equipped laboratories. For example, the assay has been used to detect human leptospirosis and mycobacteria antigens in patients with pulmonary tuberculosis.31,32 Furthermore, Dot-ELISA has been shown as a quick and reliable assay for the detection of other infectious diseases caused by bacteria, viruses, and parasites such as Chagas, syphilis, Toxocariasis canis, Neospora caninum, chronic Schistosomiasis japonica, cystic echinococcosis, malaria, and neurocysticercosis.33–38
In the present study, the method was developed based on the successful amplification, cloning, and expression of rSAG1 and rGRA7. An expression vector with a short His fusion tag to the recombinant protein was used to avoid possible nonspecific reactions of recombinant proteins with serum proteins in in-house Dot-ELISA rapid tests. Therefore, SAG1 and GRA7 genes of T. gondii RH strain were cloned using T7 promoter-based pET28a vectors and highly expressed in BL21 (DE3) pLysS E. coli systems. These vectors included multiple advantages such as common restriction sites, strong T7 promoters, medium copy numbers, reasonable yields in mini preparations, and His Tag sequences to assay expression levels and purify proteins using independent strategies. The His Tag sequence binds to divalent cations (eg, Ni2+) immobilized on His bind metal chelation resins.39,40 In the current study, recombinant proteins were successfully purified using one-step chromatography procedure and Ni-NTA resins, verified by Western blot. Previously, several Toxoplasma recombinant proteins have been prepared in E. coli and assessed for their potential as diagnostic antigens in detection of infections in humans.9,10 Selection of T. gondii antigens with conserved T/B cell epitopes is essential for the successful use of these protein epitopes in diagnosis.41 The SAG1 has been used extensively and demonstrated as a good serological marker for the detection of anti-T. gondii antibodies in acute and chronic infections.9,10 Furthermore, studies have demonstrated that GRA7 is a novel vaccine antigen and diagnostic tool.10 These explain the reasons for the selection of SAG1 and GRA7 as antigen candidates in the current study.
Results from the in-house Dot-ELISA using rSAG1 antigen showed a higher sensitivity to sera from humans with chronic toxoplasmosis than sensitivity to sera from humans in AIS group (83.7 instead of 81.2%; Table 4,5). Studies have shown that rSAG1 can detect IgG reactivity in chronic phase of the infection. However, in other studies, no reactivity was seen in chronic but seen in acute phases of the infection.5,9 These differences in detection levels of rSAG1 in human sera possibly occur due to differences in selected gene fragments, cloning strategies, and preparations of this complex molecule in various studies. Therefore, various epitopes may present depending on the cloning and preparation techniques. The SAG1 protein includes a native structure of six intramolecular cysteine bridges that form immunologically conformational epitopes.42,43 However, the rSAG1 antigen was expressed in the form of insoluble inclusion bodies. This phenomenon possibly occurred because of saturated host cell folding machinery, cofactor deficiency, or rare codons affecting rapid mRNA decay, which could downregulate the expression rate of foreign genes in heterologous systems.44,45 Furthermore, irregular interactions of the multiple disulfide bonds might result in misfolded insoluble proteins as well as persistence of rSAG1 C-terminal hydrophobic region (acceptor of GPI group).46,47 In the current study and after one-step chromatographic purification of rSAG1 under denaturing conditions, a cost-saving dialysis procedure was used for refolding the proteins.23 Therefore, the rSAG1 ELISA was more sensitive than other assays, as sensitive as soluble rSAG1-based assays.7,47,48 The correct refolding procedure used in the current study may improve the specific immunoreactivity of this highly complex molecule. In this study, in-house Dot-ELISA using rGRA7 antigen showed a considerably higher sensitivity to sera from patients with IgM acute toxoplasmosis than to sera from CIS group (87.5% instead of 66.2%; Table 4,5). When GRA7 is secreted from tachyzoites and bradyzoites, it directly contacts with the host immune system inducing strong antibody and cell-mediated responses in acute and chronic infections and can hence be used to detect anti-T. gondii antibodies in early and late stages of the infection, more associated to acute infections.12,49–51 In humans with T. gondii, GRA7 can be detected much earlier than other antigens such as SAG1 and MAG1. Therefore, some epitopes of GRA7 seem to play important roles in human antibody responses in acute toxoplasmosis.52,53
In most studies, recombinant proteins are used as ELISA antigens to detect specific IgG and IgM; mostly coated on ELISA plates alone. However, these proteins may be coated in various combinations including 2–3 proteins.9,10 In a study by Johnson et al (1992), ELISAs based on the combination of two recombinant antigens of H4/GST and H11/GST resulted in a higher sensitivity (81.3%) than that IgM ELISA did with either H4/GST or H11/GST (54% and 61%, respectively).54,55 After initial introductions, a combination of recombinant proteins including GRA7, GRA8, and ROP1 was then reported as an antigen preparation for the detection of IgM against T. gondii in human sera. Similarly, a combination of rGRA7, rGRA8, and rSAG1 was studied for the detection of IgG antibodies.5 Data showed the potential use of two or three complementary recombinant antigens to achieve sensitivity rates comparable to that achieved with a crude antigen preparation. Therefore, these recombinant proteins can be used together for the serodiagnosis of toxoplasmosis. Moreover, several combinations of the recombinant proteins have been suggested for the detection of IgG antibodies against T. gondii. Some of these combinations include GRA7, GRA8, and SAG1; GRA7, GRA8, SAG2, and H4; SAG1, GRA1, and GRA7; SAG1 and GRA5 mixed with MAG1, GRA2, or ROP1.9,10 Results from many studies have revealed that combining complementary recombinant T. gondii antigens improves relative sensitivity of the test. The highlighted combinations of the recombinant antigens which included at least one of GRA7, GRA8, SAG2, and H4 proteins could be used in the differentiation of recent and past infections.56 Results of the in-house Dot-ELISA strips in CIS group showed that the highest sensitivities achieved using STAg and combined rSAG1 and rGRA7 antigens for the Toxo IgG included 88.7% and 86.2%, respectively. However, the highest specificity, validity, relative agreement, and kappa value were achieved using a combination of rSAG1 and rGRA7 antigens. In AIS group, the highest sensitivity was seen for STAg (90.6%) and a combination of rSAG1 and rGRA7 (90.6%) antigens for the detection of Toxo IgM. However, GRA7 or SAG1 alone showed 87.5% and 81.2% of sensitivity, respectively. In the present study, the overall diagnostic performance observed for rSAG1 and rGRA7 combination in in-house Dot-ELISA strips was achieved in AIS at maximum values of 90.6%, 92%, 76.3%, 97.2%, 91.3%, and 91.7% for diagnostic sensitivity, specificity, PPV, NPV, validity, and relative agreement, respectively. In general, STAg and a combination of rSAG1 and rGRA7 were more sensitive (89.3% and 87.5%, respectively) for the detection of Toxo IgG and IgM. The current results have shown that no serologic cross-reactivity is seen between the rSAG1 and rGRA7 of in-house Dot-ELISA strips and the non-Toxo parasitic infection sera. Some epitopes featured in the native antigen seem that have not been presented in recombinant proteins and hence cannot be recognized by T. gondii or cross-react with other antibodies. Furthermore, immune diversity can affect epitope diagnostic values.57
Whole sequences of SAG1 and GRA7 used in this work were assessed using ELISA in previous studies by the authors with good results.21,22 Results showed sensitivity and specificity values of rSAG1 as 87% and 95% for acute phase and 93% and 95% for chronic phase sera, respectively. Sensitivity and specificity of rGRA7 included 96% and 90% for acute and 89% and 90% for chronic phase sera, respectively. Based on results from the current study, it can be concluded that the transfer of the recombinant antigens from ELISA microplates to this new diagnostic procedure (in-house Dot-ELISA) was successful. In the previous study, recombinant antigens were coated separately in ELISA microplates for the detection of T. gondii antibodies in human sera.21,22 In the current study, antigens were used separately and in combination forms for the detection of specific antibodies against T. gondii in human sera. Using other antigens (separately or combined) and/or deepening the optimization of the technique can increase sensitivity and specificity values. Moreover, assays including use of NC membranes have multiple advantages over the classic ELISA since protein absorption is higher in membranes.58,59 The current study was a pilot study for designing a diagnostic kit using the highlighted protocol. The Dot-ELISA method is easily used as all incubation steps can be carried out at room temperature or 37°C with mild agitation. Moreover, results can be verified by naked eyes and the method does not need the use of expensive equipment or highly trained researchers. The Dot-ELISA assay greatly simplifies diagnostic procedures for the detection of T. gondii infections, compared to that commercially available ELISA kits do. Furthermore, the protocol used in this study is less expensive and can produce much recombinant proteins. Stability of the in-house Dot-ELISA strips has suggested that the validity period of these strips includes at least 6 months at room temperature with no loss of sensitivity and specificity. Stability of antigens in NC strips has been assessed in other studies. The NC membranes with antigens can be stored for up to 5 months at room temperature without any reactivity changes.38
Conclusion
The rSAG1 and rGRA7 antigens can be described as valuable diagnostic markers of toxoplasmosis. However, rGRA7 was more appropriate for IgM Dot-ELISA than IgG Dot-ELISA. For the detection of Toxo IgG and IgM, the highlighted parameters of combined recombinant proteins were more significant than those of single recombinant proteins in in-house Dot-ELISA with a similar sensitivity to the native antigens of T. gondii. Therefore, combination of these recombinant proteins can replace soluble extracts of tachyzoites in serologic tests. Indeed, the Dot-ELISA rapid test kit described in this study is fast and simple to use. The prepared antigen NC strips can be stored at room temperature for at least 6 months without antigenic changes. Therefore, strips can be used for the serodiagnosis of toxoplasmosis in fields as well as less equipped laboratories, where the use of major immunodiagnostic assays is limited by their dependency on electricity, refrigeration, or laboratory-grade water. In conclusion, data from the current study suggest that the in-house Dot-ELISA rapid test based on rSAG1 and rGRA7 combination is a promising diagnostic tool, which can be used as a serological screening test for toxoplasmosis.
Data availability statement
All data generated during this study are included in this published article and the datasets analyzed during the current study are publicly available in the Figshare repository, https://figshare.com/s/bad4c70e468cb86aedea. Furthermore, the SAG1 and GRA7 sequencing data are available at GenBank under accession numbers MK250980 and MK250981, respectively.
Acknowledgment
This research project was supported by the Tehran University of Medical Sciences and Health Services (Project Number: 29999), Tehran, Iran. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We would like to acknowledge all staff from the toxoplasmosis laboratory (Department of Medical Parasitology and Mycology, Tehran University of Medical Sciences, Tehran, Iran) for their useful collaboration. The authors would like to express gratitude to Dr Sara Ayazian Mavi and Dr Vahid Jajarmi for their technical assistance. We also thank all the heads of Department and staff that directly or indirectly helped us in rendering technical support.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Dubey JP. Toxoplasmosis of Animals and Humans.
2. Tenter AM, Heckeroth AR, Weiss LM. Toxoplasma gondii: from animals to humans. Int J Parasitol. 2000;30(12–13):1217–1258.
3. Montoya J, Liesenfeld O. Toxoplasmosis. Lancet. 2004;363:1965–1976. doi:10.1016/S0140-6736(04)16412-X
4. Liu Q, Wang ZD, Huang SY, Zhu XQ. Diagnosis of toxoplasmosis and typing of Toxoplasma gondii. Parasit Vectors. 2015;8(1):292. doi:10.1186/s13071-015-0902-6
5. Aubert D, Maine GT, Villena I, et al. Recombinant antigens to detect Toxoplasma gondii-specific immunoglobulin G and immunoglobulin M in human sera by enzyme immunoassay. J Clin Microbiol. 2000;38:1144–1150.
6. Beghetto E, Spadoni A, Bruno L, Buffolano W, Gargano N. Chimeric antigens of Toxoplasma gondii: toward standardization of toxoplasmosis serodiagnosis using recombinant products. J Clin Microbiol. 2006;44:2133–2140. doi:10.1128/JCM.00390-06
7. Pfrepper KI, Enders G, Gohl M, et al. Seroreactivity to and avidity for recombinant antigens in toxoplasmosis. Clin Diagn Lab Immunol. 2008;12:977–982.
8. Costa JG, Vilarino MJ. Antigens to detect the acute phase of toxoplasmosis in pregnant women: standardized comparison. Biomark Med. 2018;12(5):517–534.
9. Kotresha D, Noordin R. Recombinant proteins in the diagnosis of toxoplasmosis. APMIS. 2010;118:529–542. doi:10.1111/j.1600-0463.2010.02655.x
10. Holec-Gasior L. Toxoplasma gondii recombinant antigens as tools for serodiagnosis of human toxoplasmosis: current status of studies. Clin Vac Immunol. 2013;20(9):1343–1351. doi:10.1128/CVI.00117-13
11. Kasper LH, Bradley MS, Pfefferkorn ER. Identification of stage-specific sporozoite antigens of Toxoplasma gondii by monoclonal antibodies. J Immunol. 1984;132:443.
12. Jacobs D, Vercammen M, Saman E. Evaluation of recombinant dense granule antigen 7 (GRA7) of Toxoplasma gondii for detection of immunoglobulin G antibodies and analysis of a major antigenic domain. Clin Diagn Lab Immunol. 1999;6:24–29.
13. Gamble HR, Andrews CD, Dubey JP, Webert DW, Parmley SF. Use of recombinant antigens for detection of Toxoplasma gondii infection in swine. J Parasitol. 2000;86:459–462. doi:10.1645/0022-3395(2000)086[0459:UORAFD]2.0.CO;2
14. Revello MG, Gerna G. Human cytomegalovirus infection in the mother, fetus, and newborn infant. Clin Microbiol Rev. 2002;15:680–715. doi:10.1128/CMR.15.4.680-715.2002
15. Pappas MG, Lunde MN, Hajkowski R, McMahon J. Determination of IgM and IgG antibodies to Toxoplasma using the IFA test, ELISA, and Dot-ELISA procedures. Vet Parasitol. 1986;20:31–42. doi:10.1016/0304-4017(86)90090-7
16. Pappas MG, Hajkowski R, Hockmeyer WT. Dot enzyme-linked immunosorbent assay (Dot-ELISA): a micro technique for the rapid diagnosis of visceral leishmaniasis. J Microbiol Methods. 1983;64:205–214.
17. Gupta A, Dixit AK, Dixit P. Dot-ELISA in the diagnosis of parasitic diseases of animals. J Parasit Dis. 2008;32(1):10–14.
18. Azami SJ, Keshavarz H, Rezaian M, Mohebali M, Shojaee S. Rapid detection of Toxoplasma gondii antigen in experimentally infected mice by dot—ELISA. Iranian J Parasitol. 2011;6:28–33.
19. Teimouri A, Modarressi MH, Shojaee S, et al. Detection of toxoplasma-specific immunoglobulin G in human sera: performance comparison of in house Dot-ELISA with ECLIA and ELISA. Eur J Clin Microbiol Infect Dis. 2018. doi:10.1007/s10096-018-3266-y
20. Lebech M, Joynson DH, Seitz HM, et al. Classification system and case definitions of Toxoplasma gondii infection in immunocompetent pregnant women and their congenitally infected offspring. European research network on congenital toxoplasmosis. Eur J Clin Microbiol Infect Dis. 1996;15:799–805.
21. Selseleh M, Keshavarz H, Mohebali M, Shojaee S, Modarressi MH, Eshragian M. Production and evaluation of Toxoplasma gondii recombinant surface antigen 1 (SAG1) for serodiagnosis of acute and chronic Toxoplasma infection in human sera. Iranian J Parasitol. 2012;7:1–9.
22. Selseleh M, Keshavarz H, Mohebali M, et al. Production and evaluation of Toxoplasma gondii recombinant GRA7 for serodiagnosis of human infections. Korean J Parasitol. 2012;50(3):233–238. doi:10.3347/kjp.2012.50.3.233
23. Burgess RR. Refolding solubilized inclusion body proteins. Methods Enzymol. 2009;17:259–282.
24. Cabrita LD, Bottomley SP. Protein expression and refolding—a practical guide to getting the most out of inclusion bodies. Biotechnol Annu Rev. 2004;10:31–54.
25. Teimouri A, Azami SJ, Keshavarz H, et al. Anti-Toxoplasma activity of various molecular weights and concentrations of chitosan nanoparticles on tachyzoites of RH strain. Int J Nanomedicine. 2018;13:1341–1351. doi:10.2147/IJN.S177627
26. Hawkes R, Niday E, Gordon J. A Dot-Immunobinding assay for monoclonal and other antibodies. Anal Biochem. 1982;119(1):142–147.
27. Landis JR, Koch GG. The measurement of observer agreement for categorical data. Biometrics. 1977;33(1):159–174. doi:10.2307/2529310
28. Jacobson RH. Validation of serological assays for diagnosis of infectious diseases. Rev Sci Tech. 1998;17(2):469–526. doi:10.20506/rst.17.2.1119
29. Dard C, Fricker-Hidalgo H, Brenier-Pinchart MP, Pelloux H. Relevance of and new developments in serology for toxoplasmosis. Trends Parasitol. 2016;32:492–506. doi:10.1016/j.pt.2016.04.001
30. Sonaimuthu P, Fong MY, Kalyanasundaram R, Mahmud R, Lau YL. Serodiagnostic evaluation of Toxoplasma gondii recombinant Rhoptry antigen 8 expressed in E. coli. Parasit Vectors. 2014;7:297. doi:10.1186/1756-3305-7-23
31. El-Masry S, El-Kady I, Zaghloul MH, Al-Badrawey MK. Rapid and simple detection of a mycobacterium circulating antigen in serum of pulmonary tuberculosis patients by using a monoclonal antibody and Fast-Dot-ELISA. Clin Biochem. 2008;41:145–151. doi:10.1016/j.clinbiochem.2008.04.011
32. Blanco RM, Takei K, Romero EC. Leptospiral glycolipo protein as a candidate antigen for serodiagnosis of human leptospirosis. Lett Appl Microbiol. 2009;49:267–273. doi:10.1111/j.1472-765X.2009.02665.x
33. Coelho JS, Soares IS, Lemos EA, et al. A multianalyte Dot-ELISA for simultaneous detection of malaria, Chagas disease, and syphilis-specific IgG antibodies. Diagn Microbiol Infect Dis. 2007;58:223–230. doi:10.1016/j.diagmicrobio.2006.12.011
34. Cai SF, Li WG, Wang M. Detection of specific IgG in the sera of patients with chronic Schistosomiasis japonica by dot-ELISA with the recombinant Sj26-Sj32 fusion protein. Chin J Parasitol Parasit Dis. 2011;29:21–24.
35. Pina R, Gutierrez AH, Gilman RH, et al. A dot-ELISA using a partially purified cathepsin-L-like protein fraction from Taenia solium cysticerci, for the diagnosis of human neurocysticercosis. Ann Trop Med Parasitol. 2011;105:311–318. doi:10.1179/136485911X12987676649782
36. Sedaghat F, Sadjjadi SM, Hosseini SV, Kazemian S, Sarkari B. Evaluation of a simple Dot-ELISA in comparison with counter current immunoelectrophoresis for diagnosis of human hydatidosis. Clin Lab. 2011;57:201–205.
37. Bojanich MV, Marino GL, Lopez MA, Alonso JM. An evaluation of the dot-ELISA procedure as a diagnostic test in an area with a high prevalence of human Toxocara canis infection. Mem Inst Oswaldo Cruz. 2012;107:194–197. doi:10.1590/S0074-02762012000200007
38. Yunuen Cervantes-Landína A, Martínez-Martínez I, Reyes PA, Shabibc M, Espinoza Gutiérrez B. Estandarización de la técnica Dot-ELISA para la detección de anticuerpos anti-Trypanosoma cruzi y su comparación con ELISA y Western blot. Enferm Infecc Microbiol Clin. 2014;32:363–368.
39. Harning D, Spenter J, Metsis A. Recombinant Toxoplasma gondii surface antigen 1 (P30) expressed in Escherichia coli is recognized by human Toxoplasma-specific immunoglobulin M (IgM) and IgG antibodies. Clin Diagn Lab Immunol. 1996;3(3):355–357.
40. Quail MA. DNA Cloning. Encyclopedia of Life Sciences. New York: John Willey and Sons Ltd; 2010. doi:10.1038/npg.els.0005344
41. Khan AM, Miotto O, Heiny A, et al. A systematic bioinformatics approach for selection of epitope-based vaccine targets. Cell Immunol. 2006;244:141–147. doi:10.1016/j.cellimm.2007.02.005
42. Burg JL, Perelman D, Kasper LH, Ware PL, Boothroyd JC. Molecular analysis of the gene encoding the major surface antigen of Toxoplasma gondii. J Immunol. 1988;141:3584–3591.
43. Cesbron-Delauw MF, Tomavo S, Beauchamps P, et al. Similarities between the primary structures of two distinct major surface proteins of Toxoplasma gondii. J Biol Chem. 1994;26:16217–16222.
44. Idicula-Thomas S, Balaji PV. Understanding the relationship between the primary structure of proteins and its propensity to be soluble on over expression in Escherichia coli. Protein Sci. 2005;14:582–592.
45. Rosano GL, Ceccarelli EA. Rare codon content affects the solubility of recombinant proteins in a codon bias-adjusted Escherichia coli strain. Microb Cell Fact. 2009;24:8–41.
46. Lilie H, Schwarz E, Rudolph R. Advances in refolding of proteins produced in E. coli. Curr Opin Biotechnol. 1998;9:497–501. doi:10.1016/S0958-1669(98)80035-9
47. Wu K, Chen XG, Li H, et al. Diagnosis of human toxoplasmosis by using the recombinant truncated surface antigen 1 of Toxoplasma gondii. Diagn Microbial Infect Dis. 2009;64:261–266. doi:10.1016/j.diagmicrobio.2009.02.009
48. Pietkiewicz H, Hiszczyn ska-Sawicka E, Kur J, et al. Usefulness of Toxoplasma gondii-specific recombinant antigens in serodiagnosis of human toxoplasmosis. J Clin Microbiol. 2004;42:1779–1781. doi:10.1128/JCM.42.4.1779-1781.2004
49. Jacobs D, Dubremetz JF, Loyens A, Bosman F, Saman E. Identification and heterologous expression of a new dense granule protein (GRA7) from Toxoplasma gondii. Mol Biochem Parasitol. 1998;91:237–249.
50. Nigro M, Gutierrez A, Hoffer AM, et al. Evaluation of Toxoplasma gondii recombinant proteins for the diagnosis of recently acquired toxoplasmosis by an immunoglobulin G analysis. Diagn Microbiol Infect Dis. 2003;47:609–613. doi:10.1016/S0732-8893(03)00156-1
51. Kotresha D, Poonam D, Muhammad Hafiznur Y, et al. Recombinant proteins from new constructs of SAG1 and GRA7 sequences and their usefulness to detect acute toxoplasmosis. Trop Biomed. 2012;29:129–137.
52. Fischer HG, Stachelhaus S, Sahm M, Meyer HE, Reichmann G. GRA7, an excretory 29 kDa Toxoplasma gondii dense granule antigen released by infected host cells. Mol Biochem Parasitol. 1998;91:251–262.
53. Neudeck A, Stachelhaus S, Nischik N, Striepen B, Reichmann G, Fischer HG. Expression variance, biochemical and immunological properties of Toxoplasma gondii dense granule protein GRA7. Microbes Infect. 2002;4:581–590. doi:10.1016/S1286-4579(02)01576-9
54. Tenter AM, Johnson AM. Recognition of recombinant Toxoplasma gondii antigens by human sera in an ELISA. Parasitol Res. 1991;77:197–203. doi:10.1007/BF00930858
55. Johnson AM, Roberts H, Tenter AM. Evaluation of a recombinant antigen ELISA for the diagnosis of acute toxoplasmosis and comparison with traditional antigen ELISAs. J Med Microbiol. 1992;37:404–409. doi:10.1099/00222615-37-6-404
56. Li S, Galvan G, Araujo FG, Suzuki Y, Remington JS, Parmley S. Serodiagnosis of recently acquired Toxoplasma gondii infection using an enzyme-linked immunosorbent assay with a combination of recombinant antigens. Clin Diagn Lab Immunol. 2000;7:781–787.
57. Gondim L, Mineo JR, Schares G. Importance of serological cross-reactivity among Toxoplasma gondii, Hammondia spp., Neospora spp., Sarcocystis spp. and Besnoitia besnoiti. Parasitology. 2017;144(7):851–868. doi:10.1017/S0031182017000063
58. Lyashchenko KP, Singh M, Colangeli R, Gennaro ML. A multi-antigen print immunoassay for the development of serological diagnosis of infectious diseases. J Immunol Methods. 2000;242:91–100.
59. Costa JG, Vilarino MJ. Semi quantitative Dot Blot with the GRA8 antigen to differentiate the stages of toxoplasmosis infection. J Microbiol Methods. 2018;149:9–13. doi:10.1016/j.mimet.2018.04.015
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.