Back to Journals » International Journal of Nanomedicine » Volume 11
Development of self-nanoemulsifying drug delivery systems for the enhancement of solubility and oral bioavailability of fenofibrate, a poorly water-soluble drug
Authors Mohsin K ![]() , Alamari R, Ahmad A, Raish M
, Alamari R, Ahmad A, Raish M ![]() , Alanzi F, Hussain MD
, Alanzi F, Hussain MD ![]()
Received 13 January 2016
Accepted for publication 14 April 2016
Published 14 June 2016 Volume 2016:11 Pages 2829—2838
DOI https://doi.org/10.2147/IJN.S104187
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Kazi Mohsin,1 Rayan Alamri,1 Ajaz Ahmad,2 Mohammad Raish,3 Fars K Alanazi,1 Muhammad Delwar Hussain4
1Kayyali Chair for Pharmaceutical Industry, Department of Pharmaceutics, 2Department of Clinical Pharmacy, 3Department of Pharmaceutics, College of Pharmacy, King Saud University, Riyadh, Saudi Arabia; 4Department of Pharmaceutical and Biomedical Sciences, College of Pharmacy, California Health Science University, Clovis, CA, USA
Background: Self-nanoemulsifying drug delivery systems (SNEDDS) have become a popular formulation option as nanocarriers for poorly water-soluble drugs. The objective of this study was to investigate the factor that can influence the design of successful lipid formulation classification system (LFCS) Type III SNEDDS formulation and improve the oral bioavailability (BA) of fenofibrate.
Materials and methods: LFCS Type III SNEDDS were designed using various oils, water-soluble surfactants, and/or cosolvents (in considering the polarity of the lipids) for the model anticholesterol drug, fenofibrate. The developed SNEDDS were assessed visually and by measurement of the droplet size. Equilibrium solubility of fenofibrate in the SNEDDS was conducted to find out the maximum drug loading. Dynamic dispersion studies were carried out (1/100 dilution) in water to investigate how much drug stays in solution after aqueous dispersion of the formulation. The BA of SNEDDS formulation was evaluated in the rat.
Results: The results from the characterization and solubility studies showed that formulations containing mixed glycerides were highly efficient SNEDDS as they had higher solubility of the drug and produced nanosized droplets. The dispersion studies confirmed that SNEDDS (containing polar mixed glycerides) can retain >98% drug in solution for >24 hours in aqueous media. The in vivo pharmacokinetics parameters of SNEDDS formulation in comparison with pure drug showed significant increase in Cmax and AUC0–t, ~78% and 67%, respectively. The oral BA of fenofibrate from SNEDDS in rats was ~1.7-fold enhanced as compared with the BA from pure drug.
Conclusion: Fenofibrate-loaded LFCS Type III SNEDDS formulations could be a potential oral pharmaceutical product for administering the poorly water-soluble drug, fenofibrate, with an enhanced oral BA.
Keywords: lipid-based formulation, self-nanoemulsifying drug delivery systems, fenofibrate, solubility improvement, oral bioavailability
Introduction
The discovery and development of a safe and effective drug involve balancing efficacy with factors such as bioavailability (BA), toxicity, and disposition within the body. If the drug is hydrophobic or poorly water soluble, oral administration may lead to poor systemic exposure, presenting a considerable technical challenge and possible requirement of parenteral administration. In recent years, two-thirds of all the new chemical entities identified in drug discovery programs have been emerging as poorly aqueous-soluble compounds (solubility, 100 μg/mL).1 These new chemical entities, which are lipophilic molecules, present great challenge to the formulators due to their poor solubility and erratic absorption from the gastrointestinal (GI) tract following oral administration.
Lipid-based dosage forms have gained high priority and become more prominent in recent years in pharmaceutical industries. Among several approaches, which are currently available to incorporate drugs into lipid vehicles resulting in a variety of dosage forms, self-emulsifying drug delivery systems (SEDDS), self-microemulsifying drug delivery systems, and self-nanoemulsifying drug delivery systems (SNEDDS) have proved to be the most successful approaches in improving the solubility and BA of drugs belonging to Biopharmaceutical Classification Systems (BCS) Classes II and IV.2 These systems advantageously present the drug in dissolved form, and their relatively smaller droplet size provides a large interfacial area enhancing the activity of pancreatic lipase to hydrolyze triglycerides, thereby promoting faster release of the drug and/or formation of mixed micelles of bile salts containing the drug.3–5 The development of Neoral® (cyclosporin A) as a commercial product exhibits an excellent example of the utilization of these systems.6,7
Fenofibrate is a lipid-regulating agent that has chemical, pharmacological, and clinical similarities to the other fibrate drugs, such as clofibrate and gemfibrozil.8 The chemical structure of fenofibrate is shown in Figure 1. Fenofibrate is a BCS Class II drug with various available doses (45 mg, 54 mg, 100 mg, 145 mg, 160 mg, and 200 mg).9 The low oral BA of fenofibrate may be due to its solubility and dissolution limitations. It is a nonelectrolyte, small lipophilic ester molecule (molecular weight 360.8) with low aqueous solubility (<3 μg/mL) and has a fairly high octanol/water partition coefficient (log P=4.6).10
 | Figure 1 Chemical structure of fenofibrate. |
A lipid formulation classification system (LFCS) with four categories has been proposed for lipid-based formulations based on the aqueous dispersion and water solubility of the excipient blends.11 The information of LFCS is quite substantial, to gain knowledge of excipient selection and compare the performance of various lipid-based drug delivery systems. However, the key point of the LFCS is to identify the most suitable formulations for specific drugs relating to their physicochemical properties. Previous studies have shown that Type III formulation systems are the most efficient formulations for hydrophobic drugs such as cyclosporin A, ritonavir, saquinavir, etc.12,13 Therefore, in this study, LFCS IIIA and IIIB systems, particularly SNEDDS, have been used for the formulation of the hydrophobic model drug, fenofibrate.
We investigated the development aspects of Type III LFCS lipid formulation, issues, and the fate of fenofibrate in lipid systems. Significant effort in this research study was aimed at increasing the solubilization of fenofibrate by the formation of successful SNEDDS (transparent micellar systems) with improved oral BA.
Materials and methods
Materials
All chemicals used in this study were obtained from commercial suppliers. Fenofibrate, (2-[4-(4-chlorobenzoyl) phenoxy]-2-methylpropionic acid 1-methylethyl ester) and its metabolite, fenofibric acid (FA, purity >99.5%), were supplied by Sigma-Aldrich Co. (St Louis, MO, USA). The internal standard fluvastatin was obtained from Riyadh Pharma Industry Ltd. (Riyadh, Saudi Arabia). Details of the lipids (oils and nonionic surfactants), their compositions, and suppliers are provided in Table 1. All excipients were used without further purification. High performance liquid chromatography (HPLC) grade methanol, sodium dihydrogen phosphate, and sodium chloride were purchased from BDH Laboratory Supplies (Poole, UK). The 1 M HCl, which was diluted to obtain 0.1 M solution, was provided by Avonchem (Macclesfield, Cheshire, UK). Rat plasma containing ethylenediaminetetraacetic acid as anticoagulant was collected in-house. Water used in this study was obtained from a Milli-Q water purification system (Sartorius, Geottingen, Germany). All other chemicals and solvents were of analytical purity.
 | Table 1 Description of the lipids and their suppliers, used for the formulation development |
Animals
Male Wistar albino rats weighting 180–220 g were obtained from Experimental Animal Care Center, College of Pharmacy, King Saud University. The animals were maintained under controlled conditions of temperature (22°C±1°C), humidity (50%–55%), light (12-hour light/12-hour dark cycle) with free access to rat chow (Grain Silos & Flour Mills Organization, Riyadh, Saudi Arabia), and drinking water. Animals were acclimatized to the laboratory conditions for 7 days prior to experiments. All experimental procedures including handling, treatment, and euthanasia were conducted in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH Publications no 80-23; 1996) as well as the Ethical committee of Experimental Animal Care Center, College of Pharmacy, King Saud University (approval 0389-EACC, December 2014). The animals were divided into two groups of six rats each. Group 1 served as a control and was administered fenofibrate in normal saline. Group II was administered an SNEDDS formulation diluted with saline.
Experimental methods
Design of LFCS IIIA and IIIB SNEDDS
A number of oils and surfactants were blended to prepare SNEDDS lipid-based formulations. The formulations were prepared with varying concentrations of oil, surfactant, and hydrophilic cosolvents by a simple preparation method. Initially, a primary oil mixture was prepared with different oils. Then, a surfactant and a cosolvent were added to the oil at various ratios. The final mixture was vortexed until homogeneity was achieved. The preconcentrate was kept in an airtight 3 mL glass tube until use. Subsequently, the most interesting LFCS Type IIIA and IIIB formulations, which are likely to produce SNEDDS, have been investigated carefully using the model drug fenofibrate. Table 2 shows the formulations used in this study.
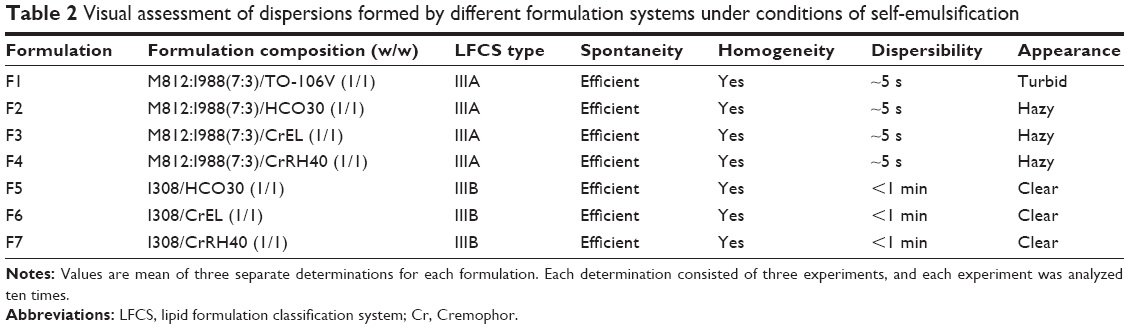 | Table 2 Visual assessment of dispersions formed by different formulation systems under conditions of self-emulsification |
Visual assessment of the formulations
Within the context of self-emulsifying efficiency, a visual assessment is very common and able to minimize the excess usage of chemicals by trial and error. In this study, visual assessment method was used to examine the self-emulsification properties of the formulation.14,15 For sample preparation, 100 μL of each formulation was diluted with 50 mL of water in a 50 mL volumetric flask (1:500 dilution) and agitated gently for 1 minute at room temperature. The miscibility, homogeneity, and the appearance were evaluated visually.
Determination of droplet size and polydispersity index
The droplet size of the emulsion is a crucial factor to its performance because it could determine the rate and extent of drug release as well as absorption. The droplet size and polydispersity index of the diluted LFCS Type III SEDDS/SNEDDS were measured by laser diffraction analysis using Zetasizer Nano (Model ZEN3600; Malvern Instruments, Malvern, UK) particle sizing systems. The formulations were diluted at a ratio of 1:1,000 v/v (SEDDS/SNEDDS:distilled water) and mixed for 1 minute before testing. The diluted samples were placed directly into the cuvette, and the data were collected ten times. All experiments were performed in triplicates.
Equilibrium solubility of fenofibrate in LFCS IIIA and IIIB formulations
The solubility of fenofibrate within the SEDDS/SNEDDS was determined using a shake flask method to observe how the drug solubility is changed as water is incorporated into the system. The samples were prepared by adding an excess amount of drug to the formulation, which was then shaken and thoroughly mixed with a vortex mixer. Three to six replicates were performed for each formulation. The samples were incubated in a dry heat incubator at 37°C for 7 days. The samples were centrifuged in 1.5-mL microfuge tubes at 2,500× g to separate excess solid drug from the dissolved drug. An aliquot of the supernatant was weighed and diluted in an appropriate solvent. The dissolved fenofibrate was analyzed by using an ultrahigh-performance liquid chromatography (UHPLC) method developed by our group.10
Influence of pH on fenofibrate solubility
Although the solubility of the drug in water is the underlying driver for solubility in the GI fluids, the solubility in the GI tract may additionally be influenced by the pH profile of the GI tract. The pH of the GI tract may have a significant influence on the regional absorption rate for drugs that ionize in this range. To investigate the fate of ester drug fenofibrate in GI tract on dispersion, one of the SNEDDS formulations, F5, was investigated. The solubility experiments were conducted following the solubility method described previously, by diluting with water (pH 6.0), 0.1 M HCl (pH 1.1), and phosphate-buffered saline (PBS, pH 7.5). The influence of pH solubility of fenofibrate was examined in the formulations of I308/HCO30/aqueous system.
Dynamic dispersion studies
Fenofibrate was dissolved in each SEDDS/SNEDDS at 80% saturation level based on its equilibrium solubility studies in the relevant anhydrous formulation. All of the formulations investigated in the equilibrium solubility studies were included in the corresponding dynamic dispersion studies to examine whether the drug will precipitate during dispersion in aqueous media and the rate of precipitation. One gram of each formulation was dropped into 100 mL of water in a glass jar and kept in a dry heat incubator at 37°C for 24 hours. During this 24-hour period, 1 mL of the dispersed sample from each container was withdrawn periodically (0–24 hours) and centrifuged at 2,500× g. A 100 μL aliquot of the resulting clear supernatant was assayed by the UHPLC method10 to find out the amount of the drug that remained dissolved in the sample. All of the experiments were performed in triplicates.
BA studies
The BA study was performed in male albino Wistar rats. The BA of SNEDDS formulation of fenofibrate, F1, M812:I988(7:3)/TO-106V (1/1), was compared with fenofibrate powder. The rats were fasted overnight before dosing but allowed free access to water during the whole experiment and were randomly allocated to two groups: fenofibrate SNEDDS (group A) and fenofibrate raw powder (group B) in a crossover design.16 Both SNEDDS formulations of fenofibrate (diluted with normal saline, 1:10 ratio) and fenofibrate powder (suspended in saline, 10.6 mg/mL) were administered orally at a dose equivalent to 9 mg/kg of fenofibrate. Blood samples (0.5 mL) were taken from fossa orbital’s vein at 0.0 hour, 0.5 hour, 1 hour, 1.5 hours, 3 hours, 4.5 hours, 6 hours, 12 hours, 24 hours, and 36 hours after drug administration, in heparinized tubes. Plasma was separated from the blood samples by centrifugation at 2,500× g for 10 minutes and stored at −20°C until analysis.
UHPLC analysis of plasma samples
Fenofibrate is a prodrug that is biotransformed by tissue and plasma esterases to the active metabolite FA. Therefore, no fenofibrate is detectable in the plasma after oral administration. Accordingly, the pharmacokinetic assessment of fenofibrate is based on the concentration of FA in the plasma. Liquid–liquid extraction procedure was used for the extraction of FA from the rat plasma.16,17 The plasma samples were transferred into a series of 1.5 mL centrifugation tubes. A fixed amount of internal standard (fluvastatin) solution (25 μg/mL) was added to the plasma sample and vortexed. Plasma precipitation was carried out using methanol (1 mL) and vortexed for 5 minutes. The tubes were centrifuged for 10 minutes at 2,500× g. The whole supernatant (organic layer) was transferred into clean centrifuge tubes and was evaporated to dryness under nitrogen gas at 45°C–50°C. Dry residues were then reconstituted in 225 μL of mobile phase and vortexed. The concentration of FA in the reconstituted residue was determined by a modified UHPLC method reported earlier.18
Briefly, the UHPLC system consisted of a Dionex® solvent manager equipped with a Dionex® automatic sample manager and a photodiode array detector (Thermo Fisher Scientific, Waltham, MA, USA). The mobile phase was an isocratic mixture of methanol and water in a ratio of 65%:35% (v/v). Freshly prepared mobile phase was filtered through an online 0.20 μm filter and degassed continuously by an online degasser within the UHPLC system. The flow rate of the mobile phase was 0.3 mL/min. An Acquity® UPLC BEH C18 column (2.1×50 mm2, 1.7 μm) kept at 25°C was used for the analysis. The total run time was 2.5 minutes. The detector wavelength was set at 284 nm, and the injection volume was 1.0 μL. The developed method was validated as per International Conference on Harmonization guidelines. The linearity of the method was found to be suitable in the range of 0.1–10 μg/mL (r2=0.9993).
Pharmacokinetic data analysis
A noncompartmental pharmacokinetic analysis was used to determine the pharmacokinetic behavior of fenofibrate. The pharmacokinetic parameters were calculated using PK solver program (Microsoft Excel). Area under plasma concentration–time curve (AUC) was calculated using linear trapezoid method. The elimination rate constant (Kel) was calculated from the slope of the logarithm of the plasma concentration versus time. The relative BA of the SNEDDS to the control was calculated as follows: relative BA% = AUCSNEDDS/AUCcontrol. The apparent elimination half-life (T1/2) was calculated as 0.693/Kel. The maximum plasma concentration (Cmax) and time to maximum concentration (Tmax) after oral administration were determined directly from the time–concentration curve.
Statistical analysis of pharmacokinetic data
Differences in pharmacokinetic parameters (eg, oral clearance, volume of distribution at steady state, and AUC0–∞) of SNEDDS formulation of fenofibrate and control fenofibrate powder were assessed by paired t-test using Graph Pad Prism Version 3.00 for Windows (GraphPad Software, Inc., La Jolla, CA, USA). Statistical significance was assumed when P≤0.05.
Results and discussion
Visual assessment of the formulation
The following visual assessments were chosen for optimization of formulations: miscibility of the oil/surfactant mixture, homogeneity, dispersion time, and appearance upon dilution with water. The ratio of dilution 1:500 was maintained for all the formulations. Within the scope of this study, if the formulations were homogeneous and took less time to disperse (ie, <1 minute and well dispersed), they were considered to be efficient. The results from the efficiency assessment (Table 2) showed that all the seven formulations were found to be promising in terms of their assessment criteria and were used for further optimization. Most of these formulations were considered as SNEDDS due to their hazy and transparent appearances. Visual observation, which is a primary means of assessment to differentiate good and poor formulations, may be enough for an experienced formulator.
Determination of droplet size and polydispersity index
The droplet size analysis of all the seven formulations indicated that they form nanosized droplets upon dilution with water. All the formulations formed droplets with average size ranges of 40–67 nm except formulation F1, which formed droplets with an average size of 220 nm (Table 3). Although the formulation F1 (contained 50% surfactant TO-106V) formed larger particle size, it was found to be a stable formulation upon dilution.
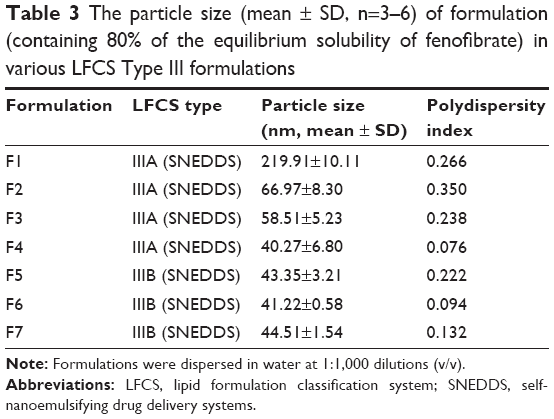 | Table 3 The particle size (mean ± SD, n=3–6) of formulation (containing 80% of the equilibrium solubility of fenofibrate) in various LFCS Type III formulations |
There are two criteria commonly being proposed to describe the efficiency of self-emulsifying formulation: 1) the rate of emulsification and 2) the particle size distribution of the resultant emulsion. Particle size plays a key role in oral absorption of the drug in vivo. The smaller the droplet size, the larger the interfacial surface area that will be provided for drug absorption, although it should be recognized that the dispersion may be modified substantially by digestion. The performance of lipid-based delivery systems is governed by their fate in the GI tract, rather than the particle size of the initial dispersion.11,19,20
Photon correlation spectroscopy (PCS) is useful and a common method for the determination of emulsion droplet size.19,21,22 The PCS technique can best fit if the emulsion properties are not changed following the substantial aqueous dilutions necessary for applying this method. SNEDDS can be easily distinguished by PCS, based on the droplet size and dispersion, which are stable, isotropic, and clear oil in water (O/W) dispersions.23,24
Effect of surfactant on the droplet size of the formulation
Surfactants are critical factors, which can determine the formation of SNEDDS during formulation development and stability of the nanosize of the droplets after the aqueous dilution of self-emulsifying formulations.25
Figure 2 demonstrated that different surfactants (from lipophilic to hydrophilic: TO-107V, HCO30, Cremophor EL, and Cremophor RH40) with the same lipid components showed significant differences in droplet sizes. The hydrophilic surfactant (HCO30, Cremophor EL, Cremophor RH40) with lipid mix M812/I988 (F2–F4) produced lower droplet sizes around 40–67 nm compared with the lipophilic surfactant TO-106V in formulation F1 (droplet size around 220 nm). These results strongly support the previous findings that a surfactant of high hydrophilic lipophilic balance (HLB) forms the finest droplet sizes.11 The self-emulsifying efficiency of any formulation (SEDDS/SNEDDS) is strongly associated with the mean droplet size of the produced emulsion.14,26 The results in Figure 2 suggest that the surfactant has a significant role on the formation of SNEDDS and its stability in the GI tract during aqueous dispersion.
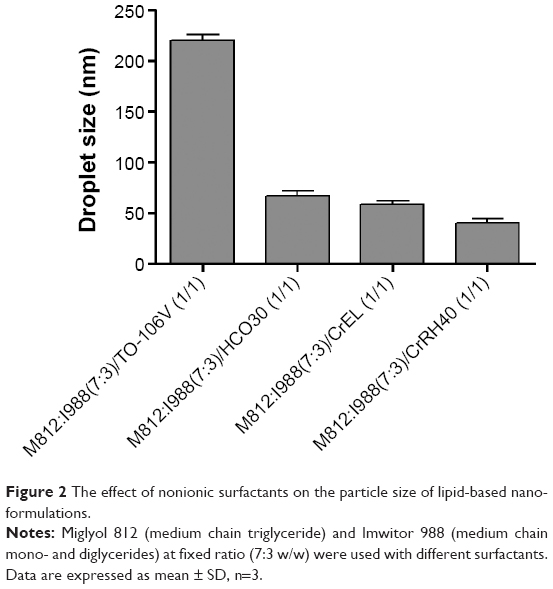 | Figure 2 The effect of nonionic surfactants on the particle size of lipid-based nano-formulations. |
Equilibrium solubility studies
Equilibrium solubility is an important element for any drug compound because it provides the necessary information for maximum dose that can be incorporated in a single unit capsule/tablet. All anhydrous formulations were stored in this study for 7 days at 37°C temperature, as a typical period, to make sure that the equilibrium had been achieved.10 Equilibrium solubility of fenofibrate in various alternative Type III lipid-based formulations (seven formulations) is presented in Table 4.
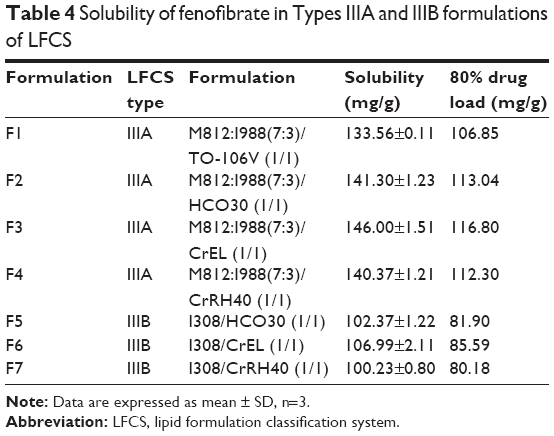 | Table 4 Solubility of fenofibrate in Types IIIA and IIIB formulations of LFCS |
Among Type IIIA and IIIB formulations, fenofibrate was significantly soluble in Type IIIA systems (solubility was between 133 mg/g and 140 mg/g in all F1–F4 formulations). In Type IIIB formulations, fenofibrate solubility dropped down from 100 mg/g to 107 mg/g in all F5–F7 formulations. The higher solubility in Type IIIA suggests that fenofibrate can be dissolved in a higher amount in the formulation containing mix mono-, di-, and triglycerides (ie, M812/I988) than the formulation containing monoglycerides itself (ie, I308).
Influence of pH on the solubility of fenofibrate
Physicochemical properties of a drug and physiological factors in the GI tract greatly affect the solubility and the GI absorption of the drug. The physicochemical properties include lipophilicity, ionization, and chemical stability of the drug in the GI tract. As many drugs are weak electrolytes (acids or bases), their solubility is dependent on their ionization constant (usually denoted by the pKa) and the pH of the dissolution media. The pH of the GI fluids widely varies with location in the GI tract. In the fasted stomach, typically, the pH values are in the range of 1–2, while in the upper small intestine, the pH values lie between 5 and 6.5. It is advisable to choose formulation pH that is close to the pH environment at the targeted dosing site. To investigate the effect of pH on the solubility of fenofibrate, a selected formulation, F5 (I308/HCO30, 1/1 w/w), was diluted with PBS (pH 7.5) and 0.1 M HCl (pH 1.1) and water (pH 6.0).
The solubility of fenofibrate was high (~102.37 mg/g) in the anhydrous formulation, but decreased in all the aqueous media – water, PBS, and 0.1 M HCl. However, the solubilities of fenofibrate were considerably higher after dilution with PBS of the F5 formulation, in comparison with water and 0.1 M HCl. The solubility profile in Figure 3 shows that pH change in the formulation had negligible effect on the solubility of fenofibrate, which was expected for this nonelectrolyte drug. The pH of the microenvironment within the diluted formulations may not have drastic effect on the solubility of fenofibrate during equilibration or dispersion.
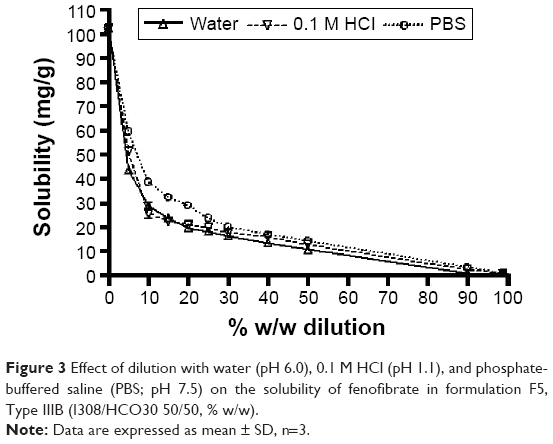 | Figure 3 Effect of dilution with water (pH 6.0), 0.1 M HCl (pH 1.1), and phosphate-buffered saline (PBS; pH 7.5) on the solubility of fenofibrate in formulation F5, Type IIIB (I308/HCO30 50/50, % w/w). |
Dynamic dispersion and drug precipitation studies
Results from the fenofibrate precipitation experiment over 24 hours in aqueous media show that formulations F1 and F3 maintained almost 90% drug in solution whereas F5, F6, and F7 precipitated 70% out of the solution (Figure 4). The overall dispersion studies confirmed that the mixed glycerides can retain a high percentage of drugs in solution for >24 hours in the intestinal media. Thus, the BA of fenofibrate can be significantly increased if it stays in solubilized form during the digestion time (around 4 hours) in vivo.
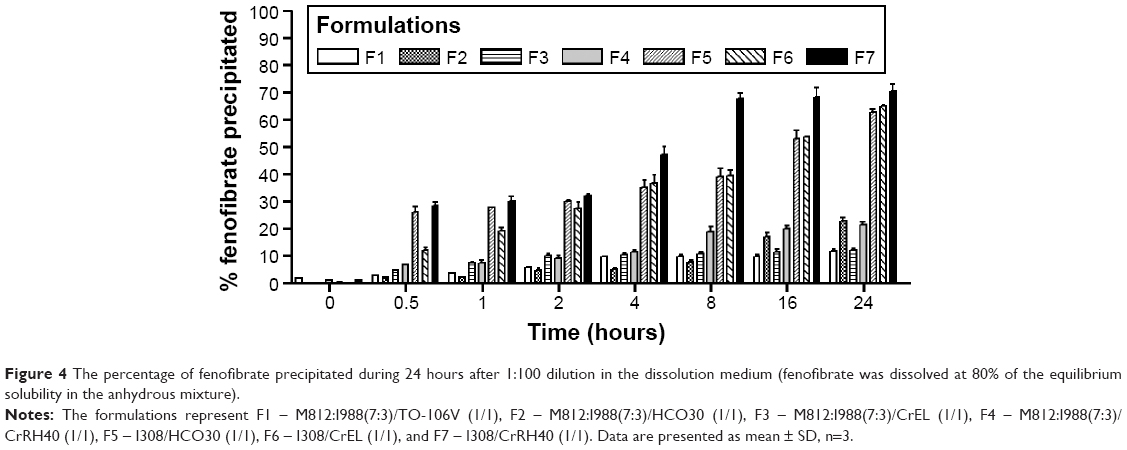 | Figure 4 The percentage of fenofibrate precipitated during 24 hours after 1:100 dilution in the dissolution medium (fenofibrate was dissolved at 80% of the equilibrium solubility in the anhydrous mixture). |
In vitro dispersion tests assess the ability of lipid-based vehicles to disperse into various types of media and to assess whether the drug partitions from the vehicle into the aqueous medium. There are multiple roles for in vitro dispersion tests, which are employed to guide drug development and selection of appropriate formulations for further in vivo studies. A range of biorelevant dissolution test media and experimental methodologies have been developed that have application in drug release studies from lipid-based oral formulations.27,28 However, an in vitro dispersion test in aqueous media can estimate how much drug will be in solution before absorption and thus may predict the fate of the drug in vivo.
Dispersion testing can be carried out using a standard dissolution apparatus but, assuming that the drug is initially in solution in the anhydrous formulation, the emphasis should be on detecting unwanted precipitation of the drug rather than dissolution. This is why, the dynamic dispersion test was important in the current studies. In vitro dispersion tests are appropriate for the prediction of whether precipitation is likely to occur prior to digestion. In this study, samples were removed from the dispersion vessel at various intervals within 24 hours and analyzed to determine the likelihood of precipitation during GI transit.
To avoid precipitation of the drug upon dispersion is the desired goal for pharmaceutical applications. Correlations between the investigations of the equilibrium solubility of the drug in the aqueous diluted formulation and corresponding dynamic precipitation tests could help to predict whether precipitation is likely to take place and whether it would affect BA.10 Increasing the solubilization capacity of the formulation extensively over the desired drug concentration could help avoid in vivo drug precipitation. Another possible method to slow down or prevent drug precipitation is to formulate with hydrophilic polymeric ingredients in the formulation that act as precipitation inhibitors.29–31
In vivo absorption study
The in vivo pharmacokinetic behavior of a selected SNEDDS formulation (F1) was studied to quantify FA in the plasma after oral administration of fenofibrate. The aim was to correlate the enhancement of solubility and percentage of solubilized drug with the enhancement of its BA. The pharmacokinetics of SNEDDS containing fenofibrate on comparison with pure drug suspension demonstrated the enhancement of BA of the drug (Table 5 and Figure 5). Fenofibrate is reported to be well absorbed after oral administration, with peak plasma levels attained in 6–8 hours.32 The Cmax of FA after oral administration of fenofibrate powder was 549.39±10.21 ng/mL and the Tmax was 6 hours, while in the oral administration SNEDDS formulation of fenofibrate the Cmax and Tmax were 977.35±13.09 ng/mL and 6 hours, respectively. The Cmax value of FA from the SNEDDS formulation was significantly increased (78%, P<0.0001). The AUC0–t of FA was also significantly increased in the SNEDDS-treated group as compared to only fenofibrate-treated group (67%, P<0.0001), from 7,419.50±78.13 ng h/mL to 12,414.46±86.28 ng h/mL, respectively. The improvement in BA of fenofibrate from SNEDDS formulation may be due to decreased particle size and increased solubility of fenofibrate. The increase in relative BA was found to be 1.7-fold. The calculated oral clearance was significantly decreased (41%, P<0.05) from 0.79±0.12 mL/kg to 0.56±0.09 mL/kg, while the estimated oral volume of distribution at steady state was significantly decreased (98%, P<0.001) from 29.23±5.36 mL/kg to 14.78±3.25 mL/kg. The BA study in rats shows that the BA of fenofibrate is improved by SNEDDS formulation. The enhanced oral BA of fenofibrate from SNEDDS formulation may be due to the increased solubility and dispersion rate of the drug from the SNEDDS formulation.
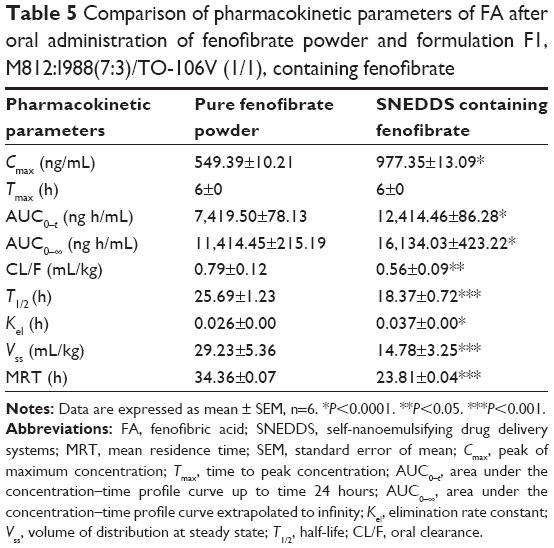 | Table 5 Comparison of pharmacokinetic parameters of FA after oral administration of fenofibrate powder and formulation F1, M812:I988(7:3)/TO-106V (1/1), containing fenofibrate |
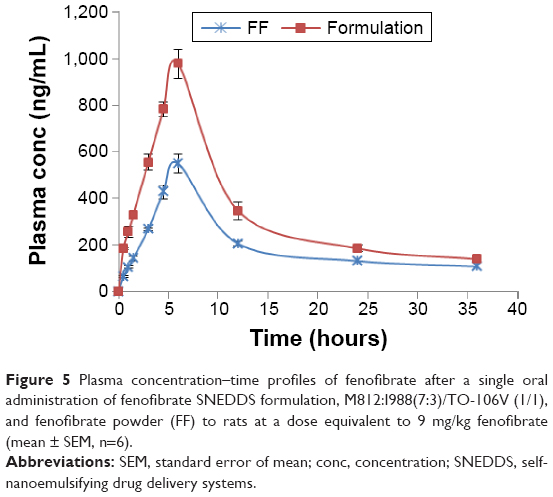 | Figure 5 Plasma concentration–time profiles of fenofibrate after a single oral administration of fenofibrate SNEDDS formulation, M812:I988(7:3)/TO-106V (1/1), and fenofibrate powder (FF) to rats at a dose equivalent to 9 mg/kg fenofibrate (mean ± SEM, n=6). |
There are several possible mechanisms that could increase the BA of SNEDDS. It is assumed that the faster uptake of drug-loaded formulations (Type III systems) from the resultant emulsion by the enterocytes at the absorption site could initiate rapid absorption of fenofibrate from the SNEDDS.33 High drug solubilization capacity and self-emulsifying ability of the formulation F1 may have contributed to the increased BA of fenofibrate. The formulation F1 contains TO-106V (PEG-6 sorbitan oleate) as surfactant, which may have also modified the permeability by disturbing the cell membrane. The SNEDDS may also increase lymphatic transport of fenofibrate through transcellular pathway.34
The formulation F1 may minimize the variable effect of food (food effect) on the absorption of fenofibrate. The food effect is an important factor for the BA of many poorly water-soluble drugs, and SNEDDS formulations can avoid this food effect.35 Thus, the formulation F1 has potential for enhancing the BA and also minimizing the food effect during oral administration of fenofibrate in humans.
Our studies showed that LFCS Type IIIA formulations are efficient for the hydrophobic drug fenofibrate. The particle sizes of formulation F1 were much larger (220 nm) than the other formulations studied, such as F2, F3, and F4 of the Type IIIA. These formulations have particle sizes of 40–67 nm (Table 3) and showed higher solubility of fenofibrate (140–146 mg/g, Table 4). Formulations F2, F3, and F4 maintained almost 90% drug in solution up to 4 hours during dynamic dispersion and drug precipitation studies (Figure 4). Thus, in addition to formulation F1, these formulations (F2, F3, and F4) have potential to significantly enhance the BA of fenofibrate if it stays in solubilized form during the digestion time (around 4 hours) in vivo. Further studies are needed for the optimized SNEDDS formulation to further increase the BA of fenofibrate.
Conclusion
The SNEDDS have high potential to improve the BA of poorly soluble lipophilic compounds. Nevertheless, the unique characteristics of this lipid-based dosage form present significant challenges to scientists in many ways. For example, the safety issues need to be considered, particularly when a new excipient is used in the formulation. The research in this study shows that, despite the diversity and complexity, there are general considerations that one can follow in selecting and developing SNEDDS formulations for poorly water-soluble drugs. These include investigation of 1) the LFCS and the role of lipid compositions, 2) dynamic dispersion studies that provide the rationale for minimizing the tendency of drug precipitation from the lipid formulation systems, and 3) the BA studies of the SNEDDS.
The in vitro and in vivo studies indicated that the solubility and ultimately the oral BA of fenofibrate were increased by the SNEDDS formulation.
Acknowledgments
This project was funded by National Plan for Science, Technology and Innovation (MAARIFAH) and King Abdulaziz City for Science and Technology, Kingdom of Saudi Arabia, Award Number (12MED2524-02).
Disclosure
The authors report no conflicts of interest in this work.
References
Hauss DJ. Oral lipid-based formulations. Adv Drug Deliv Rev. 2007;59(7):667–676. | ||
Kohli K, Chopra S, Dhar D, Arora S, Khar RK. Self-emulsifying drug delivery systems: an approach to enhance oral bioavailability. Drug Discov Today. 2010;15(21–22):958–965. | ||
Shah NH, Phuapradit W, Zhang Y-E, Ahmed H, Malick AW. Lipid-based isotropic solutions: design considerations. In: Hauss DJ, editor. Oral Lipid-Based Formulations: Enhancing the Bioavailability of Poorly Water-Soluble Drugs. New York: Informa Healthcare; 2007:129–148. | ||
Pouton CW. Self-emulsifying drug delivery systems – assessment of the efficiency of emulsification. Int J Pharm. 1985;27(2–3):335–348. | ||
Shah NH, Carvajal MT, Patel CI, Infeld MH, Malick AW. Self-emulsifying drug delivery systems (SEDDS) with polyglycolyzed glycerides for improving in vitro dissolution and oral absorption of lipophilic drugs. Int J Pharm. 1994;106(1):15–23. | ||
Hörter D, Dressman JB. Influence of physicochemical properties on dissolution of drugs in the gastrointestinal tract. Adv Drug Deliv Rev. 2001;46(1–3):75–87. | ||
Chono S, Takeda E, Seki T, Morimoto K. Enhancement of the dissolution rate and gastrointestinal absorption of pranlukast as a model poorly water-soluble drug by grinding with gelatin. Int J Pharm. 2008;347(1–2):71–78. | ||
Devraj R, Williams HD, Warren DB, Mohsin K, Porter CJ, Pouton CW. In vitro assessment of drug-free and fenofibrate-containing lipid formulations using dispersion and digestion testing gives detailed insights into the likely fate of formulations in the intestine. Eur J Pharm Sci. 2013;49(4):748–760. | ||
Ashok RP, Pradeep RV. Preparation and in vivo evaluation of SMEDDS (self-microemulsifying drug delivery system) containing fenofibrate. AAPS J. 2007;9(3):344–352. | ||
Mohsin K, Long MA, Pouton CW. Design of lipid-based formulations for oral administration of poorly water-soluble drugs: precipitation of drug after dispersion of formulations in aqueous solution. J Pharm Sci. 2009;98(10):3582–3595. | ||
Pouton CW. Lipid formulations for oral administration of drugs: non-emulsifying, self-emulsifying and ‘self-microemulsifying’ drug delivery systems. Eur J Pharm Sci. 2000;11(suppl 2):S93–S98. | ||
Lei Y, Lu Y, Qi J, Nie S, Hu F, Pan W. Solid self-nanoemulsifying cyclosporin A pellets prepared by fluid-bed coating: preparation, characterization and in vitro redispersibility. Int J Nanomedicine. 2011;6:795–805. | ||
Fatouros DG, Karpf DM, Nielsen FS, Mullertz A. Clinical studies with oral lipid based formulations of poorly soluble compounds. Ther Clin Risk Manag. 2007;3(4):591–604. | ||
Kommuru TR, Gurley B, Khan MA, Reddy IK. Self-emulsifying drug delivery systems (SEDDS) of coenzyme Q10: formulation development and bioavailability assessment. Int J Pharm. 2001;212(2):233–246. | ||
Craig DQM, Barker SA, Banning D, Booth SW. An investigation into the mechanisms of self-emulsification using particle size analysis and low frequency dielectric spectroscopy. Int J Pharm. 1995;114(1):103–110. | ||
Masnatta LD, Cuniberti LA, Rey RH, Werba JP. Determination of bezafibrate, ciprofibrate and fenofibric acid in human plasma by high-performance liquid chromatography. J Chromatogr B Biomed Appl. 1996;687(2):437–442. | ||
Abe S, Ono K, Mogi M, Hayashi T. High-performance liquid chromatographic method for the determination of fenofibric acid and reduced fenofibric acid in human blood, plasma and urine. Yakugaku Zasshi. 1998;118(10):447–455. | ||
Weng T, Qi J, Lu Y, et al. The role of lipid-based nano delivery systems on oral bioavailability enhancement of fenofibrate, a BCS II drug: comparison with fast-release formulations. J Nanobiotechnology. 2014;12(1):39. | ||
Gershanik T, Benita S. Self-dispersing lipid formulations for improving oral absorption of lipophilic drugs. Eur J Pharm Biopharm. 2000;50(1):179–188. | ||
Kang BK, Lee JS, Chon SK, et al. Development of self-microemulsifying drug delivery systems (SMEDDS) for oral bioavailability enhancement of simvastatin in beagle dogs. Int J Pharm. 2004;274(1–2):65–73. | ||
Wakerley MG, Pouton CW, Meakin BJ. Evaluation of the self-emulsifying performance of a non-ionic surfactant-vegetable oil mixture. J Pharm Pharmacol. 1987;39(6):b70. | ||
Gursoy RN, Benita S. Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomed Pharmacother. 2004;58(3):173–182. | ||
Constantinides PP. Lipid microemulsions for improving drug dissolution and oral absorption: physical and biopharmaceutical aspects. Pharm Res. 1995;12(11):1561–1572. | ||
Vonderscher J, Meinzer A. Rationale for the development of Sandimmune Neoral. Transplant Proc. 1994;26(5):2925–2927. | ||
Pouton CW, Porter CJ. Formulation of lipid-based delivery systems for oral administration: materials, methods and strategies. Adv Drug Deliv Rev. 2008;60(6):625–637. | ||
Atef E, Belmonte A. Formulation and in vitro and in vivo characterization of a phenytoin self-emulsifying drug delivery system (SEDDS). Eur J Pharm Sci. 2008;35(4):257–263. | ||
Klein S, Stippler E, Wunderlich M, Dressman J. Development of dissolution tests on the basis of gastrointestinal physiology. In: Dressman J, Krämer J, editors. Pharmaceutical Dissolution Testing. Boca Raton: Taylor and Francis; 2005:193–228. | ||
Dressman JB, Alsenz J, Schamp K, Beltz K. Characterizing release from lipid-based formulations. In: Hauss DJ, editor. Oral Lipid-Based Formulations: Enhancing the Bioavailability of Poorly Water-Soluble Drugs. New York: Informa Healthcare; 2007:241–256. | ||
Gao P, Morozowich W. Design and development of supersaturatable self-emulsifying drug delivery systems for enhancing the gastrointestinal absorption of poorly soluble drugs. In: Hauss DJ, editor. Oral Lipid-Based Formulations: Enhancing the Bioavailability of Poorly Water-Soluble Drugs. New York: Informa Healthcare; 2007:303–328. | ||
Gao P, Guyton ME, Huang T, Bauer JM, Stefanski KJ, Lu Q. Enhanced oral bioavailability of a poorly water soluble drug PNU-91325 by supersaturatable formulations. Drug Dev Ind Pharm. 2004;30(2):221–229. | ||
Gao P, Rush BD, Pfund WP, et al. Development of a supersaturable SEDDS (S-SEDDS) formulation of paclitaxel with improved oral bioavailability. J Pharm Sci. 2003;92(12):2386–2398. | ||
Najib J. Fenofibrate in the treatment of dyslipidemia: a review of the data as they relate to the new suprabioavailable tablet formulation. Clin Ther. 2002;24(12):2022–2050. | ||
Zhang X, Chen G, Zhang T, Ma Z, Wu B. Effects of PEGylated lipid nanoparticles on the oral absorption of one BCS II drug: a mechanistic investigation. Int J Nanomedicine. 2014;9:5503–5514. | ||
Chen Y, Lu Y, Chen J, et al. Enhanced bioavailability of the poorly water-soluble drug fenofibrate by using liposomes containing a bile salt. Int J Pharm. 2009;376(1–2):153–160. | ||
Christiansen ML, Holm R, Kristensen J, et al. Cinnarizine food-effects in beagle dogs can be avoided by administration in a self nano emulsifying drug delivery system (SNEDDS). Eur J Pharm Sci. 2014;57:164–172. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.